
Imaging of HIV entry and egress
Anupriya Aggarwal A and Stuart G Turville A BA Laboratory of HIV Biology
Immunovirology and Pathogenesis Program
The Kirby Institute
University of New South Wales
NSW 2010, Australia
B Laboratory of HIV Biology
Immunovirology and Pathogenesis Program
The Kirby Institute
University of New South Wales
NSW 2010, Australia
Tel: +61 2 8382 4950
Fax: + 61 2 8382 4945
Email: sturville@kirby.unsw.edu.au
Microbiology Australia 35(2) 107-109 https://doi.org/10.1071/MA14035
Published: 15 May 2014
During the early stages of HIV research, imaging of HIV was confined to the ultrastructural level1. These early images gave us glimpses of the viral life cycle from the early stages of entry, with HIV detection at the plasma membrane and within endocytic/vesicular compartments through to different sites of viral assembly/budding in CD4 T cells and macrophages1–5. Whilst these previous snapshots of fixed specimens were seminal in nature, the increasing use of fluorescent proteins (FP) and key advances in fluorescent microscope technologies now give us the tools to test hypotheses in not only live cells, but also with live virus that could also be tracked in real-time. Herein we review the advances in HIV tracking, with an emphasis on recent observations that link HIV to the cortical F-actin network during HIV egress.
Imaging tags and viral entry
One of the first comprehensive live imaging studies of HIV focused solely on entry and movement towards the nucleus. Led by Hope and McDonald6,7, labeling and tracking of HIV particles entering cells was largely based around two key methods. First was the ability to make virions and supply FP via separate constructs and thus allow labeling of virus at the protein level. Second was the ability of the viral protein Vpr to not only package into budding virions through its non covalent association with the p6 region of HIV Gag8, but also its ability to remain associated with the Reverse Transcriptase complex (RTC) post entry9. Thus the relatively small size of Vpr and its ability to be fused with one of many FP and its ability to be delivered into virions in trans, made it indispensible for HIV entry trafficking from the membrane to the nucleus. Labeling of virions has now expanded beyond HIV Vpr and now includes HIV Gag fluorescent fusion proteins and lipid markers10. Whilst FP have been used extensively to date, the emergence of small genetic tags in various key HIV proteins (such as HIV Integrase and Nucleocapsid11,12), now enables investigators to track several HIV products en route to the nucleus.
The pitfalls and challenges of imaging HIV egress
As highlighted above, labeling of mature HIV particles for entry studies was primarily achieved using a system of supplying markers in trans. Thus post one round of infection, the ability to track virions was lost. For laboratories like our own, the study of viral spread between cells was our major aim and any marker that was lost in a single round provided limitations to these studies. Thus viral genomic incorporation of a visible tag was the next challenge for researchers. Indeed success in genomic incorporation ensured multiple rounds could be detected and viral egress in physiologically relevant cells could potentially be tracked. Initial attempts at introducing FP within the viral genome, although successful, came at the cost of severe viral attenuation13. Attenuation, we hypothesised, was the result of the size of the insertion, so ourselves and others examined alternatives that would lower the genomic load of the tag. The ‘smaller’ tag of choice as an alternative to FP was a system known as genetic tags and represented peptides, that once fused to protein of interest, could be detected by a membrane permeable biarsenical dye14. Whilst genetic tags were one solution, our ultimate aim was to use this as a complementary approach by developing a system of genetically tagged and FP containing viruses to ensure that any labeling specific artifacts were accounted for.
Tracking of HIV within infected cells
The biggest obstacle to the use of genetic tags was the poor signal to noise ratios11,15. Indeed early staining procedures observed significant background staining that was only overcome if the signal of the fusion protein was significantly above background or in cellular areas that were not prone to dye accumulation (i.e. mitochondria). For those wishing to use this system for viral entry events, the issue was ingeniously addressed by either cell free viral labeling or arguably the least invasive method known as metabolic labeling11,12. Whilst this enabled studies of viral entry by tagging many different HIV proteins11, studies of viral egress still remained a challenge due to background noise. Refinement of the tag sequence through library screening and increased stringency in the staining procedures increased the feasibility of using this technique in infected cells16. Indeed both ourselves and others applied this technology to arguably the most difficult to stain cells (largely due to the abundance of mitochondria) and subsequently successfully achieved staining of viral particles within infected cells15,17,18. At the same time both ourselves and others found the solution of working with the large FPs in the context of the major structural protein HIV Gag. The key was based on two levels of rescuing the virus. The first level by introducing flanking HIV protease cleavage sites around FPs when they were placed in the context of HIV Gag. The second by diluting the FPs within viral preparations through the supply of wild type HIV Gag and Gag-Pol only at the protein level18–20.
Tracking viral pathways that were previously undetectable
Given our ability to work with both genetic tags and FPs within the HIV genome, we had an unprecedented foundation to control for any labeling specific artifacts. That is, given the diverse nature of the two types of viral labeling, the probability of artifacts in both techniques would be intrinsically low. Whilst this approach ensured imaging would be representative of the virus life cycle, each approach had key features that enabled us to look at the virus in a completely new light.
The benefits of imaging virions using genetic tags
The power of genetic tags was initially at four key levels. Firstly the low genetic burden on the virus, secondly the fact the tag can remain with key viral proteins, thirdly that the virus did not need to be rescued and finally that the tag was retained after weeks of viral passaging. That said, these advantages were instantly counter balanced by the staining procedure and the fact the resulting fluorescence was still limiting and as a consequence would limit its versatility for live imaging at either high frame rates or over prolonged periods of time. Whilst these features have been known since the development of the technique, what was intriguing was what the system actually detected in live HIV infected cells. To be brief, the system is based on key spaced cysteine residues within the peptide fused to the protein of interest. The cysteines however need to be reduced to ensure they recognise the dye. Thus the genetic tag system represents a REDOX sensor, where depending on the REDOX state of the tag, it is turned ‘on’ or turned ‘off’ for dye recognition. Initially this did provide confusion when interpreting staining of HIV particles that were either newly forming at the membrane or had already undergone fission at the plasma membrane and/or were in vesicular compartments of neighboring cells. The confusion can be summarised by the simple fact the virus phases through reduced and non-reduced conditions during the assembly and fission process respectively. Indeed the majority of retroviruses21, curiously encode a sulfur-containing amino acid at the Protease dimer interface that enables viral protease to be regulated by a REDOX switch. That is, when reduced within the infected cell cytoplasm viral protease is active, but post egress/fission, protease is rendered inactive. Whilst the latter describes a viral enzymatic switch based on REDOX, this in turn translated to a visible switch when utilising the genetic tag system, ‘on’ when virions were forming and ‘off’ when they had undergone fission at the plasma membrane (Figure 1). Thus the additional power of this technique is its ability to detect only newly forming virions at the membrane that have yet to cycle through the fission process15.
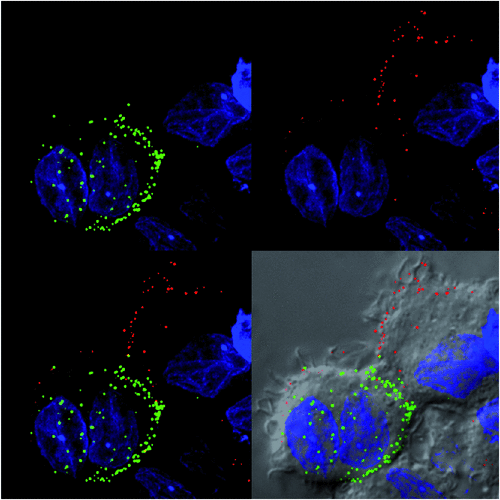
|
Living cells reveal previously unknown pathways and structures
The most important feature of both FPs and genetic tag approaches is the ability to keep cells alive. This enables preservation of cellular-viral structures that would otherwise be lost in the harsh conditions of chemical fixation and/or immuno-labeling. As a working example of this, the combined use of genetic tags and FPs within the HIV genome led to first observation and characterisation of cellular/viral structures known as HIV filopodia (Figure 2)18. Briefly, cells of the immune system promiscuously probe their external environment using membrane extensions known as filopodia. Filopodia can be further defined as slender extensions formed at the plasma membrane by the active polymerisation of F-Actin by Arp2/3 and/or a family formin member. It was the ability to actually observe these structures that led us to critically rethink how we view viral egress at the plasma membrane at several levels. Firstly, the high-jacking by the virus of highly dynamic and promiscuously contacting structures would indeed be predictably important for the spread of the virus. Secondly, HIV filopodia as a culminating phenotype supports hard-wiring of HIV egress to the underlying cortical F-Actin network in every infected cell type. Whilst these initial observations were critiqued as an in vitro phenotype, cryoelectron microscopy of HIV infection in vivo has recently independently observed HIV filopodia22.
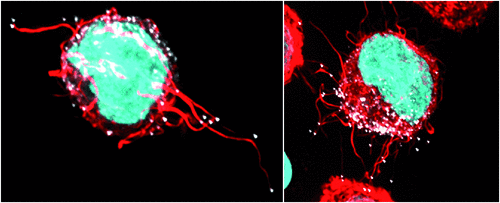
|
Conclusions
The process of developing labeling techniques for HIV virions has created many challenges for molecular virologists, especially in keeping the virus close to wild type. Through key advances in labeling of virus for live cell microscopy and the exponential increase in fluorescence microscope technology, we are currently observing biology that was previously in the dark. Given the power of each labeling and detection technique gives scientists, not only in virology and also cellular biology, the ability to test new hypotheses, further iterations will refine what we observe and likely provide more instances where we observe biological events that are not only unknown but potentially pivotal in understanding the underlying pathogenesis to the disease.
Acknowledgement
This work was outlined and supported by project grant APP1046703 of the National Health and Medical Research Council of Australia (SGT).
References
[1] Phillips, D.M. (1995) Images in clinical medicine. Human immunodeficiency virus. N. Engl. J. Med. 332, 233.| Images in clinical medicine. Human immunodeficiency virus.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DyaK2M7gslelsQ%3D%3D&md5=8afd4cfc46c279b8af8a368fd643e3deCAS | 7808490PubMed |
[2] Orenstein, J.M. et al. (1988) Cytoplasmic assembly and accumulation of human immunodeficiency virus types 1 and 2 in recombinant human colony-stimulating factor-1-treated human monocytes: an ultrastructural study. J. Virol. 62, 2578–2586.
| 1:STN:280:DyaL1c3nsFShsw%3D%3D&md5=9e7d2faead91503f7ee8371e7489ce51CAS | 3260631PubMed |
[3] Filice, G. et al. (1987) Human immunodeficiency virus (HIV): an ultrastructural study. Microbiologica 10, 209–216.
| 1:STN:280:DyaL2s3ivVelsg%3D%3D&md5=0d55dd725e6d15c3cff43ba6c442b502CAS | 3647212PubMed |
[4] Goto, T. et al. (1988) Entry of human immunodeficiency virus (HIV) into MT-2, human T cell leukemia virus carrier cell line. Arch. Virol. 102, 29–38.
| Entry of human immunodeficiency virus (HIV) into MT-2, human T cell leukemia virus carrier cell line.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DyaL1M%2FmtFelug%3D%3D&md5=12728e8d5e063ab023fb7e37180f78d3CAS | 2904253PubMed |
[5] Grigoriev, V.B. et al. (1992) Localization by immunogold labelling of HIV-1 structural proteins on Lowicryl embedded HIV-1 infected cell ultrathin sections. J. Submicrosc. Cytol. Pathol. 24, 163–167.
| 1:STN:280:DyaK383ovVyltw%3D%3D&md5=f27fd6df8f06240f94f040f33ce4e7e6CAS | 1600507PubMed |
[6] McDonald, D. et al. (2002) Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 159, 441–452.
| Visualization of the intracellular behavior of HIV in living cells.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XoslKmtrc%3D&md5=28d464dfceb57ee1e78f33f3f47a9c2dCAS | 12417576PubMed |
[7] McDonald, D. et al. (2003) Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science 300, 1295–1297.
| Recruitment of HIV and its receptors to dendritic cell-T cell junctions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXktFGkt7o%3D&md5=2f2ae62339f4e8058da5c120b6ed0386CAS | 12730499PubMed |
[8] Paxton, W. et al. (1993) Incorporation of Vpr into human immunodeficiency virus type 1 virions: requirement for the p6 region of gag and mutational analysis. J. Virol. 67, 7229–7237.
| 1:CAS:528:DyaK2cXkvFyrsA%3D%3D&md5=b8b63df612bf6cd0e122e5ef6cefb740CAS | 8230445PubMed |
[9] Fassati, A. and Goff, S.P. (2001) Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1. J. Virol. 75, 3626–3635.
| Characterization of intracellular reverse transcription complexes of human immunodeficiency virus type 1.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXisVSnu7Y%3D&md5=eab216611740bfa61e41afb32276050eCAS | 11264352PubMed |
[10] Campbell, E.M. et al. (2007) Labeling HIV-1 virions with two fluorescent proteins allows identification of virions that have productively entered the target cell. Virology 360, 286–293.
| Labeling HIV-1 virions with two fluorescent proteins allows identification of virions that have productively entered the target cell.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXjs1Ojtrs%3D&md5=4a81a1017ae8e1454af96de6ccb6344fCAS | 17123568PubMed |
[11] Pereira, C.F. et al. (2011) Labeling of multiple HIV-1 proteins with the biarsenical-tetracysteine system. PLoS ONE 6, e17016.
| Labeling of multiple HIV-1 proteins with the biarsenical-tetracysteine system.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXisFSmsrg%3D&md5=9f8e7e987e552d9be32cef795bedd3a6CAS | 21347302PubMed |
[12] Arhel, N. et al. (2006) Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat. Methods 3, 817–824.
| Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XpvVCmtLk%3D&md5=2737111dbf694860571a435d9a6c488bCAS | 16990814PubMed |
[13] Muller, B. et al. (2004) Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative. J. Virol. 78, 10803–10813.
| Construction and characterization of a fluorescently labeled infectious human immunodeficiency virus type 1 derivative.Crossref | GoogleScholarGoogle Scholar | 15367647PubMed |
[14] Adams, S.R. et al. (2002) New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J. Am. Chem. Soc. 124, 6063–6076.
| New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xjt1Krsbs%3D&md5=0e58ee1a3810b8bd228bdd79c02f149fCAS | 12022841PubMed |
[15] Turville, S.G. et al. (2008) Resolution of de novo HIV production and trafficking in immature dendritic cells. Nat. Methods 5, 75–85.
| Resolution of de novo HIV production and trafficking in immature dendritic cells.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXht1Srtg%3D%3D&md5=cbe289b4243d868ec1d78ceff6f46441CAS | 18059278PubMed |
[16] Martin, B.R. et al. (2005) Mammalian cell-based optimization of the biarsenical-binding tetracysteine motif for improved fluorescence and affinity. Nat. Biotechnol. 23, 1308–1314.
| Mammalian cell-based optimization of the biarsenical-binding tetracysteine motif for improved fluorescence and affinity.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtVOhu7jL&md5=e59a063a832e04b3985077c81b55d0a4CAS | 16155565PubMed |
[17] Gousset, K. et al. (2008) Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog. 4, e1000015.
| Real-time visualization of HIV-1 GAG trafficking in infected macrophages.Crossref | GoogleScholarGoogle Scholar | 18369466PubMed |
[18] Aggarwal, A. et al. (2012) Mobilization of HIV spread by diaphanous 2 dependent filopodia in infected dendritic cells. PLoS Pathog. 8, e1002762.
| Mobilization of HIV spread by diaphanous 2 dependent filopodia in infected dendritic cells.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XovFSqs7c%3D&md5=fca557455c2e851463933323fda05774CAS | 22685410PubMed |
[19] Hubner, W. et al. (2007) Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J. Virol. 81, 12 596–12 607.
| Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlKku73L&md5=9534b950a023c53cc76a3e1e4924abe3CAS |
[20] Dale, B.M. et al. (2011) Cell-to-cell transfer of HIV-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion. Cell Host Microbe 10, 551–562.
| Cell-to-cell transfer of HIV-1 via virological synapses leads to endosomal virion maturation that activates viral membrane fusion.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1CrsbbE&md5=184ab01402370265b0a927fda061234aCAS | 22177560PubMed |
[21] Davis, D.A. et al. (2003) Reversible oxidative modification as a mechanism for regulating retroviral protease dimerization and activation. J. Virol. 77, 3319–3325.
| Reversible oxidative modification as a mechanism for regulating retroviral protease dimerization and activation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXhsVOgsrk%3D&md5=460153f7e7b5125f27741efd19935890CAS | 12584357PubMed |
[22] Ladinsky, M.S. et al. (2014) Electron tomography of HIV-1 infection in gut-associated lymphoid tissue. PLoS Pathog. 10, e1003899.
| Electron tomography of HIV-1 infection in gut-associated lymphoid tissue.Crossref | GoogleScholarGoogle Scholar | 24497830PubMed |
Biographies
Stuart Turville is the head of the HIV Biology laboratory at the Kirby Institute and a NHMRC CDF Research Fellow at the UNSW, Australia. His research focuses on the molecular understanding of the HIV spread between permissive cells at the early stages of transmission. Recently his laboratory is now moving towards gene therapy strategies in treating HIV infection and improving gene therapy delivery.
Anupriya Aggarwal is doing her Postdoctoral training in Dr Stuart Turville’s lab at the Kirby Institute, UNSW, Australia. She completed her PhD at University of Sydney where she studied the post entry trafficking of HSV-1 in primary neurons. Her current research focuses on understanding virus-host cytoskeletal interactions in context of HIV infection of permissive cells.