

Characterisation of tracers for aging of α-pinene secondary organic aerosol using liquid chromatography/negative ion electrospray ionisation mass spectrometry
Farhat Yasmeen A D , Reinhilde Vermeylen A , Nicolas Maurin B , Emilie Perraudin B C , Jean-François Doussin B and Magda Claeys A EA Department of Pharmaceutical Sciences, University of Antwerp, Universiteitsplein 1, BE-2610 Antwerp, Belgium.
B LISA, Universités Paris-Est-Créteil et Paris Diderot, CNRS UMR 7583, 61 Avenue du Général de Gaulle, F-94010, Créteil, France.
C EPOC, University of Bordeaux, CNRS UMR 5805, 351 Cours de la Libération, F-33400 Talence, France.
D Present address: Chemistry Department, University of Engineering and Technology, G.T. Road, Lahore 54840, Pakistan.
E Corresponding author. Email: magda.claeys@ua.ac.be
Environmental Chemistry 9(3) 236-246 https://doi.org/10.1071/EN11148
Submitted: 1 December 2011 Accepted: 23 February 2012 Published: 3 May 2012
Journal Compilation © CSIRO Publishing 2012 Open Access CC BY-NC-ND
Environmental context. Ambient fine aerosol from forested sites contains secondary organic aerosol from the oxidation of monoterpenes that are emitted by the vegetation, mainly by conifers. These biogenic aerosols can have varying lifetimes in the atmosphere because they contain first-generation oxidation products of α-pinene as well as aged products formed through further photooxidation, fragmentation, hydrolysis, and dimerisation reactions. We focus on the structural characterisation of secondary organic aerosol products that are simulated in a smog chamber experiment and can serve as potential tracers for aging processes in biogenic aerosols.
Abstract. Secondary organic aerosol (SOA) from the oxidation of α-pinene is a very complex and dynamic mixture containing products with a different chemical nature and physicochemical properties that are dependent on chemical evolution or aging processes. In this study, we focus on the chemical characterisation of major products that are formed upon α-pinene ozonolysis SOA and subsequent aging through OH-initiated reactions in the absence of NOx, which include known as well as unknown tracers. The mass spectrometric data obtained for selected unknown compounds that show an increased relative abundance upon aging are interpreted in detail and tentative structures for them are proposed taking into account their formation through photooxidation of α-pinene. Known tracers for α-pinene SOA aging that were identified include norpinic acid, 10-hydroxypinonic acid, diaterpenylic acid acetate, and diesters formed by esterification of pinic acid with terpenylic acid or 10-hydroxypinonic acid. Novel tracers for α-pinene SOA aging that were tentatively identified include dinorpinic acid and 8-hydroxypinonic acid. In addition, reaction mechanisms are proposed to explain the formation of the observed α-pinene SOA tracers.
Introduction
It is becoming increasingly recognised that secondary organic aerosol (SOA) from the oxidation of α-pinene is a very complex and dynamic mixture containing products with a different chemical nature and physicochemical properties that are dependent on chemical evolution or aging processes.[1–3] These processes comprise oxidation reactions of semivolatile precursors in the gas phase that result in more polar oxygenated products with an increased O/C ratio, which are less volatile and more hydrophilic, and thus may enhance the capability of SOA particles to serve as cloud condensation nuclei. On the other hand, oxidation reactions of semivolatile precursors in the gas phase may also result in fragmentation to smaller molecules. In addition, aging processes occur in the particle phase where heterogeneous reactions take place; examples of the latter reactions are esterification with sulfuric acid of hydroxy- or epoxy-containing SOA products,[4–7] esterification of pinic acid with hydroxy-containing terpenoic acids[8–10] and OH-initiated oxidation reactions.[11]
Limited information is available on the oxidation processes that form the molecular basis of α-pinene SOA aging processes.
A useful marker compound for aged α-pinene SOA is the C8-tricarboxylic acid 3-methyl-1,2,3-butanetricarboxylic acid (MBTCA), which is a higher-generation photooxidation product of α-pinene generated in the presence of NOx and is formed through further reaction of pinonic acid, a first-generation α-pinene SOA tracer.[12–14] MBTCA has been reported in several field studies to occur at significant concentrations, supporting its atmospheric relevance (for a review, see Hallquist et al.[15]; see recent field studies[16,17]).
In this study, we focus on the chemical characterisation of major products that are formed upon formation of α-pinene ozonolysis SOA and subsequent aging through OH-initiated reactions in the absence of NOx. These conditions are relevant to pristine forests such as the Amazonian rainforest, where NO/HO2 and NO/RO2 ratios are very low.[18] The targeted SOA products include known as well as unknown compounds, and are denoted as tracers. We also discuss differences in their relative abundances (RAs) as this parameter gives information about their evolution during the aging process. The mass spectrometric data obtained for selected unknown compounds is interpreted in detail and tentative structures for them are proposed taking into account that they are formed through photooxidation of α-pinene and likely have retained the dimethylbutane ring. With regard to analytical methodology, we resort to high-performance liquid chromatography (LC) in combination with negative ion electrospray ionisation mass spectrometry, (–)ESI-MS. The use of high-performance liquid chromatography offers the advantage in that it allows the separation of isobaric and isomeric terpenoic acids that are formed during α-pinene SOA formation and aging, whereas the use of (–)ESI-MS is suitable for the sensitive detection of terpenoic acids containing at least one carboxylic acid group, which is readily deprotonated during the ionisation process. On the other hand, the combination with linear ion trap MS[19] allows one to obtain good-quality first-order mass spectra and second-order (MS2) and third-order (MS3) product ion spectra during the elution of a chromatographic peak. In previous work, we have applied LC/(–)ESI-linear ion trap MS methodology and detailed interpretation of the mass spectrometric data to structurally characterise tracers formed from the photooxidation of isoprene and monoterpenes.[5,10,20–22] Caution, however, should be taken with regard to the structural elucidation of unknown compounds using the latter approach as the assignments remain tentative. However, they are important to make advances in our current understanding of the chemical processes that are on the molecular basis of SOA aging.
Experimental
SOA production and aging experiments
α-Pinene SOA was generated in the CESAM chamber (French acronym for Experimental Multiphasic Atmospheric Simulation Chamber) at the University of Paris-Est at Créteil. The CESAM chamber is designed to allow research in multiphase atmospheric (photo-)chemistry which involves both gas-phase and condensed-phase processes including aerosol and cloud chemistry and has been described in detail elsewhere.[23] Briefly, the apparatus consists of a cylindrical 4.2-m3 stainless steel chamber. Light is provided by three xenon arc lamps (4 kW, XPO 4000 W/HS, OSRAM), located on the top of the simulation chamber. Borosilicate filters are located in front of each lamp to reduce actinic UV radiation below 300 nm. Both temperature and relative humidity are measured with a HMP234 Vaisala humidity and temperature transmitter equipped with a capacitive thin-film polymer sensor Humicap (Vaisala, Helsinki, Finland). The chamber does not have any cooling system; hence, the temperature gradually increased during the irradiation period from 292 to ~297 K.
The parent hydrocarbon (i.e. α-pinene) and photooxidation products were monitored using a Fourier-transform infrared spectrometer (FTIR) from Bruker GmbH (Ettlingen, Germany). The total optical path length for the in-situ FTIR measurement was set to 192 m. Ozone was measured using a commercial Horiba APOA 370 instrument (Kyoto, Japan). Particle size distributions (20–980 nm in diameter) were measured with a TSI 3080 scanning mobility particle sizer (SMPS) and a TSI 3010 condensation particle counter (TSI, St Paul, MN, USA).
Two photooxidation experiments were performed; the initial starting conditions for the experiments are given in Table 1. The experiments were carried out under dry conditions (i.e. at a relative humidity below 1 %), and at room temperature and atmospheric pressure, in the absence of seed aerosol. Furthermore, no OH radical scavenger was used.

|
Between each experiment the chamber was cleaned by maintaining a secondary vacuum (~8 × 10–4 hPa) overnight. The chamber was then filled with synthetic air produced from a mixture of ~200 hPa of O2 (Air Liquide, Alphagaz15 class 1) and ~800 hPa of nitrogen produced from the evaporation of pressurised liquid nitrogen. Finally, known amounts of α-pinene were evaporated in a glass bulb and introduced in a flow of oxygen. The mixture was allowed to stay for at least 20 min before injecting ozone. Ozone was produced by passing a flow of pure O2 through a commercial dielectric ozone generator (MBT 802N, Messtechnik GmbH, Stahnsdorf, Germany).
Less than 2 min after ozone injection a monomodal SOA formation could be detected using a SMPS measurement. This aerosol grew in number of particles and mass for the first 15 min and then reached a maximum (Fig. 1), where no α-pinene was left in the chamber according to FTIR data. The lighting system was then turned on in order to mimic photochemical aging of the produced aerosol. For each of the experiments a sample was taken before injection in the chamber (blank filter) and immediately after the maximum SOA concentration was reached, and a third sample was then taken after a few hours of irradiation.
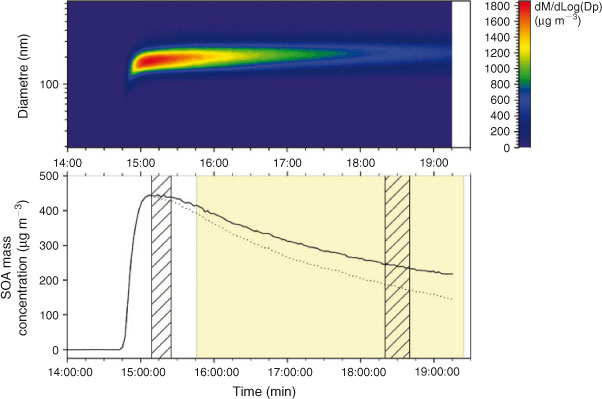
|
SOA sampling and sample workup
SOA collection was achieved using 47-mm glass fibre filters (Glass Microfibre filters GF/F Whatman). A standard cleaning procedure was applied to the filters before use: the filters were extracted three times with CH2Cl2 (99.8 % HPLC grade) in an ultrasonic bath for 10 min, dried under a flow of nitrogen and then baked for 5 h at 450 °C. The filter holder was installed just after an active charcoal denuder, which was used to trap reactive gases such as ozone (which could induce chemical evolution of the sample if they would adsorb on the filter) and to reduce positive sampling artefacts. However, this setup has the disadvantage that there is a risk of adsorption of semivolatile organic compounds and thus may result in negative sampling artefacts. The samplings were performed at a flow rate of 3 L min–1. This value was chosen after having checked the optimal trapping of ozone and the total transmission of particles. For each sample the SOA load was calculated from the sample volume and the aerosol mass concentration averaged over the sampling time as determined by SMPS, assuming an aerosol density of 1 g cm–3.
Complete filters were used and spiked with 150 ng of sebacic acid as an internal recovery standard, and then extracted three times with 20 mL of methanol. The combined methanol extracts were evaporated with a rotary evaporator at 30 °C to ~1 mL and then completely dried under a nitrogen stream. The dried residues were redissolved in 150 µL of water and an aliquot of 10 µL was injected for LC/MS analysis.
Mass spectrometric analysis
The LC/MS system comprised a Surveyor Plus system (pump and autosampler), a linear ion trap mass spectrometer (LXQ) equipped with an electrospray ionisation source, and a data system using Xcalibur version 2.0 software (Thermo Scientific, San Jose, CA, USA). A T3 Atlantis C18 column (3 μm; 2.1 × 150 mm2) (Waters, Milford, MA, USA) was employed. The mobile phases consisted of acetic acid 0.1 % (v/v) (A) and methanol (B). The applied 80-min gradient elution program was as follows: the concentration of eluent B was maintained at 3 % for 2 min, increased to 90 % in 18 min, maintained at 90 % for 43 min, decreased to 3 % in 5 min, and maintained at 3 % for 12 min. The flow rate was 0.2 mL min–1. A shift in the retention times (RTs) for the same chromatographic peaks up to 0.3 min was noted, which is likely a result of temperature effects (the column was operated at room temperature). The linear ion trap was operated under the following conditions: sheath gas flow (nitrogen), 0.75 L min–1; auxiliary gas flow (nitrogen), 1.5 L min–1; source voltage, –4.5 kV; capillary temperature, 350 °C; and maximum ion injection time, 200 ms. The ion optics of the LXQ instrument were optimised for maximum [M – H]– signal intensity using direct introduction of an aqueous malic acid solution of 50 μg mL–1 at a flow rate of 200 μL min–1. For MS2 and MS3 experiments, an isolation width of 2 m/z units and a normalised collision energy of 35 % were applied.
The RAs discussed in the text were calculated using extracted ion chromatograms (EICs) of the deprotonated molecules of the terpenoic compounds and by comparing the concentrations in the samples obtained before and after irradiation, thereby using the same response factor as for sebacic acid that was added in the same amount to both samples (i.e. 150 ng).
Results and discussion
Two α-pinene ozonolysis experiments were carried with irradiation (Table 1); as the results were quite similar for the two experiments but the effects of the irradiation more pronounced for the second one owing to the longer irradiation time (230 min v. 160 min), the results will only be discussed in detail for the second experiment (E0208). For the first experiment (E0202), only the most pronounced changes in the RAs of α-pinene-related terpenoic acids will be mentioned; no conclusions could be drawn about the molecular weight (MW) 358 and 368 diesters because of their low signal intensities. The mass concentration–time curve indicating the periods of sample collection for the second experiment are shown in Fig. 1. The corresponding selected chromatographic data (base peak chromatograms (BPCs) and EICs) for α-pinene ozonolysis SOA obtained before and after irradiation are shown in Fig. 2. The BPCs reveal distinct signals in the terpenoic acid region (RT 10–27 min). EICs are given for the m/z values corresponding to the deprotonated forms ([M – H]–) of the various tracers that are discussed below. The IUPAC names of the known and unknown α-pinene SOA tracers are given between parentheses the first time they are discussed.

|
With regard to the use of α-pinene ozonolysis SOA for aging experiments, it should be noted that this model has limitations for simulating the processes that occur in ambient conditions, where fresh SOA can also be formed through OH-initiated reactions. More specifically, SOA formed through photooxidation of α-pinene contains tracers that are present in different RAs and concentrations compared with α-pinene ozonolysis SOA.[24] Nonetheless, the α-pinene ozonolysis SOA (without OH scavenger) allows us to study aging processes at a molecular level as most of the tracers detected in the latter type of SOA and fresh SOA obtained through photooxidation are the same. The reason behind the formation of the same SOA tracers is the formation of hydroxyl radicals through the photolysis of ozone in the presence of water vapour.
MW 158 compounds
The α-pinene ozonolysis SOA before irradiation contains two m/z 157 compounds eluting at RTs 12.0 and 14.4 min (Fig. 2, Abi), of which the compound eluting at 12.0 min is a known tracer corresponding to terebic acid (2,2-dimethyl-5-oxooxolane-3-carboxylic acid). In a previous study,[10] the formation of terebic acid has been explained from terpenylic acid [2- (2,2-dimethyl-5-oxooxalan-3-yl)acetic acid] through further OH-initiated reactions. In the sample obtained after irradiation, terebic acid was below detection level (Fig. 2, Aai) in the second experiment, whereas an increase in its RA by 70 % was noted in the first one. The RA of the second-eluting compound (RT 14.2 min) decreased by 11.5 % in the second experiment, whereas no change was noted in the first one. A possible reason for the disappearance of terebic acid upon irradiation in the second experiment is complete hydrolysis to diaterebic acid [3-(1-hydroxy-1-methylethyl)butanedionic acid], which in turn reacts with pinic acid [3-(carboxymethyl)-2,2-dimethylcyclobutaneacetic acid] and leads to a high-MW 344 diester containing diaterebic acid and pinic acid as monomeric units.[10] The unknown m/z 157 compound (RT 14.4–14.2 min) was tentatively attributed to dinorpinic acid (3-carboxy-2-methyl-cyclobutylmethanoic acid), based on detailed interpretation of the (–)ESI-MS data (Fig. 3; Scheme 1). The m/z 157 MS2 spectrum shows diagnostic product ions at m/z 155, 113, 111 and 71, whereas the m/z 157 → 111 MS3 spectrum reveals additional product ions at m/z 93, 83 and 69, which are all in agreement with the proposed dinorpinic acid structure.
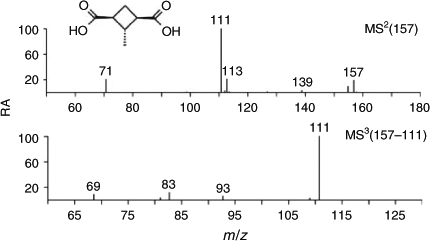
|

|
The m/z 157 compound eluting at RT 24.4 min in the sample obtained before irradiation also corresponds to an unknown compound that increased in RA after irradiation (RT 24.3 min) ~1.4 times. This compound was identified as a MW 312 compound and could only be partially characterised. Its (–)ESI (first-order) mass spectrum (not shown) reveals a deprotonated molecule at m/z 311 and fragment ions at m/z 293 (loss of water), 267 (loss of CO2) and 157. However, its m/z 157 MS2 spectrum was distinctly different from that obtained for terebic and dinorpinic acids in that m/z 157 was stable upon MS2 under the experimental conditions (i.e. normalised collision energy of 35 %).
MW 172 compounds
The α-pinene ozonolysis SOA before irradiation contains two m/z 171 compounds eluting at RTs 13.4 and 15.9 min (Fig. 2, Bbi), which are both known α-pinene SOA tracers, i.e. terpenylic acid[21] and norpinic acid (3-carboxy-2,2-dimethyl-cyclobutylmethanoic acid).[25–27] With respect to the assignment of the m/z 171 compound eluting at 15.9 min to norpinic acid, for which no authentic standard was available, the MS2 fragmentation behaviour was exactly the same as that reported in a recent study of our laboratory for an unknown compound present in α-pinene ozonolysis SOA,[10] which was tentatively identified as norpinic acid based on detailed interpretation of its MS data. The RA of terpenylic acid (RT 13.2 min) decreased after irradiation (Fig. 2, Bai) with 93 %, whereas that of norpinic acid (RT 15.7 min) increased by 60 %. These results suggest that norpinic acid is a potential tracer for aged α-pinene SOA. The increase in the RA of norpinic acid upon irradiation was even more pronounced in the first experiment (E0202), where it was enhanced ~7-fold. A likely reason why the RA of terpenylic acid decreased upon irradiation is hydrolysis to diaterpenylic acid [3-(1-hydroxy-1-methylethyl)pentanedionic acid], a hydroxy-containing terpenoic acid which in turn reacts with pinic acid and leads to a high-MW 358 diester containing pinic acid and diaterpenylic acid as monomeric units,[10] similar to observations outlined above for terebic acid. Evidence for the hydrolysis of terpenylic acid to diaterpenylic acid under the experimental conditions used will be discussed below.
MW 184 and 186 compounds
The m/z 183 and 185 compounds are well known α-pinene SOA tracers and correspond to pinonic acid (3-acetyl-2,2-dimethylcyclobutaneacetic acid) and pinic acid. They are major products of the ozonolysis and OH-initiated oxidation of α-pinene.[24–32] The RAs of both pinonic and pinic acid were found to decrease substantially upon irradiation, by 69 and 36 % (Fig. 2). The decrease in the RA of pinonic acid can be readily explained by further OH-initiated reactions, as, for example, to MBTCA and the mono-hydroxylated pinonic acids, 8- and 10-hydroxypinonic acid (see below). In addition, degradation of pinonic acid to norpinic acid also appears to be an important route, as the RA of norpinic acid was found to increase upon irradiation (60 % in the second experiment v. ~7-fold in the first one). The decrease in the RA of pinic acid, on the other hand, can be partly explained by incorporation into higher MW 344, 358 and 368 diesters, as discussed (above and below). It may also be explained in part by further OH-initiated reactions, as for example, to norpinic and dinorpinic acids. As for the first experiment (E0202), the decrease in the RA of pinonic acid was very comparable upon irradiation, i.e. 42 %, whereas that of pinic acid did not change.
MW 190 compound
The m/z 189 compound (Fig. 2, Ebi; RT 11.7 min) was assigned to diaterpenylic acid, the hydrolysis product of terpenylic acid. (–)ESI-MS data of diaterpenylic acid are presented in Fig. 4. Its RA was found to decrease (57 %) upon irradiation (Fig. 2, Eai), consistent with its consumption in the formation of the high-MW 358 diester containing diaterpenylic acid and pinic acid as monomeric units. The m/z 189 product ion MS2 spectrum shows ions at m/z 171 (loss of water), 145 (loss of CO2) and 127 (combined loss of water and CO2). The m/z 171 product ion MS3 spectrum is similar to the m/z 171 product ion MS2 spectrum of terpenylic acid.[21,22] In addition, the m/z 189 → 127 MS3 spectrum shows a product ion at m/z 83 also observed in the m/z 171 → 127 product ion MS3 spectrum of terpenylic acid.[21,22] These data allowed us to infer that the unknown MW 190 compound is diaterpenylic acid. As to the first experiment (E0202), a similar decrease in the RA of diaterpenylic acid (41 %) could be noted upon irradiation.
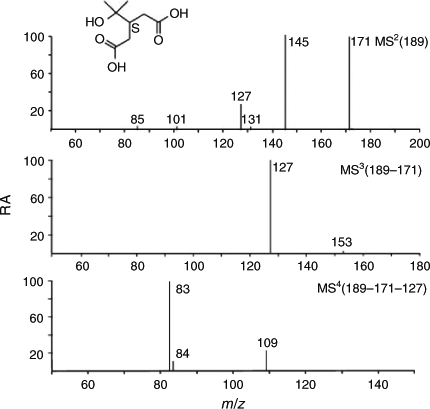
|
MW 200 compounds
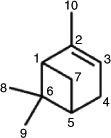
The m/z 199 EIC obtained before irradiation (Fig. 2, Fbi) shows three distinct peaks at RTs 14.8, 15.5 and 19.7 min. It can be seen that after irradiation (Fig. 2, Gai) the first-eluting peak has substantially decreased (91 %), suggesting that this MW 200 compound further reacts and is consumed upon irradiation (see below). Its (–)ESI-MS data are given in Fig. 5. This unknown MW 200 compound was tentatively assigned as 10-hydroxypinonic acid (2,2-dimethyl-3-hydroxyacetylcyclobutylethanoic acid) based on detailed interpretation of the mass spectrometric data (see below). To denote the various carbon positions in α-pinene and oxidation products such as pinonic acid, the numbering relating to the structure of pinane proposed in previous work by Larsen et al.[33] has been followed (Fig. 6). It should be noted that 10-hydroxypinonic acid has been previously reported in the literature[26,27] but so far no synthesised reference compound is available. It was tentatively identified as an α-pinene ozonolysis SOA product by Yu et al.[26] who resorted to gas chromatography/electron ionisation (EI)-MS with prior derivatisation to pentafluorobenzyloxime trimethylsilyl derivatives. Using interpretation of the EI mass spectrum of the derivatised unknown MW 200 compound, evidence was obtained for the position of the hydroxy group at the terminal C10 position. In another study, Glasius et al.[27] reported that a MW 200 hydroxypinonic acid isomer present in α-pinene ozonolysis SOA could also be generated from myrtenol, consistent with the presence of the hydroxy group at the C10 position. The m/z 199 MS2 spectrum shows diagnostic product ions at m/z 181, 155 and 137, whereas the m/z 199 → 181 MS3 spectrum reveals additional product ions at m/z 163, 153, 123 and 81, which can all, except m/z 81, be readily explained with the proposed 10-hydroxypinonic acid structure (Scheme 2). For the formation of m/z 81 starting from the m/z 181 precursor ion, a complex rearrangement reaction involving the dimethylcyclobutane part must be invoked.
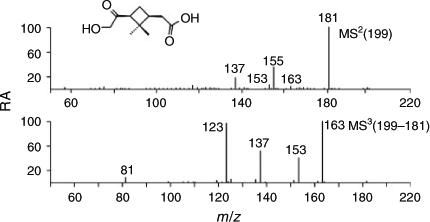
|
|

|
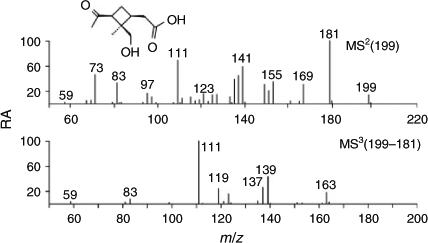
The second-eluting unknown MW 200 compound (RT 15.5 min) was tentatively identified as 8-hydroxypinonic acid (2-hydroxymethyl-2-methyl-3-acetylcyclobutylethanoic acid; containing a hydroxymethyl group cis relative to the carboxy and carboxymethyl groups) based on detailed interpretation of the (–)ESI-MS data and mechanistic considerations (see below) (Fig. 7; Scheme 3). The m/z 199 MS2 spectrum shows product ions at m/z 181, 169, 155, 141, 139, 137 and 111, whereas the m/z 199 → 181 MS3 spectrum reveals an additional product ion at m/z 163, which can all be readily explained with the proposed 8-hydroxypinonic acid structure. The m/z 169 product ion formed by the loss of formaldehyde (30 units) can be regarded as a diagnostic ion because it supports the presence of a terminal hydroxymethyl group. On the other hand, the loss of ketene (42 units) from m/z 181 giving rise to m/z 139 is consistent with the presence of a carboxymethyl group.[34]
|

|
The third-eluting unknown MW 200 compound (RT 19.7 min) appeared to be distinctly different from a hydroxypinonic acid isomer and could only be partially characterised. Interpretation of the (–)ESI-MS data (not shown) did not allow us to propose a tentative structure for this unknown compound. Its [M – H]– ion shows the loss of 32 units (methanol) (m/z 167; RA 100 %) and 42 units (ketene) (m/z 157; RA 27 %), pointing to a methyl ester and a carboxymethyl group in the unknown molecule. The second- and third-eluting MW 200 compounds were found to decrease in RA upon irradiation, with 54 and 62 % less than for the first-eluting MW 200 compound (91 %) tentatively attributed to 10-hydroxypinonic acid, suggesting that these compounds also further react upon aging. With respect to hydroxypinonic acids, it is worth mentioning that different isomers have also been noted by Larsen et al.[35] for the OH-initiated photooxidation of α-pinene.
As to the first experiment (E0202), the decrease in the RA of the first-eluting MW 200 compound, tentatively identified as 10-hydroxypinonic acid, upon irradiation was only 14.3 %, whereas the second- and third-eluting MW 200 compounds increased by 50 and 55 %.
MW 204 compounds
Several m/z 203 compounds with a low signal intensity could be detected in the α-pinene ozonolysis SOA sample before irradiation (Fig. 2, Gbi); of these, the peak at RT 18.7 min corresponds to MBTCA.[12] After irradiation an increase in its RA could be noted (45 %) (Fig. 2, Gai), which was more pronounced in the first experiment (E0202; 169 %). MBTCA was only a minor α-pinene SOA product under the conditions used in the present study before and after irradiation; a likely reason is that the experiments were carried out in the absence of NOx, which is required for the formation of MBTCA.[12,13] Recent chamber experiments demonstrated that pinonic acid serves as a precursor for the gas-phase OH-initiated formation of MBTCA,[14] supporting a previous proposal.[12]
MW 232 compounds
Three m/z 231 compounds could be clearly detected in the α-pinene ozonolysis SOA sample before irradiation (Fig. 2, Hbi) with RTs 12.6, 13.4 and 16.7 min. Of these peaks, the second-eluting one corresponds to a known non-covalent adduct of terpenylic acid (RT 13.4 min; Fig. 2, Bbi) with the acetate anion attributed to acetic acid present in the mobile phase[12]; it can be noted that this peak can no longer be detected after irradiation (Fig. 2, Hai) owing to terpenylic acid being consumed in the formation of a higher MW 358 diester, as discussed in detail above. The third-eluting m/z 231 compound corresponds to diaterpenylic acid acetate [3-(1-acetyloxy-1-methylethyl)pentanedionic acid], which is a known 1,8-cineole and α-pinene tracer.[21,36] Its RA was found to increase upon irradiation ~2-fold in the second experiment (33 % in the first experiment), suggesting that diaterpenylic acid acetate is also a potential tracer for aged α-pinene SOA. The formation of diaterpenylic acid acetate has in previous work been directly explained from α-pinene through OH-initiated reactions or ozonolysis.[21] However, considering that it is increased upon aging it is more likely that it is formed by esterification of diaterpenylic acid with acetic acid, which may also be formed from α-pinene during oxidation reactions.[37,38] The first-eluting m/z 231 compound (RT 12.6 min) could only be partially characterised based on interpretation of the (–)ESI-MS data (not shown). Its m/z 231 MS2 spectrum shows product ions at m/z 213 (RA 25 %; loss of water) and 183 (RA 100 %; combined loss of water and CH2O), pointing to the presence of a hydroxy and a terminal hydroxymethyl group respectively, whereas its m/z 231→ 183 MS3 spectrum reveals a product ion at m/z 137 (loss of 46 units; combined loss of H2 and CO2, or formic acid), consistent with the presence of a carboxy group or a formate ester group. Its RA was found to increase by 54 % upon irradiation.
MW 358 and 368 compounds
The m/z 357 compound (Fig. 2, Ibi; RT 20.0 min) is a known α-pinene SOA tracer, which has recently been structurally elucidated as a high-MW 358 diester formed between pinic acid and diaterpenylic acid.[9,10] Its RA abundance was found to substantially increase upon irradiation (Fig. 2, Iai; RT 20.2 min), namely ~2.4 times, suggesting that it is a useful tracer for α-pinene SOA aging.
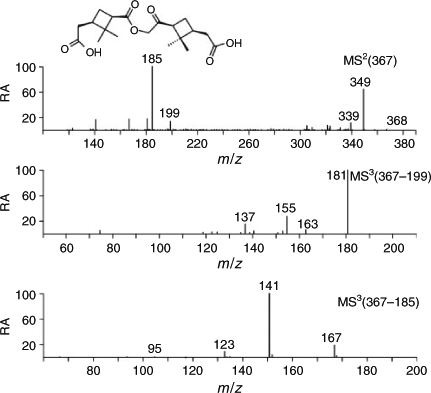
The major m/z 367 compound (Fig. 2, Jbi; RT 21.1 min) corresponds to a known α-pinene SOA tracer, which has structurally been elucidated as a high-MW 368 diester formed between pinic acid and 10-hydroxypinonic acid.[8] (–)ESI-MS data of the MW 368 compound present in the sample before irradiation are given in Fig. 8. The m/z 367 MS2 spectrum shows major product ions at m/z 349 (loss of water) and 185. The m/z 367 → 199 MS3 spectrum is similar to the m/z 199 MS2 spectrum obtained for the MW 200 compound tentatively identified as 10-hydroxypinonic acid (Fig. 5), whereas the m/z 367 → 185 MS3 spectrum is similar to the m/z 185 MS2 spectrum obtained in a previous study for a pinic acid reference compound.[22] It can be seen that the MW 368 diester is more abundant (approximately twice) than the MW 358 compound (RT 20.0 min) before irradiation. However, in contrast to the MW 358 diester formed between pinic acid and diaterpenylic acid the MW 368 diester is found to decrease upon irradiation (Fig. 2, Jai) with 59 %. A possible reason for this decrease is that 10-hydroxypinonic acid is not stable upon irradiation. Degradation reactions are likely taking place in the particle phase considering the low volatility of the MW 368 diester. In this respect, recent evidence has been provided for OH-initiated heterogeneous oxidation of α-pinene ozonolysis SOA.[11]
|
Chemical reactions involved in α-pinene SOA aging
The novel tracers for α-pinene SOA aging that were tentatively identified can be explained through different types of chemical reactions, including:
-
OH-initiated hydroxylation of pinonic acid; examples of this process are the formation of 8- and 10-hydroxypinonic acids (Scheme 4a). This reaction is thought to take place mainly in the gas phase, considering the vapour pressure of pinonic acid.[39] However, heterogeneous oxidation in the particle phase cannot be ruled out.
-
OH-initiated hydroxylation and fragmentation of pinic acid; examples of this process are the formation of norpinic and dinorpinic acids (Scheme 4b). These reactions are also thought to take place in the gas phase, considering the vapour pressure of pinic acid.[39] As in the case of pinonic acid, heterogeneous oxidation in the particle phase cannot be ruled out.
-
hydrolysis of lactone-containing terpenoic acids; an example of this process is the formation of diaterpenylic acid through hydrolysis of terpenylic acid (Scheme 4c), a reaction that is believed to take place in the particle phase as it involves acid catalysis.
-
esterification of pinic acid with a hydroxy-containing terpenoic acid such as diaterpenylic (Scheme 4c) and 10-hydroxypinonic acids, an acid-catalysed reaction that is believed to occur in the particle phase.
As to the mechanistic pathways of products formed through OH-initiated hydroxylation of pinonic acid, i.e. 8- and 10-hydroxypinonic acids (Scheme 4a), the initial H radical abstraction likely occurs at the theoretically favoured C4 position (referring to pinane (Fig. 6); note that this position corresponds to the C2 position in pinonic acid).[40] Subsequently, the created radical site may migrate to other positions, resulting in hydroxylation at carbon positions 8 or 10. The same rationale can be followed for the formation of dinorpinic acid through OH-initiated hydroxylation and fragmentation of pinic acid (Scheme 4b), which can proceed through hydroxylation at the C8 position, again following an initial H abstraction at the C4 position. With respect to migration of the initial radical site, evidence has been obtained for migration upon OH radical-initiated oxidation of α-pinene in the presence of NO and highly acidic sulfuric acid-containing seed particles; more specifically, upon formation of the MW 295 nitrooxy organosulfates the initial radical site on the cyclohexane ring created by reaction of the OH radical on the double bond migrates to a ring position and the methyl group of the dimethylcyclobutane ring.[5]

|
Conclusions and perspectives
Several potential tracers for α-pinene SOA aging have been tentatively identified in the present study using detailed interpretation of LC/(–)ESI-MS data. Their formation has been explained by OH-initiated hydroxylation and fragmentation, hydrolysis, and esterification of pinic acid with a hydroxy-containing terpenoic acid. SOA aging can thus be regarded as a very complex process involving different types of chemical reactions resulting in a complex array of products with different MWs and polarities. Experiments are currently in progress in our laboratories to precisely determine the evolution of these tracers during α-pinene SOA aging. Further research is warranted to determine whether the novel tracers (i.e. dinorpinic and 8-hydroxypinonic acids) that were tentatively identified in the present study and the corresponding sulfate ester (in the case of 8-hydroxypinonic acid) occur in ambient fine aerosol. Furthermore, efforts should also be made to synthesise the tentatively identified novel tracers both for unambiguous identification and quantification purposes.
Acknowledgements
Research at the University of Antwerp was supported by the Belgian Federal Science Policy Office (BIOSOA project) and the Research Foundation–Flanders (FWO) (SOAGAPS project). Research at LISA was supported by the European Community within the I3 projects ‘Integrating of European Simulation Chambers for Investigating Atmospheric Processes’ (EUROCHAMP-2, contract number 228335) and by the French Agency for Environment and Energy Management (ADEME) through the grant of N. Maurin.
References
[1] J. H. Kroll, J. H. Seinfeld, Chemistry of secondary organic aerosol: formation and evolution of low-volatility organics in the atmosphere. Atmos. Environ. 2008, 42, 3593.| Chemistry of secondary organic aerosol: formation and evolution of low-volatility organics in the atmosphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXls1Kksbs%3D&md5=e4081486255a7e6de6be34deb1620aecCAS |
[2] J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prévôt, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger, D. R. Worsnop, Evolution of organic aerosols in the atmosphere. Science 2009, 326, 1525.
| Evolution of organic aerosols in the atmosphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFensbjE&md5=d9c7580ed2340a3d184f8f5b2249b226CAS |
[3] J. H. Kroll, N. M. Donahue, J. L. Jimenez, S. H. Kessler, M. R. Canagaratna, K. R. Wilson, K. E. Altieri, L. R. Mazzoleni, A. S. Wozniak, H. Bluhm, E. R. Mysak, J. D. Smith, C. E. Kolb, D. R. Worsnop, Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol. Nat. Chem. 2011, 3, 133.
| Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXovVGhuw%3D%3D&md5=1dab8e3c16047688989051db2562c7a6CAS |
[4] J. D. Surratt, J. H. Kroll, T. E. Kleindienst, E. O. Edney, M. Claeys, A. Sorooshian, N. L. Ng, J. H. Offenberg, M. Lewandowski, M. Jaoui, R. C. Flagan, J. H. Seinfeld, Evidence for organosulfates in secondary organic aerosol. Environ. Sci. Technol. 2007, 41, 517.
| Evidence for organosulfates in secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xht1OmsLzM&md5=3ba1496dcab5f18f744e84387c36ab24CAS |
[5] J. D. Surratt, Y. Gómez-González, A. W. H. Chan, R. Vermeylen, M. Shahgholi, T. E. Kleindienst, E. O. Edney, J. H. Offenberg, M. Lewandowski, M. Jaoui, W. Maenhaut, M. Claeys, R. C. Flagan, J. H. Seinfeld, Organosulfate formation in biogenic secondary organic aerosol. J. Phys. Chem. A 2008, 112, 8345.
| Organosulfate formation in biogenic secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXpvFOgsrw%3D&md5=8ed1709bc00dac5ed4673e4b498bad19CAS |
[6] J. D. Surratt, A. W. H. Chan, N. C. Eddingsaas, M. N. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg, J. H. Seinfeld, Reactive intermediates revealed in secondary organic aerosol formation from isoprene. Proc. Natl. Acad. Sci. USA 2010, 107, 6640.
| Reactive intermediates revealed in secondary organic aerosol formation from isoprene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXltFSjsL0%3D&md5=0c09f16899742e34501fe30fbdaddb76CAS |
[7] Y. Iinuma, C. Müller, T. Berndt, O. Böge, M. Claeys, H. Herrmann, Evidence for the existence of organosulfates from beta-pinene ozonolysis in ambient secondary organic aerosol. Environ. Sci. Technol. 2007, 41, 6678.
| Evidence for the existence of organosulfates from beta-pinene ozonolysis in ambient secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXpvVCntLo%3D&md5=ee5b0887594bf9e8f467cab9f1b8db9bCAS |
[8] L. Müller, M.-C. Reinnig, J. Warnke, T. Hoffmann, Unambiguous identification of esters as oligomers in secondary organic aerosol formed from cyclohexene and cyclohexene/α-pinene ozonolysis. Atmos. Chem. Phys. 2008, 8, 1423.
| Unambiguous identification of esters as oligomers in secondary organic aerosol formed from cyclohexene and cyclohexene/α-pinene ozonolysis.Crossref | GoogleScholarGoogle Scholar |
[9] Y. Gao, W. A. Hall, M. V. Johnston, Molecular composition of monoterpene secondary organic aerosol at low mass loading. Environ. Sci. Technol. 2010, 44, 7897.
| Molecular composition of monoterpene secondary organic aerosol at low mass loading.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFOksbbI&md5=0ddd3d291d2376e1af8dd4a80a9089e8CAS |
[10] F. Yasmeen, R. Vermeylen, R. Szmigielski, Y. Iinuma, O. Böge, H. Herrmann, W. Maenhaut, M. Claeys, Terpenylic acid and related compounds: precursors for dimers in secondary organic aerosol from the ozonolysis of α- and β-pinene. Atmos. Chem. Phys. 2010, 10, 9383.
| Terpenylic acid and related compounds: precursors for dimers in secondary organic aerosol from the ozonolysis of α- and β-pinene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1ait7jM&md5=b1d18dbae61484d25a937b3e8c8def2eCAS |
[11] I. J. George, J. P. D. Abbatt, Chemical evolution of secondary organic aerosol from OH-initiated heterogeneous oxidation. Atmos. Chem. Phys. 2010, 10, 5551.
| Chemical evolution of secondary organic aerosol from OH-initiated heterogeneous oxidation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVCjsbzN&md5=4ebf61f7682e06ef89cd4881f47031a9CAS |
[12] R. Szmigielski, J. D. Surratt, Y. Gómez-González, P. Van der Veken, I. Kourtchev, R. Vermeylen, F. Blockhuys, M. Jaoui, T. E. Kleindienst, M. Lewandowski, J. H. Offenberg, E. O. Edney, J. H. Seinfeld, W. Maenhaut, M. Claeys, 3-methyl-1,2,3-butanetricarboxylic acid: an atmospheric tracer for terpene secondary organic aerosol. Geophys. Res. Lett. 2007, 34, L24811.
| 3-methyl-1,2,3-butanetricarboxylic acid: an atmospheric tracer for terpene secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar |
[13] N. L. Ng, P. S. Chhabra, A. W. H. Chan, J. D. Surratt, J. H. Kroll, A. J. Kwan, D. C. McCabe, P. O. Wennberg, A. Sorooshian, S. M. Murphy, N. F. Dalleska, R. C. Flagan, J. H. Seinfeld, Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes. Atmos. Chem. Phys. 2007, 7, 5159.
| Effect of NOx level on secondary organic aerosol (SOA) formation from the photooxidation of terpenes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlOnsrvL&md5=da8699ff1c75702b18eda2c0655cd695CAS |
[14] L. Müller, M.-C. Reinnig, K. H. Naumann, H. Saathoff, T. F. Mentel, N. Donahue, T. Hoffmann, Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid – a mass spectrometric study of SOA aging. Atmos. Chem. Phys. 2012, 12, 1483.
| Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid – a mass spectrometric study of SOA aging.Crossref | GoogleScholarGoogle Scholar |
[15] M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, Th. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski, J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155.
| The formation, properties and impact of secondary organic aerosol: current and emerging issues.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFGhs77M&md5=0437ae491e64eca2afaa26d3b70e085eCAS |
[16] Y. Y. Zhang, L. Müller, R. Winterhalter, G. K. Moortgat, T. Hoffmann, U. Pöschl, Seasonal cycle and temperature dependence of pinene oxidation products, dicarboxylic acids and nitrophenols in fine and coarse air particulate matter. Atmos. Chem. Phys. 2010, 10, 7859.
| Seasonal cycle and temperature dependence of pinene oxidation products, dicarboxylic acids and nitrophenols in fine and coarse air particulate matter.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlWls7nN&md5=98e7b8c58cf9949e4e19be9222480710CAS |
[17] Y. Gómez-González, W. Wang, R. Vermeylen, X. Chi, J. Neirynck, I. A. Janssens, W. Maenhaut, M. Claeys, Chemical characterization of atmospheric aerosols during a 2007 summer field campaign at Brasschaat, Belgium: sources and source processes, time series, diel variations, and temperature dependences. Atmos. Chem. Phys. 2012, 12, 125.
| Chemical characterization of atmospheric aerosols during a 2007 summer field campaign at Brasschaat, Belgium: sources and source processes, time series, diel variations, and temperature dependences.Crossref | GoogleScholarGoogle Scholar |
[18] J. Lelieveld, T. M. Butler, J. N. Crowley, T. J. Dillon, H. Fischer, L. Ganzeveld, H. Harder, M. G. Lawrence, M. Martinez, D. Taraborrelli, J. Williams, Atmospheric oxidation capacity sustained by a tropical forest. Nature 2008, 452, 737.
| Atmospheric oxidation capacity sustained by a tropical forest.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXksFSisrc%3D&md5=82b80e6866bf8e3505279f816a8c1510CAS |
[19] D. J. Douglas, A. J. Frank, D. Mao, Linear ion traps in mass spectrometry. Mass Spectrom. Rev. 2005, 24, 1.
| Linear ion traps in mass spectrometry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmsFWlsA%3D%3D&md5=6591423835e95e43feb4c3669c8b6573CAS |
[20] Y. Gómez-González, J. D. Surratt, F. Cuyckens, R. Szmigielski, R. Vermeylen, M. Jaoui, M. Lewandowski, J. H. Offenberg, T. E. Kleindienst, E. O. Edney, F. Blockhuys, C. Van Alsenoy, W. Maenhaut, M. Claeys, Characterization of organosulfates from the photooxidation of isoprene and unsaturated fatty acids in ambient aerosol using liquid chromatography/(–)electrospray ionization mass spectrometry. J. Mass Spectrom. 2008, 43, 371.
| Characterization of organosulfates from the photooxidation of isoprene and unsaturated fatty acids in ambient aerosol using liquid chromatography/(–)electrospray ionization mass spectrometry.Crossref | GoogleScholarGoogle Scholar |
[21] M. Claeys, Y. Iinuma, R. Szmigielski, J. D. Surratt, F. Blockhuys, C. Van Alsenoy, O. Böge, B. Sierau, Y. Gómez-González, R. Vermeylen, P. Van der Veken, M. Shahgholi, A. W. H. Chan, H. Herrmann, J. H. Seinfeld, W. Maenhaut, Terpenylic acid and related compounds from the oxidation of α-pinene: implications for new particle formation and growth above forest. Environ. Sci. Technol. 2009, 43, 6976.
| Terpenylic acid and related compounds from the oxidation of α-pinene: implications for new particle formation and growth above forest.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVSis7nK&md5=d924fad936ba357434e464138d5e8905CAS |
[22] F. Yasmeen, R. Szmigielski, R. Vermeylen, Y. Gómez-González, J. D. Surratt, A. W. H. Chan, J. H. Seinfeld, W. Maenhaut, M. Claeys, Mass spectrometric characterization of isomeric terpenoic acids from the oxidation of α-pinene, β-pinene, d-limonene, and Δ3-carene in fine forest aerosol. J. Mass Spectrom. 2011, 46, 425.
| Mass spectrometric characterization of isomeric terpenoic acids from the oxidation of α-pinene, β-pinene, d-limonene, and Δ3-carene in fine forest aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvVWmsbc%3D&md5=56ccfd37a3596772b619d7fbac668451CAS |
[23] J. Wang, J.-F. Doussin, S. Perrier, E. Perraudin, Y. Katrib, E. Pangui, B. Picquet-Varrault, Design of a new multi-phase experimental simulation chamber for atmospheric photosmog, aerosol and cloud chemistry research. Atmos. Meas. Tech. 2011, 4, 2465.
| Design of a new multi-phase experimental simulation chamber for atmospheric photosmog, aerosol and cloud chemistry research.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xjt1Wltrw%3D&md5=dcace131f6d2aefeb609d6f241caa2d7CAS |
[24] R. Winterhalter, R. Van Dingenen, B. R. Larsen, N. R. Jensen, J. Hjorth, LC-MS analysis of aerosol particles from the oxidation of α-pinene by ozone and OH-radicals. Atmos. Chem. Phys. Discuss. 2003, 3, 1.
| LC-MS analysis of aerosol particles from the oxidation of α-pinene by ozone and OH-radicals.Crossref | GoogleScholarGoogle Scholar |
[25] T. S. Christoffersen, J. Hjorth, O. Horie, N. R. Jensen, D. Kotzias, L. Molander, P. Neeb, L. Ruppert, R. Winterhalter, A. Virkkula, K. Wirtz, B. R. Larsen, cis-Pinic acid, a possible precursor for organic aerosol formation from ozonolysis of α-pinene. Atmos. Environ. 1998, 32, 1657.
| cis-Pinic acid, a possible precursor for organic aerosol formation from ozonolysis of α-pinene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXjt1WqsLY%3D&md5=0fd990a41cbd47b1afd2844e9cc66ce3CAS |
[26] J. Z. Yu, D. R. Cocker, R. J. Griffin, R. C. Flagan, J. H. Seinfeld, Gas-phase ozone oxidation of monoterpenes: gaseous and particulate products. J. Atmos. Chem. 1999, 34, 207.
| Gas-phase ozone oxidation of monoterpenes: gaseous and particulate products.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXmt1ens7w%3D&md5=b5781d7c99e3f0d5cc0d30f8fead703eCAS |
[27] M. Glasius, M. Duane, B. R. Larsen, Determination of polar terpene oxidation products in aerosols by liquid chromatography-ion trap mass spectrometry. J. Chromatogr. A 1999, 833, 121.
| Determination of polar terpene oxidation products in aerosols by liquid chromatography-ion trap mass spectrometry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXhs1OhurY%3D&md5=f6167afb8dab659bc5a547c62e8db314CAS |
[28] S. Hatakeyama, K. Izumi, T. Fukuyama, H. Akimoto, Reactions of ozone with alpha-pinene and beta-pinene in air – yields of gaseous and particulate products. J. Geophys. Res.–Atmos. 1989, 94, 13013.
| Reactions of ozone with alpha-pinene and beta-pinene in air – yields of gaseous and particulate products.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXhtVKhsr8%3D&md5=57747d5d0cad5dd2793aaa296f4aed51CAS |
[29] T. Hoffmann, J. R. Odum, F. Bowman, D. Collins, D. Klockow, R. C. Flagan, J. H. Seinfeld, Formation of organic aerosols from the oxidation of biogenic hydrocarbons. J. Atmos. Chem. 1997, 26, 189.
| Formation of organic aerosols from the oxidation of biogenic hydrocarbons.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXjvVKmsLg%3D&md5=205688de84956f44aeb070d75650cee9CAS |
[30] T. Hoffmann, R. Bandur, U. Marggraf, M. Linscheid, Molecular composition of organic aerosols formed in the α-pinene/O3 reaction: implications for new particle formation processes. J. Geophys. Res.–Atmos. 1998, 103, 25569.
| Molecular composition of organic aerosols formed in the α-pinene/O3 reaction: implications for new particle formation processes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXntFSku7g%3D&md5=9f1b23c4529005440214637a15a71d38CAS |
[31] J. Z. Yu, R. C. Flagan, J. H. Seinfeld, Identification of products containing –COOH, –OH, and –C=O in atmospheric oxidation of hydrocarbons. Environ. Sci. Technol. 1998, 32, 2357.
| Identification of products containing –COOH, –OH, and –C=O in atmospheric oxidation of hydrocarbons.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXksFSrurk%3D&md5=be5ef5f6b2e38846b7d01ea5d7f87c70CAS |
[32] M. Glasius, M. Lahaniati, A. Calogirou, D. Di Bella, N. R. Jensen, J. Hjorth, M. Duane, D. Kotzias, B. R. Larsen, Carboxylic acids in secondary aerosols from oxidation of cyclic monoterpenes by ozone. Environ. Sci. Technol. 2000, 34, 1001.
| Carboxylic acids in secondary aerosols from oxidation of cyclic monoterpenes by ozone.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXotlyntA%3D%3D&md5=47e40f45cc0b573b20e087f2b2c00bd4CAS |
[33] B. R. Larsen, M. Lahaniati, A. Calogirou, D. Kotzias, Atmospheric oxidation products of terpenes: a new nomenclature. Chemosphere 1998, 37, 1207.
| Atmospheric oxidation products of terpenes: a new nomenclature.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXlt1ehs7w%3D&md5=b3a94bb81b0da3382060ac0d3e1768b2CAS |
[34] K. A. Harrison, K. L. Clay, R. C. Murphy, Negative ion electrospray and tandem mass spectrometric analysis of platelet activating factor (PAF) (1-hexadecyl-2-acetyl-glycerophosphocholine). J. Mass Spectrom. 1999, 34, 330.
| Negative ion electrospray and tandem mass spectrometric analysis of platelet activating factor (PAF) (1-hexadecyl-2-acetyl-glycerophosphocholine).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXislGktL8%3D&md5=167e264137b69233190756ad8dcbbee6CAS |
[35] B. R. Larsen, D. Di Bella, M. Glasius, R. Winterhalter, N. R. Jensen, J. Hjorth, Gas-phase OH oxidation of monoterpenes: gaseous and particulate products. J. Atmos. Chem. 2001, 38, 231.
| Gas-phase OH oxidation of monoterpenes: gaseous and particulate products.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXivVaiurw%3D&md5=633b15f22db88d1b9d7377356b85ff78CAS |
[36] Y. Iinuma, O. Böge, M. Keywood, T. Gnauk, H. Herrrmann, Diaterebic acid acetate and diaterpenylic acid acetate: atmospheric tracers for secondary organic aerosol formation from 1,8-cineole oxidation. Environ. Sci. Technol. 2009, 43, 280.
| Diaterebic acid acetate and diaterpenylic acid acetate: atmospheric tracers for secondary organic aerosol formation from 1,8-cineole oxidation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsV2rtbrF&md5=4626458ec6c531af85d525abd5da2ebcCAS |
[37] A. Lee, A. H. Goldstein, M. D. Keywood, S. Gao, V. Varutbangkul, R. Bahreini, N. L. Ng, R. C. Flagan, J. H. Seinfeld, Gas-phase products and secondary aerosol yields from the ozonolysis of ten different terpenes. J. Geophys. Res.–Atmos. 2006, 111, D07302.
| Gas-phase products and secondary aerosol yields from the ozonolysis of ten different terpenes.Crossref | GoogleScholarGoogle Scholar |
[38] Y. Yu, M. J. Ezell, A. Zelenyuk, D. Imre, L. Alexander, J. Ortega, B. D’Anna, C. W. Harmon, S. N. Johnson, B. J. Finlayson-Pitts, Photooxidation of α-pinene at high relative humidity in the presence of increasing concentrations of NOx. Atmos. Environ. 2008, 42, 5044.
| Photooxidation of α-pinene at high relative humidity in the presence of increasing concentrations of NOx.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXntFGluro%3D&md5=bbd2eda78f05035c8a700fcc3d5f9930CAS |
[39] M. Bilde, S. N. Pandis, Evaporation rates and vapor pressures of individual aerosol species formed in the atmospheric oxidation of alpha- and beta-pinene. Environ. Sci. Technol. 2001, 35, 3344.
| Evaporation rates and vapor pressures of individual aerosol species formed in the atmospheric oxidation of alpha- and beta-pinene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXltF2lu7o%3D&md5=9893500782d9b13a13dd55c3cfebd234CAS |
[40] L. Vereecken, J. Peeters, Enhanced H-atom abstraction from pinonaldehyde, pinonic acid, pinic acid, and related compounds: theoretical study of C–H bond strengths. Phys. Chem. Chem. Phys. 2002, 4, 467.
| Enhanced H-atom abstraction from pinonaldehyde, pinonic acid, pinic acid, and related compounds: theoretical study of C–H bond strengths.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xmtlehtg%3D%3D&md5=385993e1ec742b168fcc743bd5235ccdCAS |