

Determination of inorganic As, DMA and MMA in marine and terrestrial tissue samples: a consensus extraction approach
Zuzana Gajdosechova A * , Patricia Grinberg A , Kevin Kubachka B , Mesay Wolle C , Andrea Raab D , Joerg Feldmann D , Rebecca Sim E F , Ásta H. Pétursdóttir E , Tomáš Matoušek G , Stanislav Musil G , Ben Wozniak H , Stephen Springer H , Nausheen W. Sadiq H , Hakan Gurleyuk H , Calvin H. Palmer A , Indumathi Pihillagawa Gedara A and Zoltan Mester A
A * , Patricia Grinberg A , Kevin Kubachka B , Mesay Wolle C , Andrea Raab D , Joerg Feldmann D , Rebecca Sim E F , Ásta H. Pétursdóttir E , Tomáš Matoušek G , Stanislav Musil G , Ben Wozniak H , Stephen Springer H , Nausheen W. Sadiq H , Hakan Gurleyuk H , Calvin H. Palmer A , Indumathi Pihillagawa Gedara A and Zoltan Mester A
A National Research Council Canada, Metrology, 1200 Montreal Road, Ottawa, ON K1A 0R6, Canada.
B Forensic Chemistry Center, Office of Regulatory Science, Office of Regulatory Affairs, U.S. Food and Drug Administration, Cincinnati, OH 45237, USA.
C Division of Bioanalytical Chemistry, Office of Regulatory Science, Center for Food Safety and Applied Nutrition, US Food and Drug Administration, 5001, Campus Drive, College Park, MD 20740, USA.
D Institute for Chemistry, TESLA - Analytical Chemistry, University of Graz, Universitätsplatz 1/I, 8010, Graz, Austria.
E Matís, Research and Innovation, Vinlandsleid 12, 113, Reykjavik, Iceland.
F University of Iceland, School of Engineering and Natural Sciences, Dunhagi 3, 107 Reykjavik, Iceland.
G Institute of Analytical Chemistry of the Czech Academy of Sciences, Veveří 97, 602 00, Brno, Czech Republic.
H Brooks Applied Labs, 18804 North Creek Parkway, Suite 100, Bothell, WA 98011, USA.
Environmental Chemistry 20(2) 5-17 https://doi.org/10.1071/EN23006
Submitted: 13 January 2023 Accepted: 3 April 2023 Published: 4 May 2023
© 2023 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Environmental context. Arsenic can be found in all environmental compartments in a large number of chemical forms of varying toxicity. We performed an inter-laboratory comparison study focusing on quantitation of some of the most toxic arsenic forms in seven different biological materials and found very good agreement among the submitted results. Certification of the studied materials will provide suitable quality control samples for environmentally relevant concentrations of arsenic in food products and biota.
Rationale. Arsenic (As) speciation analysis in biological matrices has been performed for several decades; however, there are very few matrix certified reference materials available for the validation of analytical methods. The literature data on the mass fractions of As species in the existing certified reference materials are inconsistent and suggest method extraction dependency.
Methodology. In the present study, an international round-robin study was organised to identify the possible sources of discrepancies in quantitation of several As species in different matrices of biological reference materials: one plant tissue, three marine and three terrestrial biological tissues. Each participating laboratory was provided with a set of identical calibration standard solutions, and analysed the samples by following a common group extraction method as well as using an in-house protocol.
Results. The results showed that significant biases can be introduced by insufficient verification of the analyte’s mass fractions in the calibration standard solutions. The choice of extraction method seems to have very little impact on the quantitation of As species in the studied plant and terrestrial biological tissues. However, following a prescribed extraction method led to significant reduction of uncertainties in more complex samples such as marine animal tissues.
Discussion. Some differences in the mass fractions of As species extracted from marine animal tissues in water (with and without H2O2) were observed and should be further investigated. Despite the variety of extraction methods used, very good agreement between reported mass fractions was achieved and the combined consensus values will be published as certified reference values of As species in the studied materials.
Keywords: Arsenic speciation, certified reference material, extraction method, high performance liquid chromatography, hydride generation, inorganic arsenic, marine tissue, mass spectrometry.
Introduction
The toxicity of As depends on its oxidation state but also whether it is present in its organic or inorganic chemical forms. In general, trivalent arsenic compounds are more toxic than pentavalent forms, and inorganic species are more toxic than organic species (Hughes 2002). However, a few studies have observed that trivalent monomethylarsonous acid (MMAIII) exhibits higher cytotoxicity than trivalent inorganic As (iAs) (Petrick et al. 2000; Styblo et al. 2000). Within the group of organoarsenic species, the toxicity generally decreases with increasing degree of methylation (Leermakers et al. 2006). For instance, although still potentially carcinogenic, studies have suggested the toxicity of pentavalent dimethylarsinic acid (DMAV) is lower than pentavalent monomethylarsonic acid (MMAV) (Irvine et al. 2006). Organic compounds such as arsenobetaine (AsBet), arsenocholine (AsChol) and As containing sugars (arsenosugars) are considered non-toxic (Kaise et al. 1985; Francesconi 2010; Luvonga et al. 2020); however, some arseno-lipids (As-lipids), e.g. arseno-hydrocarbons, have shown considerable cytotoxicity and an ability to cross the blood–brain barrier in some studies (Meyer et al. 2014a, 2014b; Müller et al. 2018).
The current regulations of maximum levels of As in food are trying to address this complexity by targeting specific As species. For instance, the maximum allowable level of As in rice was changed from total As to inorganic arsenic (iAs (sum of AsIII and AsV)) levels (EU 2015; USFDA 2016b; Health Canada 2021), which have been shown to vary greatly among rice samples surveyed in the literature (Bralatei et al. 2015; USFDA 2016a; Menon et al. 2020). However, in order for regulatory maximum levels to be established for foods, laboratories must have the capability to measure the contaminant accurately. For this purpose, several proficiency test and round-robin studies for iAs in rice were organised (de la Calle et al. 2012; Pétursdóttir et al. 2014a; Cordeiro et al. 2016). Depending on the specific design of the study, the general purpose of a round-robin study is to determine the reproducibility of a tested method and/or verify a new analytical method through an interlaboratory comparison. The consistency in the reported values of iAs in rice studies led to the conclusion that quantitation of iAs in rice is achievable, independent of the analytical methods evaluated, thus regulation of iAs is feasible (de la Calle et al. 2011). However, the quantitation of iAs in other foodstuffs, such as seafood, is proving to be more challenging (Baer et al. 2011; Fiamegkos et al. 2016). While As in rice is usually distributed between DMA and iAs, marine animal tissues and algae have been shown to contain larger numbers of organic As species.
Taking into consideration the differences in the toxic properties of organic and inorganic As species, method extraction efficiencies, chromatographic recoveries and species inter-conversion may complicate the quantitation of iAs in biological matrices. Matrix certified reference materials (CRMs) can serve as powerful quality control tools and are commonly used for accuracy evaluation of analytical methods. Although a large number of CRMs are available on the market for total As content, very few are certified for As species other than AsBet. The limited choices consist primarily of plant tissue matrices such as rice and seaweed. There are no available CRMs with certified iAs in animal tissue, whether it is of a terrestrial or marine origin. In such instances, speciation data on CRMs published in the peer-reviewed literature are often used as information values. However, the published data of major As species (MMA, DMA and iAs) showed very little agreement between the values in dogfish muscle (DORM-2), lobster hepatopancreas (TORT-2) and fish protein (DOLT-4) CRMs from the National Research Council Canada (NRC). For example, in DORM-2, the reported values (mg kg−1) of iAs ranged between 0.004 ± 0.001 (Wang et al. 2007) and 0.64 ± 0.49 (Hirata et al. 2006), DMA between 0.16 ± 0.03 (Karthikeyan and Hirata 2004) and 0.66 ± 0.04 (Pizarro et al. 2003) and MMA between 0.015 ± 0.004 (Wahlen et al. 2004) and 0.31 ± 0.12 (Karthikeyan and Hirata 2004). Similar inconsistencies have been reported for iAs in TORT-2 (ranging between 0.186 ± 0.03 and 4.46 ± 0.03 mg kg−1) (Pétursdóttir et al. 2014b) and DOLT-4 (ranging between 0.011 ± 0.002 and 0.253 ± 0.019 mg kg−1) (Llorente-Mirandes et al. 2017). Clearly, there is a large inconsistency in the published values of As species in the selected CRMs and they should not be used as indicators of trueness of the results without rigorous verification.
The aim of the present study was to identify possible sources of discrepancies in measured mass fractions of iAs, DMA and MMA in different matrix CRMs (one plant tissue, three marine and three terrestrial biological tissues) from the NRC and define a simple and reproducible extraction protocol for several As species. The CRMs were certified for total As but not for As species. For this purpose, a round-robin study was designed with an international team of researchers in As speciation analysis from eight laboratories.
Experimental
Round-robin campaign design
NRC Canada supplied each laboratory with existing NRC CRMs, i.e. three marine biological tissues [DORM-5 (fish protein) (Grinberg et al. 2021), DOLT-5 (dogfish liver) (Yang et al. 2014) and TORT-3 (lobster hepatopancreas) (Willie et al. 2013)], one plant material [CAME-1 (canola meal – Brassica napus) (Grinberg et al. 2023a)] and three terrestrial biological tissues [KRIK-1 (cricket powder – Gryllodes sigillatus) (LeBlanc et al. 2023a), BFLY-1 (de-fatted black soldier fly larvae, Hermetia illucens) (Grinberg et al. 2023b) and VORM-1 (mealworm meal – Tenebrio molitor) (LeBlanc et al. 2023b)]. Besides the CRMs, NRC also provided, to each laboratory, standard solutions of MMA, DMA and AsV of an accurate determined concentration, as described later. Samples were extracted using a common extraction method selected by the participating researchers, called the group extraction method (GEM), and each laboratory was also asked to use their in-house extraction method. Each laboratory used the analytical instrument of their choice for the As species measurement.
Dry weight determination
Accurately weighed 0.5 g aliquots of each CRM were placed in a vacuum oven in triplicate, and the oven was subsequently evacuated and maintained below 25 mm Hg to dry the sample to a constant mass. Samples were left in the vacuum until a stable final dry weight was reached. Once dried, the mass loss was determined for the individual CRMs.
Sample extraction methods
The conditions for the GEM were agreed by the participating laboratories with an aim to reflect the current knowledge of As species integrity under various extraction conditions and simplicity, so the method could be applied by a large and diverse number of laboratories. Accurately weighed 0.25 g samples were extracted in 10 mL of 1% v/v H2O2 (prepared by diluting 16.6 mL of 30% H2O2 to a final volume of 500 mL) in deionised water using a hot block which was pre-heated. The extraction temperature was set to 95°C in the extraction solution and a 60 min timer was started when the monitored samples placed in the hot block reached 95°C. Following the extraction, samples were centrifuged at 3000×g and the supernatant was analysed for As species by the instrumentation of choice (Table 1). In addition, six participating laboratories carried out another preparation of the CRMs using their preferred, in-house extraction method (Table 1).
| Lab | In-house extraction method | Analytical instrumentation | Calibration method |
| Lab 1 | 0.25 g + 10 mL of 1% H2O2, heated in water bath at 95°C for 60 min; centrifuged after extraction | HPLC-ICP-MS/MS | SA/EC |
| Lab 2 | 0.25 g of sample + 15 mL of water, heated at 90°C for a total of 75 min; centrifuged and filtered using PDVF 2-stage filters after extraction | HPLC-ICP-MS/MS | EC |
| Lab 3 | 0.25 g sample + 10 mL of water, heated in hot block at 90°C for 30 min with a ramp time of 45 min; centrifuged and filtered through a 0.45 μm pore size PVDF filter after extraction | HPLC-ICP-MS | EC |
| Lab 4 | 0.5 g sample + 10 mL 0.02 M TFA + 1% H2O2, heated in water bath at 95°C, 60 min (shaking); centrifuged after extraction | HPLC-ICP-MS/MS | EC |
| Lab 5 | 0.2 g + 10 mL of 2% HNO3 and 3% H2O2, heated in quartz digestion tubes using an Ultrawave digestion system (10 min ramp to 90°C and 50 bar, held for 30 min); centrifuged after extraction | HPLC-ICP-MS | EC |
| Lab 6 | Same as group extraction methodA | HG-CT-ICP-MS/MSB | EC |
| Lab 7 | Same as group extraction methodA | HPLC-HG-ICP-MS/MSC | EC |
| Lab 8 | 0.5 g + 10 mL of 0.4% HNO3 and 3% H2O2, heated and stirred at 90°C for 60 min; filtered via 0.45 µM PES filters after extraction | HPLC-ICP-MS | EC |
ALabs 6 and 7 shared the same extract.
BFor details see Matoušek et al. (2017).
CFor details see Marschner et al. (2016, 2019).
PVDF, polyvinylidene difluoride membrane; TFA, trifluoroacetic acid; HG, hydride generation; CT, cryo-trapping; EC, external calibration; SA, standard addition; PES, polyethersulfone membrane; HPLC, high performance liquid chromatography; ICP-MS, inductively coupled plasma–mass spectromtery.
Considering that the analysed samples were measured by high performance liquid chromatorgaphy (HPLC) where significant dilution occurs, the differences between sample and calibration standard matrices became negligible and it was not pertinent to prescribe the calibration method. Regardless, Lab 1 carried out quantitation using both external calibration and standard addition methods. The details of the chromatographic conditions are listed in Supplementary Table S1 and a description of chemicals used by participating laboratories can be found in the Supplementary material.
Calibration standards preparation
Solutions of MMA and DMA, respectively, were prepared by dissolving salts of disodium monomethyl arsenic acid (purity 99%, Sigma Aldrich) and cacodylic acid (purity 99%, Sigma Aldrich) in water to an approximate As mass fraction of 20 mg kg−1. Similarly, the AsV standard solution was prepared by dilution of a 1000 mg kg−1 stock solution (Inorganic Ventures) to approximately 20 mg kg−1. The exact mass fraction of As in each stock solution was determined against NRC HIAS-1 high purity As CRM (Methven et al. 2020). For this purpose, 1 mL of the MMA and DMA standard solutions were digested in 6 mL of concentrated HNO3 in five replicates using a Multiwave 7000 digestion system, with a 15 min ramp to 200°C and held at 200°C for 15 min. The digested solution of MMA and DMA, together with the solution of AsV, were measured as unknown samples using an Agilent 5110 ICP-OES (Agilent Technologies, Canada). Each participant was provided with 10 mL of each standard solution.
Total As determination
Accurately weighed 0.25 g samples were digested in 7 mL of concentrated HNO3 and 0.5 mL of 30% H2O2 using a Multiwave 3000 microwave system (Anton Paar) set to 1400 W with a 15 min ramp and a 30 min holding time. Two procedural blanks and CRM were included in each digestion cycle. After cooling, the digests were transferred into Teflon tubes and evaporated to approximately 1 mL. The final residues were quantitatively transferred to 25 mL polyethylene bottles and diluted to 25 g with water. All samples were analysed using two inductively coupled plasma–mass spectrometry (ICP-MS) instruments, an Element XR (ThermoFisher Scientific Inc., Bremen, Germany) and an Agilent 8900 (Agilent Technologies, Santa Clara, CA, USA). On the Element XR, As was measured at mass 75 using high resolution, while on the Agilent 8900, O2 was used as a reactive gas to mass shift As from m/z 75 to 91. Quantitation was performed by one-point standard addition.
Consensus value assignment
Although the ISO document ‘Guide to the expression of uncertainty in measurement’ (JCGM 1995) states that the uncertainty associated with a measured value should reflect the contributions of all sources of uncertainty; the uncertainty provided by laboratories is often underestimated.
Consensus values were obtained using the NIST Consensus Builder and Hierarchical Bayes (Laplace) as the method of analysis (Koepke et al. 2020). The NIST Consensus Builder combines measurement results provided by participating laboratories into a consensus estimate. The consensus estimate calculates the measurement uncertainty comprised of stated uncertainties associated with the individual measured values and any additional component of uncertainty that manifests itself only when these measured values are inter-compared. It uses a random effects model and is capable of detecting, evaluating and propagating an uncertainty component that accounts for the mutual inconsistency of the measurement results, called dark uncertainty (Thompson and Ellison 2011).
For detailed chromatograms, chromatographic conditions, summary of measured mass fraction and databases containing mass fractions from each participating laboratory please see the Supplementary material.
Results and discussion
Extraction method
The reviewed publications (Pizarro et al. 2003; Karthikeyan and Hirata 2004; Wahlen et al. 2004; Hirata et al. 2006; Wang et al. 2007; Pétursdóttir et al. 2014b; Llorente-Mirandes et al. 2017) of As species mass fractions in the CRMs suggested that quantitation of As species in biological tissues is likely extraction method dependent. In a previous study, the results from proficiency tests of iAs quantitation in rice (de la Calle et al. 2011) showed that if equal extraction efficiency is achieved among labs then, regardless of the analytical instrumentation used for the detection, agreement among values can be found. Therefore, when designing the present study, one GEM was established which was followed by each participant. A large number of extraction methods have been described in the literature and some rigorous testing has been performed on CRMs and biological tissues (Pétursdóttir et al. 2014b; Wolle and Conklin 2018b). Some methods are able to extract a larger proportion of total As; however, it may be at the expense of As species integrity (Wolle and Conklin 2018a). Our preliminary experiments using marine animal tissue (data not shown) showed a linear relationship between the degradation of phosphate arsenoribose (arsenosugar-482) and extraction time when performed under acidic conditions. Similarly, with increasing extraction time, an increase in DMA mass fraction was recorded; most likely the result of arsenosugars degradation (data not shown). An extensive study of extraction methods on a large number of biological tissues showed that acidic (HNO3, HCl) and alkali (TMAH) solutions have a higher extraction recovery for total As than water under the same heating conditions (Wolle and Conklin 2018a). However, further investigation of As species stability under acidic and alkali extraction conditions showed degradation of the arsenosugars, which was also observed in other studies (Gamble et al. 2003; Foster et al. 2007; Narukawa et al. 2012; Wolle and Conklin 2018a). Additionally, degradation of non-polar As species into DMA, AsChol and other unknown compounds was also observed when the lipid fraction was extracted in acidic or alkali solution (Wolle and Conklin 2018b). Importantly, no degradation of arsenosugars or lipids was observed when extractions were performed in water (Wolle and Conklin 2018a). Therefore, the GEM extraction was carried out in water. To eliminate binding of AsIII to thiols, which may hinder the extraction process, as well as to facilitate oxidation of AsIII to AsV, the extraction water contained 1% H2O2.
Our preliminary experiments also have shown a higher extraction yield when the extraction was carried out in vials of larger diameter, i.e. 100 mL versus 18 mL microwave vessels (data not shown). At the set extraction temperature (95°C), the samples are not boiling, hence there is no continuous agitation of the extraction mixture. Therefore, the GEM used by all participants employed 50 mL vials which have a distinctly larger diameter than the 15 mL alternative. The GEM did not prescribe filtration of the supernatants; however, some participants filtered the extracts. As can be seen in Table 2, no considerable difference between the mass fraction of filtered and unfiltered samples was observed.
| Condition tested | n | Range |
| 0.45 µm PES | 5 | 0.0160–0.020 |
| 0.45 µm PVDF | 2 | 0.0176–0.0179 |
| 0.2 µm Nylon | 2 | 0.0181–0.0183 |
| Unfiltered | 2 | 0.0178–0.0181 |
PES, polyethersulfone membrane; PVDF, polyvinylidene difluoride membrane.
For the in-house extraction methods, each participating lab selected a method routinely used for analysis of similar samples. Lab 1 used 1% H2O2 in water, Lab 2 and 3 used water without H2O2, Lab 4 used 20 mM TFA with 1% H2O2 and Lab 5 used 2% HNO3 with 3% H2O2. Lab 8 closely followed an established CEN method for the determination of iAs in foodstuffs (European Committee for Standardization 2016) which also prescribed a mixture of HNO3 and H2O2 but with a lower HNO3 concentration (0.4%) (Table 1). The extraction apparatus used were water bath (Lab 1 and 4), hot block (Lab 2, 3 and 8) and microwave using 18 mL vessels (Lab 5).
Quantitation method
The choice of quantitation method and quality of calibration standards can have significant impact on the accuracy of the results. Lab 1 carried out quantitation using both external calibration and standard addition methods and reported no significant differences between the calculated values. However, the commercially available salts of As species for preparation of calibration standard solutions can be of varying purity and their exact concentration and purity should be verified using National Metrology Institute (NMI) traceable standards. To determine an exact concentration, the calibration standards were digested in closed vessels to achieve complete oxidation of As species to AsV. It was previously observed, although not widely discussed in the literature, that the ICP-MS response differs among individual As species (Narukawa et al. 2006, 2007). Therefore, to avoid any systematic errors caused by calibration standards, each participant was provided with identical calibration standard solutions of DMA, MMA and AsV prepared in water. One participant reported values quantified by NRC provided calibration standards as well as by their own calibration standards. Both results, from group and in-house extraction methods, were higher when quantified using NRC provided calibration stock standards (data not published). The systematic under-estimation can be explained by an incorrect As mass fraction in the in-house DMA stock solution used for quantitation, which reiterates the importance of cross-calibrating calibration standards with NMI traceable stock solutions.
Poor chromatographic separation can lead to a significant quantitation bias. The majority of the studied samples contained only a few As species resulting in good baseline separation. However, more complex samples, such as marine biological tissues, presented several challenges. AsBet elutes in the void volume of anion exchange columns on which AsIII is also poorly retained, and therefore can elute very close to AsBet. Since AsBet is present at a several orders of magnitude higher mass fraction than AsIII, in marine tissue samples, AsIII may be unidentified due to a broad elution peak of AsBet (Supplementary Fig. S1). This of course, can be eliminated by the addition of H2O2 either prior or post extraction to convert AsIII into AsV. DOLT-5 contained an unknown compound eluting close to AsV under the chromatographic conditions used by Lab 1 (Fig. 1a). However, the chromatographic conditions selected by Lab 4 did not provide sufficient separation between the unknown compound and AsV (Fig. 1b) and the initially reported mass fraction of AsV by Lab 4 was the sum of both species. The unknown compound co-eluted with AsV, as shown in Fig. 1b, and although the peak of AsV is relatively broad, there is no visible shoulder which would indicate insufficient separation.
Chromatograms of DOLT-5 extract showing separation between unknown As species and AsV (a) using a PRP-X100 anion exchange column, 40 mM ammonium carbonate mobile phase with 5% methanol at 1 mL min–1 flow rate and co-elution of unknown compound and AsV (b) using a PRP-X100 anion exchange column, 60 mM ammonium carbonate mobile phase at 1 mL min–1 flow rate. For more details on chromatographic conditions see Supplementary Table S1.

As mass fraction determination
Plant tissue matrix
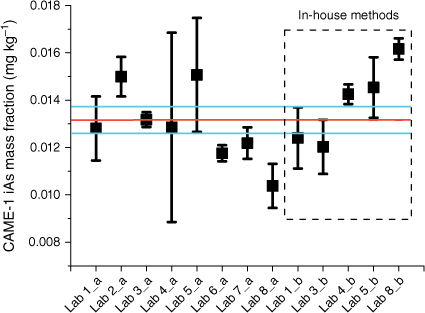
The plant tissue used in this study was CAME-1, canola meal, the high protein residue left over after extraction of oil. The residual material was a coarse powder with large sized particles, and is used in animal feed formulations. Several participants reported the need to filter the supernatant due to difficulties in sedimentation of the plant tissue. Most of the participants reported iAs (Fig. 2) as the only species present in the material, with a consensus value (Koepke et al. 2020) of 0.013 ± 0.001 mg kg−1 when GEM was used and 0.014 ± 0.001 mg kg−1 obtained by in-house methods (Table 3). The uncertainty associated with the consensus values was reduced from 7 to 5% by following the GEM.
Mean mass fractions of iAs (mg kg−1) in NRC CRM CAME-1, canola meal. Mass fractions were measured by the GEM (a) and in-house methods (b). Error bars indicate standard deviation (n ≥ 3), the red line marks the consensus value and the blue lines mark the standard uncertainty of the consensus value (k = 1) (Koepke et al. 2020).
| iAs | DMA | MMA | ||||||||
| CRM (AsT, mg kg−1) | Extraction method | n | Consensus value (mg kg−1) | u% | n | Consensus value (mg kg−1) | u% | n | Consensus value (mg kg−1) | u% |
| CAME-1 (0.03 ± 0.003) | GEM | 58 | 0.013 ± 0.001 | 5 | – | NQ | – | – | NQ | – |
| In-house | 28 | 0.014 ± 0.001 | 7 | – | NQ | – | – | NQ | – | |
| Combined | 86 | 0.013 ± 0.001 | 4 | – | NQ | – | – | NQ | – | |
| BFLY-1 (0.104 ± 0.005) | GEM | 61 | 0.052 ± 0.002 | 4 | 55 | 0.007 ± 0.0003 | 4 | 61 | 0.003 ± 0.0002 | 7 |
| In-house | 28 | 0.054 ± 0.006 | 10 | 22 | 0.007 ± 0.001 | 12 | 28 | 0.003 ± 0.0004 | 11 | |
| Combined | 89 | 0.052 ± 0.002 | 4 | 77 | 0.007 ± 0.0003 | 4 | 89 | 0.003 ± 0.0002 | 5 | |
| VORM-1 (0.129 ± 0.004) | GEM | 58 | 0.113 ± 0.005 | 4 | – | NQ | – | – | NQ | – |
| In-house | 28 | 0.104 ± 0.005 | 4 | – | NQ | – | – | NQ | – | |
| Combined | 86 | 0.110 ± 0.003 | 3 | – | NQ | – | – | NQ | – | |
| KRIK-1 (0.129 ± 0.004) | GEM | 46 | 0.006 ± 0.001 | 14 | 52 | 0.021 ± 0.001 | 4 | – | NQ | – |
| In-house | 18 | 0.006 ± 0.001 | 11 | 25 | 0.022 ± 0.001 | 6 | – | NQ | – | |
| Combined | 64 | 0.006 ± 0.001 | 8 | 77 | 0.021 ± 0.001 | 4 | – | NQ | – | |
| TORT-3 (59.5 ± 3.8) | GEM | 57 | 0.624 ± 0.023 | 4 | 54 | 3.79 ± 0.352 | 9 | 56 | 0.360 ± 0.032 | 9 |
| In-house | 36 | 0.510 ± 0.076 | 15 | 18 | 2.41 ± 0.837 | 35 | 33 | 0.208 ± 0.036 | 17 | |
| Combined | 93 | 0.596 ± 0.029 | 5 | 72 | 3.44 ± 0.412 | 12 | 89 | 0.307 ± 0.034 | 11 | |
| DOLT-5 (34.6 ± 2.4) | GEM | 55 | 0.033 ± 0.003 | 9 | 51 | 3.31 ± 0.222 | 7 | 55 | 0.249 ± 0.034 | 13 |
| In-house | 36 | 0.037 ± 0.005 | 14 | 30 | 2.55 ± 0.536 | 21 | 33 | 0.157 ± 0.033 | 21 | |
| Combined | 91 | 0.034 ± 0.002 | 7 | 81 | 3.09 ± 0.188 | 6 | 88 | 0.201 ± 0.024 | 12 | |
| DORM-5 (13.3 ± 0.7) | GEM | 58 | 0.015 ± 0.001 | 4 | 60 | 0.281 ± 0.030 | 11 | 54 | 0.014 ± 0.002 | 13 |
| In-house | 36 | 0.019 ± 0.002 | 12 | 30 | 0.280 ± 0.060 | 21 | 28 | 0.011 ± 0.003 | 24 | |
| Combined | 94 | 0.016 ± 0.001 | 7 | 90 | 0.288 ± 0.027 | 9 | 82 | 0.013 ± 0.001 | 11 |
AN, the number of laboratories; AsT, total arsenic; GEM, group extraction method; NQ, not quantified.
Lab 6 reported both DMA and MMA at a low mass fraction (0.00028 ± 0.00002 and 0.00008 ± 0.00001 mg kg−1, respectively, Supplementary Tables S3, S4) as well as traces of some other unknown species. Lab 8 also reported the presence of DMA; however, the mass fraction was below the limit of quantitation of their methods (LOQ > 0.0008 mg kg−1). The mass fraction of iAs accounted for 40% of total As. A low overall extraction efficiency of As species from plant materials is common (Kuehnelt et al. 2000; Mattusch et al. 2000; Geiszinger et al. 2002; Mir et al. 2007). There are several factors that may contribute to this observation. Arsenic may be present in a number of chemical forms with concentrations below the LOD of the analytical methods. Besides the iAs identified in this study, plants may accumulate other As species: DMA, MMA, tetramethylarsonium ion (TETRA), AsBet, AsChol and trimethylarsine oxide (TMAO) as reported by other publications (Kuehnelt et al. 2000; Geiszinger et al. 2002). Owing to lower detection limits, one of the participants, Lab 6, reported several unknown compounds at trace levels which may be a result of either their partial extractability from the tissue or low natural concentrations.
Terrestrial animal tissue matrix
There is a very good agreement between the levels of iAs extracted with the group and in-house extraction methods from terrestrial biological tissues (BFLY-1, VORM-1, KRIK-1). It can be seen in Table 3 that the uncertainty associated with the consensus value for iAs in BFLY-1 was significantly lower when GEM was applied and no significant difference (P > 0.05) was observed in the uncertainties for iAs in VORM-1 and KRIK-1.
In VORM-1, the consensus value following the GEM for iAs was 0.113 ± 0.002 mg kg−1, and following in-house extraction methods was 0.104 ± 0.005 mg kg−1. The combined consensus value of iAs (0.107 ± 0.003 mg kg−1) represented 83% of total As, with iAs being the only As species detected by the majority of participants. Lab 6 was the only lab that reported low levels of DMA (0.00027 ± 0.00001 mg kg−1, Supplementary Table S3) and MMA (0.00032 ± 0.00002 mg kg−1, Supplementary Table S4). The use of a defined GEM did not reduce the associated uncertainty, which was relatively low, 4% for both extraction methods.
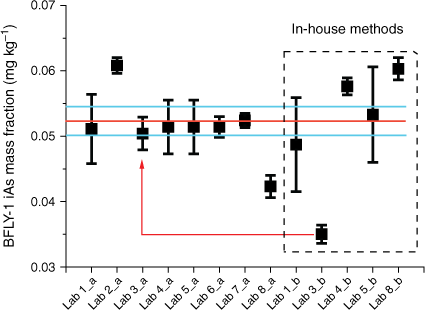
The group extraction method provided good agreement among the iAs values in BFLY-1 with an associated uncertainty of 4%, however, the results from in-house extraction methods are skewed by a single lab result (Fig. 3; red line and arrow) which increased the associated uncertainty to 10% (Table 3). Lab 3 reported a significantly lower iAs mass fraction (0.035 ± 0.001 mg kg−1, Supplementary Table S2) using their in-house extraction method (water with no added H2O2) in comparison with other labs. The Lab’s result using the GEM was 0.050 ± 0.003 mg kg−1 (Supplementary Table S2). The combined consensus mass fraction of iAs with the GEM was 0.052 ± 0.002 mg kg−1 (Table 3), which was equivalent to 0.0013 mg kg−1 of iAs in the extraction solution. If AsIII was present in this sample, its mass fraction would be below the LOD in the extraction solution and without oxidation of AsIII it would be undetected, and this is what could account for the lower mass fraction of iAs reported by Lab 3. In fact, Lab 3 performing in-house extraction without H2O2 did not detect any AsIII in the chromatograms. Furthermore, AsIII binds strongly to peptides such as glutathione through cysteinyl sulfhydryl (-SH) groups (Zhao et al. 2009). These complexes are stable in water without H2O2. Other As species detected in BFLY-1 were DMA and MMA with consensus mass fractions of 0.007 ± 0.0003 mg kg−1 and 0.003 ± 0.0002 mg kg−1, respectively, determined by GEM and 0.007 ± 0.001 mg kg−1 and 0.003 ± 0.0004 mg kg−1, determined by an in-house method (Table 3). The percentage uncertainty associated with the mass fractions of both MMA and DMA increase when in-house methods were applied. The sum of As species calculated on combined mass fractions from both group and in-house methods accounted for 59% of the total As. The low recovery could be due to some As bound to chitins in the exoskeleton, or the presence of As in lipid soluble form; both cannot be extracted using the applied mild extraction conditions. To the authors’ best knowledge, in the only study reporting As species in the black soldier fly larvae, (Biancarosa et al. 2019) the major mass fraction of As was an unidentified organic As species, accounting for 61% of total As, followed by DMA, AsBet and iAs. In the current study, the distribution of As species within black soldier fly larvae (BFLY-1) is very different from the results published by Biancarosa et al. (2019). This most probably results from the diet the insects were fed. The black soldier fly larvae in Biancarosa et al. (2019) had been fed on a substrate containing 60% seaweed, which is known to be rich in arsenosugars and As-lipids, hence high fractions of organic As species. Unfortunately, the feed of the black soldier fly larvae studied in the present work was not known.
Average mass fractions of iAs (mg kg−1) in NRC CRM BFLY-1. Mass fractions were measured by the GEM (a) and in-house methods (b). Error bars indicate standard deviation (n ≥ 3), red line marks the consensus value and blue lines mark the standard uncertainty of the consensus value (k = 1) (Koepke et al. 2020). Red line with arrow indicates GEM and in-house results from a single lab with low in-house results.
The percentage uncertainty in mass fractions of iAs in KRIK-1 extracted by GEM was slightly higher (14%) when compared with in-house methods (11%), and the mass fractions of iAs in this sample was the lowest from all the studied samples: 0.006 ± 0.001 mg kg−1 (GEM, Table 3) and 0.006 ± 0.001 mg kg−1 (in-house methods, Table 3). Only Lab 6 reported trace levels of MMA. However, the mass fraction of DMA, present at almost one order of magnitude higher than iAs and MMA, had a relatively low associated uncertainty for both extraction methods: 0.021 ± 0.001 mg kg−1 (4%) for GEM and 0.022 ± 0.001 mg kg−1 (6%) for in-house methods (Table 3). Thus, it is very likely that the higher uncertainties associated with iAs are driven by instrumental precision due to the low mass fraction of these species rather than extraction inconsistencies. It should be noted that the extracts from this matrix were cloudy solutions, most probably due to solubilisation of fat components. Neither additional centrifugation nor filtration improved the quality of the extracts. Although, outside of the scope of this study, the sample was analysed for organic As species using HPLC-ICP-MS (cation exchange chromatography) and LC-orbitrap-MS by Lab 1, where the largest fraction of As was found to be AsBet at 0.210 ± 0.015 mg kg−1. The sum of the As species (including AsBet) accounted for 81% of the total As for both consensus values, following group and in-house extraction methods. So far, there are no published studies on As species in edible cricket products; however, the total As concentration in the studied samples falls within the range reported in the recent survey of edible insects available on the Canadian market (0.035–0.34 mg kg−1) (Kolakowski et al. 2021).
Marine animal tissue matrix
Arsenic speciation analysis in marine animal tissue has been performed for several decades, but no suitable CRM is available for iAs to validate the measurements. In this study, existing CRMs (TORT-3, DORM-5 and DOLT-5) were used to provide a consensus value for iAs, DMA and MMA. The consensus mass fraction of iAs in TORT-3 (0.624 ± 0.023 mg kg−1, Table 3), following the GEM, had a low associated uncertainty of 4%, which nonetheless increased to 15% using the in-house extraction methods (consensus value of 0.510 ± 0.076 mg kg−1, Table 3). The percentage uncertainty from the in-house extraction methods was mainly driven by results reported by Labs 2 and 3 (Fig. 4; red lines and arrows).
Average mass fractions of iAs (mg kg−1) in TORT-3 certified reference material. Mass fractions were measured by the GEM (a) and in-house methods (b). Error bars indicate standard deviation (n ≥ 3), the red line marks the consensus value and blue lines mark the standard uncertainty of the consensus value (k = 1) (Koepke et al. 2020). Red lines with arrows indicate GEM and in-house results from Labs 2 and 3 with very low in-house results.

It should be noted that Labs 2 and 3 were not using H2O2 in their in-house extraction methods (water and heat only). A similar value of iAs (0.341 ± 0.030 mg kg−1) was reported by Wolle and Conklin (Wolle and Conklin 2018b) using the same extraction method as Lab 2. In a recently published study, Tibon et al. (2021) used water/methanol (1:1) as an extraction medium without H2O2 and reported a mass fraction of AsIII of 0.361 ± 0.012 mg kg−1 and AsV of 0.270 ± 0.024 mg kg−1, for which the sum is comparable with the mean value reported in the present study. To the best of our knowledge, there are no other published results of iAs in TORT-3 extracted without H2O2 and thus several investigations were carried out in the present study in an attempt to explain the lower mass fractions of iAs reported by Labs 2 and 3. Generally, H2O2 is used to oxidise AsIII to AsV which aids the quantitation of iAs in the presence of excess AsBet, which can co-elute with AsIII from anion exchange columns. Additionally, small mass fractions of AsIII or AsV may fall below the LOD if they are not analysed as the sum of both species after treatment with H2O2.
To identify the possible sources of lower mass fractions of iAs as reported by the water extractions, TORT-3 was extracted with H2O2 (Fig. 5 blue line) and without H2O2 (Fig. 5 black line). Subsequently after the extraction (and removal of the residual matrix), a portion of the extract without H2O2 was spiked with H2O2 (Fig. 5 red line) and analysed, whereas another portion spiked with H2O2 was subjected to heating conditions as during the extraction step (Fig. 5 green line). It was observed that, when H2O2 is added into the extract post-extraction and analysed directly, in addition to the oxidation of AsIII to AsV there is a slight increase in the mass fraction of a compound eluting in the shoulder of DMA, most probably DMAA (Supplementary Fig. S2), and low quantities of the unknown compound U2 could be detected (Fig. 5 red line). However, when H2O2 was used during extraction (Fig. 5 blue line) or after extraction and subjected to heating (Fig. 5 green line), a significantly larger number of changes in As species were observed. The peak area of iAs increased by 35%, equivalent to the 0.206 mg kg−1 of the consensus value from the group extraction method. After the mathematical correction, the results from Lab 3b would be in agreement with the rest of the labs; however, the results from Lab 2b would approach, but remain slightly outside, the confidence interval of the consensus value.
Chromatograms of extracted TORT-3 in water (black line), in 1% H2O2 (blue line), in water and spiked with H2O2 after extraction (red line) and in water and spiked with H2O2 after extraction and subjected to thermal treatment (green line).
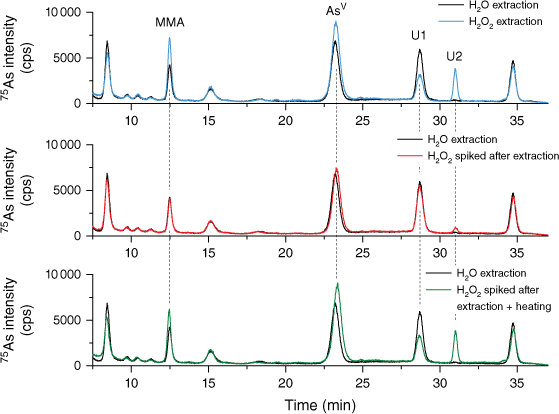
Similarly, the DMA mass fraction and the compound eluting in the shoulder of the DMA peak increased, which could be a result of the breakdown of some arsenosugars, as previously reported (Ebisuda et al. 2003; Wolle and Conklin 2018a). Interestingly, the peak area of the compound eluting at the retention time of MMA also increased. There is no evidence in the literature of MMA being a degradation product of organic As compounds, therefore it is possible that the compound is not actually MMA. Changes in peak areas were also observed for unknown compounds U1 and U2, the peak areas of the former decreased, whereas the peak area of U2 significantly increased. The unknown compound U2 was not present in the chromatogram after extraction without H2O2 suggesting that this compound is either entirely formed by the degradation of larger organo-arsenicals when H2O2 is being used or it is an oxidised analogue of thio-arsenical. Clearly, when it comes to complex matrices such as TORT-3, the choice of an extraction method is important. Presently, it is not clear whether extraction in water without H2O2 leads to underestimation of the As species mass fraction or if addition of H2O2 into the extractant causes species conversion and/or enables detection of thio-arsenicals.
Other reported values for iAs in TORT-3 were published in a study focusing on selective hydride generation (HG), where results from a reference detection technique (HPLC-HG-AFS) were compared with selective HG-ICP-MS (Marschner et al. 2019). The same HPLC-HG based method was used in the present study although it was coupled to a different detector (ICP-MS/MS); however, the results were significantly different. The published results for iAs determined by HPLC-HG-AFS were 0.428 ± 0.006 mg kg−1 in comparison with 0.645 ± 0.024 and 0.653 ± 0.020 mg kg−1 (Supplementary Table S2) in the present study for HPLC-HG-ICP-MS/MS and for HG-CT-ICP-MS/MS, respectively. The method in the Marschner et al. (2019) study employed a microwave digestion system for extraction. Interestingly, a similar in-house extraction method was used by Lab 5, with the exception of the hold time at 90°C for 10 min used by Marschner et al. (2019), instead of 30 min in the present study (Lab 5). The mass fraction reported by Lab 5 using their in-house extraction method was 0.493 ± 0.062 mg kg−1 (Supplementary Table S2), which is in good agreement with the published results (Marschner et al. 2019). Both microwave digestion systems used narrow 18 mL vessels in which the contact area between the sample and extractant is smaller compared to larger volume tubes, which could contribute to the lower extraction efficiencies.
Because of the degradation of arsenosugars into DMA, quantitation of DMA in the matrices containing arsenosugars is challenging. As it was already mentioned, the addition of H2O2 increased the peak area of DMA, and even if there was a good agreement between the reported values, the accuracy of the consensus value would be questionable. The results provided by labs that didn’t use H2O2 in their in-house method, i.e. Lab 2 (1.85 ± 0.015 mg kg−1, Supplementary Table S3) and Lab 3 (1.60 ± 0.039 mg kg−1, Supplementary Table S3), are significantly lower than the results provided by other participants. Although they are in good agreement with previously published data (1.181 ± 0.030 mg kg−1 (Tibon et al. 2021) and 1.303 ± 0.067 mg kg−1 (Wolle and Conklin 2018b)), the number of publications is very limited. Similarly, as it was in the case for iAs, values reported by HG-CT-ICP-MS/MS and HPLC-HG-ICP-MS/MS were significantly higher (Lab 6, 4.33 ± 0.210 mg kg−1 and Lab 7, 3.49 ± 0.168 mg kg−1, Supplementary Table S3) in the current study than those previously published (1.617 ± 0.072 mg kg−1 (Marschner et al. 2019)) using the HPLC-HG-AFS method.
The analysis of DOLT-5 for iAs has been challenging due to its low mass fraction; about 10-fold lower than in TORT-3. Inorganic As accounts for ˂ 0.2% of total As with a consensus mass fraction of 0.033 ± 0.003 (GEM, Table 3) and 0.037 ± 0.005 mg kg−1 (in-house methods, Table 3). The relative standard uncertainty decreased from 14 to 9% when GEM was applied. It was observed that the results provided by Lab 3 using an in-house method that does not contain H2O2 resulted in a 36% higher mass fraction than when quantified by the GEM (Supplementary Table S2). This observation is rather unusual as the trend was reversed in CAME-1 and the terrestrial animal CRMs. There are two published studies reporting values for iAs in DOLT-5 and while one of them is within the confidence interval of the consensus value reported in this study, 0.029 ± 0.007 mg kg−1 (Marschner et al. 2019), the other is significantly higher, 0.218 ± 0.009 mg kg−1 (sum of 0.125 ± 0.008 as AsIII and 0.093 ± 0.004 as AsV) (Tibon et al. 2021). The latter publication did not show a chromatogram for this CRM and hence interpretation of the observed difference can only be addressed by assumptions. Under the extraction conditions used in Tibon et al. (2021), AsIII would elute close to the void volume with AsBet and other cations. The mass fraction of AsBet is nearly 200 times higher than the reported AsIII which could potentially influence the quantitation of AsIII. Additionally, under the applied chromatographic conditions of Labs 1, 2, 3 and 5 (Supplementary Table S1), an unknown As compound was eluting close to AsV (Fig. 1). It is not clear from the published results if these two compounds were chromatographically resolved or if they could contribute to the reported mass fraction of AsV.
The presence of arsenosugars in DOLT-5 has been previously reported (Tibon et al. 2021), as well as observed in this study (Supplementary Fig. S3), and thus the quantitation using H2O2 will most probably overestimate the mass fraction of DMA in this material. Results reported from the in-house method without H2O2 were significantly lower than the consensus value from GEM (3.31 ± 0.222 mg kg−1, Table 3), 1.19 ± 0.126 mg kg−1 reported by Lab 2 and 1.64 ± 0.055 mg kg−1 by Lab 3 (Supplementary Table S3). A higher mass fraction was reported by others, 1.870 ± 0.120 mg kg−1 using water/methanol (1:1, v/v) as an extractant (Tibon et al. 2021) and 1.742 ± 0.045 mg kg−1 was reported by a HPLC-HG-AFS method following extraction in 2% HNO3 with 3% H2O2 (Marschner et al. 2019).
Similarly, in DORM-5, the iAs mass fraction accounted for <0.2% of total As with a consensus mass fraction of 0.015 ± 0.001 mg kg−1 (Table 3) reported using the GEM. The associated relative standard uncertainty of 4% was driven by results provided by Lab 1, which reported significantly higher mass fractions than the rest of the participants but could not provide any technical explanation which would justify exclusion of these results (Supplementary Table S2). Application of in-house methods increased the relative standard uncertainty to 12% with a consensus value of 0.019 ± 0.002 mg kg−1 (Table 3). The iAs mass fraction in DORM-5 is one of the lowest among the studied CRMs. There was also an added challenge of insufficient sedimentation, even after prolonged centrifugation, as reported by several labs. DORM-5 has only recently been released and thus there are no published studies of As in this CRM. However, the low mass fraction of iAs is representative of typical iAs values found in a similar matrix – fish protein (Wolle and Conklin 2018b).
The consensus values for DMA from both the group (0.281 ± 0.030 mg kg−1, Table 3) and in-house (0.280 ± 0.060 mg kg−1, Table 3) extraction methods matched very well for DORM-5, although the associated uncertainty increased from 11 to 21% when in-house methods were applied. The results using in-house methods of Labs 2 and 3 were significantly lower than the rest of the reported mass fractions, as was in the case of TORT-3 and DOLT-5 (Supplementary Table S3). From the studied samples, as well as evidence provided in the literature (Wolle and Conklin 2018b), it became clear that if the matrix contains arsenosugars, the assignment of an accurate mass fraction to DMA is very challenging. Using the various in-house extraction conditions by the participating labs, which to a certain degree represent some of the most common extraction conditions reported in the literature (Ardini et al. 2020), arsenosugars undergo degradation which leads to formation of DMA and other compounds. Additionally, tissues high in fat may contain As-lipids, which can be hydrolysed during the extraction. As previously shown (Ebisuda et al. 2003), DMA is one of the As-lipid hydrolysis products, potentially providing a significant contribution to DMA mass fraction in some samples.
Conclusion
In summary, the presented round-robin study led to several important observations. By following the steps in the prescribed group method, several variables were eliminated which led to reduced variability of reported values. As seen in Table 3, the uncertainties associated with the consensus values generated by the in-house extraction methods are greater in comparison with GEM but even with these very complex matrices, such as seafood, they are below 14% for iAs. The increased uncertainties of the consensus values generated by the in-house extraction methods imply that some inconsistences can be expected when comparing published data produced by different extraction methods and applied to similar matrices. However, only a slight reduction in the uncertainties was observed in the plant and terrestrial animal tissues, which is indicative of an extraction method independence for the tested methods in the presented study. Additionally, the low uncertainties in the consensus values generated by GEM suggest that if the extraction efficiencies between labs are comparable, the choice of analytical methods will have negligible impact on the quantitation. Nonetheless, it has to be stressed that the choice of appropriate chromatographic conditions that account for complexity and number of As species present is critical for obtaining accurate results. It was shown that the use of identical calibration standard solutions by the participating laboratories contributed to the reduction of the consensus uncertainty values. Therefore, greater attention should be paid to the verification of an accurate mass fraction in the calibration stock solutions. And lastly, the present results identified extractant dependent behaviour of As species in TORT-3 and possibly other similar matrices, where origin and implications are not clear at the moment and will require further investigation.
Conflicts of interest
Joerg Feldmann is an Editor for Environmental Chemistry and was blinded from the peer review process for this manuscript. Two of the study’s authors (K. Kubachka and M. Wolle) are US Food and Drug Administration (FDA) employees, and it must be declared that the opinions expressed by the authors do not reflect any current or future opinion or position by the FDA regarding arsenic analysis or regulation, and that the mention of manufacturers or trade names is for clarity and does not constitute an endorsement.
Acknowledgements
S. Musil and T. Matoušek gratefully acknowledge the support provided by the Czech Academy of Sciences (Institutional Research Plan RVO: 68081715). Marc Armin Preihs is acknowledged for his help with sample preparation. Phuong Mai Le is acknowledged for quantitation of AsBet in KRIK-1, Kenny Nadeau is acknowledged for measurement of total As and Christina Brophy is acknowledged for the moisture content determination.
References
Ardini F, Dan G, Grotti M (2020) Arsenic speciation analysis of environmental samples. Journal of Analytical Atomic Spectrometry 35(2), 215-237.
| Crossref | Google Scholar |
Baer I, Baxter M, Devesa V, Vélez D, Raber G, Rubio R, Llorente-Mirandes T, Sloth JJ, Robouch P, de la Calle B (2011) Performance of laboratories in speciation analysis in seafood – Case of methylmercury and inorganic arsenic. Food Control 22, 1928-1934.
| Crossref | Google Scholar |
Biancarosa I, Sele V, Belghit I, Ørnsrud R, Lock E-J, Amlund H (2019) Replacing fish meal with insect meal in the diet of Atlantic salmon (Salmo salar) does not impact the amount of contaminants in the feed and it lowers accumulation of arsenic in the fillet. Food Additives & Contaminants: Part A 36, 1191-1205.
| Crossref | Google Scholar |
Bralatei E, Lacan S, Krupp EM, Feldmann J (2015) Detection of Inorganic Arsenic in Rice Using a Field Test Kit: A Screening Method. Analytical Chemistry 87, 11271-11276.
| Crossref | Google Scholar |
Cordeiro F, Cizek-Stroh A, de la Calle B (2016) Determination of total and inorganic arsenic in rice. JRC Technical Report 102940. (European Commission: Brussels, Belgium) Available at https://joint-research-centre.ec.europa.eu/system/files/2016-10/irmm-pt-43_report.pdf
de la Calle MB, Emteborg H, Linsinger TPJ, Montoro R, Sloth JJ, Rubio R, Baxter MJ, Feldmann J, Vermaercke P, Raber G (2011) Does the determination of inorganic arsenic in rice depend on the method? TrAC Trends in Analytical Chemistry 30, 641-651.
| Crossref | Google Scholar |
de la Calle MB, Baer I, Robouch P, Cordeiro F, Emteborg H, Baxter MJ, Brereton N, Raber G, Velez D, Devesa V, Rubio R, Llorente-Mirandes T, Raab A, Feldmann J, Sloth JJ, Rasmussen RR, D’Amato M, Cubadda F (2012) Is it possible to agree on a value for inorganic arsenic in food? The outcome of IMEP-112. Analytical and Bioanalytical Chemistry 404, 2475-2488.
| Crossref | Google Scholar |
Ebisuda K-i, Kunito T, Fujihara J, Kubota R, Shibata Y, Tanabe S (2003) Lipid-soluble and water-soluble arsenic compounds in blubber of ringed seal (Pusa hispida). Talanta 61, 779-787.
| Crossref | Google Scholar |
EU (2015) Commission Regulation (EU) 2015/1006 of 25 June 2015 amending Regulation (EC) No 1881/2006 as regards maximum levels of inorganic arsenic in foodstuffs (Text with EEA relevance) (OJ L 161 26.06.2015) p. 14. Available at http://data.europa.eu/eli/reg/2015/1006/oj
Fiamegkos I, Cordeiro F, Robouch P, Vélez D, Devesa V, Raber G, Sloth JJ, Rasmussen RR, Llorente-Mirandes T, Lopez-Sanchez JF, Rubio R, Cubadda F, D’amato M, Feldmann J, Raab A, Emteborg H, de la Calle MB (2016) Accuracy of a method based on atomic absorption spectrometry to determine inorganic arsenic in food: Outcome of the collaborative trial IMEP-41. Food Chemistry 213, 169-179.
| Crossref | Google Scholar |
Foster S, Maher W, Krikowa F, Apte S (2007) A microwave-assisted sequential extraction of water and dilute acid soluble arsenic species from marine plant and animal tissues. Talanta 71, 537-549.
| Crossref | Google Scholar |
Francesconi KA (2010) Arsenic species in seafood: origin and human health implications. Pure and Applied Chemistry 82, 373-381.
| Crossref | Google Scholar |
Gamble BM, Gallagher PA, Shoemaker JA, Parks AN, Freeman DM, Schwegel CA, Creed JT (2003) An investigation of the chemical stability of arsenosugars in basic environments using IC-ICP-MS and IC-ESI-MS/MS. Analyst 128, 1458-1461.
| Crossref | Google Scholar |
Geiszinger A, Goessler W, Kosmus W (2002) Organoarsenic compounds in plants and soil on top of an ore vein. Applied Organometallic Chemistry 16, 245-249.
| Crossref | Google Scholar |
Grinberg P, Nadeau K, Brophy C, Pihillagawa Gedara I, LeBlanc K, Simon A, Yang L, Mihai O, Le PM, Gajdosechova Z, McRae G, Cauduro VH, Soares CSL, Mello PA, Flores EMM, Meija J, Mester Z (2021) ‘DORM-5: Fish Protein Certified Reference Material.’ (National Research Council of Canada) 10.4224/crm.2021.dorm-5
Grinberg P, LeBlanc K, Nadeau K, Gajdosechova Z, Brophy C, Pihillagawa Gedara I, Yang L, Simon A, Palmer C, Cauduro VH, Soares CSL, Mello PA, Flores EMM, Sim R, Pétursdóttir ÁHE, Raab A, Feldmann J, Musil S, Matousek T, Kubachka K, Wolle M, Wozniak BJ, Springer S, Gürleyük H, Meija J, Mester Z (2023a) ‘CAME-1: Canola Meal Certified Reference Material.’ (National Research Council of Canada) 10.4224/crm.2023.came-1
Grinberg P, LeBlanc K, Nadeau K, Gajdosechova Z, Brophy C, Pihillagawa Gedara I, Yang L, Simon A, Palmer C, Cauduro VH, Soares CSL, Mello PA, Flores EMM, Sim R, Pétursdóttir ÁHE, Raab A, Feldmann J, Musil S, Matousek T, Kubachka K, Wolle M, Wozniak BJ, Springer S, Gürleyük H, Meija J, Mester Z (2023b) ‘BFLY-1: Black Soldier Fly Meal Certified Reference Material.’ (National Research Council of Canada) 10.4224/crm.2023.bfly-1
Health Canada (2021) Health Canada’s proposal to add a maximum level for inorganic arsenic in rice-based foods intended specifically for infants and uoung children. Health Canada, Ottawa, ON, Canada. Available at https://www.canada.ca/en/health‐canada/services/food‐nutrition/public‐involvement‐partnerships/proposal‐add‐maximum‐level‐inorganic‐arsenic‐rice‐based‐foods/document.html [accessed 24 April 2023]
Hirata S, Toshimitsu H, Aihara M (2006) Determination of arsenic species in marine samples by HPLC-ICP-MS. Analytical Sciences 22, 39-43.
| Crossref | Google Scholar |
Hughes MF (2002) Arsenic toxicity and potential mechanisms of action. Toxicology Letters 133, 1-16.
| Crossref | Google Scholar |
Irvine L, Boyer IJ, Desesso JM (2006) Monomethylarsonic acid and dimethylarsinic acid: developmental toxicity studies with risk assessment. Birth Defects Research Part B: Developmental and Reproductive Toxicology 77, 53-68.
| Crossref | Google Scholar |
Kaise T, Watanabe S, Itoh K (1985) The acute toxicity of arsenobetaine. Chemosphere 14, 1327-1332.
| Crossref | Google Scholar |
Karthikeyan S, Hirata S (2004) Ion chromatography–inductively coupled plasma mass spectrometry determination of arsenic species in marine samples. Applied Organometallic Chemistry 18, 323-330.
| Crossref | Google Scholar |
Koepke A, Lafarge T, Possolo A, Toman B (2020) NIST Consensus Builder. Available at https://consensus.nist.gov/app/nicob [accessed 16 November 2022]
Kolakowski BM, Johaniuk K, Zhang H, Yamamoto E (2021) Analysis of Microbiological and Chemical Hazards in Edible Insects Available to Canadian Consumers. Journal of Food Protection 84, 1575-1581.
| Crossref | Google Scholar |
Kuehnelt D, Lintschinger J, Goessler W (2000) Arsenic compounds in terrestrial organisms. IV. Green plants and lichens from an old arsenic smelter site in Austria. Applied Organometallic Chemistry 14, 411-420.
| Crossref | Google Scholar |
LeBlanc K, Grinberg P, Nadeau K, Le PM, Gajdosechova Z, Brophy C, Pihillagawa Gedara I, Yang L, Simon A, Palmer C, Cauduro VH, Soares CSL, Mello PA, Flores EMM, Sim R, Pétursdóttir ÁHE, Raab A, Feldmann J, Musil S, Matousek T, Kubachka K, Wolle M, Wozniak BJ, Springer S, Gürleyük H, Meija J, Mester Z (2023a) ‘RIK-1: Cricket Flour Certified Reference Material.’ (National Research Council of Canada) 10.4224/crm.2023.krik-1
LeBlanc K, Grinberg P, Nadeau K, Gajdosechova Z, Brophy C, Pihillagawa Gedara I, Yang L, Simon A, Palmer C, Cauduro VH, Soares CSL, Mello PA, Flores EMM, Sim R, Pétursdóttir ÁHE, Raab A, Feldmann J, Musil S, Matousek T, Kubachka K, Wolle M, Wozniak BJ, Springer S, Gürleyük H, Meija J, Mester Z (2023b) ‘VORM-1: Mealworm powder Certified Reference Material.’ (National Research Council of Canada) 10.4224/crm.2023.vorm-1
Leermakers M, Baeyens W, De Gieter M, Smedts B, Meert C, De Bisschop HC, Morabito R, Quevauviller P (2006) Toxic arsenic compounds in environmental samples: Speciation and validation. TrAC Trends in Analytical Chemistry 25, 1-10.
| Crossref | Google Scholar |
Llorente-Mirandes T, Rubio R, López-Sánchez JF (2017) Inorganic Arsenic Determination in Food: A Review of Analytical Proposals and Quality Assessment Over the Last Six Years. Applied Spectroscopy 71, 25-69.
| Crossref | Google Scholar |
Luvonga C, Rimmer CA, Yu LL, Lee SB (2020) Organoarsenicals in Seafood: Occurrence, Dietary Exposure, Toxicity, and Risk Assessment Considerations – A Review. Journal of Agricultural and Food Chemistry 68, 943-960.
| Crossref | Google Scholar |
Marschner K, Musil S, Dědina J (2016) Achieving 100% efficient postcolumn hydride generation for As speciation analysis by atomic fluorescence spectrometry. Analytical Chemistry 88, 4041-4047.
| Crossref | Google Scholar |
Marschner K, Pétursdóttir ÁH, Bücker P, Raab A, Feldmann J, Mester Z, Matoušek T, Musil S (2019) Validation and inter-laboratory study of selective hydride generation for fast screening of inorganic arsenic in seafood. Analytica Chimica Acta 1049, 20-28.
| Crossref | Google Scholar |
Matoušek T, Wang Z, Douillet C, Musil S, Stýblo M (2017) Direct speciation analysis of arsenic in whole blood and blood plasma at low exposure levels by hydride generation-cryotrapping-inductively coupled plasma mass spectrometry. Analytical Chemistry 89, 9633-9637.
| Crossref | Google Scholar |
Mattusch J, Wennrich R, Schmidt AC, Reisser W (2000) Determination of arsenic species in water, soils and plants. Fresenius’ Journal of Analytical Chemistry 366, 200-203.
| Crossref | Google Scholar |
Menon M, Sarkar B, Hufton J, Reynolds C, Reina SV, Young S (2020) Do arsenic levels in rice pose a health risk to the UK population? Ecotoxicology and Environmental Safety 197, 110601.
| Crossref | Google Scholar |
Meyer S, Matissek M, Müller SM, Taleshi MS, Ebert F, Francesconi KA, Schwerdtle T (2014a) In vitro toxicological characterisation of three arsenic-containing hydrocarbons. Metallomics 6, 1023-1033.
| Crossref | Google Scholar |
Meyer S, Schulz J, Jeibmann A, Taleshi MS, Ebert F, Francesconi KA, Schwerdtle T (2014b) Arsenic-containing hydrocarbons are toxic in the in vivo model Drosophila melanogaster. Metallomics 6, 2010-2014.
| Crossref | Google Scholar |
Mir KA, Rutter A, Koch I, Smith P, Reimer KJ, Poland JS (2007) Extraction and speciation of arsenic in plants grown on arsenic contaminated soils. Talanta 72, 1507-1518.
| Crossref | Google Scholar |
Müller SM, Ebert F, Raber G, Meyer S, Bornhorst J, Hüwel S, Galla H-J, Francesconi KA, Schwerdtle T (2018) Effects of arsenolipids on in vitro blood-brain barrier model. Archives of Toxicology 92, 823-832.
| Crossref | Google Scholar |
Narukawa T, Kuroiwa T, Yarita T, Chiba K (2006) Analytical sensitivity of arsenobetaine on atomic spectrometric analysis and the purity of synthetic arsenobetaine. Applied Organometallic Chemistry 20, 565-572.
| Crossref | Google Scholar |
Narukawa T, Kuroiwa T, Chiba K (2007) Mechanism of sensitivity difference between trivalent inorganic As species [As (III)] and pentavalent species [As (V)] with inductively coupled plasma spectrometry. Talanta 73, 157-165.
| Crossref | Google Scholar |
Narukawa T, Inagaki K, Zhu Y, Kuroiwa T, Narushima I, Chiba K, Hioki A (2012) Preparation and certification of Hijiki reference material, NMIJ CRM 7405-a, from the edible marine algae hijiki (Hizikia fusiforme). Analytical and Bioanalytical Chemistry 402, 1713-1722.
| Crossref | Google Scholar |
Petrick JS, Ayala-Fierro F, Cullen WR, Carter DE, Vasken Aposhian H (2000) Monomethylarsonous acid (MMAIII) is more toxic than arsenite in Chang human hepatocytes. Toxicology and Applied Pharmacology 163, 203-207.
| Crossref | Google Scholar |
Pétursdóttir ÁH, Friedrich N, Musil S, Raab A, Gunnlaugsdóttir H, Krupp EM, Feldmann J (2014a) Hydride generation ICP-MS as a simple method for determination of inorganic arsenic in rice for routine biomonitoring. Analytical Methods 6, 5392-5396.
| Crossref | Google Scholar |
Pétursdóttir ÁH, Gunnlaugsdóttir H, Krupp EM, Feldmann J (2014b) Inorganic arsenic in seafood: Does the extraction method matter? Food Chemistry 150, 353-359.
| Crossref | Google Scholar |
Pizarro I, Gómez M, Cámara C, Palacios MA (2003) Arsenic speciation in environmental and biological samples: extraction and stability studies. Analytica Chimica Acta 495, 85-98.
| Crossref | Google Scholar |
Styblo M, Del Razo LM, Vega L, Germolec DR, Lecluyse EL, Hamilton GA, Reed W, Wang C, Cullen WR, Thomas DJ (2000) Comparative toxicity of trivalent and pentavalent inorganic and methylated arsenicals in rat and human cells. Archives of Toxicology 74, 289-299.
| Crossref | Google Scholar |
Thompson M, Ellison SLR (2011) Dark uncertainty. Accreditation and Quality Assurance 16, 483-487.
| Crossref | Google Scholar |
Tibon J, Silva M, Sloth JJ, Amlund H, Sele V (2021) Speciation analysis of organoarsenic species in marine samples: method optimization using fractional factorial design and method validation. Analytical and Bioanalytical Chemistry 413, 3909-3923.
| Crossref | Google Scholar |
Wahlen R, Mcsheehy S, Scriver C, Mester Z (2004) Arsenic speciation in marine certified reference materials Part 2. The quantification of water-soluble arsenic species by high-performance liquid chromatography-inductively coupled plasma mass spectrometry. Journal of Analytical Atomic Spectrometry 19, 876-882.
| Crossref | Google Scholar |
Wang RY, Hsu YL, Chang LF, Jiang SJ (2007) Speciation analysis of arsenic and selenium compounds in environmental and biological samples by ion chromatography-inductively coupled plasma dynamic reaction cell mass spectrometer. Analytica Chimica Acta 590, 239-44.
| Crossref | Google Scholar |
Willie S, Pihillagawa Gedara I, Maxwell P, Meija J, Mester Z, Yang L (2013) ‘TORT-3: Lobster hepatopancreas reference material for trace metals.’ (National Research Council of Canada) 10.4224/crm.2013.tort-3
Wolle MM, Conklin SD (2018a) Speciation analysis of arsenic in seafood and seaweed: Part I—evaluation and optimization of methods. Analytical and Bioanalytical Chemistry 410, 5675-5687.
| Crossref | Google Scholar |
Wolle MM, Conklin SD (2018b) Speciation analysis of arsenic in seafood and seaweed: Part II—single laboratory validation of method. Analytical and Bioanalytical Chemistry 410, 5689-5702.
| Crossref | Google Scholar |
Yang L, Willie S, Grinberg P, Pihillagawa Gedara I, Clancy V, Maxwell P, McRae G, Meija J, Mester Z (2014) ‘DOLT-5: Dogfish Liver Certified Reference Material for Trace Metals and other Constituents.’ (National Research Council of Canada) 10.4224/crm.2014.dolt-5
Zhao FJ, Ma JF, Meharg AA, Mcgrath SP (2009) Arsenic uptake and metabolism in plants. New Phytologist 181, 777-794.
| Crossref | Google Scholar |