
A spectrum of (avoidable) HIV latency?
Miles DavenportComplex Systems in Biology Group
Centre for Vascular Research
University of New South Wales
Sydney, NSW 2052, Australia
Tel: +61 2 9385 2762
Fax: +61 2 9385 1797
Email: m.davenport@unsw.edu.au
Microbiology Australia 35(2) 95-96 https://doi.org/10.1071/MA14029
Published: 28 April 2014
Long-lived latently HIV-infected cells present a major barrier to the eradication of the virus under ART. Current strategies are aimed at eliminating this reservoir of cells once it is established. However, it may be easier to prevent the formation of the reservoir rather than eliminate it.
Current anti-retroviral therapies are able to effectively suppress HIV replication and reduce viral levels. However, they are unable to eliminate a pool of cells that are infected with virus, but remain dormant after infection. This pool includes different cell types, such as CD4+ T cells and macrophages, that have integrated virus, but fail to express viral proteins for a prolonged period, and are thus designated ‘latently infected’. Our current concept of latency is very much shaped by the problems caused by these extremely long-lived infected cells, persisting at fairly stable levels for many years on therapy and requiring the continuous administration of ART.
This particular view of latency has not always been the case. The observation that some cells only expressed viral antigens after a delay has been known for many years1. However, these early in vitro reports concerned the observation that there was an inducible pool of cells that were not currently producing virus that could be later reactivated. Only in the era of highly effective ART has it become clear quite how long-lived this inducible reservoir can be2.
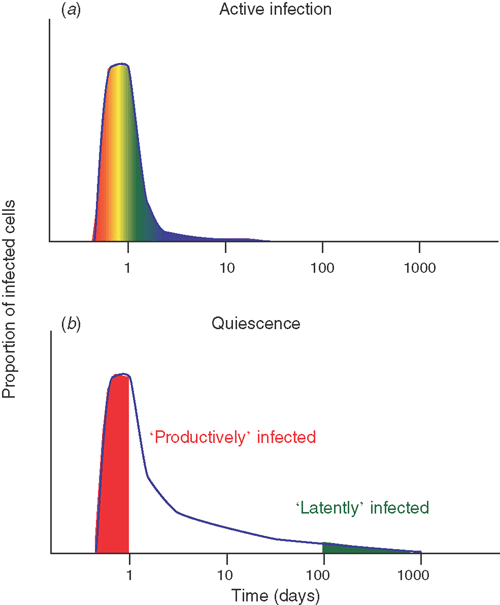
Our current understanding of latency appears heavily influenced by our observations of long-term viral persistence in the ART era. We thus tend to discuss this in terms of a dichotomous view of ‘latent’ vs ‘productively infected’ cell populations. However, studies of the dynamics of early virus infection in vitro show that infected cells undergo a wide range of ‘delays’ until they commence viral protein production3 (Figure 1). Thus, although experimental studies often measure the number of virus-producing cells at 24 hours as a measure of ‘productively infected cells’, less than half of the total cells that produce virus over the first 4 days have actually started viral production by 24 hours3. The rest of the cells have a spectrum of delays before virus production (even when only observed over only 4 days). Is long-term latency just an extreme end of this spectrum – those cells which don’t commence viral production for many years? Moreover, is this variable delay a ‘viral strategy for persistence’ (what is the benefit to virus of long-persistence in the absence of therapy?), or merely a byproduct of the virus’ need for host cell factors?
|
Separate from these short-term studies of viral production, other work has considered whether the long-lived latent cells seen under treatment are also present during active infection. Using a novel approach to identify latent virus ‘laid down’ in resting CD4+ T cells at various times after infection, it has been shown that there seems little long-lived viral DNA seen in SIV-infected macaques with active infection and high viremia4. Instead, the viral DNA seems to rapidly turn over (regularly ‘purging’ the latent pool) during active infection. However, in animals with well-controlled infection and low viral loads, SIV DNA seems to persist in the same way as observed in HIV patients on ART.
If long-lived cells aren’t present during very active infection, when do they ‘emerge’ and why do we see them during therapy? The most likely scenario seems that at the commencement of therapy, cells that would usually be short-lived in the presence of active infection instead survive and become long-lived in the environment of relative immune quiescence induced by ART5. This suggests that the period of early anti-retroviral therapy may be a unique window in which to modulate the establishment of latency.
Understanding how latency is formed, maintained and ultimately eliminated is central to many current strategies for HIV cure. If our image of latency is one of a dichotomous ‘short-lived’ vs ‘long-lived’ pool of infected cells seen under prolonged therapy, we may fail to grasp some of the important features of viral persistence. The wide diversity of delays between infection and viral production, and evidence for the ability of the environment to modify these delays (reflected as differences in viral persistence in active infection vs treatment) suggest that interventions at the start of treatment may be most successful. If our definition of latency is the existence of long-lived cells under therapy, then we will target this period of infection for intervention. However, if our goal is to modulate viral persistence, there may be many avenues to achieve this before the establishment of conventional latency.
References
[1] Leonard, R. et al. (1988) Cytopathic effect of human immunodeficiency virus in T4 cells is linked to the last stage of virus infection. Proc. Natl. Acad. Sci. USA 85, 3570–3574.| Cytopathic effect of human immunodeficiency virus in T4 cells is linked to the last stage of virus infection.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXktlKnsr0%3D&md5=99d98ab9e7ee332926d39eceffad6f9eCAS | 3259321PubMed |
[2] Ramratnam, B. et al. (2000) The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat. Med. 6, 82–85.
| The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXks1Wlug%3D%3D&md5=0399193c6855775831cf01c8baba9450CAS | 10613829PubMed |
[3] Petravic, J. et al. (2014) Intracellular dynamics of HIV infection. J. Virol. 88, 1113–1124.
| Intracellular dynamics of HIV infection.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhvFOktr0%3D&md5=0fc1fb5cdb06a2216ff302221785fbc6CAS | 24198427PubMed |
[4] Reece, J. et al. (2012) An ‘escape clock’ for estimating the turnover of SIV DNA in resting CD4+ T cells. PLoS Pathog. 8, e1002615.
| An ‘escape clock’ for estimating the turnover of SIV DNA in resting CD4+ T cells.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xlsl2qsLo%3D&md5=3de7a740452da703e444f337ff1d2a55CAS | 22496643PubMed |
[5] Strain, M.C. et al. (2003) Heterogeneous clearance rates of long-lived lymphocytes infected with HIV: intrinsic stability predicts lifelong persistence. Proc. Natl. Acad. Sci. USA 100, 4819–4824.
| Heterogeneous clearance rates of long-lived lymphocytes infected with HIV: intrinsic stability predicts lifelong persistence.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjt12nsbY%3D&md5=1e98592dc6ebe768c8c9767b80a039f2CAS | 12684537PubMed |
Biography
Miles Davenport is a Professor at UNSW in Sydney. His research team combines bioinformatic and mathematical approaches to understanding infection dynamics.