

Iron(III)-induced activation of chloride from artificial sea-salt aerosol
Julian Wittmer A D , Sergej Bleicher A B , Johannes Ofner C and Cornelius Zetzsch AA Atmospheric Chemistry Research Unit, Bayreuth Center of Ecology and Environmental Research (BayCEER), University of Bayreuth, Dr Hans-Frisch Strasse 1–3, D-95448 Bayreuth, Germany.
B Department of Forensic Toxicology, Synlab MVZ Weiden GmbH, Zur Kesselschmiede 4, D-92637 Weiden, Germany.
C Division Environmental and Process Analytics, Institute of Chemical Technologies and Analytics, Vienna University of Technology, AT-1060 Vienna, Austria.
D Corresponding author. Email: julian.wittmer@uni-bayreuth.de
Environmental Chemistry 12(4) 461-475 https://doi.org/10.1071/EN14279
Submitted: 20 December 2014 Accepted: 27 March 2015 Published: 2 July 2015
Journal Compilation © CSIRO Publishing 2015 Open Access CC BY-NC-ND
Environmental context. Inorganic, natural aerosols (sea-salt, mineral dust, glacial flour) and contributions of anthropogenic components (fly ash, dust from steel production and processing, etc.) contain iron that can be dissolved as FeIII in saline media. This study investigates photochemical processes in clouds and aerosols producing gas-phase Cl as a function of salt- and gas-phase composition employing a simulation chamber. Atomic Cl may contribute to the oxidative capacity of the troposphere, and our findings imply local sources.
Abstract. Artificial sea-salt aerosol, containing FeIII at various compositions, was investigated in a simulation chamber (made of Teflon) for the influence of pH and of the tropospheric trace gases NO2, O3 and SO2 on the photochemical activation of chloride. Atomic chlorine (Cl) was detected in the gas phase and quantified by the radical clock technique. Dilute brines with known FeIII content were nebulised until the relative humidity reached 70–90 %. The resulting droplets (most abundant particle diameter: 0.35–0.46 µm, initial surface area: up to 3 × 10–2 cm2 cm–3) were irradiated with simulated sunlight, and the consumption of a test mixture of hydrocarbons was evaluated for Cl, Br and OH. The initial rate of atomic Cl production per aerosol surface increased with FeIII and was ~1.9 × 1018 atoms cm–2 s–1 at Cl–/FeIII = 13. The presence of NO2 (~20 ppb) increased it to ~7 × 1018 atoms cm–2 s–1, the presence of O3 (630 ppb) to ~9 × 1018 atoms cm–2 s–1 and the presence of SO2 at 20 and 200 ppb inhibited the release slightly to ~1.7 and ~1.1 × 1018 atoms cm–2 s–1. The observed production of atomic Cl is discussed with respect to pH and speciation of the photolabile aqueous FeIII complexes.
Introduction
Iron-containing aerosols have natural (e.g. mineral dust, glacial flour) or anthropogenic (combustion of fossil fuel, fuel-oil fly ash, metal processing industry, etc.) sources and represent (together with upwelling) the source of dissolved iron in offshore waters.[1] The deposited dissolved iron, in the range of 0.26 Tg year–1,[2] has been suggested as being coupled to atmospheric carbon dioxide (CO2) concentration by activation of the oceanic food chain (‘Iron hypothesis’).[3–7] Apart from fertilisation of the oceans, iron is known to act as a catalyst in the photo-Fenton cycle in ferrous (Fe2+) and ferric (Fe3+) form, and it is utilised in wastewater treatment. Concerning the role of iron in the atmosphere, besides the proposed indirect CO2 reduction, the interactions between gaseous and aqueous phases containing iron and other organic and inorganic compounds are of interest.
Reactive halogen species (RHS) have a large effect on the budget of ozone (O3) and nitrogen oxides (NOx), affecting the oxidation capacity of the atmosphere[8,9] and interacting with secondary organic aerosols.[10] In particular, the atomic chlorine radical (Cl) is an important atmospheric oxidant that can considerably influence the lifetime of methane (CH4).[11] The main mechanisms responsible for the activation of halides to reactive forms are well investigated, including heterogeneous activation by NOx and O3 and autocatalytic halogen activation by HOBr and HOCl.[12,13] However, the potential role of iron in the release mechanisms of RHS remains unclear and is of special interest in the current work. Whereas a global tropospheric influence is questionable, the photochemistry of iron can significantly contribute to atomic Cl production in the gas phase, at least in some regions. Relevant iron–halide mixtures are present e.g. in coastal regions, intertidal zones, the combustion plumes of ships, volcanic plumes or brine-containing soils. In particular, hypersaline environments, such as the Dead Sea or Australian salt lakes,[14] offer conditions that promote iron-induced halogen release, as recently investigated by Wittmer et al. for modelled salt surfaces.[15] The present study, however, concentrates on the effect of Fe3+ in saline aerosols that can also be found in these regions and when mineral aerosol particles mix with sea salt. In particular, chloride (Cl–) enhances the dissolution of iron.[16] The dominant presence of dissolved Fe3+ in saline aerosols and the photoreduction to Fe2+ were observed by several studies,[17–19] but mainly with a focus on the role of the photochemistry of iron in the aqueous phase.[20] Whereas the photolysis of ferric ions in saline media has been well investigated,[21–23] less effort has been directed at the implications for the gas phase.
The photolysis of FeCl2+ and FeCl2+ directly yields chlorine atoms (Cl•) in the liquid phase of the humid saline aerosol, and these react very fast with Cl– to form Cl2•– (R1, reaction rate constant k = 2 × 1010 M–1 s–1[21]). For convenience, only aqueous-phase radicals will be marked with a dot in the following. The combination of Cl• with another Cl2•– (R2), or alternatively the combination of two Cl2•– (R3) and the dissociation of the produced Cl3– thereby (R4) leads to a degassing of Cl2.[24]

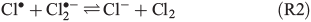
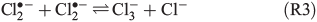

An alternative, indirect pathway for chloride activation is the photolysis of the slightly less photoactive species FeOH2+, producing OH• radicals in the liquid that again can form Cl• by ClOH•–.
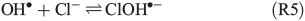

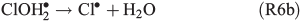
A more detailed summary and description of FeIII-induced Cl2 formation can be found in Lim et al.[24] or Wittmer et al.[15]
In addition to the photosensitive iron-induced halogen release mechanism, further trace gas species, such as O3, NOx and sulfur dioxide (SO2), possibly influence Cl2 release, mainly when dissolved in the aqueous phase where they can change the pH,[25] coordinate with Fe2+ or Fe3+,[26] scavenge Cl•[22] or additionally activate halides by the known heterogeneous mechanisms.[13] Concerning O3, Sadanaga et al.[27] observed an enhancement of the O3 uptake rate and Cl2 release in the presence of water-soluble Fe3+ and O3 in the dark. When SO2 is involved, the absorbed amount is highly pH-dependent.[28] The dissolved SO2 is mainly in bisulfite and sulfite forms, which are oxidised to sulfate depending on the pH and the availability of catalysts such as Fe3+.[29,30] The presence of sulfate can strongly inhibit the chloride activation process by scavenging Cl• and OH• or forming stable complexes with ferrous ions.[15,22] Apart from iron-influenced systems, polluted air masses considerably influence halogen activation in sea salt,[8,12,31,32] as well as the environmental oxidation of Cl– and Br– by the triplet states of chromophoric dissolved organic matter.[33] The present study aims at a deeper insight into the effects of FeIII in saline media with a focus on a better quantification of surface-related gaseous Cl production and the effects of the trace gases mentioned above.
Experimental
Smog-chamber set-up
The experiments were conducted in a cylindrical Teflon smog chamber (fluorinated ethylene propylene, FEP 200A, DuPont, Wilmington, DE, USA) with a volume of more than 3500 L (diameter: 1.33 m, height: 2.5 m, surface/volume ratio: 3.8 m–1). Here, the chamber and its analytical instrumentation are only briefly introduced. More detailed specifications can be found elsewhere.[15,31,34]
The chamber was suspended above a solar simulator that generates a mean actinic flux comparable with the summer sun at 50° latitude. The chamber was equipped with a differential pressure sensor (Kalinsky Elektronik DS1, Kalinsky Sensor Elektronik GmbH & Co. KG, Erfurt, Germany) to monitor the slight overpressure of 0.6–1 Pa, which was controlled by a continuous flow of 5–6 L min–1 of hydrocarbon-free, humidified zero air (zero-air generator, cmc instruments, <1 ppb of O3, <500 ppt NOx, <100 ppb of CH4) for all experiments. The temperature was adjusted to 20 °C and monitored by two light-shielded sensors (Rotronic, HC2-IC102) at different heights (bottom and top) to observe and avoid possible thermic layering. A Teflon fan inside the chamber assured constant mixing and kept temperature gradients below 1 °C. NO, NOx and O3 were monitored by chemiluminescence gas analysers (EcoPhysics, CLD 88p (EcoPhysics GmbH, Dürnten, Switzerland) coupled with a photolytic converter, PLC 860, for NO and NOx, and UPK 8001 (Bendix, Bad Nauheim, Germany) for O3).
Gas-phase Cl, Br and OH were indirectly quantified by the radical clock method,[35] monitoring the consumption of selected hydrocarbons (2,2-dimethylpropane, DMP, Linde AG, Munich, Germany, ≥99 %; 2,2-dimethylbutane, DMB, Sigma–Aldrich Chemie GmbH, Taufkirchen, Germany, ≥99 %; 2,2,4-trimethylpentane, TMP, Janssen-Cilag GmbH, Neuss, Germany, ≥99 %; toluene, Tol, Aldrich ≥99.9 %) and n-perfluorohexane (PFH, Aldrich, ≥99 %) as inert standard. The concentrations of the hydrocarbons was measured at time intervals of 15 min by gas chromatography (GC, Siemens Sichromat 2, 50-m Al2O3-PLOT column, 0.25 mL min–1 He as carrier gas) with a flame ionisation detector (FID), custom-built liquid nitrogen cryo-trap enrichment in glass-lined stainless-steel tubing and a Nafion tube counterflushed from outside with zero air to dry the sampling flux.[15]
Sample preparation and chemicals used
An artificial seawater stock solution with a molar Cl–/Br– ratio of 997 was prepared according to Kester et al.[36] The mixture included NaCl (23.9 g L–1), Na2SO4·10H2O (9.1 g L–1), KCl (0.68 g L–1), NaHCO3 (0.2 g L–1), KBr (0.098 g L–1), H3BO3 (0.026 g L–1), NaF (0.003 g L–1), MgCl2 (5.1 g L–1), and CaCl2·2H2O (1.5 g L–1). Depending on the experiment, a specific amount of FeCl3 was dissolved in the stock solution and diluted afterwards (1 : 28) to obtain a Cl– concentration of 15–35 mmol L–1 (Table 1) that provides optimal aerosol size distributions for long residence times of the suspended droplets. The aerosol production from the prepared solutions and the subsequent measurement are described in the following section. For the reference experiment with FeIII-doped NaCl, the FeCl3 was directly added to an 18 mmol L–1 NaCl solution. The added amount of FeCl3 and the resulting Cl–/FeIII ratios are listed in Table 1. More details of the prepared artificial seawater are included in the Supplementary material (Table S1).

|
On the basis of the different sea-salt mixtures, the effect of various gas species was tested by adding O3 from a silent ozoniser (Sorbios GSG 12, Sorbios GmbH, Berlin, Germany) with an electrical discharge applied to pure oxygen (Rießner-Gase GmbH, Lichtenfels, Germany, >99.995 %), NO2 from a gas cylinder (Rießner Gase, 104 vpm (volume parts per million) NO2 with a purity of 98 % in synthetic air), or SO2 from a gas cylinder (Rießner Gase, 0.99 % SO2 with a purity of 99.98 % in N2 with a purity of 99.999 %).
Aerosol production and measurement
The sea-salt particles were generated by nebulising the prepared solutions (Table 1) with an ultrasonic nebuliser (Quick Ohm QUV-HEV FT25/16-A, 35 W, 1.63 MHz, Quick-Ohm Küpper & Co. GmbH, Wuppertal, Germany) at a starting relative humidity (RH) of ≥40 % to avoid crystallisation.[12] During injection (taking typically 30–50 min), the RH increased to 70–90 %. Thus, we can assume that the droplets equilibrate to form a saturated solution, leading to most abundant particle diameters between 350 and 460 nm based on the concentration of the stock solution and the final RH after injection.[37] The size distributions were determined by an electrostatic classifier (TSI, 3071, TSI GmbH, Aachen, Germany) in combination with a neutraliser (85Kr) and a condensation nucleus counter (TSI, 3020). The scanning method and the multiple charge correction were applied with custom-written software.[38] Typical size distributions, determined after 30–50 min of injection, are shown in Fig. 1 for different stock compositions. To avoid the condensation of the sea salt within the transfer line (~6.4-mm tube made of copper) and thus the dripping of solution inside the chamber, the tube was additionally heated by a heating wire to ~60–70 °C. During aerosol generation, the NOx signal rose typically up to 3–5 ppb, probably originating from the sonochemical formation of nitrate and nitrite[39] and including a possible cross-sensitivity of the NOx analyser to nitrous acid (HONO).[40]

|
The chamber walls were either cleaned with deionised water (Seralpur Pro 90 CN, <0.055 µS cm–1, Seral, Ransbach-Baumbach, Germany), or the total FEP-Teflon film was exchanged after the experiment (depending on the risk of contamination expected for subsequent runs). To minimise the outgassing of gas-phase products,[41] every new chamber was conditioned by adding O3 at an RH of 50–80 % and irradiating for at least 4 h with the solar simulator and an additional UV lamp (Phillips TUV 55 W, λ = 253.7 nm).
After every experiment, the particles were collected by a Sioutas cascade impactor (SKC) in the aerodynamic diameter ranges of >2.5, 1–2.5, 0.5–1, 0.25–0.5 and <0.25 µm[42] on aluminium foil as a collection substrate. The impactor samples were analysed using an FEI Quanta 200 scanning-electron microscope (SEM), equipped with an energy-dispersive X-ray (EDX) detector for imaging (Octane Pro Silicon Drift (SDD) EDX detector from AMETEK GmbH, Wiesbaden, Germany). The hyperspectral data-cube from the SEM-EDX measurements was analysed using the software package Imagelab (Epina Software Laboratories, www.imagelab.at, accessed April 2015).[43]
Data analysis (radical clock)
The gas chromatograms were evaluated the same way as described before[15] to quantify the quasi-stationary concentrations of Clqs, OHqs, and Brqs in the gas phase, based on the measured, dilution-corrected and smoothed hydrocarbon (HCi) profiles, resulting in a system of four differential equations with three unknown variables according to:
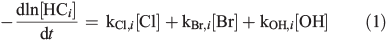
where kX,i is the reaction rate constant of HCi towards the radical X (X = Cl, Br, and OH; Table S2, Supplementary material). The corresponding total production (QX) and production rates (dQX/dt) are obtained by equalising sources and sinks in a photostationary steady-state:
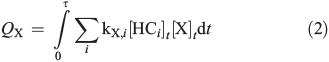
Considering a constant total reactivity of the chamber contents towards [X] on the one hand (ΣikX,i[HCi]0) and only the reactivity of the measured hydrocarbons on the other hand (ΣikX,i[HCi]t), which means either assuming that reaction products have the same reactivities as the HCi or neglecting the reactivities of the reaction products totally, we obtain an estimate of the minimum and maximum QX and dQX/dt. These assumptions are reasonably valid for Cl atoms but less reliable for Br atoms and OH radicals, where the reactivities of reaction products may exceed the reactivities of the original hydrocarbons by far. However, they are valid at the very beginning of the irradiation, where the consumption of the hydrocarbons is still low.
The measurements of the particle size distributions (number density N, cm–3) allow us to determine the available reaction surface area (A, m2 m–3) and the particle volume (Vparticle, m3 m–3), which equals the liquid water content (LWC) plus the tare volume of the ions (radius, rCl– = 181 pm, rSO42– = 230 pm, rNa+ = 102 pm, rK+ = 138 pm, rMg2+ = 72 pm, rCa2+ = 100 pm, rFe3+ = 65 pm)[44] at the adjusted RH. The measured LWC was corrected by considering the main ions as spherical. The contribution of deposited particles on the chamber walls to the activated Cl was considered by measuring the Cl activation of a totally wall-deposited aerosol load (Chamber wall effects section). The Cl– production terms are normalised to obtain absolute production rates, dQabs/dt, and absolute total production Qabs cm–2 of the aerosol surface. Therefore, the production rate (atoms cm–3 s–1) is multiplied by the chamber volume (Vchamber) to obtain the total number of atoms produced per second. The result is divided by the actual active aerosol surface, which we define as the sum of (i) the measured aerosol surface when the lights were turned on (A0,light) and its deposition rate, exp(–tlight/τs), with the aerosol surface lifetime τS; (ii) the active, deposited surface during the time of injection; and (iii) the active, deposited surface formed after the injection is finished (Eqn 3). Whereas (ii) is determined by assuming an approximately linear increase of the aerosol surface during injection and calculating the respective deposition until the injection is stopped (tinj,end), (iii) is based on the measured aerosol surface area directly after injection (A0,inj) and its deposition during the time tinj, which starts when the injection ends. Both terms are multiplied by a factor of 0.2, which is the fraction of deposited surface area that contributes to the halogen activation (Chamber wall effects section).
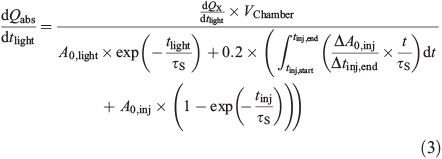
Results and discussion
Table 2 presents an overview of the experiments performed, including Cl–/FeIII ratios, pH of the diluted stock solution, gas-phase composition, initial reactivity of HCs against Cl, OH, and Br, initial aerosol surface, resulting quasi-stationary concentrations and absolute production. Based on the two-to-five orders of magnitude lower reactivity of the applied HCs against Br and OH compared with Cl, depletion was dominated by the reaction with Cl, and no significant interpretation for Br and OH was possible in most cases. Before adding FeCl3 to the samples, several FeIII-free blank experiments were performed, including pure NaCl aerosol, artificial sea-salt aerosol and artificial sea-salt aerosol with O3 and NO2 addition respectively (see Table 2). Except for the artificial sea-salt aerosol with addition of 700 ppb O3, Cl production was hardly detectable. Generally, no indication of Cl, Br or OH production was observed during the dark phases (aerosol, NO2, O3, SO2 injections and waiting periods), based on the constant HC time profiles.

|
Chamber wall effects
Owing to flashovers in the classifier throughout some experiments caused by the high RH, only the measurements at the beginning of those experiments were evaluable. Therefore, the measurements after injection and when the lights were turned on were used to calculate the dilution-corrected loss by deposition, based on the mean dilution-corrected lifetimes τN,S (N, number density; S, total aerosol surface area) over the experiments without flashovers and neglecting the loss by coagulation. The deposition velocity depends on the salt concentration in the nebulised solution and thus on the mean particle diameter after injection. For instance, the artificial sea-salt solution and the pure NaCl solution gave lifetimes of τN = 24 800 s (τS = 30 100 s) and τN = 25 000 s (τS = 31 100 s). For the higher-concentration FeIII-doped artificial seawater (29–35 mmol Cl– L–1), the most abundant particle diameters were between 430 and 460 nm, resulting in a faster sedimentation with τN = 4590 ± 240 s and τS = 6070 ± 520 s compared with τN = 6845 s and τS = 8820 s for the lower-concentration pure NaCl + FeCl3 solution (24 mmol Cl– L–1) with most abundant particle diameters between 390 and 420 nm. The respective contour plots can be found in the Supplementary material (Fig. S1).
To quantify the particle deposition and its contribution to the active surface area (and thus to Cl activation), a test measurement was performed to determine the fraction of Cl release by the active wall surface compared with the active aerosol surface: the iron-doped artificial seawater sample (Cl–/FeIII = 13) was injected and allowed to deposit totally for 17 h (<0.005 % of the surface area should have remained suspended) while keeping the RH at 80 %. Then the ‘aerosol-free’ chamber was irradiated, resulting in Cl production that was 20 ± 4 % compared with the actual production measured for the same sample in an aerosol experiment (Iron(III)-catalysed Cl atom production section). This production was evaluated by taking the mean of the quotient of each total production (deposited and not deposited) normalised by the respective initial LWC directly after injection. In Eqn 3, the contribution of deposited, active aerosol surface area is accounted for by adding 20 % of the deposited surface area since the time of injection to the surface area when the lights were turned on (corrected for deposition). The smaller active fraction when deposited on the wall can be explained by a physical surface reduction during the adhesion process of the droplets (possibly coagulating to larger droplets on the hydrophobic surface) and by an inhomogeneous irradiation of the chamber walls (especially the large fraction of wall surface perpendicular to the solar simulator), but also by a drying effect due to the heating of the Teflon walls during irradiation. Fig. 2 demonstrates the contribution of active, deposited surface on the wall to the total active surface area during the experiment with FeIII-doped, pH-adjusted (pH 2.1–2.3) artificial seawater (Cl–/FeIII = 101). The figure additionally includes dQabs/dt and Qabs, which are discussed in the Effect of pH and FeIII speciation chemistry section.

|
Iron(III)-catalysed Cl atom production
Significant Cl activation was detected for every sample with Cl–/FeIII < 101 (Table 2), whereas Cl production was close to the detection limit during the blank experiments including FeIII-free NaCl and artificial sea-salt aerosol in zero air. The effect of the constituents of the FeIII-containing artificial seawater mixture was investigated by comparing the Cl release with a pure NaCl + FeCl3 mixture. Both samples were prepared with a molar ratio of Cl–/FeIII = 13. Both experiments were conducted in the same manner and under the same conditions (21 ± 1 °C and 72 ± 2 % RH). After 45 min of aerosol injection and a 15-min waiting period, the light was turned on. The aerosol size distributions (Fig. 1) showed small differences, with a lower diameter for the most abundant particles but only a slightly lower surface-to-volume ratio for the artificial sea salt/FeIII mixture (1.29 × 107 m–1) compared with the NaCl/FeIII mixture (1.31 × 107 m–1) caused by the similar hydration behaviour at the given RH (Köhler theory).[45] The Cl production rates and corresponding total production are shown in Fig. 3. Slightly higher Cl production of the pure NaCl/FeIII mixture was observed, which was probably caused by the speciation chemistry (as photolabile FeIII-Cl complexes can form more easily in the absence of competing ions such as SO42–).[46] Considering the diluted stock solutions, the pH was between 1.9 and 2.2 for both experiments. The buffer effect of the added bicarbonate (0.2 g L–1) in the artificial seawater is very low, considering the high amount of FeCl3 added (13 g L–1). Although the radical clock method is also able to quantify Br and OH, the depletion of HCs was dominated by Cl with its much higher reaction rate constants (Table S2, Supplementary material), which prevents significant interpretation of Br and OH (in most cases), which were close to the detection limit (Br ~109 molecules cm–3; OH ~106 molecules cm–3).

|
In order to determine the dependence of the chlorine activation on the Cl–/FeIII ratio in the aerosol, a series of experiments with a Cl–/FeIII ratio of (i) 13, (ii) 53, (iii) 101, (iv) 955 and (v) a blank without iron addition was conducted. Whereas no significant Cl concentration was detected for run (iv) and (v), (iii) resulted in a total Cl production of (0.9–1.0) × 1021 atoms cm–2, run (ii) of (2.3–2.6) × 1021 atoms cm–2, and run (i) of (1.1–1.8) × 1022 atoms cm–2 in the first 100 min (Fig. 4). This demonstrates a continuous increase in produced Cl with increasing fraction of FeIII in the salt. A 1.9-times higher fraction of FeIII results in a 2–3-times higher Cl production (comparing (iii) and (ii)), and 4.1 times more FeIII results in 5–7-times higher Cl production (comparing (ii) and (i)). The higher amount of added FeCl3 cannot only explain the disproportionately higher Cl activation. In fact, the shift in pH from adding 10 times more FeCl3 (pH 4 → 2) is in addition responsible for a higher fraction of photolabile FeIII-Cl complexes (discussed in the Effect of pH and FeIII speciation chemistry section). A slight decrease was observed for (iii), indicating exhaustion of the Cl source, whereas samples (i) and (ii) resulted in a more stable Cl production rate.

|
Effects of NO2, O3 and SO2
Experiments on the typical tropospheric traces and pollutants NO2, O3 and SO2 were performed by adding 20 ppb of NO2, 630 ppb of O3, and 20 and 200 ppb of SO2. These levels were chosen with the aim to better observe their effects and to obtain aerosol to pollutant ratios approximately comparable with natural conditions (discussed in the Environmental significance section). Figs 5 and 6 show the time profiles of the trace gases (corrected for dilution) combined with the results on Clqs, dQabs/dt and Qabs obtained by the radical clock for the experiments with FeIII-doped artificial seawater (Cl–/FeIII = 13) and addition of NO2 and O3 respectively. After the aerosol injection, the addition of NO2 and a waiting period of 55 min (12 min in case of the O3 experiment), the solar simulator was turned on (indicated by 0 min in the figures). For both experiments, significantly elevated Cl production was observed compared with the experiments in zero air. The higher Cl source is related to the additional activation mechanisms in the presence of O3 and NOx,[13] which are discussed in the following text. Fast formation of O3 was observed in the NOx experiment when the light was turned on, caused by the well-known photochemical cycle of NO, NO2 and O3 (photolysis rate coefficients: JNO2 = 6.7 × 10–3 s–1, JO3 = 2 × 10–4 s–1) on the one hand and the re-oxidation of NO to NO2 and OH by HO2 (formed by the depletion of the injected HCi) on the other hand. Therefore, the O3 concentration exceeded the NOx concentration in the course of the experiment, and the O3 formation stopped when NO was depleted. Because O3 is not present during the dark period, only negligible amounts of dark-phase nitrogen oxides like dinitrogen pentoxide (N2O5) could be present at the beginning of the irradiation and potentially explain the additional Cl source by the proposed heterogeneous activation mechanisms.[12,13,32,47] During irradiation, the presence of O atoms and O3 formation can cause the formation of NO3 and thus N2O5, although NO3 is extremely short-lived under our conditions (JNO3 = 0.11 s–1). At an NOx concentration of 20 ppb, the proposed formation and photolysis of nitrosyl chloride (ClNO) by NO2 uptake of the sea-salt aerosol[48–50] is too slow to contribute significantly to gaseous Cl production. For instance, Karlsson and Ljungström[51] estimated a total ClNO production of the order of 1.1 × 107 atoms cm–3 in a flow reactor at aerosol concentrations of 3 × 105 cm–3 (surface/volume = 2 × 107 m–1) when 50 ppb NO2 was present. Owing to a photolysis rate of JClNO = 1.8 × 10–3 s–1 in our chamber, this would result in comparable Cl production, whereas the actual total production in our experiments is of the order of 1012–1013 atoms cm–3 and thus exceeds the production caused by ClNO formation by far, even if Karlsson and Ljungström[51] assume a sea-salt surface three orders of magnitude lower. The negligible influence of NO2- or N2O5-induced Cl activation was confirmed by a blank experiment (iron-free artificial sea salt with 20 ppb NO2 addition), where the artificial sea-salt aerosol without FeIII was irradiated in the presence of 20 ppb NOx and the quasi-stationary Cl concentration remained below 104 molecules cm–3 combined with a slower NOx depletion (Supplementary material, Fig. S2). Rather, the formation of XONO2 (X = Cl, Br) by XO (R7, R8), or HNO3 by OH (R9) and the subsequent uptake by the aerosol may explain the NOx loss,[12,31,52] which predominantly ends up as nitrate (with a possible back-reaction pathway to N2O5 or XNO2 by HNO3 at low pH).[13]
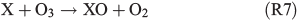
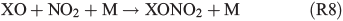
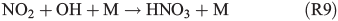

|

|
The uptake of XONO2 can result in additional X2 or BrCl release, as e.g. summarised in the review by Rossi.[13] The uptake of HNO3 leads to an acidification of the aerosol and contributes to the reoxidation of FeII to FeIII through photolysis and OH• formation in the aqueous phase[20] and thus is potentially responsible for the enhanced gaseous Cl production, according to the mechanisms described in the Introduction. Additionally, the uptake of HNO3 leads to acid displacement with a subsequent release of HCl,[53,54] whose reaction with OH is supposed to be a main Cl–atom source in the marine troposphere.[55] In general, a low pH facilitates the release of HCl and other gaseous Cl precursors.[25,31,56,57] For example, Keene and Savoie[56] determined a gaseous HCl mixing ratio in the range of 0.1 ppb, and Keene et al.[25] modelled HCl outgassing of 1 ppb day–1 for acidified sea salt (pH 3). Transferred to our conditions with typical OH concentrations of 106–107 molecules cm–3 and a much higher LWC, this leads to gaseous Cl production rates in the range of 109–1010 atoms cm–3 h–1 and thus potentially contributes less than 10 % to the observed Cl production. This accounts also for the HCl formation caused by hydrogen abstraction during the consumption of the injected HCi by Cl atoms. Furthermore, Zetzsch and Behnke[58] investigated photochemical Cl production rates from 200 to 500 ppb O3 and 300 ppb HCl in the presence of NaCl, Fe2O3 and SiO2 aerosol. They concluded that the heterogeneous Cl– activation exceeds the Cl source from the gas-phase reaction of OH and HCl by far.
Several effects were observed in the experiment where 630 ppb O3 was added (Fig. 6). The total Cl production per hour is three to five times higher than the value obtained from the FeIII-containing sample in zero air and is similar to the NOx experiment (Table 2). However, the reactivity of O3 towards Cl (~180 s–1) is comparable with the total reactivity of the injected HCi (~200 s–1) and not considered in the calculation of the total production Q, as it does not represent a final sink but rather initiates a reaction cycle by HO2 and HOCl where finally Cl2 is produced.[59,60] In addition, Sadanaga et al.[27] observed enhanced O3 uptake rates in the presence of water-soluble FeIII in synthetic sea salt without irradiation. During the short dark period, O3 depletion was hardly detectable, whereas we observed an approximately four times lower O3 lifetime for the experiment with added FeIII (~104 s) compared with the pure artificial sea-salt sample (~4 × 104). In general, the O3 destruction is related to autocatalytic halogen activation where Br especially plays a dominant role.[34] This difference possibly explains the much higher Cl and Br production (Fig. 6a, b) at the same level of quasi-stationary OH concentrations (~107 molecules cm–3) for both experiments. Concerning Br production, the quasi-stationary Br concentration was again of the order of 109 atoms cm–3 (close to the detection limit) and resulted in Qabs values of (1–3) × 1021 atoms cm–2 h–1 for the iron-free and (3–8) × 1021 atoms cm–2 h–1 for the iron-doped sea salt. Adding O3 to the iron-free aerosol, the significant enhancement in Cl production (~3–4.5 × 1021 atoms cm–2 h–1 from the iron-free sample in zero air to the iron-free sample with O3 addition) is relatively low compared with the much stronger increase in Cl production when adding O3 to the iron-containing sample (1–4 × 1022 atoms cm–2 h–1 from the iron-containing sample in zero air to the iron-containing sample with O3 addition). This indicates that we observed an O3-induced Cl and Br production that increases with addition of FeIII (or decreasing aerosol pH) and contributes markedly to the enhanced iron-induced activation. Several mechanisms come into consideration for the observed effects. At these O3 levels, a main mechanism (which is responsible for the high Cl and Br production and fast O3 depletion) is the so-called ‘Bromine explosion’[61] with the net reaction:
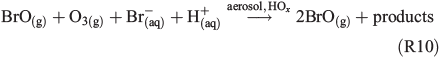
and the associated formation of OClO and ClO.[34] However, it does not sufficiently explain the increase in production rates from ~2 × 1018 to ~1019 atoms cm–2 s–1 with O3 addition when FeIII is involved (Figs 3 and 6a), because the pH (when <5) is supposed to have no large effect on the mechanism,[25,62] and the additional Cl production rate (when estimated from the iron-free experiment) is ~1018 atoms cm–2 s–1 (Fig. 6b).
A further approach is the enhanced formation of H2O2 in the gas phase (R11, R12):
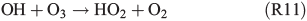
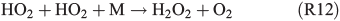
which enters the aqueous phase rapidly and oxidises FeII back to FeIII, and is able to form HOCl– or HOBr–, which further dissociate and finally form Cl2 or Br2.[63]
Knipping et al.[64] even report a direct uptake of OH and subsequent Cl2 release from NaCl aerosols. Furthermore, NOx (3–5 ppb is present after aerosol injection) forms NO3 and N2O5 by O3 during the dark phase, indicated by the loss of NOx when O3 is injected (Fig. 6a, b), and thus leads to HNO3 formation in the aqueous phase with similar consequences to those described above (R9). NO3 is able to efficiently produce atomic X from solid and humidified salts.[65,66] Moreover, the uptake of N2O5 activates Cl– by releasing ClNO2[47,67] (or even Cl2 in a multistage process by ClNO2 at acidic pH)[68] and Br– by releasing Br2 or BrNO2,[69,70] which is photolysed to atomic X (JClNO2 = 0.2 × 10–3 s–1, JBrNO2 = 3.7 × 10–3 s–1, JBr2 = 17 × 10–3 s–1). However, considering the low NOx concentrations, the dark-phase activation mechanisms by NO3 and N2O5 probably play an only minor role compared with autocatalytic light-induced activation. All given mechanisms favour the activation of bromide and could explain the high Br production rates in both O3 experiments.
In contrast to the reinforcing effects of NOx and O3, the situation changes with SO2. Two experiments were conducted with SO2 concentrations of ~20 and ~200 ppb and a waiting period of 45 min before turning the solar simulator on. Similarly to the effect of sulfate in a salt pan,[15] SO2 inhibited Cl production, because slightly lower dQabs/dt and Qabs were observed (Fig. 7). Several studies have already examined the role and the uptake of SO2 in combination with FeIII and FeII for seawater. For example, Hoppel et al.[28] report an uptake of 0.21–1.2 mmol L–1 of nebulised seawater, which is in a similar range to the sulfate concentration of 1 mmol L–1 in the artificial seawater stock solution (which is nebulised in our case). However, we used a 29-times diluted stock solution to obtain size distributions with a maximum diameter of 400–450 nm. Additionally, the dissolution of SO2 depends on further dissociation reactions in the aqueous phase that depend on pH, temperature and ion content.[71] Therefore, the uptake rate of Hoppel et al. is only transferable with caution, but considering the less-concentrated stock solution, it should only be a small amount of SO2 that dissolves in the aqueous phase of the aerosol, mainly in the form of bisulfite (HSO3–) given the pH range (2–6) of our samples. Especially at a low pH, where a high portion of dissolved Fe3+ is available, the oxidation of sulfite to sulfate is catalysed by Fe3+.[29] Moreover, a significant oxidation path is the reaction of sulfite with HOCl and HOBr, both of which can intervene in the autocatalytic halogen release.[72,73] Although sulfate can strongly inhibit halogen activation, the effect of the freshly dissolved and oxidised SO2 is probably very low with respect to the amount of sulfur that is already present in the artificial sea salt (Sample preparation and chemicals used section). A further influence of SO2 is observed, concerning the quasi-stationary OH concentrations that are close to the detection limit of 106 molecules cm–3 for both experiments, due to the oxidation of SO2 in the gas phase (SO2 + OH → HOSO2; k = 1.3 × 10–12 cm3 molecules–1 s–1),[74] which competes with Cl production by HCl + OH (k = 7.8 × 10–13 cm3 molecules–1 s–1).[75] Therefore, slightly lower Cl production is observed for the SO2 experiments. In the extreme case of 200 ppb, Cl production decreases to ~70 % of that from the SO2-free experiment.

|
Effect of pH and FeIII speciation chemistry
To estimate the effect of the aerosol pH on Cl production, the pH of the diluted stock solution (Cl–/FeIII = 101) was adjusted to pH 2.1–2.3 by gently adding 32 % HCl. The results demonstrate the immense effect of the pH (Fig. 2). The higher absolute production rate during the low-pH experiment led to eight times higher total Cl production per hour compared with the unadjusted sample (pH 3.9–4.2, Fig. 4; see also Table 2). There are multiple reasons for this effect. As evaluated in several studies for FeIII-doped salts, the speciation chemistry of FeIII strongly depends on the pH and ionic strength.[15,24,76] Fig. 8a, b show the portions of the total FeIII as FeIII complexes as a function of the pH, calculated with an equilibrium model in PHREEQC.[77] According to Wittmer et al., the model is based on the MINTEQ database,[78] and the activity coefficients are corrected by the Pitzer ion interaction approach[79] with the parameters listed in Tosca et al.[80] The main equilibrium constants involving FeIII are listed in the Supplementary material (Table S3). For some ions (e.g. F–), there are no Pitzer parameters available and the extended Debye–Hueckel equation[81,82] was applied to calculate the respective activity coefficients. To get an insight into the processes during nebulisation and evaporation, the speciation was calculated for the diluted stock solution (Fig. 8a), which the nebuliser was filled with, as well as for the final aerosol (Fig. 8b), assuming a saturation in Cl– (6.1 mol L–1) and unchanged molar ratios towards the other constituents. This assumption is extremely simplified based on the complexity of a multicomponent salt system,[45] and implies a similar hydration and solubility behaviour of NaCl and artificial sea salt but accounts for the fact that Na+ and Cl– represent by far the main ions. For example, McCaffrey et al.[83] measured similar Cl– saturation concentrations in evaporating seawater whereas the concentration may change with increasing FeIII content.

|
Whereas high fractions of Fe3+ ions and FeIII-hydroxy complexes are present in the stock solution, the decisive photosensitive FeIII-Cl complexes start to form as a considerable fraction at pH 1–4.5 with increasing ionic strength (solvent concentrations). The pH of 4.5 is a turning point for the aerosol speciation, where mainly FeIII-hydroxy complexes are present. Because FeIII-Cl complexes comprise a fraction of less than 5 % in the stock solution at the given pH of 3.9–4.2 (and it takes some time until speciation equilibrium is reached during the transition from the low-concentration stock solution to the high-concentration liquid aerosol), the smaller amount of FeIII-Cl complexes could explain the lower gaseous Cl production for the untreated sample. This situation changes when the pH is adjusted to 1.9–2.2 already in the diluted stock solution. At such low pH values, FeIII-Cl complexes are formed much more easily. Furthermore, the solubility of FeIII increases with decreasing pH, whereas more dissolved FeIII becomes available on an absolute scale.[46,84] Also, the formation of the highly soluble hypochlorous acid (HOCl; Henry’s law constant at 298 K: 2.6–9.3 × 102 M atm–1)[85] predominates at a pH between 4 and 7 over Cl2, which is favoured at a pH lower than 4 and is much more easily released into the gas phase (Henry’s law constant: 6.2–9.1 × 10–2 M atm–1).[85] Similar observations were also made by Lim et al.,[24] who ascribed the decreasing Cl2 source with decreasing pH to the speciation chemistry and solubility of the various chlorine species.
SEM-EDX results
Aerosol particles originating from the pure artificial sea water are mainly composed of NaCl, CaSO4, MgCl2 and KCl (Fig. 9a). Whereas NaCl and CaSO4 particles remain as single crystals, KCl and MgCl2 exhibit some phase-mixing and a more amorphous structure. In the case of the FeCl3-containing artificial seawater, single NaCl and CaSO4 crystals are also visible. Additionally, FeCl3 is visible in the KCl–MgCl2 phase (Fig. 9b). FeCl3 appears to cover nearly all other particles like NaCl and CaSO4, thus indicating that all aerosol particles originating from the FeCl3-enriched artificial seawater are covered by an active FeIII chloride–salt layer.

|
Fraction of active iron
Multiplying the initial LWC0 (dm3 cm–3) with the assumed saturation concentration of Cl– (6.1 mol L–1) in the aerosols produced and with Avogadro’s constant (NA = 6 × 1023 mol–1) and dividing the molar Cl–/FeIII ratio (RCl/Fe) in the stock solution yields the concentration of FeIII in the chamber. The ratio of the minimum concentration of Cl atoms produced in the first hour of the experiments (Qmin) provides an estimation of the minimum fraction of active iron λFe involved in Cl production:
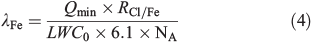
assuming that each FeIII produces 1/2Cl2 by excluding recycling effects and secondary activation mechanisms. Owing to these major assumptions and unknown uncertainties (RCl/Fe in the aerosol can differ from the bulk, or the concentration of Cl– can vary depending on the composition and thus deliquescence of the aerosol), λFe represents a rather qualitative value for relative comparisons between the experiments. The contribution of the tare volume of the ions to the measured LWC is 0.11 m3 m–3 (considered in the calculation), whereas the influence of Fe3+ ions is negligible. Fig. 10 shows the results for all FeIII-containing samples investigated, combined with the measured total minimum and maximum Cl production Qabs during the first hour. Additionally, the corresponding initial minimum production rates are displayed in the Supplementary material (Fig. S3). Again, the increase in Qabs for the artificial seawater samples with increasing FeIII concentration (Cl–/FeIII = 13, 53, 101) is highlighted, although, λFe is comparable, which indicates that similar Cl activation mechanisms are going on. This is also the case for the pure NaCl + FeIII sample where Qabs and λFe are higher, caused by the absence of competing ligands for FeIII and thus a higher fraction of photolabile FeIII–Cl complexes.

|
The presence of 20 ppb NOx in the gas phase led to a considerable increase by a factor of 3–7 for Qabs from the Cl–/FeIII = 101 and the Cl–/FeIII = 13 samples. The parallel increase of λFe (even more than 100 %) is an artefact that actually represents the additional Cl activation mechanisms induced by NOx,[13] as discussed in the Effects of NO2, O3 and SO2 section. The same applies for the experiment with 630 ppb O3. Decreasing the pH enhanced the activity of FeIII, and more than 100 % of FeIII seemed to be involved, which either is a consequence of recycling effects (re-oxidation of FeII that was formed by photolysis of FeIII complexes), or is caused by the uncertainty of λFe. For the low-pH sample as well as for the untreated samples (Cl–/FeIII = 13 and 101), an increase in Qabs and λFe was observed with the addition of 20 ppb NOx, which clearly demonstrates the NOx-induced activation of chloride. The effect is not as pronounced at low aerosol pH.
SO2 was able to slightly inhibit Cl production. In particular, high SO2 mixing ratios decreased the Qabs and thus the fraction of active iron. One can compare λFe with the salt-pan measurements, where a proportion of 0.05–0.07 % of active FeIII was determined.[15] Considering a salt pan containing 0.5 g FeCl3·6H2O and 99.5 g NaCl results in a total molar FeIII content of 1.8 × 10–3. This is quite high compared with the aerosol experiments with only ~3 × 10–6 mol FeIII at an LWC of ~2.7 × 10–9 but total Cl sources comparable with the salt-pan experiments. These large differences prove the small active surface area of the salt pans compared with the homogeneously distributed and irradiated aerosol in the chamber.
Environmental significance
The ratio of the concentrations of added pollutants and the generated aerosol concentrations (2–6 mg m–3) are approximately in agreement with the conditions for sea-salt aerosol over the ocean. Although the smog chamber helps to understand the mechanisms, it is difficult to transfer the results to large-scale atmospheric processes because the aerosol surface-to-volume ratio in the smog chamber is some orders of magnitude higher than in the atmosphere.
We investigated a very simplified system with no organic contaminants in the salt and under hydrocarbon-free conditions. Note that Fe complexes with oxygen-containing organics (such as phenolic compounds) can significantly increase the solubility of iron in seawater.[86] However, complexation can strongly inhibit iron-induced chlorine activation,[15] until the phenolic constituents are oxidised by OH• and Cl• from the photo-Fenton mechanism to the less-inhibiting but solubility-promoting oxalate. The soluble fraction of iron over the ocean ranges from 0 to 95 % as the bulk marine aerosol type reflects a mixing of multiple aerosol types, and solubility varies with the origin of the iron, aerosol size and composition.[87–89] Moreover, the solubility increases owing to the photoreduction of FeIII, which is responsible for an FeII fraction in the aerosol of up to 50 % in remote marine areas.[90] An additional crucial factor for the role of FeIII photochemistry is the aerosol pH, which varies from 1–9, mainly depending on the origin and age of the aerosol and the corresponding altering processes.[25,56]
Accounting for these effects in order to quantify iron-induced Cl production in the troposphere, a sophisticated large-scale model would be required. Based on a mean molar Cl–/Fe ratio of 100–200 in the marine aerosol (central Atlantic),[91] on the lower fraction of dissolved FeIII in the aerosol that can form photolabile complexes, and on the 20–200-times larger aerosol surface area in the chamber compared with the Atlantic aerosol,[92] the mean natural contribution of FeIII-induced Cl activation cannot compete with alternative mechanisms on a global scale (also indicated by Cl below the detection limit in our experiment with Cl–/FeIII = 997). However, it may become important on a local scale with a larger FeIII burden exposed to saline media, e.g. ship plumes or other iron-containing combustion aerosols[93] or where soil dust comes into contact with sea salt, but also in brine-containing soils[94] or salt lakes such as the Dead Sea or Australian salt lakes.[14]
Furthermore, iron-doped sea-salt aerosols have been proposed as a method for climate engineering, aiming to enhance CH4 depletion with higher Cl levels in the marine boundary layer and to simultaneously fertilise the oceans.[95] Based on our results, one may try to estimate the feasibility of such a project. For our conditions, increasing the mean molar Cl–/FeIII ratio to, say, 50 by adding FeIII would have significant effects on Cl production and thus reduce the lifetime of CH4. Transferred to the global production rate of sea salt Cl– of 1785 Tg year–1,[96] an annual contribution of 56 Tg year–1 of pure FeIII (that dissolves totally in the sea-salt aerosol) would be needed. In addition, the effect of enhanced Cl production has to be scaled down by the much lower typical aerosol surface in the marine boundary layer (~60–200 µm2 cm–3[91]) compared with our experiments (~10 000–30 000 µm2 cm–3). Thus, an increase of Cl surface production by a factor of 4 in the chamber (which is the case for Cl–/FeIII = 51 compared with the blank) would result in an increase of ~2.5 % transferred to the marine boundary layer, neglecting any effect of dissolved organics or gas-phase species. A mixture with Cl–/FeIII = 13 would need 220 Tg year–1 of FeIII and result in ~17–19 % increase in Cl production. Note that the increase of atomic Cl does not directly correlate with a reduction of the CH4 lifetime owing to the flat vertical profile of CH4 (almost evenly distributed over the total troposphere)[91] compared with the Cl-producing sea salt (confined to <1 km above the oceans, the marine sea-salt layer)[91] and thus more inefficient depletion, apart from technical challenges and other potential implications for the ecosystem.
Conclusions
In the present work, we confirmed and quantified Cl formation processes induced by the photochemistry of FeIII dissolved in the liquid aerosol phase. We found that depending on the FeIII loading in the artificial sea-salt aerosol, an enormous amount of chlorine can be activated. The initial production rates varied from ~1.9 × 1018 atoms cm–2 s–1 (Cl–/FeIII = 13) to ~2.8 × 1017 atoms cm–2 s–1 (Cl–/FeIII = 101), whereas no significantly enhanced Cl production was detected for Cl–/FeIII = 955 compared with the FeIII-free salt. The differences can mainly be explained by the amount of FeIII and the corresponding speciation of FeIII complexes that depend on the pH.
The presence of 20 ppb NOx or 630 ppb O3 considerably increased the initial Cl production rate to ~7 × 1018 atoms cm–2 s–1 (Cl–/FeIII = 13) and ~9 × 1018 atoms cm–2 s–1 (Cl–/FeIII = 13) respectively (as compared with the blank value of 2 × 1018 atoms cm–2 s–1), owing to the well-known halogen activation processes and potential pH effects. Gas-phase SO2 instead slightly inhibited Cl production to ~1.7 and ~1.1 × 1018 atoms cm–2 s–1, by adding ~20 ppb and >200 ppb SO2 respectively. The strong effect of the aerosol pH on FeIII speciation and subsequent Cl formation was demonstrated by comparing two samples with an equal Cl–/FeIII ratio of 101 but a pH adjusted to 2.1–2.3 and 3.9–4.2. Here, the production rate increased by almost an order of magnitude for the more acidic pH. An approach to calculate the active fraction of FeIII involved in Cl production confirms a higher λFe with lower pH, highlights recycling effects and identifies additional activation mechanisms where NOx and O3 are involved. In natural environments, these processes may occur in natural salty media at high dissolved FeIII concentrations and locally contribute to photochemical Cl formation.
Supplementary material
The supplementary material (available on the journal’s website at http://www.publish.csiro.au/?act=view_file&file_id=EN14279_AC.pdf) includes more details on the artificial seawater composition (Table S1), the rate constants of the applied HCs towards the radicals (Table S2), the main equilibrium constants for important FeIII complexes, the contour plots of selected experiments (Fig. S1), the NOx, NO, and O3 time profiles for the FeIII-free experiment with 20 ppb NO2 addition (Fig. S2) and an overview of the initial minimum absolute Cl production rate (Fig. S3).
Acknowledgements
We thank Franz D. Oeste, gM-Ingenieurbüro Kirchhain, Germany, for advice, Agnes Bednorz and Andrej Einhorn for technical support and Elisabeth Eitenberger and Gernot Friedbacher (Vienna University of Technology) for operating the electron microscope. This work was supported by the Deutsche ForschungsGemeinschaft (DFG) within research unit 763 (HALOPROC) and by Ries Consulting GmbH&Co Betriebs KG, Hosenfeld.
References
[1] I. Y. Fung, S. K. Meyn, I. Tegen, S. C. Doney, J. G. John, J. K. B. Bishop, Iron supply and demand in the upper ocean. Global Biogeochem. Cycles 2000, 14, 281.| Iron supply and demand in the upper ocean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXhvVGmsrc%3D&md5=fcbeed2dad9f6e9bbe7e93aeff715fa0CAS |
[2] M. S. Johnson, N. Meskhidze, Atmospheric dissolved iron deposition to the global oceans: effects of oxalate-promoted Fe dissolution, photochemical redox cycling, and dust mineralogy. Geosci. Model Dev. 2013, 6, 1137.
| Atmospheric dissolved iron deposition to the global oceans: effects of oxalate-promoted Fe dissolution, photochemical redox cycling, and dust mineralogy.Crossref | GoogleScholarGoogle Scholar |
[3] A. Martínez-García, D. M. Sigman, H. Ren, R. F. Anderson, M. Straub, D. A. Hodell, S. L. Jaccard, T. I. Eglinton, G. H. Haug, Iron fertilization of the subantarctic ocean during the last Ice Age. Science 2014, 343, 1347.
| Iron fertilization of the subantarctic ocean during the last Ice Age.Crossref | GoogleScholarGoogle Scholar | 24653031PubMed |
[4] A. Martínez-Garcia, A. Rosell-Melé, S. L. Jaccard, W. Geibert, D. M. Sigman, G. H. Haug, Southern Ocean dust–climate coupling over the past four million years. Nature 2011, 476, 312.
| Southern Ocean dust–climate coupling over the past four million years.Crossref | GoogleScholarGoogle Scholar | 21814203PubMed |
[5] J. H. Martin, S. E. Fitzwater, R. M. Gordon, Iron deficiency limits phytoplankton growth in Antarctic waters. Global Biogeochem. Cycles 1990, 4, 5.
| Iron deficiency limits phytoplankton growth in Antarctic waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXitFersLk%3D&md5=edb0e5da4a14709d8e91ad0f4aedcf8dCAS |
[6] S. Blain, B. Quéguiner, L. Armand, S. Belviso, B. Bombled, L. Bopp, A. Bowie, C. Brunet, C. Brussaard, F. Carlotti, U. Christaki, A. Corbière, I. Durand, F. Ebersbach, J.-L. Fuda, N. Garcia, L. Gerringa, B. Griffiths, C. Guigue, C. Guillerm, S. Jacquet, C. Jeandel, P. Laan, D. Lefèvre, C. Lo Monaco, A. Malits, J. Mosseri, I. Obernosterer, Y.-H. Park, M. Picheral, P. Pondaven, T. Remenyi, V. Sandroni, G. Sarthou, N. Savoye, L. Scouarnec, M. Souhaut, D. Thuiller, K. Timmermans, T. Trull, J. Uitz, P. van Beek, M. Veldhuis, D. Vincent, E. Viollier, L. Vong, T. Wagener, Effect of natural iron fertilization on carbon sequestration in the Southern Ocean. Nature 2007, 446, 1070.
| Effect of natural iron fertilization on carbon sequestration in the Southern Ocean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXksFekurg%3D&md5=059a4a0cfa5a4788a368c71cfb0a0b04CAS | 17460670PubMed |
[7] R. T. Pollard, I. Salter, R. J. Sanders, M. I. Lucas, C. M. Moore, R. A. Mills, P. J. Statham, J. T. Allen, A. R. Baker, D. C. E. Bakker, M. A. Charette, S. Fielding, G. R. Fones, M. French, A. E. Hickman, R. J. Holland, J. A. Hughes, T. D. Jickells, R. S. Lampitt, P. J. Morris, F. H. Nedelec, M. Nielsdottir, H. Planquette, E. E. Popova, A. J. Poulton, J. F. Read, S. Seeyave, T. Smith, M. Stinchcombe, S. Taylor, S. Thomalla, H. J. Venables, R. Williamson, M. V. Zubkov, Southern Ocean deep-water carbon export enhanced by natural iron fertilization. Nature 2009, 457, 577.
| Southern Ocean deep-water carbon export enhanced by natural iron fertilization.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFOmsLc%3D&md5=3bbde686cc367b3d8cf15e8d87d37edcCAS | 19177128PubMed |
[8] R. von Glasow, P. J. Crutzen, Tropospheric halogen chemistry, in Treatise on Geochemistry (Eds H. D. Holland, K. K. Turekian) 2003, Vol. 4, pp 1–67 (Elsevier-Pergamon: Oxford, UK).
[9] U. Platt, G. Hönninger, The role of halogen species in the troposphere. Chemosphere 2003, 52, 325.
| The role of halogen species in the troposphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjsVGnsLk%3D&md5=d8bbac36836ae46fea2dd356e40f1111CAS | 12738256PubMed |
[10] J. Ofner, N. Balzer, J. Buxmann, H. Grothe, P. Schmitt-Kopplin, U. Platt, C. Zetzsch, Halogenation processes of secondary organic aerosol and implications on halogen release mechanisms. Atmos. Chem. Phys. 2012, 12, 5787.
| Halogenation processes of secondary organic aerosol and implications on halogen release mechanisms.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhslSnu7zO&md5=edb951750b3288159eeb3897f5ffbe7cCAS |
[11] R. von Glasow, Atmospheric chemistry: wider role for airborne chlorine. Nature 2010, 464, 168.
| Atmospheric chemistry: wider role for airborne chlorine.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjtFSmt74%3D&md5=8abbf0b2c68ac310c691aa1f4cbdd6a1CAS | 20220828PubMed |
[12] B. J. Finlayson-Pitts, The tropospheric chemistry of sea salt: a molecular-level view of the chemistry of NaCl and NaBr. Chem. Rev. 2003, 103, 4801.
| The tropospheric chemistry of sea salt: a molecular-level view of the chemistry of NaCl and NaBr.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXosVSnt78%3D&md5=2abbf2399ee1e24d4091203311bc6dbaCAS | 14664634PubMed |
[13] M. J. Rossi, Heterogeneous reactions on salts. Chem. Rev. 2003, 103, 4823.
| Heterogeneous reactions on salts.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXovFOhtb8%3D&md5=cb19c2689eeb285c2ec313caf78d6934CAS | 14664635PubMed |
[14] T. Krause, C. Tubbesing, K. Benzing, H. F. Schöler, Model reactions and natural occurrence of furans from hypersaline environments. Biogeosciences 2014, 11, 2871.
| Model reactions and natural occurrence of furans from hypersaline environments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhs1Sju73J&md5=7cb50be77d560436d32122398576f2cbCAS |
[15] J. Wittmer, S. Bleicher, C. Zetzsch, Iron(III)-induced activation of chloride and bromide from modeled saltpans. J. Phys. Chem. A 2015, 119, 4373.. [Published online early 22 September 2014]
| Iron(III)-induced activation of chloride and bromide from modeled saltpans.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhsF2nurnO&md5=f451edc24d63a93b4cf1f56d3ecee8e5CAS | 25243918PubMed |
[16] H. Fu, D. M. Cwiertny, G. R. Carmichael, M. M. Scherer, V. H. Grassian, Photoreductive dissolution of Fe-containing mineral dust particles in acidic media. J. Geophys. Res. 2010, 115, D11304.
| Photoreductive dissolution of Fe-containing mineral dust particles in acidic media.Crossref | GoogleScholarGoogle Scholar |
[17] X. Zhu, J. M. Prospero, D. L. Savoie, F. J. Millero, R. G. Zika, E. S. Saltzman, Photoreduction of iron(III) in marine mineral aerosol solutions. J. Geophys. Res. 1993, 98, 9039.
| Photoreduction of iron(III) in marine mineral aerosol solutions.Crossref | GoogleScholarGoogle Scholar |
[18] W. L. Miller, D. King, J. Lin, D. R. Kester, Photochemical redox cycling of iron in coastal seawater. Mar. Chem. 1995, 50, 63.
| Photochemical redox cycling of iron in coastal seawater.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXotVaqurg%3D&md5=4cc1867ae46672afffe887661a10f7bbCAS |
[19] R. L. Siefert, S. O. Pehkonen, Y. Erel, M. R. Hoffmann, Iron photochemistry of aqueous suspensions of ambient aerosol with added organic acids. Geochim. Cosmochim. Acta 1994, 58, 3271.
| Iron photochemistry of aqueous suspensions of ambient aerosol with added organic acids.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXltlyjt7w%3D&md5=559add60b1ba92a60f0dfe9f4befb1a3CAS |
[20] D. Vione, V. Maurino, C. Minero, E. Pelizzetti, M. A. Harrison, R.-I. Olariu, C. Arsene, Photochemical reactions in the tropospheric aqueous phase and on particulate matter. Chem. Soc. Rev. 2006, 35, 441.
| 1:CAS:528:DC%2BD28XjslKiurg%3D&md5=1cefeedaa7e80d453f15eeff061ca8beCAS | 16636727PubMed |
[21] V. A. Nadtochenko, J. Kiwi, Photolysis of FeOH2+ and FeCl2+ in aqueous solution. Photodissociation kinetics and quantum yields. Inorg. Chem. 1998, 37, 5233.
| Photolysis of FeOH2+ and FeCl2+ in aqueous solution. Photodissociation kinetics and quantum yields.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXlslOmur8%3D&md5=c32ba5d3abe5e5ded4dee10277953e5bCAS |
[22] A. Machulek, J. E. Moraes, L. T. Okano, C. A. Silvério, F. H. Quina, Photolysis of ferric ions in the presence of sulfate or chloride ions: implications for the photo-Fenton process. Photochem. Photobiol. Sci. 2009, 8, 985.
| Photolysis of ferric ions in the presence of sulfate or chloride ions: implications for the photo-Fenton process.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotVenu7w%3D&md5=47cc76188ff051f77e632b8f6e09fc13CAS | 19582274PubMed |
[23] D. Whitney King, R. A. Aldrich, S. E. Charnecki, Photochemical redox cycling of iron in NaCl solutions. Mar. Chem. 1993, 44, 105.
| Photochemical redox cycling of iron in NaCl solutions.Crossref | GoogleScholarGoogle Scholar |
[24] M. Lim, K. Chiang, R. Amal, Photochemical synthesis of chlorine gas from iron(III) and chloride solution. J. Photoch. Photobio. A: Chemistry 2006, 183, 126.
| Photochemical synthesis of chlorine gas from iron(III) and chloride solution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xos12rtb4%3D&md5=11fa0692117fb7b1a209433102459ddcCAS |
[25] W. C. Keene, R. Sander, A. A. Pszenny, R. Vogt, P. J. Crutzen, J. N. Galloway, Aerosol pH in the marine boundary layer. J. Aerosol Sci. 1998, 29, 339.
| Aerosol pH in the marine boundary layer.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXis1aqsLk%3D&md5=51d0b0a02ffe884359e7134ef95b90abCAS |
[26] J. De Laat, T. G. Le, Kinetics and modeling of the FeIII/H2O2 system in the presence of sulfate in acidic aqueous solutions. Environ. Sci. Technol. 2005, 39, 1811.
| Kinetics and modeling of the FeIII/H2O2 system in the presence of sulfate in acidic aqueous solutions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmtVSgsQ%3D%3D&md5=05e2aad6389d9761b70fc7dabb7ef83fCAS | 15819241PubMed |
[27] Y. Sadanaga, J. Hirokawa, H. Akimoto, Formation of molecular chlorine in dark condition: heterogeneous reaction of ozone with sea salt in the presence of ferric ion. Geophys. Res. Lett. 2001, 28, 4433.
| Formation of molecular chlorine in dark condition: heterogeneous reaction of ozone with sea salt in the presence of ferric ion.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XisFahsQ%3D%3D&md5=3c09e482d10828671fcd66addd812591CAS |
[28] W. Hoppel, L. Pasternack, P. Caffrey, G. Frick, J. Fitzgerald, D. Hegg, S. Gao, J. Ambrusko, T. Albrechcinski, Sulfur dioxide uptake and oxidation in sea-salt aerosol. J. Geophys. Res. 2001, 106, 27 575.
| Sulfur dioxide uptake and oxidation in sea-salt aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXpt1ejsb8%3D&md5=bc7fe409ec7a9e7892b9ad6ee713e58fCAS |
[29] M. Novič, I. Grgić, M. Poje, V. Hudnik, Iron-catalyzed oxidation of SIV species by oxygen in aqueous solution: influence of pH on the redox cycling of iron. Atmos. Environ. 1996, 30, 4191.
| Iron-catalyzed oxidation of SIV species by oxygen in aqueous solution: influence of pH on the redox cycling of iron.Crossref | GoogleScholarGoogle Scholar |
[30] J.-Z. Zhang, F. J. Millero, The rate of sulfite oxidation in seawater. Geochim. Cosmochim. Acta 1991, 55, 677.
| The rate of sulfite oxidation in seawater.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXhvFWqt7Y%3D&md5=0ea77bf50d3d0baba2cd7c788cc94121CAS |
[31] S. Bleicher, J. C. Buxmann, R. Sander, T. P. Riedel, J. A. Thornton, U. Platt, C. Zetzsch, The influence of nitrogen oxides on the activation of bromide and chloride in salt aerosol. Atmos. Chem. Phys. Discuss. 2014, 14, 10 135.
| The influence of nitrogen oxides on the activation of bromide and chloride in salt aerosol.Crossref | GoogleScholarGoogle Scholar |
[32] W. Behnke, C. Zetzsch, Heterogeneous photochemical formation of Cl atoms from NaCl aerosol, NOx and ozone. J. Aerosol Sci. 1990, 21, S229.
| Heterogeneous photochemical formation of Cl atoms from NaCl aerosol, NOx and ozone.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXktFCnu74%3D&md5=88821dda5e5b2bbf950295eb9bad8102CAS |
[33] M. Brigante, M. Minella, G. Mailhot, V. Maurino, C. Minero, D. Vione, Formation and reactivity of the dichloride radical in surface waters: a modelling approach. Chemosphere 2014, 95, 464.
| Formation and reactivity of the dichloride radical in surface waters: a modelling approach.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhs12ltLfF&md5=22326bef2a603cc2100c2bcd7d9be8abCAS | 24383074PubMed |
[34] J. Buxmann, N. Balzer, S. Bleicher, U. Platt, C. Zetzsch, Observations of bromine explosions in smog chamber experiments above a model salt pan. Int. J. Chem. Kinet. 2012, 44, 312.
| Observations of bromine explosions in smog chamber experiments above a model salt pan.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XkvVKjtbk%3D&md5=c551be0e6328638413cf622fcf1c9199CAS |
[35] W. Behnke, W. Holländer, W. Koch, F. Nolting, C. Zetzsch, A smog chamber for studies of the photochemical degradation of chemicals in the presence of aerosols. Atmos. Environ. 1988, 22, 1113.
| A smog chamber for studies of the photochemical degradation of chemicals in the presence of aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXltFGgsro%3D&md5=612ce50072eab86049641b94f30acccaCAS |
[36] D. R. Kester, I. W. Duedall, D. N. Connors, R. M. Pytkowicz, Preparation of artificial seawater. Limnol. Oceanogr. 1967, 12, 176.
| Preparation of artificial seawater.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF2sXhtFylur0%3D&md5=a36a7ec97053b17c4a61856b69e2f00cCAS |
[37] K. Zhang, E. Knipping, A. Wexler, P. Bhave, G. Tonnesen, Size distribution of sea-salt emissions as a function of relative humidity. Atmos. Environ. 2005, 39, 3373.
| Size distribution of sea-salt emissions as a function of relative humidity.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXksV2itbk%3D&md5=8f8513ffbefce0f684f40be832216aedCAS |
[38] N. Balzer, Kinetische Untersuchungen der Halogen-Aktivierung einer simulierten Salzpfanne in einer Smogkammer 2012, Ph.D. thesis, University of Bayreuth, Germany.
[39] P. Supeno, P. Kruus, Sonochemical formation of nitrate and nitrite in water. Ultrason. Sonochem. 2000, 7, 109.
| Sonochemical formation of nitrate and nitrite in water.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXlsFSisrc%3D&md5=f3c78397ba887c95ee848c3bd3c51531CAS |
[40] T. B. Ryerson, E. J. Williams, F. C. Fehsenfeld, An efficient photolysis system for fast-response NO2 measurements. J. Geophys. Res. 2000, 105, 26 447.
| An efficient photolysis system for fast-response NO2 measurements.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXovVGrs70%3D&md5=9ae021a8a8950a76ffa4fec09fec9f0dCAS |
[41] N. A. Kelly, Characterization of fluorocarbon-film bags as smog chambers. Environ. Sci. Technol. 1982, 16, 763.
| Characterization of fluorocarbon-film bags as smog chambers.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL38XlsV2ktLo%3D&md5=3bb52c62e782504ed1e7b06cab961c32CAS | 22299783PubMed |
[42] C. Misra, M. Singh, S. Shen, C. Sioutas, P. M. Hall, Development and evaluation of a personal cascade impactor sampler (PCIS). J. Aerosol Sci. 2002, 33, 1027.
| Development and evaluation of a personal cascade impactor sampler (PCIS).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XktFOgu78%3D&md5=421fb5ffb6377367277ba41f96cf4c65CAS |
[43] H. Lohninger, J. Ofner, Multisensor hyperspectral imaging as a versatile tool for image-based chemical structure determination. Spectrosc. Eur. 2014, 26, 6.
| 1:CAS:528:DC%2BC2cXitVylt7bI&md5=8730236c021c4bf2745103043667ea15CAS |
[44] Y. Marcus, Ionic radii in aqueous solutions. Chem. Rev. 1988, 88, 1475.
| Ionic radii in aqueous solutions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXmtFKqsL8%3D&md5=50e9d8f82d0555377d11c10b46349429CAS |
[45] I. N. Tang, A. C. Tridico, K. H. Fung, Thermodynamic and optical properties of sea-salt aerosols. J. Geophys. Res. 1997, 102, 23 269.
| Thermodynamic and optical properties of sea-salt aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXntVylt7k%3D&md5=2eadd9071d224a05fb9dc3a1f98181a4CAS |
[46] F. J. Millero, Solubility of FeIII in seawater. Earth Planet. Sci. Lett. 1998, 154, 323.
| Solubility of FeIII in seawater.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXhtFyhsrY%3D&md5=a352b2728b7a51a9e56429655f4dfd69CAS |
[47] J. A. Thornton, J. P. Kercher, T. P. Riedel, N. L. Wagner, J. Cozic, J. S. Holloway, W. P. Dubé, G. M. Wolfe, P. K. Quinn, A. M. Middlebrook, B. Alexander, S. S. Brown, A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry. Nature 2010, 464, 271.
| A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjtFSmu78%3D&md5=03972d642bd308f9f8d32b728f106d1fCAS | 20220847PubMed |
[48] W. H. Schroeder, P. Urone, Formation of nitrosyl chloride from salt particles in air. Environ. Sci. Technol. 1974, 8, 756.
| Formation of nitrosyl chloride from salt particles in air.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2MXht1yrsb0%3D&md5=109c8c3e874f466612294aee2fe6501cCAS |
[49] B. J. Finlayson-Pitts, Reaction of NO2 with NaCl and atmospheric implications of NOCl formation. Nature 1983, 306, 676.
| Reaction of NO2 with NaCl and atmospheric implications of NOCl formation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXls1Wktg%3D%3D&md5=50a491c52e6a30cfd3d98e482377776fCAS |
[50] W. Behnke, H.-U. Krüger, V. Scheer, C. Zetzsch, Formation of ClNO2 and HONO in the presence of NO2, O3 and wet NaCl aerosol. J. Aerosol Sci. 1992, 23, 933.
| Formation of ClNO2 and HONO in the presence of NO2, O3 and wet NaCl aerosol.Crossref | GoogleScholarGoogle Scholar |
[51] R. Karlsson, E. Ljungström, Nitrogen dioxide and sea salt particles – a laboratory study. J. Aerosol Sci. 1995, 26, 39.
| Nitrogen dioxide and sea salt particles – a laboratory study.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXjslens78%3D&md5=d4e2711c15dadf88e2a6194e8f831b76CAS |
[52] A. Saiz-Lopez, R. von Glasow, Reactive halogen chemistry in the troposphere. Chem. Soc. Rev. 2012, 41, 6448.
| Reactive halogen chemistry in the troposphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhtlaktr7J&md5=3fb1cf9e428bf93013cfc2c1556bb7a1CAS | 22940700PubMed |
[53] D. O. De Haan, B. J. Finlayson-Pitts, Knudsen cell studies of the reaction of gaseous nitric acid with synthetic sea salt at 298 K. J. Phys. Chem. A 1997, 101, 9993.
| Knudsen cell studies of the reaction of gaseous nitric acid with synthetic sea salt at 298 K.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXhs1ahsw%3D%3D&md5=46156e8c6cc666153cb4b4318e43a149CAS |
[54] T. D. Saul, M. P. Tolocka, M. V. Johnston, Reactive uptake of nitric acid onto sodium chloride aerosols across a wide range of relative humidities. J. Phys. Chem. A 2006, 110, 7614.
| Reactive uptake of nitric acid onto sodium chloride aerosols across a wide range of relative humidities.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XkvFGltro%3D&md5=734889d806f87f09f9ac296acf768220CAS | 16774205PubMed |
[55] W. Behnke, V. Scheer, C. Zetzsch, Production of a photolytic precursor of atomic Cl from aerosols and Cl– in the presence of O3, in Naturally Produced Organohalogens (Eds A. Grimvall, E. de Leer) 1995, pp. 375–384 (Springer: Dordrecht, Netherlands).
[56] W. C. Keene, D. L. Savoie, The pH of deliquesced sea-salt aerosol in polluted marine air. Geophys. Res. Lett. 1998, 25, 2181.
| The pH of deliquesced sea-salt aerosol in polluted marine air.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXktlCltrs%3D&md5=36d7148da516de0daba989c841d043c5CAS |
[57] P. Brimblecombe, S. L. Clegg, The solubility and behaviour of acid gases in the marine aerosol. J. Atmos. Chem. 1988, 7, 1.
| The solubility and behaviour of acid gases in the marine aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXlsFGhtbg%3D&md5=bda1d47d554aa661c86c959da1ad1fe9CAS |
[58] C. Zetzsch, W. Behnke, Heterogeneous reactions of chlorine compounds, in The Tropospheric Chemistry of Ozone in the Polar Regions (Eds H. Niki, K. H. Becker) 1993, pp. 291–306 (Springer: New York).
[59] S. Pechtl, R. von Glasow, Reactive chlorine in the marine boundary layer in the outflow of polluted continental air: a model study. Geophys. Res. Lett. 2007, 34, L11813.
| Reactive chlorine in the marine boundary layer in the outflow of polluted continental air: a model study.Crossref | GoogleScholarGoogle Scholar |
[60] C. B. Faxon, D. T. Allen, Chlorine chemistry in urban atmospheres: a review. Environ. Chem. 2013, 10, 221.
| Chlorine chemistry in urban atmospheres: a review.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhtVaksLbI&md5=d8e1eee561eefe259765e066c4e3fd69CAS |
[61] M. Hausmann, U. Platt, Spectroscopic measurement of bromine oxide and ozone in the high Arctic during Polar Sunrise Experiment 1992. J. Geophys. Res. 1994, 99, 25 399.
| Spectroscopic measurement of bromine oxide and ozone in the high Arctic during Polar Sunrise Experiment 1992.Crossref | GoogleScholarGoogle Scholar |
[62] S. Fickert, J. W. Adams, J. N. Crowley, Activation of Br2 and BrCl via uptake of HOBr onto aqueous salt solutions. J. Geophys. Res. 1999, 104, 23 719.
| Activation of Br2 and BrCl via uptake of HOBr onto aqueous salt solutions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXntleht7c%3D&md5=475c6748b8e523377a2f3a68d24b7614CAS |
[63] K. W. Oum, Formation of molecular chlorine from the photolysis of ozone and aqueous sea-salt particles. Science 1998, 279, 74.
| Formation of molecular chlorine from the photolysis of ozone and aqueous sea-salt particles.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXivF2nsg%3D%3D&md5=6f044ded1eafe00e45f93b25add495edCAS | 9417027PubMed |
[64] E. M. Knipping, M. J. Lakin, K. L. Foster, P. Jungwirth, D. J. Tobias, R. B. Gerber, D. Dabdub, B. J. Finlayson-Pitts, Experiments and simulations of ion-enhanced interfacial chemistry on aqueous NaCl aerosols. Science 2000, 288, 301.
| Experiments and simulations of ion-enhanced interfacial chemistry on aqueous NaCl aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXislersr4%3D&md5=95a2b65c33e3acc3c98cf1b883f1e6f6CAS | 10764637PubMed |
[65] S. Seisel, F. Caloz, F. F. Fenter, H. van den Bergh, M. J. Rossi, The heterogeneous reaction of NO3 with NaCl and KBr: a non-photolytic source of halogen atoms. Geophys. Res. Lett. 1997, 24, 2757.
| The heterogeneous reaction of NO3 with NaCl and KBr: a non-photolytic source of halogen atoms.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXnslOlt74%3D&md5=7bcfc96298acce35b9f3ec2266ccfdaeCAS |
[66] Y. Rudich, R. K. Talukdar, A. R. Ravishankara, R. W. Fox, Reactive uptake of NO3 on pure water and ionic solutions. J. Geophys. Res. 1996, 101, 21 023.
| Reactive uptake of NO3 on pure water and ionic solutions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XmsVOhtL0%3D&md5=98e76aaf2d10abaf783608664ef74670CAS |
[67] W. Behnke, C. George, V. Scheer, C. Zetzsch, Production and decay of ClNO2 from the reaction of gaseous N2O5 with NaCl solution: bulk and aerosol experiments. J. Geophys. Res. 1997, 102, 3795.
| Production and decay of ClNO2 from the reaction of gaseous N2O5 with NaCl solution: bulk and aerosol experiments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXhvFClsLY%3D&md5=7286cf5eef80379b0ace3628f3cfdaffCAS |
[68] J. M. Roberts, H. D. Osthoff, S. S. Brown, A. R. Ravishankara, N2O5 oxidizes chloride to Cl2 in acidic atmospheric aerosol. Science 2008, 321, 1059.
| N2O5 oxidizes chloride to Cl2 in acidic atmospheric aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVSitrnE&md5=c955aee628e3485b08e177ed789a13d0CAS | 18599742PubMed |
[69] F. F. Fenter, F. Caloz, M. J. Rossi, Heterogeneous kinetics of N2O5 uptake on salt, with a systematic study of the role of surface presentation (for N2O5 and HNO3). J. Phys. Chem. 1996, 100, 1008.
| Heterogeneous kinetics of N2O5 uptake on salt, with a systematic study of the role of surface presentation (for N2O5 and HNO3).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XjvFU%3D&md5=54ef7e47722789118e074564e9e80fc5CAS |
[70] B. J. Finlayson-Pitts, F. E. Livingston, H. N. Berko, Synthesis and identification by infrared spectroscopy of gaseous nitryl bromide, BrNO2. J. Phys. Chem. 1989, 93, 4397.
| Synthesis and identification by infrared spectroscopy of gaseous nitryl bromide, BrNO2.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXisVWls74%3D&md5=094217cc8bf9d7ce53e0a12e325d36aeCAS |
[71] M. E. Gebel, B. J. Finlayson-Pitts, J. A. Ganske, The uptake of SO2 on synthetic sea salt and some of its components. Geophys. Res. Lett. 2000, 27, 887.
| The uptake of SO2 on synthetic sea salt and some of its components.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXisFSmsLk%3D&md5=9781a1d36a724c18f16bc0002b02c332CAS |
[72] R. Vogt, P. J. Crutzen, R. Sander, A mechanism for halogen release from sea-salt aerosol in the remote marine boundary layer. Nature 1996, 383, 327.
| A mechanism for halogen release from sea-salt aerosol in the remote marine boundary layer.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XlvFaisbg%3D&md5=51b21d9d084032f194a0345c4e7d63e0CAS |
[73] R. C. Troy, D. W. Margerum, Non-metal redox kinetics: hypobromite and hypobromous acid reactions with iodide and with sulfite and the hydrolysis of bromosulfate. Inorg. Chem. 1991, 30, 3538.
| Non-metal redox kinetics: hypobromite and hypobromous acid reactions with iodide and with sulfite and the hydrolysis of bromosulfate.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXlt1Wis7k%3D&md5=ee834516a73bd20d876b16fa4071f167CAS |
[74] R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – Gas-phase reactions of Ox, HOx, NOx and SOx species. Atmos. Chem. Phys. 2004, 4, 1461.
| Evaluated kinetic and photochemical data for atmospheric chemistry: Volume I – Gas-phase reactions of Ox, HOx, NOx and SOx species.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXnvFaqtrY%3D&md5=cf1e634dedfaecb4c0fb69a69a0565d5CAS |
[75] R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe, Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III – Gas-phase reactions of inorganic halogens. Atmos. Chem. Phys. 2007, 7, 981.
| Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III – Gas-phase reactions of inorganic halogens.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXjvVOhsLw%3D&md5=745cb00be41c1d087e2ea7e815125ffaCAS |
[76] V. Nadtochenko, J. Kiwi, Primary photochemical reactions in the photo-Fenton system with ferric chloride. 1. A case study of xylidine oxidation as a model compound. Environ. Sci. Technol. 1998, 32, 3273.
| Primary photochemical reactions in the photo-Fenton system with ferric chloride. 1. A case study of xylidine oxidation as a model compound.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXlvVSrsbc%3D&md5=cbe9dde05cfe484fb89484ac88459f82CAS |
[77] D. L. Parkhurst, C. A. Appelo, User’s Guide to PHREEQC (Version 2): a Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations. Water Resource Investigations Report 1999, pp. 99–4259 (US Geological Survey: Denver, CO, USA).
[78] J. D. Allison, D. S. Brown, K. J. Novo-Gradac, MINTEQA2/PRODEFA2, A Geochemical Assessment Model for Environmental Systems: Version 3.0 User’s Manual 1991 (Environmental Research Laboratory, Office of Research and Development, US Environmental Protection Agency: Athens, GA).
[79] K. S. Pitzer, Thermodynamics of electrolytes. I. Theoretical basis and general equations. J. Phys. Chem. 1973, 77, 268.
| Thermodynamics of electrolytes. I. Theoretical basis and general equations.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3sXns1Khug%3D%3D&md5=bc9c481b72feb14a0a9a6a8c5b74fcc7CAS |
[80] N. J. Tosca, S. M. McLennan, B. C. Clark, J. P. Grotzinger, J. A. Hurowitz, A. H. Knoll, C. Schröder, S. W. Squyres, Geochemical modeling of evaporation processes on Mars: insight from the sedimentary record at Meridiani Planum. Earth Planet. Sci. Lett. 2005, 240, 122.
| Geochemical modeling of evaporation processes on Mars: insight from the sedimentary record at Meridiani Planum.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1aqurrE&md5=a3d4f2bc4e3f36af21ff21fe1ae34126CAS |
[81] A. H. Truesdell, B. F. Jones, WATEQ, a computer program for calculating chemical equilibria of natural waters. US Geol. Surv. J. Res. 1974, 2, 233.
| 1:CAS:528:DyaE2MXhtVaqsrs%3D&md5=6e9d81a0d278c959a2a61c417b887c66CAS |
[82] E. Hückel, Zur Theorie konzentrierterer wässeriger Lösungen starker Elektrolyte. Phys. Z. 1925, 26, 93.
[83] M. A. McCaffrey, B. Lazar, H. D. Ho, The evaporation path of seawater and the coprecipitation of Br– and K+ with halite. J. Sediment. Petrol. 1987, 57, 928.
| 1:CAS:528:DyaL2sXmtVWgu7g%3D&md5=df540693409ec03b4b2b0e5923bc3907CAS | 11542110PubMed |
[84] X. Liu, F. J. Millero, The solubility of iron in seawater. Mar. Chem. 2002, 77, 43.
| The solubility of iron in seawater.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXptFemt7c%3D&md5=abf58b20569672ea8308a48b5a38d4d1CAS |
[85] R. Sander, Compilation of Henry’s Law Constants for Inorganic and Organic Species of Potential Importance in Environmental Chemistry. Ver. 3 1999. Available at http://www.henrys-law.org/henry-3.0.pdf [Verified May 2013].
[86] K. Kuma, J. Nishioka, K. Matsunaga, Controls on iron(III) hydroxide solubility in seawater: the influence of pH and natural organic chelators. Limnol. Oceanogr. 1996, 41, 396.
| Controls on iron(III) hydroxide solubility in seawater: the influence of pH and natural organic chelators.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XkvFKhtrY%3D&md5=c866311ba7784586af8a1af241362b3cCAS |
[87] E. R. Sholkovitz, P. N. Sedwick, T. M. Church, A. R. Baker, C. F. Powell, Fractional solubility of aerosol iron: synthesis of a global-scale data set. Geochim. Cosmochim. Acta 2012, 89, 173.
| Fractional solubility of aerosol iron: synthesis of a global-scale data set.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xos1Wqsrs%3D&md5=2587ff1d82750429da22e6a0839afb48CAS |
[88] A. R. Baker, T. D. Jickells, M. Witt, K. L. Linge, Trends in the solubility of iron, aluminium, manganese and phosphorus in aerosol collected over the Atlantic Ocean. Mar. Chem. 2006, 98, 43.
| Trends in the solubility of iron, aluminium, manganese and phosphorus in aerosol collected over the Atlantic Ocean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xht1ymsg%3D%3D&md5=d76ad5b407cffd436c999ffef364c18bCAS |
[89] R. L. Siefert, A. M. Johansen, M. R. Hoffmann, S. O. Pehkonen, Measurements of trace metal (Fe, Cu, Mn, Cr) oxidation states in fog and stratus clouds. J. Air Waste Manag. Assoc. 1998, 48, 128.
| Measurements of trace metal (Fe, Cu, Mn, Cr) oxidation states in fog and stratus clouds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXhslCku78%3D&md5=55fa1b4c462705406c59739e3dcbc5faCAS |
[90] G. Zhuang, Z. Yi, R. A. Duce, P. R. Brown, Chemistry of iron in marine aerosols. Global Biogeochem. Cycles 1992, 6, 161.
| Chemistry of iron in marine aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXps1Sk&md5=afde2f71145fcd62124283cc765057c7CAS |
[91] P. Warneck, Chemistry of the Natural Atmosphere, 2nd edn 1999 (Academic Press: San Diego, CA).
[92] C. D. O’Dowd, E. Becker, M. Kulmala, Mid-latitude North Atlantic aerosol characteristics in clean and polluted air. Atmos. Res. 2001, 58, 167.
| Mid-latitude North Atlantic aerosol characteristics in clean and polluted air.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXntVWktL0%3D&md5=c56702daaf780f28610c10e8b7fd2b4cCAS |
[93] A. Ito, Global modeling study of potentially bioavailable iron input from shipboard aerosol sources to the ocean. Global Biogeochem. Cycles 2013, 27, 1.
| Global modeling study of potentially bioavailable iron input from shipboard aerosol sources to the ocean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhtFaksbbJ&md5=d31b6785fd9c6808f01cccbea4a031dfCAS |
[94] N. M. Mahowald, S. Engelstaedter, C. Luo, A. Sealy, P. Artaxo, C. Benitez-Nelson, S. Bonnet, Y. Chen, P. Y. Chuang, D. D. Cohen, F. Dulac, B. Herut, A. M. Johansen, N. Kubilay, R. Losno, W. Maenhaut, A. Paytan, J. M. Prospero, L. M. Shank, R. L. Siefert, Atmospheric iron deposition: global distribution, variability, and human perturbations. Annu. Rev. Mar. Sci. 2009, 1, 245.
| Atmospheric iron deposition: global distribution, variability, and human perturbations.Crossref | GoogleScholarGoogle Scholar |
[95] F. D. Meyer-Oeste, Method for Cooling the Troposphere. Int. Patent CA 2748680 A1 2010.
[96] W. C. Keene, M. Khalil, K. Aslam, D. J. Erickson, A. McCulloch, T. E. Graedel, J. M. Lobert, M. L. Aucott, S. L. Gong, D. B. Harper, G. Kleiman, P. Midgley, R. M. Moore, C. Seuzaret, W. T. Sturges, C. M. Benkovitz, V. Koropalov, L. A. Barrie, Y. F. Li, Composite global emissions of reactive chlorine from anthropogenic and natural sources: reactive chlorine emissions inventory. J. Geophys. Res. 1999, 104, 8429.
| Composite global emissions of reactive chlorine from anthropogenic and natural sources: reactive chlorine emissions inventory.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjtFyluro%3D&md5=613bad9542849d000b36cbbe884fbd06CAS |