

NMR Chemical Shift and Methylation of 4-Nitroimidazole: Experiment and Theory*
Frederick Backler A , Marc Antoine Sani B , Frances Separovic B , Vladislav Vasilyev C and Feng Wang A D
B , Frances Separovic B , Vladislav Vasilyev C and Feng Wang A D
A Department of Chemistry and Biotechnology and Centre for Translational Atomaterials, Faculty of Science, Engineering and Technology, Swinburne University of Technology, Hawthorn, Vic. 3122, Australia.
B School of Chemistry, Bio21 Institute, University of Melbourne, Melbourne, Vic. 3010, Australia.
C National Computational Infrastructure, Australian National University, Canberra, ACT 0200, Australia.
D Corresponding author. Email: fwang@swin.edu.au
Australian Journal of Chemistry 74(1) 48-55 https://doi.org/10.1071/CH20199
Submitted: 18 June 2020 Accepted: 3 August 2020 Published: 1 October 2020
Abstract
Nitroimidazoles and derivatives are a class of active pharmaceutical ingredients (APIs) first introduced sixty years ago. As anti-infection agents, the structure–activity relationships of nitroimidazole compounds have been particularly difficult to study due to their low reduction potentials and unique electronic structures. In this study, we combine dynamic nuclear polarization (DNP)-enhanced solid-state (100 K), solid-state (298 K), and 1H-13C heteronuclear single quantum coherence (HSQC) solution-state NMR techniques (303 K) with density functional theory (DFT) to study the 1H, 13C, and 15N chemical shifts of 4-nitroimidazole (4-NI) and 1-methyl-4-nitroimidazole (CH3-4NI). The 4-NI chemical shifts were observed at 119.4, 136.4, and 144.7 ppm for 13C, and at 181.5, 237.4, and 363.0 ppm for 15N. The measurements revealed that methylation (deprotonation) of the amino nitrogen N(1) of 4-NI had less effect (Δδ = −4.8 ppm) on the N(1) chemical shift but was compensated by shielding of the N(3) (Δδ = 11.6 ppm) in CH3-4NI. The calculated chemical shifts using DFT for 4-NI and CH3-4NI agreed well with the experimental values (within 2 %) for the imidazole carbons. However, larger discrepancies (up to 13 %) were observed between the calculated and measured 15N NMR chemical shifts for the imidazole nitrogen atoms of both molecules, which indicate that effects such as imidazole ring resonant structures and molecular dynamics may also contribute to the nitrogen chemical environment.
Introduction
Nitroimidazoles were first in the class of compounds reported to have antimycobacterial activity in the early 1970s[1] and more recently as radiosensitizers of hypoxic tumour cells.[2] Methylation is an important pathway in the metabolism of many drugs, such as neurotransmitters and xenobiotic compounds. Although methyl conjugation of pyridine (an N-methylation) was first described over a century ago,[3] only recently have we begun to understand factors responsible for individual variations in methylation,[4] One of the challenges of modern medicine is the understanding of the biological basis for individual variation in drug response and in the occurrence of adverse drug reactions. Most of the pharmacogenetic effects that have been discovered involve inherited differences in drug metabolism.[4] Methyltransferase activities have been found to be related to individual differences in drug metabolism, effect, and toxicity.[4]
An important but not well understood genetic regulation is that of heterocyclic N-methyltransferases such as histamine N-methyltransferase (HNMT), which catalyzes the N-methylation of histamine to become N-methylhistamine.[4] 4-Nitroimidazole (4-NI) (IUPAC name 4-nitro-1H-imidazole) shares the same imidazole chemical structure as histamine (IUPAC name 2-(1H-imidazol-4-yl)ethanamine), but the C(4) position of imidazole connects an ethanamine (C(4)-CH2CH2NH2) for histamine whereas the same C(4) position of imidazole connects a nitro group (C(4)-NO2) for 4-NI (see Fig. 1). Moreover, methylation of the N(1)H for N(1)CH3 for histamine produces N-methylhistamine but for 4-NI produces 1-methyl-4-nitroimidazole (CH3-4NI). As a result, study of the chemical structure by 1H, 13C, and 15N NMR chemical shifts of the 4-NI and CH3-4NI pair will help one to better understand the genetic regulations of heterocyclic N-methyltransferases.
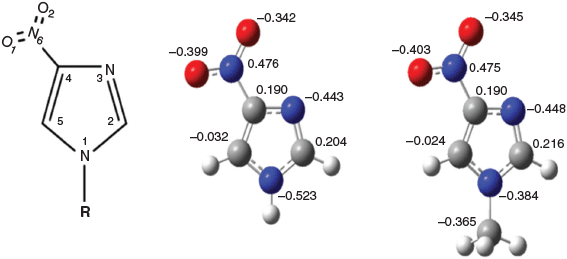
|
The structure–activity relationships (SAR) for the nitroimidazole derivatives have been established based on whole cell activity, yet the basis of this activity is not fully understood.[5] Mono-substituted nitroimidazoles and their methylation such as 4-NI and CH3-4NI are the core for a significant number of derivatives with significant chemical and pharmaceutical applications.[6–10] Studies focussed on 4-NI and its derivatives have been based on crystal structures[6,7,11] and other properties such as NMR chemical shift,[8,12] hydrogen bonding,[9] and, in recent years, state-of-the-art synchrotron-based experiments at higher temperatures. For example, C 1s and N 1s core ionization spectra of the nitroimidazoles and their derivatives measured by X-ray photoemission spectroscopy (XPS) when heated to 390 K,[13] and by mass spectrometry (MS) fragmentation at 353 K have been reported.[14] 4NI decomposed differently as negative and positive ions at high temperature above 500 K,[13] and more complex fragmentations and dynamics were seen for 2-NI.[15]
Determinations of crystal structures of many solids often use single‐crystal X‐ray diffraction (XRD). However, isolating single crystals of diffraction quality can be challenging and, in the case of amorphous active pharmaceutical ingredients (APIs), diffraction methods are less applicable.[16] NMR analysis has contributed a substantial fraction of the coordinate sets deposited in international databases of macromolecular structure (e.g. the Protein Data Bank curated at RCSB-Rutgers and the European Bioinformatics Institute).[17] Increasingly, solid-state NMR data have also been included in crystallographic studies to provide key distance measurements[18] and to clarify the tautomeric form of a material[19] or to locate regions of disorder in otherwise highly ordered solids.[20] More recently, solid-state NMR methods have advanced to refine molecular structures.[16,21–23] Often such NMR refinements result in only minor changes to structures but, in some cases, these adjustments provide new structural insights. For example, the original diffraction structure for cellulose Iα contained ambiguities at several O–H hydrogen orientations, suggesting that two significantly different hydrogen bonding arrangements were equally probable.[24] 15N NMR likely will be more sensitive to refinement than either 13C or 29Si, since 15N has a much larger chemical shift range and nitrogen atoms often contain a polarizable lone electron pair that is extraordinarily sensitive to the local electronic environment.[25] Experimental 15N tensors are at least 5 times more sensitive to crystal structure than 13C tensors due to nitrogen’s greater polarizability and larger range of chemical shifts.[22]
NMR spectroscopy has been used to screen ligands[26] and has been demonstrated to be a useful addition to conventional high-throughput screening (HTS) techniques to study the binding of small molecules to protein targets. Chemically linking these small chemical building blocks is generally expected to yield potent molecules that may evolve into a drug.[23] In particular, the positions of hydrogen atoms, which are generally determined inaccurately by XRD techniques, can be refined using solid-state NMR and quantum mechanical density functional theory (DFT) calculations to yield precise structures of the compounds.[23] Hence, NMR and XRD techniques combined with DFT calculations are powerful for refining crystal structures of drug building blocks. A large number of bioactive ligands and compounds, such as nitroimidazole and derivatives, are nitrogen containing compounds. Direct observation of the 15N NMR chemical shift of organic compounds using conventional NMR spectrometers is difficult owing to the low natural abundance (0.37 %) and the very poor receptivity of 15N.[25] As a result, insufficient information exists for 15N NMR chemical shifts of nitroimidazole derivatives such as 4-NI and CH3-4NI at various temperatures, including room temperature.
We report low temperature (100 K) dynamic nuclear polarization (DNP) enhanced NMR measurements of an API 4-NI (CAS number 3034-38-6, 97 %) and its methylated product, CH3-4NI given in Fig. 1. There is no complete 13C and 15N NMR chemical shift assignment (and accurate calculations) for all carbon (C(2), C(4) and C(5)) and nitrogen atoms (N(1), N(3) and N(6)) of this pair of compounds. The measurements are supported by accurate DFT based calculations in the present study. DNP was employed due to its impressive sensitivity and thus applicability for natural abundance 13C and 15N spectra and, in particular to enhance the sensitivity of the 15N spectrum of 4-NI, which has not been fully and accurately measured by conventional NMR spectrometers. DNP can potentially enhance the polarization and sensitivity of NMR experiments that use 1H nuclei by a factor of up to 658.[16] A satisfactory theoretical interpretation of these measurements,[9] supported by high level quantum mechanical calculations, does not exist to date.
Experimental and Computational Details
DNP-enhanced NMR measurements[27] were carried out on a 400 MHz Bruker Avance III spectrometer, equipped with a 263 GHz gyrotron and a cryogenic triple-resonance 3.2 mm magic angle spinning (MAS) probe, at a spinning rate of 8 kHz and temperature of 100 K. An equal volume to mass ratio of 10 mM AMUPol (d6-glycerol/D2O/H2O 6 : 3 : 1)[28] was added to 4-NI and packed into a 3.2 mm sapphire rotor. DNP-enhanced 13C and 15N cross polarization (CP) MAS NMR measurements were carried out using a 1H excitation pulse of 102 kHz, followed by a CP contact time of 1.5 (13C) or 3 ms (15N) using a 50 % ramped field on 1H and a 62 kHz field for 13C or 40.5 kHz for 15N and 1H SPINAL64 decoupling at 102 kHz during acquisition and a recycle delay of 30 s. Typically 16 scans for 13C and 64 scans for 15N were acquired and 50 Hz line broadening applied before Fourier transformation. The sample nitroimidazole was purchased from Alfa Aesar (purity 97 % for 4-NI) which was used without further purification and CH3-4NI was the same sample reported by Feketeova et al.[29]
13C CP MAS experiments performed on the 4-NI and CH3-4NI dry powders were carried out on a 600 MHz VnmrS spectrometer equipped with a triple-resonance 3.2 mm magic angle spinning (MAS) probe at a spinning rate of 8 kHz at room temperature. A 1H excitation pulse of 96 kHz, followed by a CP contact time of 1.0 ms using a 20 % ramped field on 1H and 52 kHz field for 13C were used with 96 kHz 1H SPINAL64 decoupling during acquisition and a recycle delay of 240 s. Typically 48 scans were acquired and 50 Hz line broadening applied before Fourier transformation.
Solution state NMR spectroscopy was achieved on a 600 MHz Bruker Avance III equipped with a 5 mm TCI cryo-probe. The 1H-13C heteronuclear single quantum coherence (HSQC) experiments were performed at 303 K in d6-DMSO, used for locking the signal. Four thousand points and 256 increments were acquired in the direct and indirect dimension, respectively. Sine-bell curve apodization and zero-filling were applied.
A DFT-based B3LYP functional with 6-311++G(d,p) basis set was employed for geometric optimization of 4-NI and CH3-4NI in the gas phase and in dimethyl sulfoxide (DMSO, ϵ = 46.7) solvent using a polarizable continuum model (PCM).[30] This DFT method is sufficient to produce reliable geometries for many organic compounds and APIs.[31–34] As pointed out by Lahiri et al.[35] there exist serious incongruities between computations performed at comparable levels of DFT and coupled cluster (CC) theory for dispersive optical properties, which is supported by polarimetric data of Stephens et al.[36] This method, however, predicts an accurate geometric structure and other properties for the target molecule, as it was employed in our earlier X-ray photoemission spectroscopic study for radiosensitizers including 4-NI with sufficient accuracy.[29] The natural bond orbital (NBO) charges are calculated using the wave function produced by B3WP91/6-311++G(d,p). The 1H, 13C, and 15N NMR isotropic shielding values were calculated using several DFT functionals, such as B3LYP, B3PW91, PBE0, M06 with two basis sets of 6-311++G(d,p) and 6-311G(d,p). The NMR chemical shifts were calculated using the invariant atomic orbital (GAIO) method.[37–41] The 1H and 13C chemical shifts are referenced to tetramethylsilane (TMS) whereas the 15N chemical shift is to NH3. The Gaussian 16 (G16)[42] computational chemistry package was used for the quantum mechanical calculations.
Results and Discussion
The 13C and 15N CP MAS NMR spectra of 4-NI and CH3-4NI are shown in Fig. 2. 4-NI displayed three 13C signals at 136.4, 144.7, and 119.4 ppm, which are assigned to C(2), C(4), and C(5) of 4-NI, respectively, in good agreement with earlier results.[43,44] The 13C chemical shifts for CH3-4NI were different for positions C(4) and C(5), 147.5 (Δδ of 1.7 ppm) and 121.3 ppm (Δδ of 1.6 ppm), respectively, while C(2) at 137.6 ppm remained within 0.5 ppm (Table 1). The methyl carbon was observed at 35.6 ppm. The 15N chemical shifts of 4-NI appeared at 180.8, 238.5, and 363.8 ppm, and were assigned to N(1), N(3), and N(6), respectively (Table 1) in good agreement with measurements for N(1) and N(3) in the crystal phase by Ueda et al.[9] at room temperature (300 K) of 180.5 and 237.4 ppm, respectively. The 15N chemical shift of the nitro nitrogen, N(6), was not determined in their measurement,[9] likely due to the low signal which was just discerned above the noise level (Fig. 2b). Note that in their measurements, Ueda et al. used 15NH4C1 (−341.2 ppm) as the external standard for the 15N chemical shift. As a result, the two measured signals due to amino (R2NH) and imino (=N–) are at approximately −200 and −135 ppm, respectively, which are referenced in the present system by adding 380.5 ppm to give the values in Table S7 (Supplementary Material). A comparison with available NMR chemical shift data for 4-NI and CH3-4NI is given in Table S7.

|

|
The methylation of N(1) produced perturbations to the 15N chemical shifts at position N(1) and N(3) while N(6) has a minor effect, with peaks observed at 176 (Δδ of −4.8 ppm), 250.1 (Δδ of 11.6 ppm), and 364.1 ppm (Δδ of 0.3 ppm), respectively. However, the 13C and 15N chemical shift of 4-NI wetted in a glycerol matrix and measured under DNP at 100 K did not show any apparent perturbation, with values maintained within 1 ppm of the dry state (Table 1). It is noteworthy that the 13C signals were significantly enhanced (ϵDNP) by a factor of 42, indicating an effective polarization transfer between the proton bath and 4-NI which supports a thorough soaking by the DNP matrix and thus a likely change in packing properties. Furthermore, 15N CP MAS of the dry state required several days of acquisition to produce a low signal to noise spectrum, while that by DNP was acquired in ~30 min with good signal to noise, in particular for N(6), which demonstrates the power of DNP. The similarity between the dry state and the frozen hydrated state for 4-NI could indicate that packing may not be relevant in modulating the chemical shifts.
Both analogues were also dissolved in d6-DMSO for further assessing the packing effects on the 13C chemical shift. The 1H-13C HSQC spectrum shown in Fig. 3 displayed two correlations for 4-NI (C(5) 118.4, H(5) 8.2 ppm and C(2) 135.4, H(2) 7.8 ppm) and three correlations for CH3-4NI (C(5) 122.0, H(5) 8.3 ppm, C(2) 137.5, H(2) 7.8 ppm, and Cmethyl 33.9, H(7) 3.8 ppm). The results are within 1 ppm of the respective dry or frozen/hydrated states, except for the methyl group (Δδ of 1.7 ppm in dry state), which may further indicate minimal impact of molecular packing on the reported 13C chemical shifts, within experimental errors.
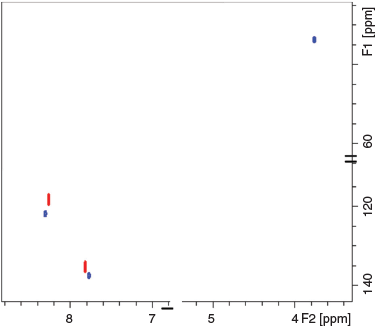
|
The chemical shift differences between 4-NI and CH3-4NI could be due to different intermolecular hydrogen bonding, as shown in Fig. 4. The X-ray crystallographic data for 4-NI indicated a chain of 4-NI molecules built by N(1)–H⋯N(3) bonding[7,11] between different 4-NI molecules stacked in the same plane. The intermolecular N(1)–H(1)⋯N(3) hydrogen bond distance was 2.1 Å[11] at room temperature but decreased to 1.9 Å at 100 K.[7] Moreover, hydrogen bonding is stronger for N(1)–H(1)⋯N(3) in the crystal phase, with a H⋯N length of 1.9 Å compared with the intermolecular hydrogen bond lengths for C(2)–H(2)⋯O(1) of 2.3 Å and C(5)–H(5)⋯O(2) of 2.4 Å. Interestingly, the more recent crystal structure for CH3-4NI[45] shows that molecules do not stack in the same plane and the intermolecular hydrogen bonds are very different. Rather than N(1)–H⋯N(3) as in 4-NI (Fig. 4a), the CH3-4NI intermolecular hydrogen bonding is between the N(3) atom and the hydrogen on C(5) of another CH3-4NI molecule in a different plane with a N(3)⋯HC(5) distance of 3.93 Å. Furthermore, in plane hydrogen bonds involving the nitro group with lengths for N(6)O⋯HCH2 of 2.57 Å and N(6)O′⋯HC(2) of 2.67 Å (Fig. 4b).
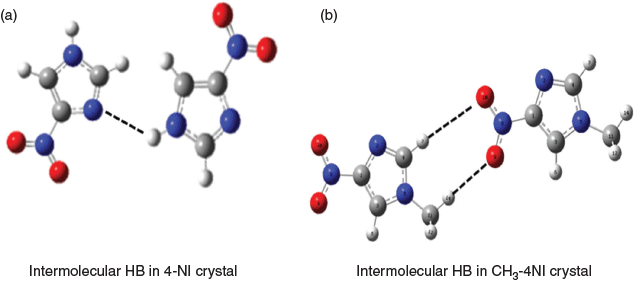
|
The DMSO solvent has the propensity to disrupt hydrogen bond networks and thus could affect the chemical shifts of 4-NI and CH3-4NI by altering formation of the N(1)–H(1)⋯N(3) or N(6)O⋯HCH2 and N(6)O′⋯HC(2) networks. By comparing the 15N and 13C chemical shifts in the dry state and in DMSO, it appeared that N(3) is the most sensitive to changes in intermolecular bonding.
In Table 2 we compare the geometric properties of crystal structures of 4-NI[7] and CH3-4NI[46] with those calculated using the B3PW91/6311++G(d,p) method. As both 4-NI and CH3-4NI are planar (imidazole ring) molecules, the agreement between the crystal structures and the calculated structures are excellent, except for small angular discrepancies which are larger in CH3-4NI than in 4-NI due to the methyl causing non-planarity. The accurately predicted geometric parameters of the compounds build confidence on other property calculations based on the geometries and the method. A comparison between the crystal and the calculated structures using different DFT functionals are given in Table S5 (Supplementary Material).

|
Table 3 compares the calculated 1H, 13C, and 15N NMR chemical shifts of 4-NI and CH3-4NI with our measurements in DMSO solution. In order to examine the accuracy of the DFT methods in producing the NMR chemical shifts, the present study used several DFT functionals, including B3LYP, B3PW91, M06, and PBE0 with slightly different basis sets of 6-311++G(d,p) and 6-311G(d,p). When the same basis set is employed, the calculated 13C NMR chemical shifts of resveratrol[32] using the B3LYP and B3PW91 DFT functionals are more accurate than the M06-2x functional. A comparison of calculated chemical shifts using several DFT functionals including the M06 DFT functional are given in Table S6 (Supplementary Material). This finding of more accurate performance of B3PW91 DFT functions for the NMR chemical shifts is in good agreement with Ramalho et al.[47] who found that, among several DFT methods, the B3LYP gives more accurate NMR chemical shifts for other nitroimidazole derivatives.[47] Moreover, we further discovered that the B3PW91 functional produced slightly better results than B3LYP (Table S6, Supplementary Material). Table 3 reports the calculations using B3PW91 and different basis sets in comparison with the present measurements in DMSO solution.

|
The 15N NMR chemical shifts of 4-NI and CH3-4NI present a challenge to both experiment and theory. Results in Table 3 show good agreement for the 1H and 13C NMR chemical shifts, which are within 2 % for the imidazole carbons for both 4-NI and CH3-4NI. The smaller basis set without diffuse functions, 6-311G(d,p) leads to more accurate chemical shifts. For example, the chemical shift of C(5) and N(1) are given by 119.3 and 183.9 ppm, which correspond well with the measurement at 118.4 and 174.9 ppm, respectively. However, the 15N NMR chemical shifts of the compounds are more sensitive to the phase and chemical structure and show greater differences between measurements under various conditions as well as between experiment and theory. In an 15N CP MAS NMR study of 4-substituted imidazole derivatives, Ueda et al.[9] found that the chemical shift difference was related to the chemical structures of the 4-position imidazole derivatives, including the nature of the substituents as well as the intermolecular hydrogen bonding of the imidazole derivatives. The chemical shifts of the imidazole nitrogens, amino N(1) and imino N(3) of 4-NI move in opposite directions, i.e. 7 ppm high field shift for N(1) but 7 ppm low field for N(3) compared with unsubstituted imidazole. The calculated 15N NMR chemical shifts of the compound exhibit large discrepancies, largely due to the effect of hydrogen bonding as identified by Ueda et al.[9]
The larger discrepancy for 15N chemical shifts for 4-NI between the calculations and the measurements suggests that other important structural information is missing in the theoretical model in addition to intermolecular hydrogen bonding. One possibility could be resonant structures for the imidazole ring as well as the ionic configuration of the nitro nitrogen (–NO2). The 15N NMR chemical shift difference is particularly large for N(3), which is nearly 10 % off the measurement for 4-NI (in solid). Such significant discrepancies between the calculated and the measured chemical shifts of 15N NMR suggest that the chemical bonding of the nitrogens in the compound pair might engage with resonant structures in solution,[48] leading to the bonding character changes for N(1) and N(3). The nitro N(6) chemical shift may be as a result of the ionic configuration[9] and the availability of the nitro (-NO2) for ionization.[49] As a result, to accurately determine and understand the electronic structures of nitroimidazole derivatives, further combined configuration and phase specific methods with molecular dynamics are needed.[50]
Conclusion
4-Nitroimidazole (4-NI) and 1-methyl-4-nitromidazole (CH3-4NI) are of importance as pharmaceutical ingredients. However, determining the structure–activity relationship of nitroimidazole and its derivatives is challenging as imidazole exhibits a unique electronic structure. In the present study, the 1H, 13C, and 15N NMR chemical shifts and the effect of methylation of 4-NI were studied using dynamic nuclear polarization (DNP)-enhanced solid-state NMR and in d6-DMSO solvent. The measurements in d6-DMSO solvent are supported by density functional theory (DFT) calculations. The complete non-hydrogen chemical shifts of 4-NI were observed in the solid-state for the first time at 136.4, 144.7, and 119.4 ppm for 13C, and at 181.5, 237.4, and 363.0 ppm for 15N. Methylation was found to have less impact on the 13C NMR chemical shifts, as the CH3- group forms an N–C bond at N(1) in CH3-4NI. However, the chemical shifts of the imidazole nitrogens, N(1) and N(3) are more affected and, in particular, the imino nitrogen N(3) changes significantly from 237.4 ppm in 4-NI to 250.1 ppm in CH3-4NI in the solid state. The amino imidazole nitrogen N(1) experienced a shielding shift of 5.5 ppm, from 181.5 ppm in 4-NI to 176.0 ppm in CH3-4NI. There was little chemical shift variation for the nitro nitrogen of N(6) with less than ± 0.3 ppm change in chemical shift as the nitro group is not within the imidazole ring.
The relationship between the electron structure and the 13C and 15N chemical shifts is complex,[44] particularly in the case of nitroimidazole and its derivatives. The sensitivity of 15N NMR chemical shifts to methylation at the N(1) position of the imidazole ring has been observed. Deprotonation of the amino N(1)–H of 4-NI to form N(1)–CH3, led to opposite chemical shift changes for N(1) and N(3) in the solid state. This may be due to a difference in intermolecular hydrogen bonding for solid 4-NI and CH3-4NI. The amino nitrogen N(1) in CH3-4NI is unable to donate a H to form the same intermolecular hydrogen bonds as in 4-NI. The NMR chemical shifts of 4-NI and CH3-4NI were supported by quantum mechanical calculations using DFT methods, including B3PW91/6-311+G(d,p), confirming that methylation of 4-NI affects the imidazole nitrogens in CH3-4NI. The calculated 1H and 13C NMR chemical shifts agreed well with measurements in DMSO, whereas the 15N chemical shift agreement between the measurements and theory differed ~13 %, suggesting that the nitrogen atoms of 4-NI and CH3-4NI may be subject to multiple configurations, hydrogen bonding, and resonant structures, that are phase dependent. In addition, theoretical considerations need further investigation, e.g. the DFT functionals, basis sets, combination of DFT functionals and basis sets,[51] implicit solvent model, and the quantum mechanical NMR GIAO approximation employed. Finally, temperature may play a role as the DFT calculations do not include temperature whereas the NMR measurements are at 100 and 298 K. Confining the nitroimidazole compounds within a single configuration may be unable to reveal the reality of the status of the compounds. Further studies, such as molecular dynamics, may provide greater insight into this direction.
As indicated by Suyama et al.[52] structural mis-assignments continue to be reported for several organic compounds, including small drug molecules and natural products. With higher-field magnets and more sensitive probes, structural mis-assignment does not necessarily attenuate, but structure elucidation of increasingly limited quantities of minor components is possible (such as 5-nitroimidazole (5-NI) for which a 400 : 1 equilibrium of 4-NI versus 5-NI is claimed[44]). Assignments can be further complicated by the greater structural complexity of larger compounds and averaging effects dependent on time scale and temperature.[32] Assigning the NMR spectra including full stereochemistry can be very challenging and combining highly accurate NMR measurements supported by computational spectroscopy is required.
Supplementary Material
4-Nitroimidazole and 1 methyl-4-nitroimidazole calculations, such as molecular properties and calculated chemicals shifts using other DFT functionals for 4NI and CH3-4NI, are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
FB acknowledges the Research Training Program Scholarships (RTPS) from the Australian Government and thanks Dr Subhojyoti Chatterjee and Dr Shawkat Islam for technical assistance. Swinburne University supercomputing (OzSTAR) and National Computational Infrastructure (NCI), supported by the Australian Government, are acknowledged for computing facilities. FS and MAS acknowledge the Australian Research Council for LIEF grant LE160100120 providing funding of DNP-NMR at the Bio21 Institute NMR facility. FW thanks University of Melbourne for hosting of sabbatical visit. The authors thank Professor Jonathan White, University of Melbourne, for gift of the CH3-4NI sample. Finally, they thank Professor Robert Metzger (SDSU) for useful discussion regarding NMR calculations.
References
[1] B. C. Chen, W. Von Philipsborn, K. Nagarajan, Helv. Chim. Acta 1983, 66, 1537.| Crossref | GoogleScholarGoogle Scholar |
[2] P. Wardman, Br. J. Radiol. 2019, 92, 20170915.
| 29303355PubMed |
[3] H. W. Ueber, Exp. Pathol. Pharmakol. 1887, 22, 253.
[4] R. Weinshilboum, Clin. Biochem. 1988, 21, 201.
| Crossref | GoogleScholarGoogle Scholar | 3044645PubMed |
[5] T. Mukherjee, H. Boshoff, Future Med. Chem. 2011, 3, 1427.
| Crossref | GoogleScholarGoogle Scholar | 21879846PubMed |
[6] P. Jiménez, J. Laynes, R. Claramunt, D. Sanz, J. P. Fayet, M. C. Vertut, J. Catalán, J. L. G. Paz, G. Pfister-Guillouzo, C. Guimon, R. Flammang, A. Maquestiau, J. Elguero, New J. Chem. 1989, 13, 151.
[7] H. L. De Bondt, E. Ragia, N. M. Blaton, O. M. Peeters, C. J. De Ranter, Acta Crystallogr. Sect. C 1993, 49, 694.
[8] A. I. Vokin, L. V. Sherstyannikova, I. G. Krivoruchka, T. N. Aksamentova, O. V. Krylova, V. K. Turchaninov, Russ. J. Gen. Chem. 2003, 73, 973.
| Crossref | GoogleScholarGoogle Scholar |
[9] T. Ueda, S. Nagatomo, H. Masui, N. Nakamura, S. Hayashi, Z. Naturforsch. A 1999, 437.
[10] T. M. T. Carvalho, L. M. P. F. Amaral, V. M. F. Morais, M. D. M. C. Ribeiro da Silva, J. Chem. Thermodyn. 2017, 105, 267.
| Crossref | GoogleScholarGoogle Scholar |
[11] I. Ségalas, J. Poitras, A. L. Beauchamp, Acta Crystallogr. Sect. C 1992, 48, 295.
| Crossref | GoogleScholarGoogle Scholar |
[12] G. A. Worth, P. M. King, W. G. Richards, Biochim. Biophys. Acta 1989, 993, 134.
| Crossref | GoogleScholarGoogle Scholar | 2804121PubMed |
[13] L. Feketeová, J. Postler, A. Zavras, P. Scheier, S. Denifl, R. A. J. O’Hair, Phys. Chem. Chem. Phys. 2015, 17, 12598.
| Crossref | GoogleScholarGoogle Scholar | 25899156PubMed |
[14] P. Bolognesi, A. R. Casavola, A. Cartoni, R. Richter, P. Markus, S. Borocci, J. Chiarinelli, S. Tošić, H. Sa’adeh, M. Masič, B. P. Marinković, K. C. Prince, L. Avaldi, J. Chem. Phys. 2016, 145, 191102.
| Crossref | GoogleScholarGoogle Scholar | 27875881PubMed |
[15] J. Chiarinelli, A. R. Casavola, M. C. Castrovilli, P. Bolognesi, A. Cartoni, F. Wang, R. Richter, D. Catone, S. Tosic, B. P. Marinkovic, L. Avaldi, Front Chem. 2019, 7, 329.
| Crossref | GoogleScholarGoogle Scholar | 31157205PubMed |
[16] L. Zhao, A. C. Pinon, L. Emsley, A. J. Rossini, Magn. Reson. Chem. 2018, 56, 583.
| Crossref | GoogleScholarGoogle Scholar | 29193278PubMed |
[17] P. C. Driscoll, in Encyclopedia of Biophysics (Ed. G. C. K. Roberts) 2013, pp. 2488–2495 (Springer: Berlin).
[18] A. Hofstetter, M. Balodis, F. M. Paruzzo, C. M. Widdifield, G. Stevanato, A. C. Pinon, P. J. Bygrave, G. M. Day, L. Emsley, J. Am. Chem. Soc. 2019, 141, 16624.
| Crossref | GoogleScholarGoogle Scholar | 31117663PubMed |
[19] F. Rossi, N. T. Duong, M. K. Pandey, M. R. Chierotti, R. Gobetto, Y. Nishiyama, Magn. Reson. Chem. 2019, 57, 294.
| Crossref | GoogleScholarGoogle Scholar | 30684385PubMed |
[20] M. G. Jain, K. R. Mote, P. K. Madhu, Crystals 2019, 9, 231.
| Crossref | GoogleScholarGoogle Scholar |
[21] S. T. Holmes, R. W. Schurko, J. Phys. Chem. C 2018, 122, 1809.
| Crossref | GoogleScholarGoogle Scholar |
[22] K. Kalakewich, R. Iuliucci, K. T. Mueller, H. Eloranta, J. K. Harper, J. Chem. Phys. 2015, 143, 194702.
| Crossref | GoogleScholarGoogle Scholar | 26590548PubMed |
[23] S. T. Holmes, O. G. Engl, M. N. Srnec, J. D. Madura, R. Quiñones, J. K. Harper, R. W. Schurko, R. J. Iuliucci, J. Phys. Chem. A 2020, 124, 3109.
| Crossref | GoogleScholarGoogle Scholar | 32233483PubMed |
[24] J. Powell, K. Kalakewich, F. J. Uribe-Romo, J. K. Harper, Phys. Chem. Chem. Phys. 2016, 18, 12541.
| Crossref | GoogleScholarGoogle Scholar | 27087051PubMed |
[25] E. Kolehmainen, B. Ośmiałowski, Int. Rev. Phys. Chem. 2012, 31, 567.
| Crossref | GoogleScholarGoogle Scholar |
[26] K. A. Mercier, M. D. Shortridge, R. Powers, Comb. Chem. High Throughput Screen. 2009, 12, 285.
| Crossref | GoogleScholarGoogle Scholar | 19275534PubMed |
[27] M.-A. Sani, S. Zhu, V. Hofferek, F. Separovic, FASEB J. 2019, 33, 11021.
| 31284743PubMed |
[28] C. Sauvee, M. Rosay, G. Casano, F. Aussenac, R. Weber, O. Ouari, P. Tordo, Angew. Chem. Int. Ed. Engl. 2013, 52, 10858.
| Crossref | GoogleScholarGoogle Scholar | 23956072PubMed |
[29] L. Feketeová, O. Plekan, M. Goonewardane, M. Ahmed, A. Albright, J. White, R. A. J. O’Hair, M. R. Horsman, F. Wang, K. C. Prince, J. Phys. Chem. A 2015, 119, 9986.
| Crossref | GoogleScholarGoogle Scholar | 26344652PubMed |
[30] Y. Takano, K. N. Houk, J. Chem. Theory Comput. 2005, 1, 70.
| Crossref | GoogleScholarGoogle Scholar | 26641117PubMed |
[31] A. Ganesan, F. Wang, J. Chem. Phys. 2009, 131, 044321.
| Crossref | GoogleScholarGoogle Scholar | 19655881PubMed |
[32] F. Wang, S. Chatterjee, J. Phys. Chem. B 2017, 121, 4745.
| Crossref | GoogleScholarGoogle Scholar | 28402662PubMed |
[33] S. Islam, A. Ganesan, R. Auchettl, O. Plekan, R. G. Acres, F. Wang, K. C. Prince, J. Chem. Phys. 2018, 149, 134312.
| Crossref | GoogleScholarGoogle Scholar | 30292192PubMed |
[34] H. Sa’adeh, F. Backler, F. Wang, S. Piccirillo, A. Ciavardini, R. Richter, M. Coreno, K. C. Prince, J. Phys. Chem. A 2020, 124, 4025.
| Crossref | GoogleScholarGoogle Scholar | 32336097PubMed |
[35] P. Lahiri, K. B. Wiberg, P. H. Vaccaro, M. T. Caricato, D. Crawford, Angew. Chem. Int. Ed. 2014, 53, 1386.
[36] D. C. Stephens, S. K. Leggett, M. C. Cushing, M. S. Marley, D. Saumon, T. R. Geballe, D. A. Golimowski, X. Fan, K. S. Noll, Astrophys. J. 2009, 702, 154.
[37] F. London, J. Phys. Radium 1937, 8, 397.
| Crossref | GoogleScholarGoogle Scholar |
[38] R. McWeeny, Phys. Rev. 1962, 126, 1028.
[39] R. Ditchfield, Mol. Phys. 1974, 27, 789.
| Crossref | GoogleScholarGoogle Scholar |
[40] K. Wolinski, J. F. Hilton, P. Pulay, J. Am. Chem. Soc. 1990, 112, 8251.
| Crossref | GoogleScholarGoogle Scholar |
[41] J. R. Cheeseman, G. W. Trucks, T. A. Keith, M. J. Frisch, J. Chem. Phys. 1996, 104, 5497.
| Crossref | GoogleScholarGoogle Scholar |
[42] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, D. J. Fox, Gaussian 16, Revision C.01 2016 (Gaussian, Inc.: Wallingford, CT).
[43] A. McKillop, D. E. Wright, M. L. Podmore, R. K. Chambers, Tetrahedron 1983, 39, 3797.
| Crossref | GoogleScholarGoogle Scholar |
[44] E. Lippmaa, M. Mägi, S. S. Novikov, L. I. Khmelnitski, A. S. Prihodko, O. V. Lebedev, L. V. Epishina, Magn. Reson. Chem. 1972, 4, 153.
[45] H. A. Shahid, S. Jahangir, S. Yousuf, M. Hanif, S. K. Sherwani, Arab. J. Chem. 2016, 9, 668.
| Crossref | GoogleScholarGoogle Scholar |
[46] J. Wang, P. Lian, L. Chen, Z. Kristallogr. – New Cryst. Struct. 2017, 232, 699.
[47] T. C. Ramalho, M. Buhl, Magn. Reson. Chem. 2005, 43, 139.
| Crossref | GoogleScholarGoogle Scholar | 15558660PubMed |
[48] W. F. Reynolds, I. R. Peat, M. H. Freedman, J. R. Lyerla, J. Am. Chem. Soc. 1973, 95, 328.
| Crossref | GoogleScholarGoogle Scholar | 4687673PubMed |
[49] A. Cartoni, A. R. Casavola, P. Bolognesi, M. C. Castrovilli, D. Catone, J. Chiarinelli, R. Richter, L. Avaldi, J. Phys. Chem. A 2018, 122, 4031.
| Crossref | GoogleScholarGoogle Scholar | 29652141PubMed |
[50] S. Hashem, L. Cupellini, F. Lipparini, B. Mennucci, Mol. Phys. 2020, e1771449.
| Crossref | GoogleScholarGoogle Scholar |
[51] D. O. Samultsev, V. A. Semenov, L. B. Krivdin, Magn. Reson. Chem. 2014, 52, 222.
| Crossref | GoogleScholarGoogle Scholar | 24573615PubMed |
[52] T. L. Suyama, W. H. Gerwick, K. L. McPhail, Bioorg. Med. Chem. 2011, 19, 6675.
| Crossref | GoogleScholarGoogle Scholar | 21715178PubMed |
* Professor Frances Separovic is the recipient of the RACI 2019 Margaret Sheil Women in Chemistry Leadership Award.