
Do pathogens contribute to multiple sclerosis aetiology?
David BoothInstitute for Immunology and Allergy Research
Westmead Millennium Institute
University of Sydney
Darcy Road, Westmead
NSW 2145, Australia
Tel: +61 2 9845 8498
Fax: +61 2 9891 3889
Email: david.booth@sydney.edu.au
Microbiology Australia 34(3) 144-146 https://doi.org/10.1071/MA13048
Published: 4 September 2013
Multiple sclerosis (MS) is a common neurological disease characterised by sclerotic plaques of dead and dying oligodendrocytes and neurons in the central nervous system, usually with evidence of accompanying inflammatory activity. Infectious agents might cause or contribute to this cell death, but none have yet been established as doing so. Instead, MS is regarded as predominantly an autoimmune disease, responsive to immunomodulators. However, there is compelling epidemiological and empirical evidence implicating Epstein–Barr virus (EBV), with minor support for other viruses, as contributing to the pathogenic immune response in MS. Now the recent dramatic advances in identifying the genetic risk factors for MS have provided some tantalising leads to microbe involvement. If conclusive evidence is found, vaccines, antivirals/antibiotics or pathogens themselves might prove useful therapeutics.
Do pathogens contribute to MS aetiology?
Multiple sclerosis is one of the most common neurological diseases of young adults. It is primarily an inflammatory disorder of the brain and spinal cord in which focal lymphocytic infiltration leads to damage of myelin and axons. It is triggered by environmental factors, especially in individuals with genetic susceptibility1. Many of the genetic variants increasing risk have now been identified2,3, and these point to variation in cell mediated immunity as driving susceptibility. These genes also potentially point to specific pathogens.
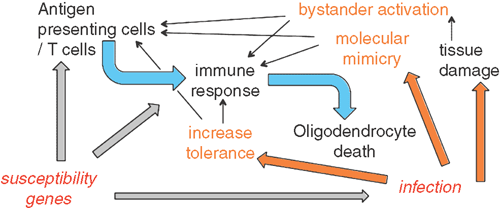
There is empirical and epidemiological evidence that pathogens could be environmental triggers, but the distinction between association and causation is problematic. Two likely mechanisms for causation have been described: bystander activation, where self-reactive T cells are activated due to tissue damage by a pathogen; and molecular mimicry, where T cells that recognize pathogen epitopes are also self-reactive and are activated by the pathogens (Figure 1). Alternatively, pathogens might be protective in autoimmune disease, by inducing tolerance4.
|
The sudden arrival and subsequent epidemics of MS in the Faroe Islands during and after WWII was most easily explained as facilitated by the arrival of British troops and accompanying pathogens in 19435. But if MS here and elsewhere was caused by an infection, the pathogen has remained elusive. From the geographic and ethnic distribution, the pathogen is likely to be ubiquitous in western countries, but also in Asia and elsewhere (because MS is described worldwide), infect children (because MS risk reflects childhood environment), cause chronic infection (because the disease is usually late onset and persistent) and is more likely in the higher latitudes (because MS prevalence increases with latitude).
The strongest candidate to date is EBV, which fulfils all these criteria6. Although typically 90% of the general population carry EBV, virtually 100% of people with MS do. Notably, though, other common chronic viruses, such as HHV6, chicken pox, measles and cytomegalovirus have also been described as more prevalent in MS7, such that exposure to these viruses might also render people susceptible to MS and the virus is not causative but associative8. More compellingly, late infection or severe infection with EBV increases risk of MS by up to 20-fold9, an increase not yet found for other pathogens. People with MS have poor CD8 responses to EBV6, T cells recognizing myelin proteins can be activated by EBV10, high titres of EBV antibodies are associated with relapses and paediatric MS11. A prospective study of 915 individuals found the 10 who were EBV negative and later developed MS were all EBV positive before they developed MS, whereas of the 28 EBV negative controls only 35% became EBV positive over the same time period12. High EBV antibody titres, in combination with the first MS susceptibility gene, and variant of largest effect yet known, HLA DRB1*1501, increases the risk of MS, additively and independently, not interactively13. Oligoclonal bands found in the CSF of many with MS can be reactive to EBV and other herpesviruses14.
EBV infects B cells through CD216, and possibly CD3515. Both of these genes are not among those known to affect susceptibility to MS. The infected B cells proliferate through signaling through EBVs own analogue of human CD40. Strikingly, genetic variants of CD40 are associated with MS, and the genotype with higher expression of CD40 decreases MS risk16. Because CD40 is a costimulatory molecule required for T cell activation, it would be expected that higher expression would increase the risk of MS. A potential explanation for this paradox is that low host CD40 expression on B cells favours proliferation/survival of EBV infected B cells, using their EBV CD40. In support of this, anti-CD20 B-cell-depleting drugs such as Rituximab, which are used as a therapies to reduce EBV infection17, are effective in MS, but those targeting terminally differentiated B cells are not18. The effect of more specific depletion of EBV infected B cells on MS needs to be investigated.
Genome wide analysis studies have implicated specific viruses for some autoimmune conditions. Individuals homozygous for the Crohn’s diseases risk allele rs601338 of FUC2, the receptor for noroviruses, are protected against norovirus infection19, but are more likely to develop Crohn’s and other autoimmune diseases such as type 1 diabetes and inflammatory bowel disease20, consistent with the ‘hygiene hypothesis’4. The MS risk gene TNFSF14 (also known as herpesvirus entry mediator ligand or LIGHT)2,3 is an entry receptor for Herpes simplex 1 (HSV1)21, a neurotropic virus. A ligand for TNFSF14, TNFRSF6B, is also a MS risk gene. SLAM family proteins are used by morbilliviruses to facilitate cell entry22. These viruses then circumvent the interferon response by blocking Tyk2 signalling23. Both SLAMF7 and TYK2 are risk genes for MS3. Measles and canine distemper virus, both implicated in the Faroe Island MS ‘epidemics’, are morbilliviruses. Further viral receptors and genes important in their infection might be in the list of MS genes. But as TNFSF14/TNFRSF6B and SLAMF7/TYK2 also have roles affecting other aspects of the immune response, further implication of a HSV1/morbillivirus contribution to MS might follow if the MS susceptibility genotype can be shown to increase tissue damage or other infectious consequences due to these viruses.
The genes affecting MS susceptibility support a role for immune response in MS, but although they, as yet, provide no smoking gun for particular viruses, the evidence that most of these genes are under recent positive selection24 is consistent with the genetic variants affecting selection against pathogens. Most of these genes are also predominantly expressed in T cells or antigen-presenting cells, consistent with a role for molecular mimicry or bystander activation contributing to disease aetiology, but also with excessive reaction to self antigen independently of pathogen effect.
A ‘smoking gun’ for one environmental trigger was found. This is vitamin D. Low vitamin D has been implicated as contributing to MS development and progression from epidemiological and empirical studies25. It now seems that genetic variants of the two genes regulating its activation in antigen presenting cells, CYP27B1 and CYP24A1, affect MS risk2. As well as regulating DC tolerisation, vitamin D can be antimicrobial, through its upregulation of cathellicidin6,26.
Current successful therapies for MS alter leukocyte survival, modulation or trafficking18. Further manipulation of the immune response might be beneficial if a microbial effect on MS aetiology is defined. Vaccination might be a useful strategy whether viruses such as EBV increase susceptibility (especially if late infection) or protect (especially if early infection) against MS27. This might include use of the pathogen itself28. To support such approaches, experimental studies are needed to determine if genes associated with MS function through their effect on infections. Novel approaches to establishing if viruses are pathogenic include sequencing of non-host DNA/RNA, especially in plaques, and amplified endogenous retroviruses. Such studies could use historic samples from the Faroe islands and elsewhere. If pathogens contribute to aetiology, even if in only a subset of patients, it is highly likely that their identification will provide significant clinical benefits.
References
[1] Compston, A. and Coles, A. (2008) Multiple sclerosis. Lancet 372, 1502–1517.| Multiple sclerosis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXht12gtLvN&md5=6eb26b8814e57f610c9e82202d4ed3a5CAS | 18970977PubMed |
[2] International Multiple Sclerosis Genetics Consortium (2011) Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature 476, 214–219.
| Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis.Crossref | GoogleScholarGoogle Scholar | 21833088PubMed |
[3] McCauley J. and International Multiple Sclerosis Genetics Consortium (2012) Immunochip: redefining the genetic architecture of multiple sclerosis (abstract 151). Presented at the 62nd Annual Meeting of The American Society of Human Genetics, San Francisco, California.
[4] Kivity, S. et al. (2009) Infections and autoimmunity – friends or foes? Trends Immunol. 30, 409–414.
| Infections and autoimmunity – friends or foes?Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXps1Wqu74%3D&md5=94b8148fc9aa0e66eabe0499a5962366CAS | 19643667PubMed |
[5] Kurtzke, J.F. and Heltberg, A. (2001) Multiple sclerosis in the Faroe Islands: an epitome. J. Clin. Epidemiol. 54, 1–22.
| Multiple sclerosis in the Faroe Islands: an epitome.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BD3M7lsFaltg%3D%3D&md5=53e57e6c1dc016c205a54d800b802a5fCAS | 11165464PubMed |
[6] Pender, M.P. (2012) CD8+ T-cell deficiency, Epstein-Barr virus infection, vitamin D deficiency, and steps to autoimmunity: a unifying hypothesis. Autoimmune Dis. , 189096.
| 22312480PubMed |
[7] Waubant, E. et al. (2011) Common viruses associated with lower pediatric multiple sclerosis risk. Neurology 76, 1989–1995.
| Common viruses associated with lower pediatric multiple sclerosis risk.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BC3MjgvV2guw%3D%3D&md5=4432a1c2f189c961cf3413806b16376eCAS | 21646624PubMed |
[8] Pakpoor, J. et al. (2013) Epstein–Barr virus and multiple sclerosis: association or causation? Expert Rev. Neurother. 13, 287–297.
| Epstein–Barr virus and multiple sclerosis: association or causation?Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXjtlGru7w%3D&md5=fe11bd0dc79777c172491e77cb93c8edCAS | 23448218PubMed |
[9] Tselis, A. (2012) Epstein–Barr virus cause of multiple sclerosis. Curr. Opin. Rheumatol. 24, 424–428.
| Epstein–Barr virus cause of multiple sclerosis.Crossref | GoogleScholarGoogle Scholar | 22617821PubMed |
[10] Lünemann, J.D. et al. (2008) EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-γ and IL-2. J. Exp. Med. 205, 1763–1773.
| EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-γ and IL-2.Crossref | GoogleScholarGoogle Scholar | 18663124PubMed |
[11] Lünemann, J.D. et al. (2008) Broadened and elevated humoral immune response to EBNA1 in pediatric multiple sclerosis. Neurology 71, 1033–1035.
| Broadened and elevated humoral immune response to EBNA1 in pediatric multiple sclerosis.Crossref | GoogleScholarGoogle Scholar | 18809840PubMed |
[12] Levin, L.I. et al. (2010) Primary infection with the Epstein–Barr virus and risk of multiple sclerosis. Ann. Neurol. 67, 824–830.
| 20517945PubMed |
[13] De Jager, P.L. et al. (2008) Integrating risk factors: HLA-DRB1*1501 and Epstein–Barr virus in multiple sclerosis. Neurology 70, 1113–1118.
| Integrating risk factors: HLA-DRB1*1501 and Epstein–Barr virus in multiple sclerosis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXjtlKju7g%3D&md5=420c530c4a6ec1f4bac9ac1ea168e3afCAS | 18272866PubMed |
[14] Virtanen, J. et al. (2013) Oligoclonal bands in multiple sclerosis reactive against two herpesviruses and association with magnetic resonance imaging findings. Mult. Scler. , .
| Oligoclonal bands in multiple sclerosis reactive against two herpesviruses and association with magnetic resonance imaging findings.Crossref | GoogleScholarGoogle Scholar | 23722324PubMed |
[15] Ogembo, J.G. et al. (2013) Human complement receptor type 1/CD35 is an Epstein–Barr virus receptor. Cell Rep. 3, 371–385.
| Human complement receptor type 1/CD35 is an Epstein–Barr virus receptor.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXis1Olt70%3D&md5=6ebbb3bd1fdea0a31f457d5a17b241e4CAS | 23416052PubMed |
[16] Gandhi, K.S. et al. (2010) The multiple sclerosis whole blood mRNA transcriptome and genetic associations indicate dysregulation of specific T cell pathways in pathogenesis. Hum. Mol. Genet. 19, 2134–2143.
| The multiple sclerosis whole blood mRNA transcriptome and genetic associations indicate dysregulation of specific T cell pathways in pathogenesis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXlvVWktro%3D&md5=b60fe350b3bdbbc94bb1e0acae7689e8CAS | 20190274PubMed |
[17] Cohen, J.I. et al. (2011) Characterization and treatment of chronic active Epstein–Barr virus disease: a 28-year experience in the United States. Blood 117, 5835–5849.
| Characterization and treatment of chronic active Epstein–Barr virus disease: a 28-year experience in the United States.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnsl2nur8%3D&md5=35de620d6e18152268a9598825497720CAS | 21454450PubMed |
[18] Buzzard, K.A. et al. (2012) What do effective treatments for multiple sclerosis tell us about the molecular mechanisms involved in pathogenesis? Int. J. Mol. Sci. 13, 12 665–12 709.
| What do effective treatments for multiple sclerosis tell us about the molecular mechanisms involved in pathogenesis?Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhvVaqtbjE&md5=d2d1d67475f00e1482ac6eb9bacd39bfCAS |
[19] Carlsson, B. et al. (2009) The G428A nonsense mutation in FUT2 provides strong but not absolute protection against symptomatic GII.4 norovirus infection. PLoS ONE 4, e5593.
| The G428A nonsense mutation in FUT2 provides strong but not absolute protection against symptomatic GII.4 norovirus infection.Crossref | GoogleScholarGoogle Scholar | 19440360PubMed |
[20] Smyth, D.J. et al. (2011) FUT2 nonsecretor status links type 1 diabetes susceptibility and resistance to infection. Diabetes 60, 3081–3084.
| FUT2 nonsecretor status links type 1 diabetes susceptibility and resistance to infection.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhsVyis7zN&md5=6cf80569daacea7899e27368290bb059CAS | 22025780PubMed |
[21] Mauri, D.N. et al. (1998) LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 8, 21–30.
| LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXptVOjsg%3D%3D&md5=9cf1f83a02f3a76facbd281a06564320CAS | 9462508PubMed |
[22] Erlenhoefer, C. et al. (2001) CD150 (SLAM) is a receptor for measles virus but is not involved in viral contact-mediated proliferation inhibition. J. Virol. 75, 4499–4505.
| CD150 (SLAM) is a receptor for measles virus but is not involved in viral contact-mediated proliferation inhibition.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BD3M3it1ejsQ%3D%3D&md5=53ef963c091dd8e249aef8c3828b4395CAS | 11312320PubMed |
[23] Chinnakannan, S.K. et al. (2013) Morbillivirus v proteins exhibit multiple mechanisms to block type 1 and type 2 interferon signalling pathways. PLoS ONE 8, e57063.
| Morbillivirus v proteins exhibit multiple mechanisms to block type 1 and type 2 interferon signalling pathways.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXjsF2jsrs%3D&md5=713e8d64056bbe48e66589a7ebed568dCAS | 23431397PubMed |
[24] Raj, T. et al. (2013) Common risk alleles for inflammatory diseases are targets of recent positive selection. Am. J. Hum. Genet. 92, 517–529.
| Common risk alleles for inflammatory diseases are targets of recent positive selection.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXksFyjsLo%3D&md5=8fb49781af5a13cea977f71383801c17CAS | 23522783PubMed |
[25] Hewer, S. et al. (2013) Vitamin D and multiple sclerosis. J. Clin. Neurosci. 20, 634–641.
| Vitamin D and multiple sclerosis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXltVemu7g%3D&md5=8ae4a92c2ebf3dd0a0b839a2f5983669CAS | 23540892PubMed |
[26] Muehleisen, B. et al. (2012) PTH/PTHrP and vitamin D control antimicrobial peptide expression and susceptibility to bacterial skin infection. Sci Transl Med. 4, 135ra66.
| PTH/PTHrP and vitamin D control antimicrobial peptide expression and susceptibility to bacterial skin infection.Crossref | GoogleScholarGoogle Scholar | 22623742PubMed |
[27] Loebermann, M. et al. (2012) Vaccination against infection in patients with multiple sclerosis. Nat Rev Neurol. 8, 143–151.
| Vaccination against infection in patients with multiple sclerosis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xjs1Ckt74%3D&md5=048e759bfc5716e4c7fbeec6d278062fCAS | 22270022PubMed |
[28] Hasseldam, H. et al. (2013) Immunomodulatory effects of helminths and protozoa in multiple sclerosis and experimental autoimmune encephalomyelitis. Parasite Immunol. 35, 103–108.
| Immunomodulatory effects of helminths and protozoa in multiple sclerosis and experimental autoimmune encephalomyelitis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXit1Wnsb4%3D&md5=d524121e7229ae6d607cdfc802cba443CAS | 23227936PubMed |
Biography
David Booth is an Associate Professor and the Hunt Family Senior MS Research Fellow of the University of Sydney.