

Ru/MgO-catalysed selective aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid
Priya Lokhande A B C , Paresh L. Dhepe A B * , Karen Wilson D * and Adam F. Lee D *
D *
A
B
C
D
Handling Editor: Curt Wentrup
Abstract
Biomass valorisation through the selective oxidation of carbohydrate and lipid derivatives offers access to an array of platform chemicals through energy- and atom-efficient catalytic processes. Supported metal nanoparticles are promising catalysts for the aerobic selective oxidation of 5-hydroxymethylfurfural (HMF) to 2,5-furandicarboxylic acid (FDCA), but typically require strong liquid base to achieve high selectivity. Here, we explore the utility of MgO as a solid base support for the Ru-catalysed aerobic oxidation of HMF, obtaining 68% FDCA yield at 160°C and 1.5 MPa of O2 using <1 mol-% metal.
Keywords: bifunctional, biomass, catalysis, MgO, monomer, oxidation, platform chemical, ruthenium.
Introduction
Climate change mitigation and the desire for improved resource utilisation and materials circularity is driving global demand for sustainable fuels and chemicals, particularly those sourced from non-fossil carbon.1,2 Although there are many routes to liquid organic hydrocarbons as transportation fuels, lignocellulosic biomass is the most energy- and atom-efficient route to oxygenated organic molecules3,4 that are the basis of many commodity, fine and speciality chemicals, and pharmaceuticals.5,6 Of biorefinery carbohydrates identified by the US Department of Energy as likely to deliver high-value biobased products,7–9 5-hydroxymethylfurfural (HMF)10,11 and its derivative 2,5-furandicarboxylic acid (FDCA), are approaching commercialisation, notably through the efforts of Avantium (and BASF) to manufacture polyethylene furanoate (PEF) as a replacement for fossil-derived polyethylene terephthalate (PET, Scheme 1).12,13
Synthesis of biomass-derived PEF from FDCA monomer v. fossil-derived PET synthesis from terephthalic acid (TPA) monomer and comparative physicochemical properties of the polymers (where X is the value for PET). MEG, monoethylene glycol.
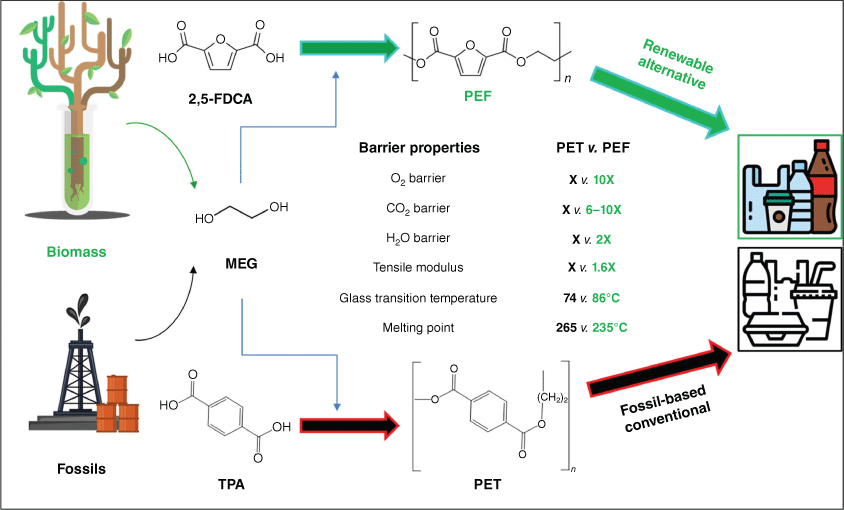
PEF offers enhanced barrier properties and increased mechanical strength relative to PET (Scheme 1), and the FDCA monomer from which it is obtained can theoretically be produced by the direct aerobic oxidation of cheap fructose, glucose or cellulose feedstocks.14 However, to date, the direct oxidation of carbohydrates requires multiphasic systems15 that hinder separation from levulinic co-products, or multiple unit operations and hazardous oxidants that challenge process safety and efficiency.16 Academic and pre-commercial studies of FDCA production thus favour the (more selective) aqueous phase, cascade oxidation of (albeit more expensive) HMF in water (Scheme 2) over heterogeneous catalysts, which can proceed by 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) or diformylfuran (DFF) intermediates.17–22 HMF itself can be prepared by the one-pot isomerisation and dehydration of cellulosic hexoses over solid acid catalysts.23–29 Note that HMF oxidation to FDCA can be readily achieved using homogeneous catalysts, e.g. metal bromides in an acidic medium (~60% yield),30 but such approaches are not amenable to process intensification through continuous flow reactors and generate copious aqueous waste streams.
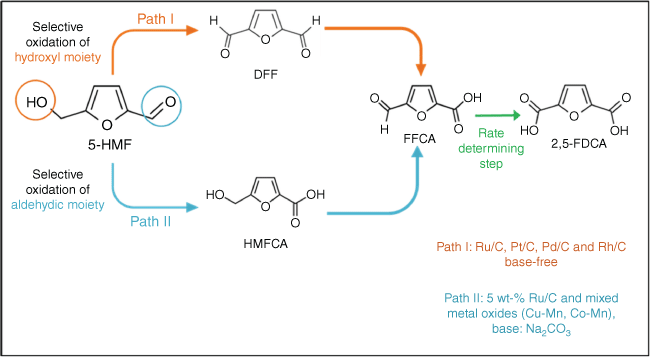
Noble metals (Pt, Pd, Au and Ru) are the most widely studied for the aerobic oxidation of HMF over carbon and oxide supports,33–36 with electrocatalytic routes emerging in recent years that exploit noble metals and non-noble metal phosphides, sulfides, borides, nitrides and hydroxides at close to ambient temperature.37–39 Many thermal and electrocatalytic oxidations employ soluble base (typically NaOH, KOH, Na2CO3 or K2CO3) to accelerate HMF activation and formation of the geminal diol precursor to HMFCA (Scheme 2, Path II), which is often considered rate-determining for FDCA production. Unfortunately, soluble acids and bases can also promote HMF degradation to humins,27,40,41 hindering FDCA separation and resulting in copious alkali-contaminated wastewater; hence, the use of (chemically robust) solid base supports is attractive. Metal oxide and hydroxide supports prepared by alkali precipitation of nitrate precursors were reported as potential solid base supports for noble metal-catalysed HMF oxidation to FDCA42; however, their mode of action is likely through leaching of soluble alkalis.43,44 A systematic study of Au nanoparticles on an alkali-free hydrotalcite, prepared using ammonium hydroxide/carbonate, confirmed that soluble base was essential to obtain a high FDCA yield when using low metal loadings.43 Strong adsorption of HMF at Au sites necessitates a homogeneous base-catalysed initial oxidation of HMF to HMFCA, unless high metal loadings are used that permit oxidation of reactively formed HMFCA to FFCA and FDCA (78% yield for 10 wt-% Au/hydrotalcite (HT) at 90°C and 0.1 MPa of O2 in the absence of soluble base). Although Gupta et al. report 99% FDCA yield over a 2 wt-% Au/HT at 95°C, this reflects a homogeneous contribution from sodium entrained during the HT synthesis.19 Gold deposited on Mg(OH)2 achieved a 93% FDCA yield at 110°C and 0.5 MPa of O2 within 2 h, although potential leaching of Mg in subcritical water was not discussed.45 Bimetallic nanoparticles (Ni, Co, or Cu with Pd) on Mg(OH)2 nanoflakes have also been studied for HMF oxidation, with Ni0.90Pd0.10/Mg(OH)2 delivering 86% isolated FDCA yield at 100°C under 0.1 MPa of air and 16 h; note a potassium tetrachloropalladate precursor was used and hence soluble base catalysis cannot be discounted. Han et al. described a solid base Pt/C–O–Mg catalyst for HMF oxidation, prepared by carbonising the surface of MgO particles and then dissolving the majority of Mg using HCl before Pt deposition; although a 75% isolated yield of high-purity FDCA resulted at 110°C and 1 MPa of O2 after 6-h reaction, the catalyst synthesis again involved alkali reagents (NaBH4 reductant).46 Mani et al. explored the use of ‘concentrated’ (2–10 wt-%) HMF feedstreams for large-scale FDCA production over MnO₂ and MnCo2O4-supported Ru catalysts. Catalysts were prone to Mn leaching in the absence of soluble base, resulting in the formation of Mn-FDCA complexes and corresponding deactivation and poor mass balances.47 Weak soluble bases mitigated Mn leaching, enabling almost quantitative conversion of HMF to FDCA at 100°C, albeit at a modest HMF:Ru molar ratio of 32 and requiring 3 MPa of O2.
Photocatalytic conversion of biomass-derived molecules has emerged as a promising approach for producing high-value chemicals, with HMF oxidation to FDCA demonstrated using Au-Ag/TiO2 catalysts under visible light. Synergies between the bimetals and plasmonic effects promoted the selective oxidation pathway, with the optimum Ag0.33Au0.66/TiO2 catalyst delivering 43% HMF conversion and 98% FDCA selectivity over 5 h under >420-nm irradiation.48 However, as for most thermocatalytic studies, a soluble base (Na2CO3) was used and hence contributions from homogeneous base catalysis cannot be discounted. Plasmonic Au nanoparticles supported on ZnO have also been studied for HMF oxidation under similar conditions, i.e. in the previous of excess Na2CO3 that may have compromised the resulting catalytic performance of 56% HMF conversion at 97% FDCA selectivity.49 The true potential of such photocatalytic systems remains to be established, but issues such as light penetration and oxidation of plasmonic metals such as Ag and Cu may prove challenging to overcome.
Ruthenium and its oxides are widely reported for the catalytic selective oxidation of amines50 and alcohols,51–55 such as benzyl alcohol to benzaldehyde21,56 and HMF to FDCA.16,31,36,57,58 Kobayashi and co-workers demonstrated the efficacy of supported ruthenium catalysts for the oxidation of alcohols to aldehydes and ketones in batch and flow reactors.54 Gorbanev et al. achieved a quantitative yield of FDCA over 2 wt-% Ru(OH)x/MgO at 0.25 MPa of O2 and 140°C in ~6 h (Table S1) from a catalyst synthesised using NaOH, but observed significant Mg leaching57; by contrast, 2 wt-% Ru(OH)x/MgAlO4 exhibited negligible leaching at this temperature, but required 0.5 MPa of O2 and 18-h reaction for ~57% FDCA yield. Antonyraj et al. obtained 95% FDCA yield over 2 wt-% Ru/MgO at 160°C and 0.6 MPa of O2 in 4 h, with good stability observed over five reaction cycles.58 MgAl2O4 spinel, MgO-La2O3, CeO2 and hydrotalcite supports have also been explored in conjunction with Ru(OH)x in a range of methylimidazolium ionic liquids, of which 1-ethyl-3-methylimidazolium acetate ([EMIm][OAc]) proved optimal.59 Dispersing Ru(OH)x over hydrotalcite, La2O3 and spinal supports afforded >90% HMF conversion at 140°C in air, but with <15% FDCA yield and required 24-h reaction. Oxidation of HMFCA appeared rate-limiting for all supports. Even at O2 pressures of 1 and 3 MPa, the maximum FDCA yield was only 31% over the most promising La2O3 support, and Ru leaching was problematic (5–10 wt-% of initial metal lost after 6-h reaction in [EMIm][OAc]).
In toluene, Ru/MgO selectively oxidised HMF to DFF (96% yield) at 110°C and 2 MPa of O2 in toluene, ∼30% HMF conversion.32 Selective DFF production is also reported for 4 wt-% Ru supported on a covalent triazine framework under mild conditions of 80°C and 2 MPa of air (64% yield), with only low (16%) HMF conversion for 6 wt-% Ru over an MgO support.60 The latter studies evidence a strong mechanistic sensitivity to the reaction solvent. Weakly basic supports also favour DFF v. FDCA.61,62 Challenges in commercialising FDCA production from HMF persist, highlighted by a Strengths, Weaknesses, Opportunities and Threats (SWOT) analysis (Supplementary Fig. S1), notably the impact of reaction conditions on catalyst performance.
Here, we report the synthesis, characterisation and application of Ru/MgO catalysts for the aqueous phase selective aerobic oxidation of HMF to FDCA, noting the significant effect of support calcination temperature, and reaction temperature and pressure.
Experimental
Material and reagents
All chemicals employed in this work were of AR grade and used as received. Magnesium oxide light, MgO (96%), was purchased from Loba Chemicals, India. Ruthenium(III) chloride hydrate (45–55 wt-% metal) was obtained from Sigma–Aldrich. Deionised water was used to prepare solutions. 5-Hydroxymethylfurfural (HMF, ≥99%), 5-hydroxymethyl-2-furancarboxylic acid (HMFCA), 5-formyl-2-furoic acid (FFCA), 2,5-furandicarboxylic acid (FDCA, 99%) and 2,5-furandicarboxaldehyde (DFF), were purchased from Sigma–Aldrich and Alfa Aesar. 4-Methylpentan-2-one (methyl isobutyl ketone, GR) and ethyl acetate (GR) were purchased from Loba Chemie, India. All reagents were used as received without any further purification.
Catalyst synthesis
Heterogeneous Ru/MgO catalysts were synthesised by the wet impregnation of RuCl3 over different MgO supports. Supports were prepared by heating commercial MgO at 60°C for 16 h, and subsequently 150°C for 6 h under vaccum. The resulting white powders were then calcined in a muffle furnace at 300, 400, 500 or 600°C for 2 or 4 h, and stored in a vacuum desiccator before use. Appropriate concentrations of aqueous solutions of RuCl3 in deionised water (0.098 mmol in 2 mL for 0.5 wt-% Ru) were added dropwise to calcined MgO supports, and the resulting slurries stirred for 16 h at 35°C. Water was then removed using a rotary evaporator, and the obtained solid dried at 60°C for 16 h, and subsequently at 150°C under vaccum for 6 h. Ru-impregnated MgO powders were then reduced under flowing H2 (10 mL min−1) at 150°C for 2 h. The light grey catalysts were stored in a desiccator to prevent water and CO2 adsorption. Sample designations reflect the Ru metal loading and calcination temperature and time, e.g. 0.5 wt-% Ru/MgO, 300°C, 2 h.
HMF oxidation
All the reactions were carried out in a 100-mL Teflon-lined Parr steel autoclave. A known quantity of HMF, deionised water and catalyst were added to the reactor. The reactor was subsequently flushed multiple times with oxygen, filled with oxygen to the desired pressure, and heated to the desired temperature with stirring at 300 rpm to promote oxygen dissolution. Once the desired temperature was achieved, stirring was increased to 800 rpm to initiate reaction while the oxygen inlet was regulated to maintain a constant pressure. The solution pH was measured before and after reaction. The analytical protocol was as previously decsribed.36 Reactions were typically performed at 160°C at which the vapour pressure of water is 0.7 MPa, far less than the 1.5 MPa of O2 pressure typically maintained in reactions. Considering the polar nature of the reactants and products, we anticipate that all organic molecules remained in the aqueous phase under such conditions. Note that one experiment was performed at 160°C and 0.5 MPa of O2, under which partitioning of HMF, HMFCA, DFF, FFCA and FDCA between liquid and vapour water phases is anticipated (this could account in part for the low reactivity discussed later).
Results and discussion
Catalyst characterisation
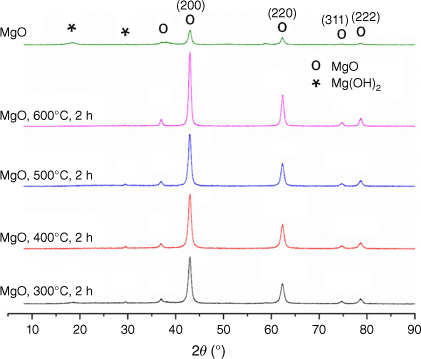
Bulk and surgface physicochemical properties of catalysts were investigated by X-ray diffraction (XRD), scanning electron microscopy (SEM), transmission electron microscopy (TEM), temperature-programmed desorption (TPD) and nitrogen porosimetry. The XRD patterns of MgO calcined at different temperatures (300, 400, 500 and 600°C) are shown in Fig. 1. The XRD patterns of MgO show main peaks corresponding to the MgO periclase phase (Joint Committee on Powder Diffraction Standards, JCPDS: 00-045-0946), and weak peaks corresponding to Mg(OH)2 or the brucite phase (JCPDS: 84-2163) can also be seen. The formation of Mg(OH)2 is due to the reaction of active MgO with water (moisture in the environment). The average crystallite size obtained from XRD is seen to increase with an increase in calcination temperature (Table 1). To understand the morphology and nature of the ruthenium loading, TEM was carried out. TEM images of the freshly prepared catalyst 0.5 wt-% Ru/MgO, 300°C, 2 h are provided in Supplementary Fig. S2. The presence of small black particles of ruthenium on the MgO with an average particle size of 20nm was confirmed using ImageJ software (ver. 1.54, W. S. Rasband, US National Institutes of Health, Bethesda, MD, USA, see https://imagej.nih.gov/ij/). N2 physisorption was performed to examine the pore diameter and surface area of the catalysts (Table 1). The main goal of varying the calcination temperature was to study the redistribution of active catalytic sites and their effects on catalytic reactions. This is because redistribution can affect the accessibility of sites on the catalyst support. As seen, when MgO was calcined at 300°C, it showed a larger specific surface area of 72 m2 g−1 (determined by the Brunauer–Emmett–Teller method) than MgO calcined at higher temperatures. For structure-insensitive reactions, higher surface areas increase the concentration of active sites. Higher calcination temperatures can cause the fusion of support particles, thereby reducing the surface area of the support. With this, it was expected that Ru supported on these supports could form larger particles; pore volume and pore size data show collapse or restructuring of pores, diminishing the effective surface area available for catalysis with an increase in temperature. These results are in line with the XRD results, wherein with an increase in calcination temperature, average crystallite size increased.
| Catalyst | Mean crystallite size (nm) | Surface area (m2 g−1) | Mean pore volume (cm3 g−1) | Mean mesopore diameter (nm) | |
|---|---|---|---|---|---|
| MgO | 10 | ||||
| MgO, 300°C, 2 h | 5 | 72 | 0.16 | 4.3 | |
| MgO, 400°C, 2 h | 7 | 71 | 0.15 | 4.4 | |
| MgO, 500°C, 2 h | 6 | 65 | 0.16 | 4.5 | |
| MgO, 600°C, 2 h | 11 | 45 | 0.19 | 4.9 |
TPD analysis with CO2 as a probe molecule indicated the total amount of basic sites present on the MgO support and catalyst (Table 2). The basicity of MgO primarily arises from cation vacancies (Lewis base sites) and hydroxyl groups (Brønsted base sites).63–65 Extended calcination times and higher temperatures promote the formation of more stable (less defective) and dehydroxylated MgO surfaces, and hence typically confer lower basicity. Higher calcination temperatures also influence the morphology and structure of the MgO support, from N2 sorption study. At elevated temperatures, transformations such as sintering may occur, affecting the distribution and strength of basic sites. Prolonged or high-temperature calcination can induce the formation of less basic phases on the MgO surface, such as periclase (MgO in a highly crystalline form), which may contribute to the observed decrease in basicity. Structural changes, such as particle growth or phase transitions, can decrease the number of active basic sites on the MgO surface. TPD measurements confirmed that higher calcination temperatures and longer calcination times decreased the total basic site density, consistent with an increase in MgO crystallite size and loss of active surface area, but had little effect on the strength (CO2 desorption temperature) of basic sites.
Catalytic HMF oxidation
The effect of calcination temperature on 0.5 wt-% Ru/MgO catalysts was subsequently studied (Table 3). The catalytic results for reactions carried out at 140°C for 6.5 h suggest that elevating the calcination temperature translates into a reduction in yield of FDCA. When the results for entries 1 and 2 (samples calcined at 300°C for 2 or 4 h respectively) are compared, it is seen that the Ru/MgO, 300°C, 2 h catalyst formed more unidentified products. Note that based on the catalyst characterisation, with an increase in calcination temperature of MgO, decrease in overall basicity, surface area and pore diameter were observed, which play an important role in deciding the activity of the catalyst. Nevertheless, solutions of all catalysts exhibited a similar pH of ~9.5. The optimal calcination temperature for achieving maximum conversion of HMF (51%) and FDCA yield (23%) after 4 h was 300°C. As HMFCA and DFF were observed, it appears that both Pathways I and II operate (Scheme 2); the latter was not observed for Ru/C catalysts in our previous work.66 From the results in Tables 2–3, FDCA production requires a support with good basicity and high surface area.
| Catalyst | HMF conversion (%) | FDCA yield (%) | HMFCA yield (%) | FFCA yield (%) | DFF yield (%) | Unidentified products (%) | Initial pH | Final pH | |
|---|---|---|---|---|---|---|---|---|---|
| 0.5 wt-% Ru/MgO, 300°C, 2 h | 48 | 19 | 3 | 10 | 1 | 15 | 9.1 | 10.3 | |
| 0.5 wt-% Ru/MgO, 300°C, 4 h | 51 | 23 | 5 | 13 | 5 | 5 | 9.3 | 10.6 | |
| 0.5 wt-% Ru/MgO, 400°C, 2 h | 35 | 15 | 5 | 7 | 2 | 6 | 9.5 | 10.8 | |
| 0.5 wt-% Ru/MgO, 500°C, 2 h | 34 | 10 | 4 | 6 | 3 | 21 | 9.6 | 10.9 | |
| 0.5 wt-% Ru/MgO, 600°C, 2 h | 28 | 12 | 3 | 4 | 1 | 8 | 9.8 | 10.9 |
Reaction conditions: 4 mmol HMF, 1.5 MPa of O2, 0.5 wt-% catalyst (substrate:metal molar ratio = 120), 30 mL of deionised water, 140°C, 6.5 h.
The most active and selective 0.5 wt-% Ru/MgO, 300°C, 4 h catalyst was subsequently chosen to optimise reaction conditions. The duration of the reaction was tested over a range of 4–10 h and a favourable yield of FDCA (68%) was achieved in 6.5 h (Fig. 2). It was observed that the rate-determining step in the reaction is the conversion of FFCA to FDCA. Therefore, prolonging the reaction time can lead to a higher yield of FDCA with a decrease in the FFCA yield. The time study also indicated that both mass balance and product distribution are dependent on reaction time. As the reaction time increased the total mass balance also fell associated with the formation of undetected products (likely HMF oligomers or humins). It is also interesting to note that though time was increased to 10 h, DFF and HMFCA still did not yield FFCA as their yields remained almost constant. This may be due to greater interaction between FFCA and the catalyst that did not allow DFF and HMFCA to interact with the catalyst surface. Alternatively, this may also suggest that the catalyst is no longer active for oxidising the alcohol group (in HMFCA) to an aldehyde group. However, at the same time, the aldehyde group in FFCA could be oxidised to a carboxylic acid group. From this trend, subsequent reactions were conducted for 6.5 h, identified as the optimum point because the highest mass balance of detected products was seen at this reaction time.
HMF conversion and product selectivity as a function of reaction time. Reaction conditions: 4 mmol HMF, 1.5 MPa of O2, 0.5 wt-% Ru/MgO, 300°C, 4 h (substrate:metal molar ratio = 120), 30 mL of deionised water, 160°C.
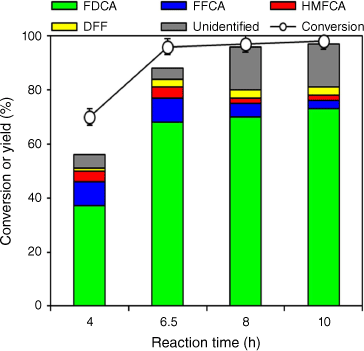
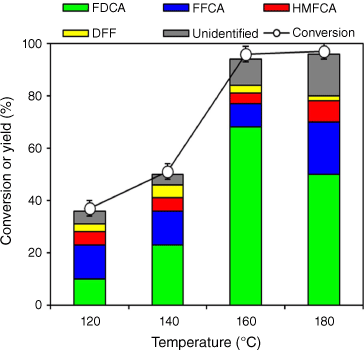
Fig. 3 shows the results on the HMF conversion and product yields of a change in reaction temperature from 120 to 160°C. At 140°C, HMF conversion reached 51% with the predominant formation of FFCA (13%) and FDCA (23%), and formation of DFF (5%) with HMFCA (5%) was also seen. Reactions performed at higher temperatures of 160 and 180°C increased HMF conversion, but the maximum FDCA yield of 68% was obtained at 160°C with 96% HMF conversion. As the temperature increased to 180°C, the yield of FDCA decreased, mostly owing to HMF degradation being a temperature-sensitive reaction and thus this reaction taking precedence over the oxidation reaction, as evidenced by an increase in unidentified products. The same observation was also made when HMF was subjected to isolation from an organic solvent in the laboratory (Fig. S3–S6) and with an increase in rotary evaporator temperature, HMF degradation increased.
HMF conversion and product selectivity as a function of reaction temperature. Reaction conditions: 4 mmol HMF, 1.5 MPa of O2, 0.5 wt-% Ru/MgO, 300°C, 4 h (substrate:metal molar ratio = 120), 30 mL of deionised water, 6.5 h.
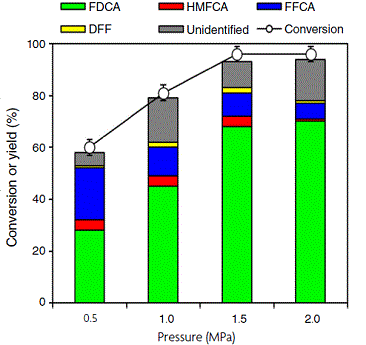
The effect of oxygen pressure on FDCA production was also investigated (Fig. 4), with 1.5 MPa of O2 the optimum. If the pressure is higher, FFCA gets converted to FDCA because higher pressure can enhance reaction rates, especially in gas–liquid reactions. It is well known that elevated pressures increase the concentration of oxygen molecules in the reaction system (water), promoting faster reaction rates. HMF conversion was 100% in the O2 pressure range of 1.5–2.0 MPa. Increasing the O2 pressure from 1.0 to 1.5 MPa, the FDCA yield increased from 45 to 68%, and unidentified products decreased from 17 to 10%; this then remained the same up to 2.0 MPa. This is because with lower availability of oxygen for the reaction (at lower pressures, owing to lower solubility), HMF instead of undergoing oxidation reactions forms degradation products, which is likely under elevated temperatures, as seen from the temperature study. Under pressure conditions between 1.0 and 2.0 MPa, the reaction may favour the conversion of FFCA to FDCA. When higher pressures were employed, complete conversion of intermediates such as DFF, HMFCA and FFCA to FDCA was still not possible. This may reflect reversible site-blocking due to adsorption of products or catalyst deactivation. Higher oxygen pressures can increase the oxidation of FFCA to FDCA up to certain limit because the thermodynamics of the oxidation reaction are influenced by the partial pressure of oxygen. Thus, understanding and manipulating thermodynamic aspects are crucial for achieving optimal FDCA yields. At elevated oyxgen, pressures are anticipated to increase oxygen solubility and hence circumvent mass-transport limitations that will otherwise lower the measured catalyst activity. Careful control of oxygen pressure is necessary to prevent undesired side reactions, mitigate the risk of explosive conditions and ensure the overall safety of the oxidation process.
HMF conversion and product selectivity as a function of oxygen pressure. Reaction conditions: 4 mmol HMF, 0.5 wt-% Ru/MgO, 300°C, 4 h (substrate:metal molar ratio = 120), 30 mL of deionised water, 160°C, 6.5 h.
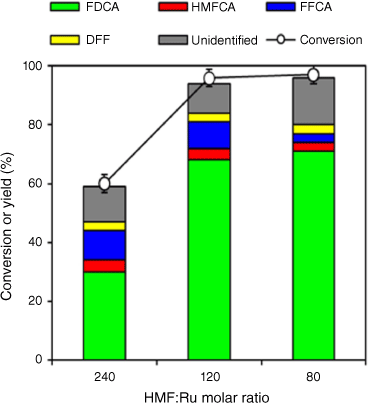
From an industrial perspective, reducing the catalyst loading, and particularly precious metal usage (termed ‘thrifting’) is critical to demonstrate technoeconomic feasibility. The molar ratio of substrate:Ru was therefore varied between 240 and 80 (Fig. 5). Although increasing the mass of catalyst from 0.1 g (substrate:Ru = 240) to 0.3 g (substrate:Ru = 80) doubled the yield of FDCA to ~68%, further increases offered minimal benefit, with 0.2 g (substrate:Ru = 120) optimal and delivering 96% HMF conversion and 68% FDCA yield. High catalyst loadings also increased the proportion of undesired side products and hence compromised the carbon mass balance.
HMF conversion and product selectivity as a function of HMF:Ru molar ratio. Reaction condition: 4 mmol HMF, 1.5 MPa of O2, 0.5 wt-% Ru/MgO, 300°C, 4 h, 30 mL of deionised water, 160°C, 6.5 h.
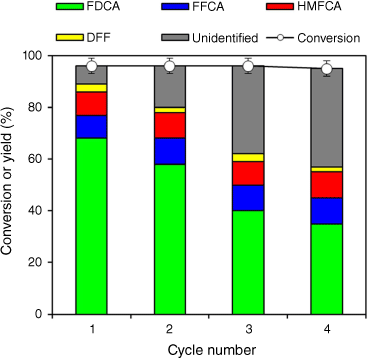
Catalyst stability is an important aspect of their potential for commercialisation, and hence reuse of the 0.5 wt-% Ru/MgO, 300°C, 4 h catalyst was examined (Fig. 6). Post-reaction catalysts were recovered from the reaction mixture by centrifugation, then washed three times with deionised water to remove any soluble organic compounds and returned to a freshly prepared reaction mixture without drying or adjusting the catalyst mass. HMF conversion remained >95% over four reaction cycles, evidencing that the catalyst retained significant activity post reaction without the need for an aggressive regeneration protocol (such as calcination to remove heavy organic residues). Deactivation of base sites would be expected to suppress HMF conversion, which is generally accepted to react through the gem-diol precursor to form HMFCA. However, these recycle measurements do not permit quantification of the degree of deactivation, for which such studies must be performed at low HMF conversion where reactions are free from potential mass-transfer limitations.67 The FDCA yield progressively decreased with each reaction cycle (from 68 to 35% over four cycles), accompanied by an increase in unidentified degradation products, suggesting Ru site blocking by water-insoluble organic residues, probably HMF oligomers. Quantitative studies of catalyst lifetime and reactivation will be explored in future studies.
HMF conversion and product selectivity over 0.5 wt-% Ru/MgO, 300°C, 4 h as a function of re-use. Reaction condition: 4 mmol HMF, 1.5 MPa of O2, 30 mL of deionised water, 160°C, 6.5 h.
Increasing the Ru loading from 0.1 to 5 wt-% offered only a small increase (10%) in HMF conversion (Supplementary Table S2); complete conversion was achieved for loadings ≥1 wt-% under our reaction conditions (160°C, substrate:metal molar ratio = 120, 1.5 MPa of O2 and 6.5 h). However, product selectivity was far more sensitive to Ru loading, with the FDCA yield rising from only 44 to 88% across the Ru loading range (Supplementary Table S2), accompanied by a corresponding fall in the yields of all reactive intermediates (HMFCA, FFCA and DFF). Higher loadings also improved the carbon mass balance, which reached 95% for Ru loadings ≥1 wt-% wherein HMF oxidation was heavily favoured over, for example, competing oligomerisation pathways. The increase in FDCA selectivity with Ru loading is analogous to that observed for Au/MgAl-hydrotalcite catalysts,43 wherein strong HMF adsorption over metal nanoparticles site-blocked oxidation of its reactively formed products and, hence, HMFCA oxidation to FDCA (by FFCA) was only efficient at >90% HMF conversion. Control reactions in the absence of catalyst or oxygen (Supplementary Table S3) evidenced modest HMF conversion (≤25%) and minimal FDCA formation (≤12%); the poorer mass balances indicate that slow thermal oxidation competes against significant oligomerisation. It is notable that no DFF was observed without catalyst and oxygen, suggesting that HMF oxidation is favoured through an HMFCA intermediate (Path B in Fig. 2). After reaction, the catalyst was filtered from the reaction mixture and the filtrate analysed by inductively coupled plasma optical emission spectroscopy (ICP-OES) to check for metal leaching. Ruthenium leaching was not observed, but low concentrations of Mg were detected, indicative of support instability and consistent with the observed increase in solution pH from hydroxide formation (Table 4).
Reaction pathways for HMF oxidation to FDCA
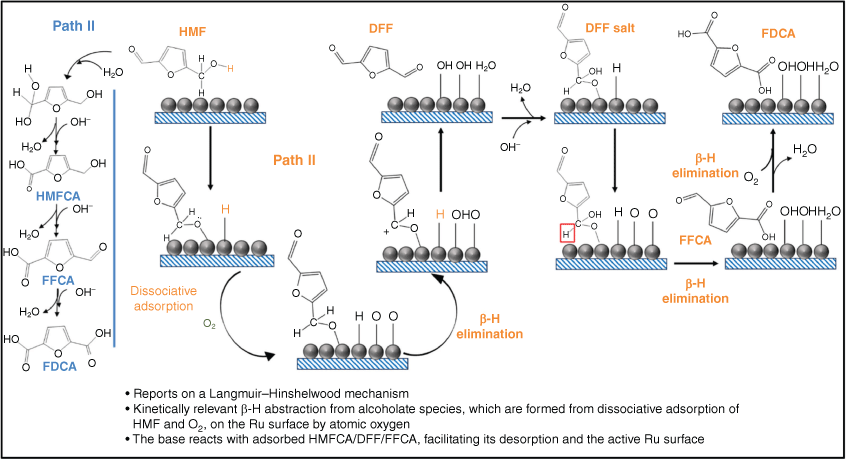
The selective oxidation of HMF to FDCA may occur through two pathways: initial oxidation of the hydroxyl group of HMF to form DFF, or of the aldehyde group to form HMFCA, and their subsequent oxidation to FFCA and then FDCA.68–72 In almost all reactions, HMFCA was formed in larger amounts than DFF after 6 h. Reaction profiles also evidence that oxidation of these intermediates to FFCA was rapid, whereas the oxidation of FFCA to FDCA was slower and hence rate-determining. Considering various literature reports for supported Ru catalysts, it is likely that HMFCA is the reactive intermediate with DFF a spectator, as for 5 wt-% Ru over carbon and mixed metal oxide supports36 (albeit in the presence of a homogeneous base such as sodium carbonate). Zheng et al. reached the same conclusion by studying the individual reaction steps in HMF oxidation to FDCA over Ru/C.31 Elementary steps for the oxidation of HMF to DFF over Ru/C in aqueous solution were proposed by Xie et al,32 HMF and O2 being dissociatively chemisorbed over Ru to form an alcoholate (R–CH2O*; R hereafter denotes the furfural CHO–C4H2O‐–part of HMF) species and atomic H* and O* (Scheme 3). Subsequent β‐H elimination of R–CH2O* by reaction with O* is irreversible and forms DFF (R–CHO*) and hydroxyl (OH*) species, with the reactively formed R–CHO* desorbing as DFF. Note that OH* can also be formed by the reaction of O* and H* adatoms. Where the transformation of HMF to DFF requires elimination of two hydrogen atoms, oxidation of the formyl group of DFF to FFCA and FDCA requires the insertion of one and two oxygen atoms respectively, from either O2 or H2O. By contrast, kinetic studies of HMF and HMFCA oxidation over Au/MgAl-hydrotalcite catalysts evidence similar kinetics with HMFCA as the key reactive intermediate, and DFF never observed.43 Davis and co-workers also propose an HMFCA intermediate to FDCA over Au/TiO2 and Pt/C catalysts, with oxygen atoms originating from water,73,74 in accordance with Nie et al.,32 who showed that HMF oxidation in toluene over Ru/C, Ru/Al2O3 and Ru/ZrO2 produced mainly DFF (>96% selectivity).
Conclusions
Selective oxidation of 5-hydroxymethylfurfural (HMF) to 2,5-furandicarboxylic acid (FDCA) as a bio-derived monomer to replace terephtalic acid was explored in the aqueous phase over Ru/MgO catalysts in the absence of added soluble base. Thermal processing of the MgO support has a significant effect on both HMF conversion and FDCA yield, with an extended moderate-temperature calcination (300°C for 4 h) optimal at 140°C and 1.5 MPa of O2, affording 51% conversion and 23% yield. Increasing the Ru loading slightly improved HMF conversion but was more beneficial for enhancing FDCA selectivity and the carbon mass balance, attributed to competitive adsorption between HMF and the reactively formed HMFCA intermediate for metal active sites at conversions <95%. Reaction optimisation for the 0.5 wt-% Ru/MgO, 300°C, 4 h catalyst delivered 96% HMF conversion and 68% FDCA yield at 160°C and 1.5 MPa of O2 in 6.5 h. Recycling studies evidenced retention of catalytic activity for HMF conversion, but a progressive decrease in selectivity to oxidation products, which likely reflects the accumulation of HMF oligomers at Ru active sites, accompanied by some leaching of Mg from the support. Judicious control over the catalyst support and metal active phase, and process optimisation, permit the effective conversion of HMF to FDCA with low concentrations of precious metal.
Data availability
The data that support this study are available on request from the corresponding author.
Declaration of funding
P. Lokhande thanks the CSIR-UGC for a Junior Research Fellowship. A. F. Lee and K. Wilson thank the Australian Research Council (DP200100204, DP200100313 and LP190100849) and CRC-P (CRCPPEIGHT000194) for financial support.
References
1 Tickner J, Geiser K, Baima S. Transitioning the chemical industry: the case for addressing the climate, toxics, and plastics crises. Environ Sci Pollut Res Dev 2021; 63(6): 4-15.
| Crossref | Google Scholar |
2 Vogt ETC, Weckhuysen BM. The refinery of the future. Nature 2024; 629(8011): 295-306.
| Crossref | Google Scholar | PubMed |
3 Zhang X, Wilson K, Lee AF. Heterogeneously catalyzed hydrothermal processing of C5–C6 sugars. Chem Rev 2016; 116(19): 12328-12368.
| Crossref | Google Scholar | PubMed |
4 Gallezot P. Conversion of biomass to selected chemical products. Chem Soc Rev 2012; 41(4): 1538-1558.
| Crossref | Google Scholar |
5 Mitchell S, Martín AJ, Guillén-Gosálbez G, Pérez-Ramírez J. The future of chemical sciences is sustainable. Angew Chem Int Ed 2024; 63: e202318676.
| Crossref | Google Scholar |
6 Chheda JN, Huber GW, Dumesic JA. Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew Chem Int Ed 2007; 46(38): 7164-7183.
| Crossref | Google Scholar |
7 Bozell JJ, Petersen GR. Technology development for the production of biobased products from biorefinery carbohydrates – the US Department of Energy’s ‘top 10’ revisited. Green Chem 2010; 12(4): 539-554.
| Crossref | Google Scholar |
8 Gandini A, Silvestre AJ, Neto CP, Sousa AF, Gomes M. The furan counterpart of poly(ethylene terephthalate): an alternative material based on renewable resources. J Polym Sci A Polym Chem 2009; 47(1): 295-298.
| Crossref | Google Scholar |
9 Gandini A. Furans as offspring of sugars and polysaccharides and progenitors of a family of remarkable polymers: a review of recent progress. Polym Chem 2010; 1(3): 245-251.
| Google Scholar |
10 van Putten R-J, van der Waal JC, de Jong E, Rasrendra CB, Heeres HJ, de Vries JG. Hydroxymethylfurfural, a versatile platform chemical made from renewable resources. Chem Rev 2013; 113(3): 1499-1597.
| Crossref | Google Scholar | PubMed |
12 de Jong E, Visser HA, Dias AS, Harvey C, Gruter G-JM. The road to bring FDCA and PEF to the market. Polymers 2022; 14(5): 943.
| Crossref | Google Scholar | PubMed |
13 Eerhart A, Faaij A, Patel MK. Replacing fossil-based PET with biobased PEF; process analysis, energy and GHG balance. Energy Environ Sci 2012; 5(4): 6407-6422.
| Crossref | Google Scholar |
14 Zhang Z, Deng K. Recent advances in the catalytic synthesis of 2,5-furandicarboxylic acid and its derivatives. ACS Catal 2015; 5(11): 6529-6544.
| Crossref | Google Scholar |
15 Yi G, Teong SP, Zhang Y. The direct conversion of sugars into 2,5-furandicarboxylic acid in a triphasic system. ChemSusChem 2015; 8(7): 1151-1155.
| Crossref | Google Scholar |
16 Wang S, Zhang Z, Liu B. Catalytic conversion of fructose and 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid over a recyclable Fe3O4–CoOx magnetite nanocatalyst. ACS Sustain Chem Eng 2015; 3(3): 406-412.
| Crossref | Google Scholar |
17 Casanova O, Iborra S, Corma A. Biomass into chemicals: aerobic oxidation of 5-hydroxymethyl-2-furfural into 2,5-furandicarboxylic acid with gold nanoparticle catalysts. ChemSusChem 2009; 2(12): 1138-1144.
| Crossref | Google Scholar | PubMed |
18 Davis SE, Houk LR, Tamargo EC, Datye AK, Davis RJ. Oxidation of 5-hydroxymethylfurfural over supported Pt, Pd and Au catalysts. Catal Today 2011; 160(1): 55-60.
| Crossref | Google Scholar |
19 Gupta NK, Nishimura S, Takagaki A, Ebitani K. Hydrotalcite-supported gold-nanoparticle-catalyzed highly efficient base-free aqueous oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid under atmospheric oxygen pressure. Green Chem 2011; 13(4): 824-827.
| Crossref | Google Scholar |
20 Gorbanev YY, Klitgaard SK, Woodley JM, Christensen CH, Riisager A. Gold-catalyzed aerobic oxidation of 5-hydroxymethylfurfural in water at ambient temperature. ChemSusChem 2009; 2(7): 672-675.
| Crossref | Google Scholar | PubMed |
21 Pasini T, Piccinini M, Blosi M, Bonelli R, Albonetti S, Dimitratos N, Lopez-Sanchez JA, Sankar M, He Q, Kiely CJ. Selective oxidation of 5-hydroxymethyl-2-furfural using supported gold–copper nanoparticles. Green Chem 2011; 13(8): 2091-2099.
| Google Scholar |
22 Diamond G, Hagemeyer A, Murphy V, Sokolovskii V. Catalytic conversion of biorenewable sugar feedstocks into market chemicals. Comb Chem High T Scr 2018; 21(9): 616-630.
| Crossref | Google Scholar |
23 Kuster B. 5‐Hydroxymethylfurfural (HMF). A review focussing on its manufacture. Starch‐Stärke 1990; 42(8): 314-321.
| Crossref | Google Scholar |
24 Moreau C, Durand R, Razigade S, Duhamet J, Faugeras P, Rivalier P, Ros P, Avignon G. Dehydration of fructose to 5-hydroxymethylfurfural over H-mordenites. Appl Catal A 1996; 145(1–2): 211-224.
| Crossref | Google Scholar |
25 Chheda JN, Román-Leshkov Y, Dumesic JA. Production of 5-hydroxymethylfurfural and furfural by dehydration of biomass-derived mono- and poly-saccharides. Green Chem 2007; 9(4): 342-350.
| Google Scholar |
26 Moliner M, Román-Leshkov Y, Davis ME. Tin-containing zeolites are highly active catalysts for the isomerization of glucose in water. Proc Natl Acad Sci USA 2010; 107(14): 6164-6168.
| Crossref | Google Scholar | PubMed |
27 Patil SK, Heltzel J, Lund CR. Comparison of structural features of humins formed catalytically from glucose, fructose, and 5-hydroxymethylfurfuraldehyde. Energ Fuel 2012; 26(8): 5281-5293.
| Crossref | Google Scholar |
28 Osatiashtiani A, Lee AF, Brown DR, Melero JA, Morales G, Wilson K. Bifunctional SO4/ZrO2 catalysts for 5-hydroxymethylfufural (5-HMF) production from glucose. Catal Sci Technol 2014; 4(2): 333-342.
| Google Scholar |
29 Osatiashtiani A, Lee AF, Granollers M, Brown DR, Olivi L, Morales G, Melero JA, Wilson K. Hydrothermally stable, conformal, sulfated zirconia monolayer catalysts for glucose conversion to 5-HMF. Acs Catal 2015; 5(7): 4345-4352.
| Google Scholar |
30 Partenheimer W, Grushin VV. Synthesis of 2,5-diformylfuran and furan-2,5-dicarboxylic acid by catalytic air-oxidation of 5-hydroxymethylfurfural. Unexpectedly selective aerobic oxidation of benzyl alcohol to benzaldehyde with metal bromide catalysts. Adv Synth Catal 2001; 343(1): 102-111.
| Crossref | Google Scholar |
31 Zheng L, Zhao J, Du Z, Zong B, Liu H. Efficient aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid on Ru/C catalysts. Sci China Chem 2017; 60(7): 950-957.
| Crossref | Google Scholar |
32 Nie J, Xie J, Liu H. Efficient aerobic oxidation of 5-hydroxymethylfurfural to 2,5-diformylfuran on supported Ru catalysts. J Catal 2013; 301: 83-91.
| Crossref | Google Scholar |
33 Sahu R, Dhepe PL. Synthesis of 2,5-furandicarboxylic acid by the aerobic oxidation of 5-hydroxymethyl furfural over supported metal catalysts. React Kinet Mech Catal 2014; 112(1): 173-187.
| Crossref | Google Scholar |
34 Ait Rass H, Essayem N, Besson M. Selective aerobic oxidation of 5-HMF into 2,5-furandicarboxylic acid with Pt catalysts supported on TiO2- and ZrO2-based supports. ChemSusChem 2015; 8(7): 1206-1217.
| Crossref | Google Scholar | PubMed |
35 Lolli A, Maslova V, Bonincontro D, Basile F, Ortelli S, Albonetti S. Selective oxidation of HMF via catalytic and photocatalytic processes using metal-supported catalysts. Molecules 2018; 23(11): 2792.
| Crossref | Google Scholar | PubMed |
36 Lokhande P, Sonone K, Dhepe PL. Industry-oriented method for the aqueous phase oxidation of crude 5-hydroxymethyl furfural (HMF) to 2,5-furandicarboxylic acid (FDCA). New J Chem 2023; 47(32): 15325-15335.
| Crossref | Google Scholar |
37 Jiang X, Li W, Liu Y, Zhao L, Chen Z, Zhang L, Zhang Y, Yun S. Electrocatalytic oxidation of 5-hydroxymethylfurfural for sustainable 2,5-furandicarboxylic acid production – from mechanism to catalysts design. SusMat 2023; 3(1): 21-43.
| Crossref | Google Scholar |
38 Lie WH, Yang Y, Yuwono JA, Tsounis C, Zubair M, Wright J, Thomsen L, Kumar P, Bedford N. Identification of catalytic activity descriptors for selective 5-hydroxymethyl furfural electrooxidation to 2,5-furandicarboxylic acid. J Mater Chem 2023; 11(11): 5527-5539.
| Crossref | Google Scholar |
39 Ma Z, Wang L, Li G, Song T. Recent advances in electrocatalytic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid by heterogeneous catalysts. Catal 2024; 14(2): 157.
| Google Scholar |
40 Schade OR, Dannecker P-K, Kalz KF, Steinbach D, Meier MAR, Grunwaldt J-D. Direct catalytic route to biomass-derived 2,5-furandicarboxylic acid and its use as monomer in a multicomponent polymerization. ACS Omega 2019; 4(16): 16972-16979.
| Crossref | Google Scholar | PubMed |
41 Li L, Hu Y, Li H, Hu P, Xue Z, Wu D, Hu C, Zhu L. Probing the formation of soluble humins in catalytic dehydration of fructose to 5-hydroxymethylfurfural over HZSM-5 catalyst. Fuel 2023; 344: 128133.
| Crossref | Google Scholar |
42 Gupta NK, Nishimura S, Takagaki A, Ebitani K. Hydrotalcite-supported gold-nanoparticle-catalyzed highly efficient base-free aqueous oxidation of 5-hydroxymethylfurfural into 2,5-furandicarboxylic acid under atmospheric oxygen pressure. Green Chem 2011; 13(4): 824-827.
| Crossref | Google Scholar |
43 Ardemani L, Cibin G, Dent AJ, Isaacs MA, Kyriakou G, Lee AF, Parlett C, Parry SA, Wilson K. Solid base catalysed 5-HMF oxidation to 2,5-FDCA over Au/hydrotalcites: fact or fiction? Chem Sci 2015; 6(8): 4940-4945.
| Crossref | Google Scholar | PubMed |
44 Yuan Q, Hiemstra K, Meinds TG, Chaabane I, Tang Z, Rohrbach L, Vrijburg W, Verhoeven T, Hensen EJM, van der Veer S, et al. Bio-based chemicals: selective aerobic oxidation of tetrahydrofuran-2,5-dimethanol to tetrahydrofuran-2,5-dicarboxylic acid using hydrotalcite-supported gold catalysts. ACS Sustain Chem Eng 2019; 7(5): 4647-4656.
| Crossref | Google Scholar |
45 Cai C, Xu J, Wang H, Xin H, Zhang Q, Wang C, Ma L, Liu Q. Homogeneous base-free oxidation of 5-hydroxymethyfufural to 2,5-furandicarboxylic acid over Au/Mg(OH)2 catalysts. ChemistrySelect 2020; 5(41): 12785-12790.
| Crossref | Google Scholar |
46 Han X, Geng L, Guo Y, Jia R, Liu X, Zhang Y, Wang Y. Base-free aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over a Pt/C–O–Mg catalyst. Green Chem 2016; 18(6): 1597-1604.
| Crossref | Google Scholar |
47 Mani M, Mariyaselvakumar M, Tothadi S, Panda AB, Srinivasan K, Konwar LJ. Base free HMF oxidation over Ru-MnO2 catalysts revisited: evidence of Mn leaching to Mn-FDCA complexation and its implications on catalyst performance. Mol Catal 2024; 554: 113811.
| Crossref | Google Scholar |
48 Zhang W, Li Q, Xia H. Photocatalytic oxidation of 5-hydroxymethylfurfural to furandicarboxylic acid over the Au-Ag/TiO2 catalysts under visible light irradiation. Appl Surf Sci 2023; 613: 156036.
| Crossref | Google Scholar |
49 Zhu P, Zhang W, Li Q, Xia H. Visible-light-driven photocatalytic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over plasmonic Au/ZnO catalyst. ACS Sustain Chem Eng 2022; 10(27): 8778-8787.
| Crossref | Google Scholar |
50 Yamaguchi K, Mizuno N. Efficient heterogeneous aerobic oxidation of amines by a supported ruthenium catalyst. Angew Chem Int Ed 2003; 42(13): 1480-1483.
| Crossref | Google Scholar |
51 Berkowitz LM, Rylander PN. Use of ruthenium tetroxide as a multi-purpose oxidant. J Am Chem Soc 1958; 80(24): 6682-6684.
| Crossref | Google Scholar |
52 Matsumoto M, Kobayashi K, Hotta Y. Acid-catalyzed oxidation of benzaldehydes to phenols by hydrogen peroxide. J Org Chem 1984; 49(24): 4740-4741.
| Crossref | Google Scholar |
53 Madhavaram H, Idriss H, Wendt S, Kim YD, Knapp M, Over H, Aßmann J, Löffler E, Muhler M. Oxidation reactions over RuO2: a comparative study of the reactivity of the (110) single crystal and polycrystalline surfaces. J Catal 2001; 202(2): 296-307.
| Crossref | Google Scholar |
54 Kobayashi S, Miyamura H, Akiyama R, Ishida T. Highly active, immobilized ruthenium catalysts for oxidation of alcohols to aldehydes and ketones. preparation and use in both batch and flow systems. J Am Chem Soc 2005; 127(25): 9251-9254.
| Crossref | Google Scholar | PubMed |
55 Pagliaro M, Campestrini S, Ciriminna R. Ru-based oxidation catalysis. Chem Soc Rev 2005; 34(10): 837-845.
| Crossref | Google Scholar | PubMed |
56 Yu H, Fu X, Zhou C, Peng F, Wang H, Yang J. Capacitance dependent catalytic activity of RuO2·xH2O/CNT nanocatalysts for aerobicoxidation of benzyl alcohol. Chem Commun 2009; 2009(17): 2408-2410.
| Crossref | Google Scholar | PubMed |
57 Gorbanev YY, Kegnæs S, Riisager A. Selective aerobic oxidation of 5-hydroxymethylfurfural in water over solid ruthenium hydroxide catalysts with magnesium-based supports. Catal Lett 2011; 141(12): 1752-1760.
| Crossref | Google Scholar |
58 Antonyraj CA, Huynh NTT, Lee KW, Kim YJ, Shin S, Shin JS, Cho JK. Base-free oxidation of 5-hydroxymethyl-2-furfural to 2,5-furandicarboxylic acid over basic metal oxide-supported ruthenium catalysts under aqueous conditions. J Chem Sci 2018; 130(11): 156.
| Crossref | Google Scholar |
59 Ståhlberg T, Eyjólfsdóttir E, Gorbanev YY, Sádaba I, Riisager A. Aerobic oxidation of 5-(hydroxymethyl)furfural in ionic liquids with solid ruthenium hydroxide catalysts. Catal Lett 2012; 142(9): 1089-1097.
| Crossref | Google Scholar |
60 Artz J, Mallmann S, Palkovits R. Selective aerobic oxidation of HMF to 2,5-diformylfuran on covalent triazine frameworks-supported Ru catalysts. ChemSusChem 2015; 8(4): 672-679.
| Crossref | Google Scholar |
61 Ferraz CP, Zieliński M, Pietrowski M, Heyte S, Dumeignil F, Rossi LM, Wojcieszak R. Influence of support basic sites in green oxidation of biobased substrates using Au-promoted catalysts. ACS Sustain Chem Eng 2018; 6(12): 16332-16340.
| Crossref | Google Scholar |
62 Wojcieszak R, Itabaiana I. Engineering the future: perspectives in the 2,5-furandicarboxylic acid synthesis. Catal Today 2020; 354: 211-217.
| Crossref | Google Scholar |
63 Wang JA, Novaro O, Bokhimi X, López T, Gómez R, Navarrete J, Llanos ME, López-Salinas E. Structural defects and acidic and basic sites in sol–gel MgO. J Phys Chem B 1997; 101(38): 7448-7451.
| Crossref | Google Scholar |
64 Branda MM, Belelli PG, Ferullo RM, Castellani NJ. Proton abstraction ability of MgO: a DFT cluster model study of the role of surface geometry. Catal Today 2003; 85(2): 153-165.
| Crossref | Google Scholar |
65 Spoto G, Gribov EN, Ricchiardi G, Damin A, Scarano D, Bordiga S, Lamberti C, Zecchina A. Carbon monoxide MgO from dispersed solids to single crystals: a review and new advances. Prog Surf Sci 2004; 76(3): 71-146.
| Crossref | Google Scholar |
66 Lokhande P, Dhepe PL. Selective and robust Ru catalyst for the aqueous phase aerobic oxidation of furfural to 2-furoic acid. ACS Appl Mater Interfaces 2023; 15(40): 47004-47015.
| Crossref | Google Scholar | PubMed |
67 Schüth F, Ward MD, Buriak JM. Common pitfalls of catalysis manuscripts submitted to chemistry of materials. Chem Mater 2018; 30(11): 3599-3600.
| Crossref | Google Scholar |
68 Zope BN, Davis SE, Davis RJ. Influence of reaction conditions on diacid formation during Au-catalyzed oxidation of glycerol and hydroxymethylfurfural. Top Catal 2012; 55(1): 24-32.
| Crossref | Google Scholar |
69 Albonetti S, Lolli A, Morandi V, Migliori A, Lucarelli C, Cavani F. Conversion of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over Au-based catalysts: optimization of active phase and metal–support interaction. Appl Catal B 2015; 163: 520-530.
| Crossref | Google Scholar |
70 Vuyyuru KR, Strasser P. Oxidation of biomass derived 5-hydroxymethylfurfural using heterogeneous and electrochemical catalysis. Catal Today 2012; 195(1): 144-154.
| Crossref | Google Scholar |
71 Chadderdon DJ, Xin L, Qi J, Qiu Y, Krishna P, More KL, Li W. Electrocatalytic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid on supported Au and Pd bimetallic nanoparticles. Green Chem 2014; 16(8): 3778-3786.
| Crossref | Google Scholar |
72 Zhou C, Deng W, Wan X, Zhang Q, Yang Y, Wang Y. Functionalized carbon nanotubes for biomass conversion: the base-free aerobic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over platinum supported on a carbon nanotube catalyst. ChemCatChem 2015; 7(18): 2853-2863.
| Crossref | Google Scholar |
73 Davis SE, Zope BN, Davis RJ. On the mechanism of selective oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid over supported Pt and Au catalysts. Green Chem 2012; 14(1): 143-147.
| Crossref | Google Scholar |
74 Zope BN, Hibbitts DD, Neurock M, Davis RJ. Reactivity of the gold/water interface during selective oxidation catalysis. Science 2010; 330(6000): 74-78.
| Crossref | Google Scholar |