

New particle formation above a simulated salt lake in aerosol chamber experiments
K. A. Kamilli A E , J. Ofner B , B. Lendl B , P. Schmitt-Kopplin C and A. Held A DA Atmospheric Chemistry, University of Bayreuth, Dr-Hans-Frisch-Straße 1-3, D-95448 Bayreuth, Germany.
B Institute of Chemical Technologies and Analytics, Vienna University of Technology, Getreidemarkt 9, AT-1060 Vienna, Austria.
C Research Unit Analytical BioGeoChemistry, Helmholtz Centre Munich, Ingolstädter Landstraße 1, D-85764 Neuherberg, Germany.
D Bayreuth Center of Ecology and Environmental Research BayCEER, Dr-Hans-Frisch-Straße 1-3, D-95448 Bayreuth, Germany.
E Corresponding author. Email: katharina.kamilli@uni-bayreuth.de
Environmental Chemistry 12(4) 489-503 https://doi.org/10.1071/EN14225
Submitted: 16 October 2014 Accepted: 2 January 2015 Published: 29 June 2015
Environmental context. Deforestation in Western Australia beginning in the mid-19th century led to a considerable change of the land surface, and Western Australia is now suffering more often from droughts. Particle formation induced by salt lakes has been identified as a potential control factor for changed precipitation patterns. This study aims to determine key factors involved in the particle formation process by simulating a simplified salt lake in an aerosol chamber in the laboratory.
Abstract. In recent field experiments, particle formation has been observed above salt lakes in Western Australia and related to changes in regional precipitation patterns. This work investigates the particle formation potential above a simulated salt lake in aerosol chamber experiments under various conditions. The salt lake mixture comprised fixed concentrations of NaBr, NaCl and Na2SO4, and varying concentrations of FeSO4 and FeCl3. Further, an organic mixture of 1,8-cineol and limonene was added under dark and light conditions. Both the presence of organic compounds and of light were found to be essential for new particle formation in our experiments. There were clear indications for conversion of FeII to FeIII, which suggests a Fenton-like reaction mechanism in the system. Contrary to the idea that a Fenton-like reaction mechanism might intensify the oxidation of organic matter, thus facilitating secondary organic aerosol formation, the observed particle formation started later and with lower intensity under elevated FeII concentrations. The highest particle number concentrations were observed when excluding FeII from the experiments. Chemical analysis of the formed aerosol confirmed the important role of the Fenton-like reaction for particle formation in this study. Ultrahigh-resolution mass spectrometry and Raman spectroscopy provide analytical proof for the formation of organosulfates and halogenated organic compounds in the experiments presented. Even though halogens and organic precursors are abundant in these experimental simulations, halogen-induced organic aerosol formation exists but seems to play a minor overall role in particle formation.
Introduction
Secondary aerosol formation from gaseous precursors is not completely understood because of the complex interplay of a vast number of chemical reactions, potential precursors and gas–particle partitioning.[1,2] Within the last years, a research focus has been placed on secondary organic aerosol (SOA) formation. In several field experiments, it has been shown that SOA contributes to a major fraction of the aerosol mass in the free troposphere. Contrary to global chemistry transport models predicting organic aerosol formation in the free troposphere,[3] and forecasting an increasing fraction of SOA from oxidation of monoterpenes in the future,[4] the experimentally measured SOA mass was typically one to two orders of magnitude larger than the modelled SOA mass.[5]
On a global scale, SOA released from anthropogenic sources plays a minor role.[6] Not considering methane, biogenic volatile organic compounds (VOC) emissions are estimated to range from 491 to 1150 Tg carbon per year, exceeding the emissions from anthropogenic sources by up to an order of magnitude.[7] The VOC emissions from trees alone exceed the level from man-made sources by a factor of 6.2,[8] and are therefore a crucial source for organic aerosol precursors. Several studies have identified SOA coming from VOCs emitted by conifer forests.[9–13] Also, the emission from eucalyptus trees is the topic of some studies.[6,14–16] To evoke new particle formation, the reactive VOCs are oxidised primarily by ozone, OH and NO3 radicals.[14] Some studies have also investigated halogen-induced SOA formation.[17–19] Important sources for reactive halogen species (RHS) are halogen release from sea-salt aerosols (e.g. Finlayson-Pitts[20]), heterogeneous reactions on aerosol surfaces[20] and salt lakes.[21,22] Saline soils and salt lakes will become even more important terrestrial sources for atmospheric chlorine and other halogens with the increase in human-caused salinisation.[23] Thus, in regions with high concentrations of RHS and VOC, the halogen-induced formation of SOA may contribute considerably to particle concentrations. However, the relative contribution of halogen-induced SOA formation has not been quantified in the natural environment.
The aqueous phase has been established as an additional source for SOA over the past decade.[24–27] For example, the oxidation of organic compounds is catalysed by FeII as well as hydrogen peroxide (H2O2) in the so-called Fenton reaction, first described by Fenton[28] on the basis of the oxidation of tartaric acid in the presence of H2O2 and a low concentration of a ferrous salt. High levels of solar radiation, which is important for a constant supply of H2O2 in surface waters and topsoils,[29] is typical for Western Australia. Indeed, both H2O2 as well as FeII were found in Australian salt-lake water samples.[30] The Fenton reaction can contribute to the production of OH radicals,[31] even though the importance of OH production by this pathway is still controversial.[32] Aqueous-phase chemistry may also slow down or inhibit atmospheric SOA formation if oxidation in the aqueous phase leads to non-volatile products that cannot escape to the atmosphere. Here, simulation experiments of new particle formation above a salt lake model were conducted in a 700-L aerosol chamber in order to investigate aerosol formation processes in salt lake environments.
Measurements and methods
Aerosol chamber setup
The main experiments were conducted in a 700-L aerosol chamber. A detailed description of this chamber can be found in Ofner et al.[33] In this glass chamber, experiments can be performed under close-to-ambient light conditions using a solar simulator.[19] Contamination of the chamber is prevented by a constant flow of particle-free zero air during the experiments, generating a slight excess pressure inside the chamber. Also, the chamber was flushed with zero air between the experiments.
The general chamber set-up consists of a cylindrical 700-L Duran glass body closed with Teflon film at the top and bottom, transparent to radiation coming from the solar simulator at the bottom.[33] This set-up was supplemented with a custom-made structure to position the simulated salt lake inside the chamber (Fig. 1). A stainless-steel rod was fixed between two opposite glass ports to support the three-point mounting of a transparent Teflon bowl inside the chamber. This structure was built in order to avoid contamination of the chamber bottom made of Teflon film, and to increase the distance between the salt lake mixture and the solar simulator. Consequently, warming and potential vaporisation of the salt lake mixture due to the solar simulator is reduced. The salt lake mixture was injected into the Teflon bowl in the chamber through a separating funnel outside the chamber and Teflon tubing fed through a glass port of the chamber (Fig. 1). This allowed the introduction of the salt lake mixture and sampling from it without contaminating the chamber with ambient air. Placed on a transparent Teflon film, the simulated salt lake mixture was irradiated from below by the solar simulator, creating irradiation conditions comparable with those above Australian salt lakes after sunrise.

|
Instrumentation
The aerosol particle number size distribution inside the chamber was measured with a custom-built scanning mobility particle sizer (SMPS) (Tropos, Leipzig, Germany) following the design recommended by Wiedensohler et al.[34] After achieving bipolar charge equilibrium, the aerosol population was size-segregated in a differential mobility analyser (DMA), and the particles were detected with a condensation particle counter (CPC Model 3772, TSI Inc., Shoreview, MN, USA). With a time resolution of 5 min, a full scan of the size distribution was measured in the mobility diameter range from particle diameter (Dp) = 10 to 772 nm in 71 bins or from Dp = 8 to 325 in 68 bins. In order to determine the onset of new particle formation with a better time resolution, and for comparison with the SMPS measurements, total particle number concentrations were measured with an additional condensation particle counter (CPC Model 5.400, Grimm Aerosol Technik, Ainring, Germany).
For subsequent chemical analysis, aerosol particles were collected on aluminium foils with a Sioutas impactor (SKC; Eighty Four, PA, USA) for 60 min at the end of the experiments. The flow rate was set to 9 L min–1 in order to sample the four separate stages of the impactor in the aerodynamic diameter range from 250 nm to 10 µm. In addition, aerosol samples were collected on quartz-fibre filters (Whatman QMA, 25 mm, GE Healthcare, Little Chalfont, UK) with a stainless-steel inline filter holder at a flow rate of 9 L min–1 for 80 min.
The impactor samples were analysed by Raman microscopy and scanning electron microscopy with energy-dispersive X-ray spectroscopy (SEM-EDX). EDX of single aerosol particles was done using an Octane Pro Silicon Drift (SDD) EDX detector from AMETEK, coupled to the FEI Quanta 200 scanning electron microscope (FEI Europe, Eindhoven, Netherlands). Raman spectra of single aerosol particles were obtained using a Horiba LabRam 800 HR Raman microscope (Horiba, Bensheim, Germany) at an excitation wavelength of 532 nm with a 300-lines mm–1 grating at an initial laser power of 10 % (~5 mW). By combining Raman spectroscopy and EDX, a detailed characterisation of the collected aerosol particles with respect to elemental composition and vibrational behaviour of the aerosol sample was obtained.[35] In addition, the quartz-fibre filters were analysed by ultrahigh-resolution mass spectrometry. The SolariX Fourier-transform ion cyclotron resonance mass spectrometer (FT-ICR/MS; Bruker, Bremen, Germany) was operated with a 12-T superconducting magnet and an Apollo II electrospray source in negative mode.[36] Because there were high salt concentrations in the sample, chlorine adducts[37] and organohalogens could not be clearly distinguished if the SOA sample was dissolved in methanol and directly injected into the ultrahigh-resolution mass spectrometer. Additionally, high salt-cluster concentrations would suppress some signals resulting from CHNOS compounds in the spectra. Therefore, the samples were desalted before analysis by extracting the filters with water and enriching the organic matter by solid phase extraction (SPE) using using BAKERBOND spe Octadecyl (C18) Disposable Extraction Columns (Avantor, Center Valley, PA, USA).[38] A blank SPE extract of a non-loaded filter was also analysed by FT-ICR/MS and did not show significant contamination from the filter material.
Ozone mixing ratios were measured during all chamber experiments using a photometric O3 analyser (Model 49i, Thermo Scientific, Franklin, MA, USA) with a lower detection limit of 1 ppb.
Simulated salt lake
A simplified salt lake mixture was prepared based on the chemical composition of various Australian salt lake samples determined in 2011 and 2012 by the Institute of Earth Science at the University of Heidelberg. With respect to inorganic salts, average concentrations of the main inorganic ions Na+, Fe2+, SO42–, Cl– and Br– were calculated. Combining these ions in four inorganic salts, the standard salt lake mixture included NaBr (0.66 g L–1), NaCl (307.71 g L–1), Na2SO4 (32.11 g L–1) and FeIISO4 (0.52 g L–1). In addition to the inorganic mixture, commercial eucalyptus oil (Primavera, Oy-Mittelberg, Germany) distilled from leaves and boughs of Portuguese Eucalyptus globulus trees was used as the organic model compound. The oil is a mixture of 85 % 1,8-cineol and 15 % limonene corresponding to the typical emission of Eucalyptus globulus. Owing to its moderate solubility, it was expected that the oil would be volatilised from the salt lake mixture in the course of the experiment, providing the gaseous organic precursor for SOA formation.
Experimental design
Three different series of chamber experiments were conducted studying the influence of light and the presence of an organic precursor (series A), the presence of FeII and FeIII (series B), and the concentration of FeII (series C) in the simulated salt lake (cf. Table 1). Experimental conditions were varied by excluding the organic precursor, by using FeCl3 instead of FeSO4, by using a mixture of FeSO4 and FeCl3, or by changing the concentration of FeSO4.

|
At first, the salt lake mixture was studied under dark and light conditions with and without the organic precursor to validate the role of light and the organic precursor in particle formation (Table 1, A1–2). The next set of experiments (Table 1, B1–4) was conducted using FeIII instead of FeII, a mixture of FeII and FeIII, and half of the concentration of FeII and FeIII in the mixture. In this set of experiments, liquid samples were extracted with a syringe. The samples of the salt lake mixture were analysed with test strips for H2O2 (Quantofix, Peroxyde 25, Machery-Nagel, Düren, Germany), for the total concentration of FeIII (Quantofix, Fer total 100, Machery-Nagel), and for FeII (MQuant, Merck, Darmstadt, Germany). In a third set of experiments (Table 1, C1–3), the concentration of FeIISO4 was varied between zero, 0.52 and 3.64 g L–1. The maximum concentration is based on the highest FeII concentration observed in Lake Boats in Western Australia. This concentration corresponds to seven times the average FeII concentration observed in the field, represented by a concentration of 0.52 g L–1 FeIISO4.
The pH of the salt lake mixture was typically adjusted to a value of 2.5 using HCl after analysing the natural pH of several salt lakes in Western Australia (T. Krause, unpubl. data). In 11 samples of natural salt lakes taken in 2013, the pH varied from 2.5 to 7.1. Particle formation was strongest at lakes in the pH range 2–4, with a maximum particle number concentration of Nmax = 2.50 × 105 cm–3 measured at Lake Dune with a daily average pH of 2.9. Four lakes were analysed in detail, showing that the pH is not constant over the course of the day but fluctuates slightly. No clear trend can be observed in the field data, but the pH values seem to influence particle formation above the salt lakes. In most cases, new particle formation (NPF) started directly after a drop in the pH. For example, at Lake Boats, the pH was between 2.7 and 2.9, and NPF started on two occasions when the pH fell below 2.85. Therefore, a pH of 2.5 was chosen for the model lake in our simulation experiments in the laboratory in order to provide pH conditions favourable for NPF. However, when a mixture of FeCl3 and FeSO4 was used (Table 1, B3–4), the pH was set to 3.0 and 3.1.
Results and discussion
Influence of experimental conditions on particle formation
As a first experiment, a pure salt mixture without the organic precursor was injected into the dark chamber (Table 1, A1). Under these conditions, particle formation, i.e. an increase of particle concentrations or particle growth, was not observed in the particle number size distributions. After 80 min, the solar simulator was switched on. Similarly, under daylight conditions, particle formation was not observed, and the particle concentration remained below 100 cm–3 (cf. Fig. 2a). This ‘blank’ experiment shows that the salt mixture of NaBr, NaCl, Na2SO4 and FeIISO4 does not induce NPF in the aerosol chamber. Additionally, this experiment indicates that neither the aerosol chamber itself nor the chamber top or bottom made of Teflon film produce a significant number of particles under the influence of radiation.

|
In a second experiment (Table 1, A2), the organic precursor was added to the salt lake mixture. In order to evaluate the importance of light and photochemical reactions for particle formation in acidic, salty environments, the salt lake mixture remained in darkness for a time (<30 min). During this time, no particle formation was observed, even though the salt lake mixture including 1,8-cineol and limonene was present in the chamber. This state is comparable with an Australian salt lake before sunrise, where NPF was not observed in the dark (K. A. Kamilli, unpubl. data). After switching on the solar simulator and initiating photochemistry in our experiments, particle formation started within 5 min, reaching a maximum particle number concentration of 2.51 × 105 cm–3 (Fig. 2b). This confirms that both the organic precursor and light are essential for NPF in our experiments. No particles were formed in the illuminated chamber with only the salt mixture present (Fig. 2a), and no particles were formed under dark conditions.
During the experiments, the initial pH of 2.5 decreased continuously. On the one hand, this indicates that the pH of the simulated salt lakes changed owing to aqueous phase chemistry. On the other hand, the pH values were not a key trigger of NPF in our simulation experiments, and NPF did not significantly change aqueous-phase chemistry. Also, contrary to the field measurements, H2O2 was not found in the samples of the simulated lake. In Western Australia, H2O2 was detected in the liquid layer of the salt lakes. However, in the field observations, it was striking that particle formation started at minimum H2O2 concentrations, which leads to the conclusion that H2O2 was typically consumed before particle formation.
Chamber experiments with mixtures of FeII and FeIII
In a second series of experiments (Table 1, B1–B4), the experimental conditions concerning light, the concentration of the organic precursor and the concentrations of NaBr, NaCl and Na2SO4 were held constant. Only the concentration of iron-containing salts was varied to investigate the potential effect of iron in the aqueous phase on particle formation.
In order to investigate the effect of varying concentrations of FeII and FeIII on particle formation, experiments B1–B4 were compared with respect to Nmax, the time after switching on the solar simulator when Nmax was reached, the maximum geometric mean diameter (GMD), the maximum particle volume concentration (Vmax), the time after switching on the solar simulator when Vmax was reached, and the particle growth rate (GR) within 45 min after NPF started. For comparability, the maximum GMD was computed in the diameter range from 10 to 325 nm within 1 h after switching on the solar simulator for all experiments.
First, the standard salt lake mixture with a concentration of 520 mg L–1 of the FeII salt was used (Table 1, B1; Fig. 3a). After switching on the solar simulator, the maximum particle number concentration of 1.10 × 105 cm–3 was reached after 7 min. The GMD reached a value of 71 nm. In the second experiment, FeIII was added to the salt lake mixture instead of FeII (Table 1, B2; Fig. 3b). Particle formation started directly after switching on the light, and the particle population grew to GMDs >100 nm within 10 min. The maximum particle number concentration was 5.4 × 104 cm–3, and the maximum GMD was 183 nm. In a third experiment (Table 1, B3; Fig. 3c), a mixture of FeII and FeIII salts showed no particle formation coming from the salt lake with the original mixture. After 20 min without any particle formation, an extra 80 µL of eucalyptus oil was added to the liquid layer. After 5 min, particle formation started, but the maximum particle number concentration of 3.3 × 103 cm–3 was quite low. Also, the particles grew to very large sizes outside the upper diameter limit of the measuring range. Reducing the concentrations of FeII as well as FeIII to 50 % (Table 1, B4; Fig. 3d) led to NPF without further addition of dissolved organic compounds. However, the delay between switching on the lights and the start of NPF was longer than in experiments B1 and B2. The maximum particle number concentration was 2.8 × 103 cm–3, and the maximum GMD was 165 nm.

|
Comparing the maximum particle number concentration and the maximum GMD of the four experiments B1 to B4, it is striking that in B1, the highest particle concentration was observed whereas the GMD was smallest. In experiment B2, the maximum particle number concentration was half of the value of experiment B1, but the GMD was the largest in this series. Even with the highest amount of organic precursor added to the experiment, the mixture of FeII and FeIII in B3 led to a very low maximum particle number concentration of 3.3 × 103 cm–3. Even though the subsequent addition of an extra amount of the organic precursor led to spontaneous NPF and a fast, obvious growth, experiment B3 was excluded from further consideration because of the different starting conditions. Using only half the concentrations of the iron salts in experiment B4 led to the lowest maximum particle number concentration in this series, whereas the GMD was large.
The maximum GMD reached within 60 min of turning on the solar simulator is related to the total aerosol mass or volume formed from the precursor gas and the particle growth rate. Assuming a fixed initial particle number size distribution, a more rapid increase in particle volume leads to a higher growth rate and a large maximum GMD. However, if the concentration of large particles is already high at the beginning of the experiment, even high particle formation intensities will yield low particle growth rates. Therefore, in addition to particle number concentrations and GMD, the maximum particle volume concentration and particle growth rates will be compared in the following text. Particle volume concentrations were estimated from the measured particle number size distributions assuming spherical particles and the GMD of each size bin as the typical diameter of all particles in this size bin.
Experiment B1 with the highest FeII concentration showed the lowest GR of 70 nm h–1. The maximum particle volume concentration was reached 29 min after switching on the lights, with a value of 6 µm3 cm–3, which is quite low. Replacing FeII with FeIII in the salt lake mixture resulted in clearly faster particle growth in experiment B2 and a high maximum particle volume concentration of 57 µm3 cm–3, which was already reached 10 min after starting the solar simulator. From this individual experiment, it remains unclear if excluding FeII from the mixture, the presence of FeIII, or a combination of both have a promoting effect on particle formation and growth. Adding both FeII and FeIII to the salt lake mixture in experiment B4 led to a high GR of 162 nm h–1, and a low maximum particle volume concentration of 2.6 µm3 cm–3 reached 55 min after switching on the solar simulator.
Apparently, even though the particle GR is quite high in experiment B4, the low maximum particle volume concentration reached late in the experiment indicates slow but constant particle formation over an extended time period when both FeII and FeIII are present.
In experiments B1, B2 and B4, i.e. in experiments when NPF was observed, the colour of the salt lake mixture changed from clear to orange-brownish more than 1 h after switching on the solar simulator. This colour change is consistent with conversion of FeII to FeIII. In addition, at the end of the experiments, recrystallisation of salt inside the salt lake mixture was observed. In order to quantify this process, batch experiments studying FeII and FeIII concentrations during the evolution of the salt lake were performed. Over time, FeII concentrations clearly decreased in the solution. This suggests a Fenton-like reaction mechanism in the mixture of iron salts and organic compounds.
Effect of FeII concentration on new particle formation
In order to study the potential effect of a Fenton-like reaction on NPF, the effect of the FeII concentration in the simulated salt lake mixture on particle formation was studied in a third series of experiments (Table 1, C1–3). In a Fenton-like reaction, the oxidation of FeII by H2O2 yields FeIII, a hydroxyl anion and a highly reactive OH radical (Reaction 1).[29,39–41]
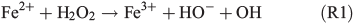
Organic matter is acting as the electron donor for the Fenton reaction in iron-rich environments.[42,43] Hydroxyl in the liquid phase may also be produced photochemically from hydrogen peroxide.[44] Herrmann et al.[45] have estimated that the main source for aqueous OH radicals is the Fenton reaction. Under the assumption that Reaction R1 actually takes place in the simulated salt lake mixture, more FeII would produce more OH radicals. Then, oxidation of the organic precursor and the formation of SOA would be facilitated. In the first experiment C1, the standard concentration of FeII was used as a reference experiment (Fig. 4a). In experiment C2, the FeII concentration was increased by a factor of seven (Fig. 5b), representing the highest FeII concentration observed in natural salt lake samples of Western Australia in 2013. Owing to the fact that an increased concentration of FeII did not lead to more abundant particle formation, a third experiment C3 excluding FeII from the salt lake mixture was run for comparison (Fig. 4c).

|

|
Fig. 4 shows that particle formation was observed quickly after switching on the solar simulator in all three experiments. Apparently, the time delay between illumination and the onset of particle formation increases when more FeII is added to the salt lake mixture. There is also a clear trend with respect to the maximum particle number concentration (cf. Table 1): With the standard FeII concentration in C1, the maximum particle number concentration was 8.3 × 104 cm–3, whereas the maximum particle number concentration was much lower for increased FeII concentrations in C2 (2.2 × 104 cm–3), and higher without FeII in C3 (1.9 × 105 cm–3). This finding is consistent with recent work by Chu et al.,[46] who observed reduced SOA formation in the aqueous phase with increased FeSO4 concentrations. Ultrahigh-resolution mass spectrometry of aerosol samples also shows a loss of CHO oligomers (CH2 homologous series) from iron-free experiment C3 to C1 and C2, and an increase of the chemical diversity in C1 and C2 with a maximum in experiment C1 (cf. Aerosol chemical composition section).
With respect to SOA formation, we have to consider oxidation of the organic precursor compounds 1,8-cineol and limonene both in the gas and in the aqueous phases. Owing to its moderate solubility in water, we assume that a certain fraction of the eucalyptus oil contained in the salt lake mixture will be released into the gas phase. In the gas phase, the OH radical is the main oxidant of 1,8-cineol,[47] and diaterebic acid acetate and diaterpenylic acid acetate have been found to contribute to particle formation as OH oxidation products of 1,8-cineol.[48] Limonene is readily oxidised by ozone and OH radicals, leading to a high aerosol yield.[49] The aqueous phase expands the possible reaction pathways for oxidation of organic compounds. For example, gas-phase ozonolysis of limonene yields water-soluble oligomers, which may be dissolved in the aqueous phase and undergo further oxidation.[50] Also, Fenton-like reactions (Reaction R1) are important aqueous-phase sources of OH radicals for further oxidation. Finally, oxidants other than ozone and OH may be produced in the aqueous phase. This includes the production of the sulfate radical (e.g. Herrmann[51]):
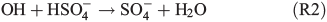
leading to the potential production of organosulfates.[36,52] Aqueous-phase ozone may also react with dissolved chloride to form reactive chlorine species like HOCl:
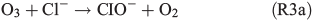
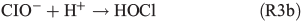
HOCl may further react with dissolved chloride to produce molecular chlorine, which is quickly released to the gas phase and, in the presence of light, photolysed yielding Cl radicals. Previous studies by Lim et al.[53] and Wittmer et al.[54] suggest that FeIII in NaCl solution promotes the release of chlorine from the aqueous phase. In our experiments, the presence of FeII and organic compounds may lead to the production of OH and FeIII by the Fenton-like Reaction R1, subsequently facilitating the release of chlorine. HOCl also reacts with the organic precursor in the aqueous phase to form chlorinated organic matter (cf. Aerosol chemical composition section). Volatile organohalogens (VOX) may be released into the gas phase. By photolytic cleavage of the C–Cl bond, a free halogen radical and a free organic radical are formed. VOX production in the aqueous phase and release into the gas phase are therefore a source for atmospheric reactive halogen species, which could oxidise SOA precursors in the gas phase.[19] It should be noted that the potential oxidation of 1,8-cineol by chlorine radicals is not well quantified yet.
In our experiments C1–C3, when the solar simulator was switched on, illumination initiated the oxidation of VOCs and the simultaneous production of photochemical oxidants and ozone.[14] Ozone production was similar in all three experiments C1 to C3. Maximum ozone mixing ratios were reached ~5 min after illumination, ranging from 45 to 55 ppb (cf. Fig. 4). The lowest ozone production was observed in experiment C3 without any FeII in the simulated salt lake. In comparison with α-pinene and limonene, 1,8-cineol tends to produce higher ozone mixing ratios, as found in previous experiments comparing 1,8-cineol, α-pinene and limonene. The ozone mixing ratios observed in experiments with monoterpenes not carrying an oxygen atom were typically only 10 % of the ozone mixing ratios observed in experiments with 1,8-cineol. Ozone can either act as the primary oxidant of limonene, which is the minor constituent of the eucalyptus oil used in this study, or alternatively, under light conditions, ozone may produce OH radicals, which react with both limonene and 1,8-cineol.
In experiment C1 with the standard FeII concentration, particle formation is observed (Fig. 4a). Increasing the FeII concentration in the salt lake mixture is expected to enhance the production of OH radicals from H2O2 in the aqueous phase. Higher OH concentrations would suggest faster oxidation of the organic precursor compounds, and thus, an increased number and faster formation of particles compared with standard FeII conditions. However, with elevated FeII concentrations in experiment C2 (Fig. 4b), the observed particle formation starts later and with lower intensity. One possible reason is the complexing of organic compounds by irradiated iron oxide.[40] Joshi et al.[55] found a moderate to good chelating activity of FeII and mainly 1,8-cineol-containing oils. This suggests that more organic matter is potentially bound by additional FeII in the aqueous phase than without FeII, and less organic matter may be released into the gas phase for subsequent participation in particle formation. Without the addition of FeII to the salt lake mixture in experiment C3, strong particle formation and CH2 homologous series of oligomers are observed. In this case, it is expected that the organic precursors are mainly oxidised in the gas phase by OH and ozone, and competitive reactions in the aqueous phase are negligible.
Aerosol chemical composition
Analysing the chemical composition of aerosol particles formed in experiments C1 to C3 by EDX, Raman spectroscopy and ultrahigh-resolution mass spectrometry may help to evaluate the relative importance of some of the potential particle formation pathways discussed above.
Electron-dispersive X-ray spectroscopy (Fig. 5) reveals CaSO4 and CaCO3 as the main inorganic species in the aerosol phase. These inorganic species are confirmed by Raman spectroscopy (Fig. 6) also indicating the presence of CaSO4 and CaCO3. The Ca2+ ions appear to originate from impurities in the salt mixture, and further display a phase separation of the involved Ca and Na ions. In addition, in case of the FeII experiments C2, small contributions of haematite (Fe2O3) were identified (Fig. 6).

|
However, a large fraction of the collected aerosol particles exhibits secondary organic features, indicating particle formation based on eucalyptol. All inorganic Raman spectra were referenced to the RRUFF database.[56] To characterise significant changes of the precursor oil (derived from Eucalyptus globulus) related to the chemical process during aerosol formation, reference spectra of the eucalyptus oil and its individual components were recorded (Fig. 7). The main component of the eucalyptus oil used is 1,8-cineol. Further, small contributions of limonene were identified. Beside the dominance of the different deformation and stretching vibrations related to C–H species, the ether group of 1,8-cineol is very predominant at 740 cm–1.

|
Raman spectra were obtained of aerosol particles collected in the C2 experiments (increased FeII concentrations; Fig. 8, blue) and C3 (without FeII; Fig. 8, black). The following interpretation of the Raman spectra is based on Socrates.[57] Many particles exhibit Raman excitations related to CaCO3 at 1085 and 710 cm–1. Owing to the position of the Raman shifts of CaCO3, the interpretation of the Raman spectra is hampered in these spectral regions. All spectra examined in the present work can be related to organic species, indicated by a predominant C–H stretch vibration. The predominance of the CH3 group in the reference spectra (Fig. 7) is also present in the Raman spectra of the aerosol particles (Fig. 8), indicating organic precursors as the main origin of the secondary particles collected. This is also confirmed by the highest number of CHO formula with mainly CH2-homologous series as observed in the mass spectra of experiment C3 (Fig. 9a). Further, the related deformation vibrations at 1430–1450 cm–1 are present. The predominant δ(ring) of the ether group (Fig. 7) of 1,8-cineol was not found in the aerosol phase. Thus, ring-opening at the position of the ether group appears to be a major step in the oxidation mechanism of 1,8-cineol. Predominant species, especially in case of the Raman spectra of the aerosol samples collected in the FeII-rich experiment C2, are νs(CO2–) at 1400 cm–1 and νas(CO2–) at 1600 cm–1. The νs(CO2–) consists of several strong bands, covering other spectral features in this region. These vibrations belong to carboxylic acid salts and can explain the formation of CaCO3 by further decarboxylation. Hence, the simultaneous occurrence of carboxylic acid salts and CaCO3 can be explained. Further, weak ν(C=O) vibrations at ~1730 cm–1 suggest the presence of oxygen-containing species. Indications of various nitrogen- and sulfur-containing species and organosulfates can be found in the Raman spectra of the aerosol particles, and appear to be enhanced in the FeII-rich experiment C2 compared with the FeII-free experiment C3. Bands at 1360 cm–1 (νs(NO2)) and 1530 cm–1 (νas(NO2)) indicate the presence of R–NO2 species. Organic nitrates are represented by the νas(NO3) band at 1630 cm–1 and the νs(NO3) band at 1250 cm–1. The related basic ν(C–N) can be found at ~920 cm–1. Further, the presence of organic sulfate species is demonstrated by the Raman spectra. Organic sulphonates are indicated by νs(SO2) at 1210 cm–1 and νas(SO2) at 1400 cm–1, which is masked by the strong νs(CO2–). The related ν(C–S) can be found at ~620 cm–1. More highly oxidised sulfur species, especially sulphonic acid salts, can be found at 1050 (νs(SO3), covered by CaCO3, and 1200 (νas(SO3)) cm–1. The corresponding ν(S–O) is located at ~850 cm–1. Bands between 680 and 720 cm–1 suggest the presence of C–Cl or further C–S species. However, this spectral region is difficult to evaluate owing to the presence of CaCO3.

|

|
Ultrahigh-resolution mass spectra clearly show CH2 homologous series of CHO compounds in the SOA fraction (Fig. 9), resulting in very regular CHO profiles in the van Krevelen diagrams and continuity in the entire mass range (Fig. 10a). The addition of iron directly affects these CHO oligomers, with increasing production of CHOS compounds with iron addition as observed in the total spectra (Fig. 9c) and in the single nominal-mass signatures (Fig. 9d). Converting all exact masses into thousands of elementary compositions enabled the description of the iron addition in terms of chemical diversity of the compositional space. This is illustrated in Fig. 10b by the number of formulas of specific atomic compositions including CHO, CHOS, CHOCl and others. In a first step, the addition of iron enhances the formation of CHO and generates a series of CHOS and halogenated organic compounds. Further increase in iron (C1 to C2) decreases on average the number of formulas but increases the relative abundance of CHOS and halogenated organic compounds. This is further confirmed when determining the formulas that increase or decrease with addition of iron in the series C3 → C1 → C2 (Fig. 11a, b). It is interesting to note that particularly the number of less oxidised CHO compounds with low O/C ratios decreases when FeII is increased, whereas CHOS and halogenated organic compounds appear or increase in intensity.

|

|
In general, a larger contribution of oxidised species such as carboxylic acid salts and sulfates is visible in the Raman spectra of the FeII-enhanced experiment C2 (Fig. 8, blue spectra) compared with the FeII-free experiment C3 (Fig. 8, black spectra). This larger contribution of CHOS compounds was confirmed by ultrahigh-resolution mass spectrometry (Fig. 9, 10), and is consistent with an important contribution of the Fenton-like Reaction R1, which causes oxidation of the initial organic precursor in the aqueous phase, and formation of organosulfates potentially by the sulfate radical formed in the aqueous-phase Reaction R2.
With respect to the role of reactive halogen species potentially leading to halogen-induced organic aerosol formation or halogenation reactions of pre-existing organic aerosol,[19] the analysis by Raman spectroscopy remains inconclusive. There are no clear indications for a strong contribution of halogen-induced reactions to NPF in the experiments presented, even though organic compounds and halogen species were simultaneously present. However, halogenated organic compounds were detected by ultrahigh-resolution mass spectrometry (Fig. 10).
Finally, diaterebic acid acetate and diaterpenylic acid acetate, tracers of OH oxidation of 1,8-cineol,[48] were confirmed in all aerosol samples collected during the chamber experiments series C with ultrahigh-resolution mass spectrometry (Fig. 12). The fraction of these tracer compounds decreases with the presence of iron, showing their further implication in the formation of sulfur-containing and halogenated species. This indicates that the gas-phase oxidation of the organic precursor compounds always contributed to the particle formation process in our simulation experiments.

|
Conclusions
In the present study, particle formation was observed in an aerosol chamber above a simulated salt lake mixture containing 1,8-cineol and limonene as organic precursor compounds for SOA formation. By evaluating maximum particle number and volume concentrations, maximum geometric mean diameter and particle growth rates in various chamber experiments (cf. Table 1), the concentration of FeII in the salt lake mixture was identified to be a key control factor in the intensity of particle formation. In salt lake environments with low pH values and high solar irradiation, FeII is converted to FeIII in the presence of organic matter in a Fenton-like reaction, which is the main source of highly reactive OH radicals in the aqueous phase.[45] The fact that the variation of FeII concentrations in the salt lake mixtures affects the intensity of particle formation in our experiments suggests a coupling of aqueous-phase chemistry and particle formation.
Release of 1,8-cineol and limonene from the aqueous phase into the gas phase and subsequent oxidation of 1,8-cineol by OH radicals,[47] and of limonene by ozone and OH radicals,[49] contribute to secondary aerosol formation in the gas phase (Fig. 13). Water-soluble products formed in the gas phase, e.g. water-soluble oligomers formed during limonene ozonolysis, may be dissolved in the liquid phase and undergo further aqueous-phase oxidation.[50] The detection of diaterebic acid acetate and diaterpenylic acid acetate in aerosols collected in experiments C1–3 confirms particle formation from the oxidation of 1,8-cineol by OH radicals.[48] Fenton-like reactions suggest additional production of OH radicals in the aqueous phase, and consequently, an increased potential for oxidation of the organic precursor compounds in the aqueous phase leading to less-volatile organic compounds (cf. Fig. 13). If these low-volatility compounds are released from the aqueous phase, they may contribute to SOA formation. However, the experiments C1 to C3 show that increased FeII concentrations reduce the intensity of particle formation observed in the gas phase. The highest particle concentrations were observed when FeII was excluded from the salt lake mixture (experiment C3). This is consistent with recent work by Chu et al.,[46] who observed reduced SOA formation in the presence of FeSO4 in their experiments under wet conditions (50 % relative humidity) in α-pinene–NOx and α-pinene–HONO photooxidation systems. They propose oxidation processes on the wet surface of the SOA layer by OH radicals breaking down the organic oxidation products into smaller molecules, which may be released into the gas phase, thereby reducing the aerosol mass concentration. We suggest that the complexing of organic compounds by irradiated iron oxide,[43] as well as the chelating effect of FeII on 1,8-cineol as observed by Joshi et al.,[48] may also reduce the availability of 1,8-cineol, limonene and their first-generation oxidation products for reactions leading to SOA. The competition of FeII-controlled aqueous-phase chemistry with secondary aerosol formation in the gas phase led to reduced particle formation in our experiments.

|
Besides this competitive effect, however, aqueous-phase chemistry expands the potential oxidation pathways of 1,8-cineol and limonene. The presence of FeII clearly increases the chemical diversity of the organic aerosol (cf. Figs 10, 11). For example, formation of sulfate radicals provides an additional oxidant and the potential for organosulfate formation in the aqueous phase.[52] Contributions of organosulfates to particle formation have been found in the mass spectra and Raman spectra of aerosol collected in experiments C2 and C3. Also, ozone in the aqueous phase may react with dissolved chloride to form reactive chlorine species like HOCl. HOCl may either react with dissolved chloride to produce molecular chlorine, which can produce Cl radicals under irradiation once released into the gas phase, or HOCl may react with organic compounds in the aqueous phase to form chlorinated organic matter as observed, e.g. in the formation of chlorinated by-products in drinking-water production.[58] Volatile organohalogens (VOX) may be released into the gas phase. Both pathways are possible sources for atmospheric reactive halogen species, which may oxidise SOA precursors in the gas phase.[19] Halogenated organic compounds were detected unequivocally by ultrahigh-resolution mass spectrometry (Fig. 10). However, even though dissolved chloride as well as 1,8-cineol and limonene were abundant in our experiments, the formation of halogen-induced organic aerosol (XOA) or halogenation of pre-existing SOA (HaloSOA) as described in Ofner et al.[19] seem to play a minor overall role in NPF in our experiments.
The obvious influence of Fenton-like reactions in the present study emphasises the relevance of the interaction of gas-phase and aqueous-phase chemistry in particle formation. For a better understanding of the relative contributions of these different pathways to particle formation under various atmospherically relevant conditions, more studies have to be carried out analysing systematically the interplay of gas-phase and aqueous-phase chemistry. With respect to FeII, a more detailed and quantitative analysis of the control of FeII over competitive aqueous-phase oxidation pathways of organic compounds is a prerequisite to quantitatively assess its influence on atmospheric particle formation.
Acknowledgements
We are indebted to the HaloProc partners, especially the Western Australia field teams in 2012 and 2013, for field data, ideas and discussions leading to this study. Further, we thank Gernot Friedbacher and Elisabeth Eitenberger (Vienna University of Technology, Institute of Chemical Technologies and Analysis) for obtaining the SEM-EDX data. Funding by the German Research Foundation (DFG) grant HE 5214/5–1 and within the DFG research group HaloProc (FOR 763) is gratefully acknowledged.
References
[1] A. G. Carlton, C. Wiedinmyer, J. H. Kroll, A review of secondary organic aerosol (SOA) formation from isoprene. Atmos. Chem. Phys. 2009, 9, 4987.| A review of secondary organic aerosol (SOA) formation from isoprene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFGlsLnK&md5=394b94edb0c2c0cbf1f8eb08b6814590CAS |
[2] M. Kulmala, V.-M. Kerminen, T. Anttila, A. Laaksonen, C. D. O’Dowd, Organic aerosol formation via sulphate cluster activation. J. Geophys. Res. 2004, 109, D04205.
| Organic aerosol formation via sulphate cluster activation.Crossref | GoogleScholarGoogle Scholar |
[3] K. Tsigaridis, J. Lathière, M. Kanakidou, D. A. Hauglustaine, Naturally driven variability in the global secondary organic aerosol over a decade. Atmos. Chem. Phys. 2005, 5, 1891.
| Naturally driven variability in the global secondary organic aerosol over a decade.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtVejs73K&md5=89f309f4316f09db38f3adf7e18635c6CAS |
[4] K. Tsigaridis, M. Kanakidou, Secondary organic aerosol importance in the future atmosphere. Atmos. Environ. 2007, 41, 4682.
| Secondary organic aerosol importance in the future atmosphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmtVWht78%3D&md5=bfb0e840bc557695f25583f08846710aCAS |
[5] C. L. Heald, D. J. Jacob, R. J. Park, L. M. Russell, B. J. Huebert, J. H. Seinfeld, H. Liao, R. J. Weber, A large organic aerosol source in the free troposphere missing from current models. Geophys. Res. Lett. 2005, 32, L18809.
| A large organic aerosol source in the free troposphere missing from current models.Crossref | GoogleScholarGoogle Scholar |
[6] C. He, F. Murray, T. Lyons, Monoterpene and isoprene emissions from 15 Eucalyptus species in Australia. Atmos. Environ. 2000, 34, 645.
| Monoterpene and isoprene emissions from 15 Eucalyptus species in Australia.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXjvVOhtQ%3D%3D&md5=bfdff42a2dc4f2402208c833dd967e1aCAS |
[7] A. Guenther, C. N. Hewitt, D. Erickson, R. Fall, C. Geron, T. Graedel, P. Harley, L. Klinger, M. Lerdau, W. A. Mckay, T. Pierce, B. Scholes, R. Steinbrecher, R. Tallamraju, J. Taylor, P. Zimmerman, A global-model of natural volatile organic compound emissions. J. Geophys. Res .– Atmos. 1995, 100, 8873.
| A global-model of natural volatile organic compound emissions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXmvFKrsb0%3D&md5=405466a9ed2bb8b0e32b6e89c5c58a0aCAS |
[8] R. A. Rasmussen, What do the hydrocarbons from trees contribute to air pollution? J. Air Pollut. Contr. Assoc. 1972, 22, 537.
| What do the hydrocarbons from trees contribute to air pollution?Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE38XltFOkt78%3D&md5=6de1bd7111a59934765d550447a1c558CAS |
[9] A. Held, A. Nowak, W. Birmili, A. Wiedensohler, R. Forkel, O. Klemm, Observations of particle formation and growth in a mountainous forest region in central Europe. J. Geophys. Res. 2004, 109, D23204.
| Observations of particle formation and growth in a mountainous forest region in central Europe.Crossref | GoogleScholarGoogle Scholar |
[10] J. G. Isebrands, A. B. Guenther, P. Harley, D. Helmig, L. Klinger, L. Vierling, P. Zimmerman, C. Geron, Volatile organic compound emission rates from mixed deciduous and coniferous forests in Northern Wisconsin, USA. Atmos. Environ. 1999, 33, 2527.
| Volatile organic compound emission rates from mixed deciduous and coniferous forests in Northern Wisconsin, USA.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjtVSns7Y%3D&md5=a5667d961fe93ac9806445a2057fe006CAS |
[11] H. Hakola, V. Tarvainen, T. Laurila, V. Hiltunen, H. Hellén, P. Keronen, Seasonal variation of VOC concentrations above a boreal coniferous forest. Atmos. Environ. 2003, 37, 1623.
| Seasonal variation of VOC concentrations above a boreal coniferous forest.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXitVGjtbg%3D&md5=5c4df4fa08d9ac4d61231fc4c638563eCAS |
[12] A. Plewka, T. Gnauk, E. Brüggemann, H. Herrmann, Biogenic contributions to the chemical composition of airborne particles in a coniferous forest in Germany. Atmos. Environ. 2006, 40, 103.
| Biogenic contributions to the chemical composition of airborne particles in a coniferous forest in Germany.Crossref | GoogleScholarGoogle Scholar |
[13] I. Kourtchev, T. Ruuskanen, W. Maenhaut, M. Kulmala, M. Claeys, Observation of 2-methyltetrols and related photooxidation products of isoprene in boreal forest aerosols from Hyytiälä, Finland. Atmos. Chem. Phys. 2005, 5, 2761.
| Observation of 2-methyltetrols and related photooxidation products of isoprene in boreal forest aerosols from Hyytiälä, Finland.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1Kgsr%2FK&md5=2ae33038fc6f6e57bf43eda6d3277ae4CAS |
[14] T. V. Nunes, C. A. Pio, Emission of volatile organic compounds from Portuguese eucalyptus forests. Chemosphere, Glob. Chang. Sci. 2001, 3, 239.
| Emission of volatile organic compounds from Portuguese eucalyptus forests.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXmsValsbw%3D&md5=c536feb0ba7869e37a1dbe8661c22b38CAS |
[15] A. J. Winters, M. A. Adams, T. M. Bleby, H. Rennenberg, D. Steigner, R. Steinbrecher, J. Kreuzwieser, Emissions of isoprene, monoterpene and short-chained carbonyl compounds from Eucalyptus spp. in southern Australia. Atmos. Environ. 2009, 43, 3035.
| Emissions of isoprene, monoterpene and short-chained carbonyl compounds from Eucalyptus spp. in southern Australia.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXls1emurw%3D&md5=64a8165c28f0cfe727b7f47b05d2da97CAS |
[16] F. Loreto, S. Delfine, Emission of isoprene from salt-stressed Eucalyptus globulus leaves. Plant Physiol. 2000, 123, 1605.
| Emission of isoprene from salt-stressed Eucalyptus globulus leaves.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXmtVegs78%3D&md5=e5511f51429013d0e04fdc9829f496a5CAS | 10938376PubMed |
[17] Q. K. Timerghazin, P. A. Ariya, Kinetics of the gas-phase reaction of atomic chlorine with selected monoterpenes. PCCP Phys. Chem. Ch. Ph. 2001, 3, 3981.
| Kinetics of the gas-phase reaction of atomic chlorine with selected monoterpenes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXmvV2it74%3D&md5=cec7f183c66e1e0378fab186800bdcbfCAS |
[18] X. Cai, R. J. Griffin, Secondary aerosol formation from the oxidation of biogenic hydrocarbons by chlorine atoms. J. Geophys. Res. 2006, 111, D14206.
| Secondary aerosol formation from the oxidation of biogenic hydrocarbons by chlorine atoms.Crossref | GoogleScholarGoogle Scholar |
[19] J. Ofner, K. A. Kamilli, A. Held, B. Lendl, C. Zetzsch, Halogen-induced organic aerosol (XOA): a study on ultrafine particle formation and time-resolved chemical characterization. Faraday Discuss. 2013, 165, 135.
| Halogen-induced organic aerosol (XOA): a study on ultrafine particle formation and time-resolved chemical characterization.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvFCrt7zE&md5=3cd9c555d5627c76c141b987497329b4CAS | 24601001PubMed |
[20] M. J. Rossi, Heterogeneous reactions on salts. Chem. Rev. 2003, 103, 4823.
| Heterogeneous reactions on salts.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXovFOhtb8%3D&md5=cb19c2689eeb285c2ec313caf78d6934CAS | 14664635PubMed |
[21] W. Junkermann, J. Hacker, T. Lyons, U. Nair, Land-use change suppresses precipitation. Atmos. Chem. Phys. 2009, 9, 6531.
| Land-use change suppresses precipitation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFWisbrN&md5=a507833bdf290c3c8bf5b2a0529b8737CAS |
[22] J. Stutz, R. Ackermann, J. D. Fast, L. Barrie, Atmospheric reactive chlorine and bromine at the Great Salt Lake, Utah. Geophys. Res. Lett. 2002, 29, 18-1.
| Atmospheric reactive chlorine and bromine at the Great Salt Lake, Utah.Crossref | GoogleScholarGoogle Scholar |
[23] W. D. Williams, Salinisation: a major threat to water resources in the arid and semi-arid regions of the world. Lakes Reservoirs: Res. Manage. 1999, 4, 85.
| Salinisation: a major threat to water resources in the arid and semi-arid regions of the world.Crossref | GoogleScholarGoogle Scholar |
[24] M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, Th. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski, J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155.
| The formation, properties and impact of secondary organic aerosol: current and emerging issues.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFGhs77M&md5=cc963da69e913dd9c61b5a6dcc3af23cCAS |
[25] Y. B. Lim, Y. Tan, M. J. Perri, S. P. Seitzinger, B. J. Turpin, Aqueous chemistry and its role in secondary organic aerosol (SOA) formation. Atmos. Chem. Phys. 2010, 10, 10521.
| Aqueous chemistry and its role in secondary organic aerosol (SOA) formation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjs1Kjtb0%3D&md5=af5bab75bd92fa0e18519ab33123fc4eCAS |
[26] M. Claeys, W. Wang, A. C. Ion, I. Kourtchev, A. Gelencsér, W. Maenhaut, Formation of secondary organic aerosols from isoprene and its gas-phase oxidation products through reaction with hydrogen peroxide. Atmos. Environ. 2004, 38, 4093.
| Formation of secondary organic aerosols from isoprene and its gas-phase oxidation products through reaction with hydrogen peroxide.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXltlyhs78%3D&md5=ce7858a6b88ccd79627416fb58ffb735CAS |
[27] A. G. Carlton, B. J. Turpin, H.-J. Lim, K. E. Altieri, S. Seitzinger, Link between isoprene and secondary organic aerosol (SOA): pyruvic acid oxidation yields low-volatility organic acids in clouds. Geophys. Res. Lett. 2006, 33, L06822.
| Link between isoprene and secondary organic aerosol (SOA): pyruvic acid oxidation yields low-volatility organic acids in clouds.Crossref | GoogleScholarGoogle Scholar |
[28] H. J. H. Fenton, Oxidation of tartaric acid in presence of iron. J. Chem. Soc. 1894, 65, 899.
| Oxidation of tartaric acid in presence of iron.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaD28XmtlCnsQ%3D%3D&md5=df6c09b18b8ca208e8aafc79b1a660efCAS |
[29] W. J. Cooper, R. G. Zika, R. G. Petasne, J. M. C. Plane, Photochemical formation of H2O2 in natural waters exposed to sunlight. Environ. Sci. Technol. 1988, 22, 1156.
| Photochemical formation of H2O2 in natural waters exposed to sunlight.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXltlCmsb0%3D&md5=d607cc53fd87bd907776259808b668f5CAS | 22148607PubMed |
[30] T. Krause, C. Tubbesing, K. Benzing, H. F. Schöler, Model reactions and natural occurrence of furans from hypersaline environments. Biogeosciences 2014, 11, 2871.
| Model reactions and natural occurrence of furans from hypersaline environments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhs1Sju73J&md5=7cb50be77d560436d32122398576f2cbCAS |
[31] H. B. Dunford, Oxidation of iron(II)/(III)I by hydrogen peroxide: from aquo to enzyme. Coord. Chem. Rev. 2002, 233–234, 311.
| Oxidation of iron(II)/(III)I by hydrogen peroxide: from aquo to enzyme.Crossref | GoogleScholarGoogle Scholar |
[32] L. Deguillaume, M. Leriche, N. Chaumerliac, Impact of radical versus non-radical pathway in the Fenton chemistry on the iron redox cycle in clouds. Chemosphere 2005, 60, 718.
| Impact of radical versus non-radical pathway in the Fenton chemistry on the iron redox cycle in clouds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXltlajsb4%3D&md5=f54a4cd46e5a9660ee7cc61d355e67e7CAS | 15963810PubMed |
[33] J. Ofner, H.-U. Krüger, H. Grothe, P. Schmitt-Kopplin, K. Whitmore, C. Zetzsch, Physico-chemical characterization of SOA derived from catechol and guaiacol – a model substance for the aromatic fraction of atmospheric HULIS. Atmos. Chem. Phys. 2011, 11, 1.
| Physico-chemical characterization of SOA derived from catechol and guaiacol – a model substance for the aromatic fraction of atmospheric HULIS.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXktlanurw%3D&md5=86c248dbdc87e2d90b7627e6fe0cdab6CAS |
[34] A. Wiedensohler, W. Birmili, A. Nowak, A. Sonntag, K. Weinhold, M. Merkel, B. Wehner, T. Tuch, S. Pfeifer, M. Fiebig, A. M. Fjäraa, E. Asmi, K. Sellegri, R. Depuy, H. Venzac, P. Villani, P. Laj, P. Aalto, J. A. Ogren, E. Swietlicki, P. Williams, P. Roldin, P. Quincey, C. Hüglin, R. Fierz-Schmidhauser, M. Gysel, E. Weingartner, F. Riccobono, S. Santos, C. Grüning, K. Faloon, D. Beddows, R. Harrison, C. Monahan, S. G. Jennings, C. D. O’Dowd, A. Marinoni, H.-G. Horn, L. Keck, J. Jiang, J. Scheckman, P. H. McMurry, Z. Deng, C. S. Zhao, M. Moerman, B. Henzing, G. de Leeuw, G. Löschau, S. Bastian, Mobility particle size spectrometers: harmonization of technical standards and data structure to facilitate high-quality long-term observations of atmospheric particle number size distributions. Atmos. Meas. Tech. 2012, 5, 657.
| Mobility particle size spectrometers: harmonization of technical standards and data structure to facilitate high-quality long-term observations of atmospheric particle number size distributions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtFCmtb7N&md5=54c415ab48bb0d515640116222366df5CAS |
[35] Y. Batonneau, S. Sobanska, J. Laureyns, C. Bremard, Confocal microprobe Raman imaging of urban tropospheric aerosol particles. Environ. Sci. Technol. 2006, 40, 1300.
| Confocal microprobe Raman imaging of urban tropospheric aerosol particles.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xlt1Ogtg%3D%3D&md5=66a6cb6f672e61e30bdc7d246ec9e378CAS | 16572789PubMed |
[36] P. Schmitt-Kopplin, A. Gelencser, E. Dabek-Zlotorzynska, G. Kiss, N. Hertkorn, M. Harir, Y. Hong, I. Gebefugi, Analysis of the unresolved organic fraction in atmospheric aerosols with ultrahigh-resolution mass spectrometry and nuclear magnetic resonance spectroscopy: organosulfates as photochemical smog constituents. Anal. Chem. 2010, 82, 8017.
| Analysis of the unresolved organic fraction in atmospheric aerosols with ultrahigh-resolution mass spectrometry and nuclear magnetic resonance spectroscopy: organosulfates as photochemical smog constituents.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht1WmtrrF&md5=74a49950e19e8b8684e3c7f1b9d79d91CAS | 20879800PubMed |
[37] L. Boutegrabet, B. Kanawati, I. Gebefügi, D. Peyron, Ph. Cayot, R. D. Gougeon, Ph. Schmitt-Kopplin, Chloride anion attachment to sugars. Mechanistic investigation and discovery of a new dopant for efficient sugar ionization/detection in mass spectrometers. Chemistry 2012, 18, 13059.
| Chloride anion attachment to sugars. Mechanistic investigation and discovery of a new dopant for efficient sugar ionization/detection in mass spectrometers.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xht12js7nN&md5=719c58d48f1f2851ecc95581b8909edaCAS | 22936644PubMed |
[38] Ph. Schmitt-Kopplin, G. Liger-Belair, B. P. Koch, R. Flerus, G. Kattner, M. Harir, B. Kanawati, M. Lucio, D. Tziotis, N. Hertkorn, I. Gebefügi, Dissolved organic matter in sea spray: a transfer study from marine surface water to aerosols. Biogeosciences 2012, 9, 1571.
| Dissolved organic matter in sea spray: a transfer study from marine surface water to aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xhs1Gks7jM&md5=5890c1ee84a9825704e345ccf2257eb9CAS |
[39] F. Haber, J. Weiss, Über die Katalyse des Hydroperoxydes. Naturwissenschaften 1932, 20, 948.
| Über die Katalyse des Hydroperoxydes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA3sXhslWktg%3D%3D&md5=b6f2b23820e30169f201002f916825cfCAS |
[40] P. Mazellier, B. Sulzberger, Diuron degradation in irradiated, heterogeneous iron/oxalate systems: the rate-determining step. Environ. Sci. Technol. 2001, 35, 3314.
| Diuron degradation in irradiated, heterogeneous iron/oxalate systems: the rate-determining step.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXkvFCrsLc%3D&md5=8515892690d6773dad05025e50b192faCAS | 11529570PubMed |
[41] C. K. Remucal, D. L. Sedlak, The role of iron coordination in the production of reactive oxidants from ferrous iron oxidation by oxygen and hydrogen peroxide. ACS Symposium Series 2011, 1071, 177.
| The role of iron coordination in the production of reactive oxidants from ferrous iron oxidation by oxygen and hydrogen peroxide.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XisFaguro%3D&md5=202034d5d2e2b705fe647e219358be6dCAS |
[42] B. M. Voelker, B. Sulzberger, Effects of fulvic acid on FeII oxidation by hydrogen peroxide. Environ. Sci. Technol. 1996, 30, 1106.
| Effects of fulvic acid on FeII oxidation by hydrogen peroxide.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28Xht1ylsb8%3D&md5=79db734c9eb2bd1732322eb44a3a5472CAS |
[43] B. M. Voelker, F. M. M. Morel, B. Sulzberger, Iron redox cycling in surface waters: effects of humic substances and light. Environ. Sci. Technol. 1997, 31, 1004.
| Iron redox cycling in surface waters: effects of humic substances and light.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXhvFGgsbs%3D&md5=4b2b2c15b33a46660a891ef38f1140f9CAS |
[44] P. Warneck, The relative importance of various pathways for the oxidation of sulfur dioxide and nitrogen dioxide in sunlit continental fair weather clouds. Phys. Chem. Chem. Phys. 1999, 1, 5471.
| The relative importance of various pathways for the oxidation of sulfur dioxide and nitrogen dioxide in sunlit continental fair weather clouds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXos1aksw%3D%3D&md5=8512c5f84d388d2f32e45a6b1085275dCAS |
[45] H. Herrmann, A. Tilgner, P. Barzaghi, Z. Majdik, S. Gligorovski, L. Poulain, A. Monod, Towards a more detailed description of tropospheric aqueous phase organic chemistry: CAPRAM 3.0. Atmos. Environ. 2005, 39, 4351.
| Towards a more detailed description of tropospheric aqueous phase organic chemistry: CAPRAM 3.0.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmtlKrtb8%3D&md5=40a171b6d0366a2c60c58aefabf50784CAS |
[46] B. Chu, Y. Liu, J. Li, H. Takekawa, J. Liggio, S.-M. Li, J. Jiang, J. Hao, H. He, Decreasing effect and mechanism of FeSO4 seed particles on secondary organic aerosol in α-pinene photooxidation. Environ. Pollut. 2014, 193, 88.
| Decreasing effect and mechanism of FeSO4 seed particles on secondary organic aerosol in α-pinene photooxidation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXht1ensbjE&md5=b8a07a286871df488c6992ee652e4f3fCAS | 25014016PubMed |
[47] S. B. Corchnoy, R. Atkinson, Kinetics of the gas-phase reactions of hydroxyl and nitrogen oxide (NO3) radicals with 2-carene, 1,8-cineole, p-cymene, and terpinolene. Environ. Sci. Technol. 1990, 24, 1497.
| Kinetics of the gas-phase reactions of hydroxyl and nitrogen oxide (NO3) radicals with 2-carene, 1,8-cineole, p-cymene, and terpinolene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXltlOlu7c%3D&md5=7f597d1956fd830bd44a32a6bee117aaCAS |
[48] Y. Iinuma, O. Böge, M. Keywood, T. Gnauk, H. Herrmann, Diaterebic acid acetate and diaterpenylic acid acetate: atmospheric tracers for secondary organic aerosol formation from 1,8-cineole oxidation. Environ. Sci. Technol. 2009, 43, 280.
| Diaterebic acid acetate and diaterpenylic acid acetate: atmospheric tracers for secondary organic aerosol formation from 1,8-cineole oxidation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsV2rtbrF&md5=df4210e52caadd3a4b85cfa82e44f584CAS | 19238952PubMed |
[49] A. Calogirou, B. R. Larsen, D. Kotzias, Gas-phase terpene oxidation products: a review. Atmos. Environ. 1999, 33, 1423.
| Gas-phase terpene oxidation products: a review.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXhsVyks74%3D&md5=3e11b35b973841ceebb28540ae30981aCAS |
[50] A. P. Bateman, S. A. Nizkorodov, J. Laskin, A. Laskin, Photolytic processing of secondary organic aerosols dissolved in cloud droplets. PCCP Phys. Chem. Ch. Ph. 2011, 13, 12199.
| Photolytic processing of secondary organic aerosols dissolved in cloud droplets.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnslOnt78%3D&md5=e72f0713db97b9948459e7e6b5a93c0dCAS |
[51] H. Herrmann, Kinetics of aqueous-phase reactions relevant for atmospheric chemistry. Chem. Rev. 2003, 103, 4691.
| Kinetics of aqueous-phase reactions relevant for atmospheric chemistry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXovFOhtb0%3D&md5=2fa383f4e068e36b9f5c826e1151d343CAS | 14664629PubMed |
[52] B. Nozière, S. Ekström, T. Alsberg, S. Holmström, Radical-initiated formation of organosulfates and surfactants in atmospheric aerosols. Geophys. Res. Lett. 2010, 37, L05806.
| Radical-initiated formation of organosulfates and surfactants in atmospheric aerosols.Crossref | GoogleScholarGoogle Scholar |
[53] M. Lim, K. Chiang, R. Amal, Photochemical synthesis of chlorine gas from iron(III) and chloride solution. J. Photochem. Photobiol. Chem. 2006, 183, 126.
| Photochemical synthesis of chlorine gas from iron(III) and chloride solution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xos12rtb4%3D&md5=11fa0692117fb7b1a209433102459ddcCAS |
[54] J. Wittmer, S. Bleicher, C. Zetzsch, Iron(III)-induced activation of chloride and bromide from modeled saltpans. J. Phys. Chem. A 2015, 119, 4373.
| Iron(III)-induced activation of chloride and bromide from modeled saltpans.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhsF2nurnO&md5=f451edc24d63a93b4cf1f56d3ecee8e5CAS | 25243918PubMed |
[55] S. Joshi, C. S. Chanotiya, G. Agarwal, O. Prakash, A. K. Pant, C. S. Mathela, Terpenoid compositions, and antioxidant and antimicrobial properties of the rhizome essential oils of different Hedychium species. Chem. Biodivers. 2008, 5, 299.
| Terpenoid compositions, and antioxidant and antimicrobial properties of the rhizome essential oils of different Hedychium species.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXjtFCksLo%3D&md5=9458563ebb2d05bce022125e6b709400CAS | 18293443PubMed |
[56] R. T. Downs, The RRUFF Project: an integrated study of the chemistry, crystallography, Raman and infrared spectroscopy of minerals, in Program and Abstracts of the 19th General Meeting of the International Mineralogical Association in Kobe, Japan 2006, O03–13. Available at http://www.geo.arizona.edu/xtal/group/pdf/IMA19_O03-13.pdf [Verified 23 May 2015].
[57] G. Socrates, Infrared and Raman Characteristic Group Frequencies, 3rd edn 2011 (Wiley: Chichester, UK).
[58] E. E. Lavonen, M. Gonsior, L. J. Tranvik, Ph. Schmitt-Kopplin, S. J. Köhler, Preferential chlorination of oxidized natural organic matter – identification of new disinfection by-products. Environ. Sci. Technol. 2013, 47, 2264.
| Preferential chlorination of oxidized natural organic matter – identification of new disinfection by-products.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvFaltLs%3D&md5=4f4943d4e046aab665383f447d742371CAS | 23373647PubMed |