

Copper Complexes of Benzoylacetone Bis-Thiosemicarbazones: Metal and Ligand Based Redox Reactivity*
Jessica K. Bilyj A , Jeffrey R. Harmer B and Paul V. Bernhardt A C
A C
A School of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, Qld 4072, Australia.
B Centre for Advanced Imaging, University of Queensland, Brisbane, Qld 4072, Australia.
C Corresponding author. Email: p.bernhardt@uq.edu.au
Australian Journal of Chemistry 74(1) 34-47 https://doi.org/10.1071/CH20210
Submitted: 30 June 2020 Accepted: 30 July 2020 Published: 9 September 2020
Abstract
Bis-thiosemicarbazones derived from the β-diketone benzoylacetone (H3banR, R = Me, Et, Ph) are potentially tetradentate N2S2 ligands whose coordination chemistry with copper is reported. In the absence of oxygen and in the presence of base they form anionic CuII complexes of the fully deprotonated ligands [CuII(banR)]–. Upon exposure to atmospheric oxygen they undergo a complex series of reactions leading to two types of products; one a ligand oxidised ketone complex [CuII(banRO)] and the other an unprecedented dimeric di-CuIII complex [(CuIII(banR))2] depending on the R substituent. Time-resolved UV-vis spectroscopy, cyclic voltammetry, spectroelectrochemistry, and electron paramagnetic resonance (EPR) spectroscopy have been used to identify intermediates on the way to stable products formed under both anaerobic and aerobic conditions. It is found that both ligand-centred and Cu-centred oxidation reactions are occurring in parallel leading to this unusually complicated mixture of products.
Introduction
Bis-thiosemicarbazones derived from aliphatic dicarbonyl compounds such as diacetyl (butane-2,3-dione) and acetylacetone (pentane-2,4-dione) have proven to be versatile N2S2 chelating ligands for several transition metals with copper featuring prominently. Apart from the interesting electronic properties that they impart on the metal, they are also readily functionalised at the terminal N4-atom of the thiosemicarbazone. Donnelly and co-workers utilised this property to prepare bifunctional chelators[1–3] that have found particular use in medical imaging with radioactive Cu isotopes.
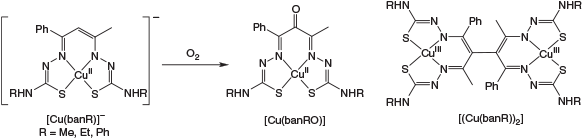
Our own interest has been on the ligands derived from acetylacetone where we have reported some remarkably facile redox reactions on the coordinated ligand; in particular conversion of the apical C-atom into a carbonyl group in the presence of oxygen as the sole oxidant.[4] These reactions do not take place in the absence of metal (Ni or Cu) and intermediates observed over the time course of the reaction suggest that both metal-centred and ligand-centred redox reactions occur in parallel. This provides a contrast between the [Cu(ATSM)] and [Cu(acacR)] (ATSM = diacetyl-bis(N4-methylthiosemicarbazone; HacacR = acetylacetone-bis(thiosemicarbazone)) families despite their structural similarities. Their divergent reactivities stem from the introduction of an acidic methylene group in the [Cu(acacR)] analogues which upon deprotonation (as in Fig. 1) introduces an extra negative charge which results in electronic delocalisation and also brings normally unstable trivalent copper within reach.

|
In this work we have studied the effects of replacing acetylacetone with the aromatic analogue benzoylacetone (1-phenyl-butane-1,3-dione) to see how this affects the structural, electrochemical, and spectroscopic properties of the resulting bis-thiosemicarbazones in complex with copper. The free ligands (H3banR, Fig. 2), which exist in their cyclised pyrazoline form, and their ring-opened copper complexes have been studied with a combination of solution spectroscopic methods (UV-vis and electron paramagnetic resonance (EPR)) in addition to coupled electrochemical measurements to explore the steric and electronic effects of this methyl→phenyl substitution.
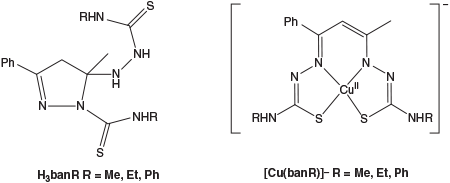
|
Experimental
Ligand Synthesis
All reagents were obtained commercially and used as received.
Proligand 1: 3,3′-(Ethane-1,2-diylbis(azaneylylidene))bis(1-phenylbutan-1-ol)
Two equivalents of benzoylacetone (2.55 g, 1.57 × 10−2 moles) and one equivalent of ethane-1,2-diamine (0.525 mL, 7.8 × 10−3 moles) were combined in ethanol (15 mL) and stirred for 16 h at room temperature. The reaction produced a colourless powder that precipitated upon addition of water (15 mL). Yield: 2.38 g, 87 %. δH (CDCl3) 2.07 (3H, s, CH3), 3.58 (4H, d, CH2), 5.7 (2H, s, CH), 7.40 (6H, m, Ar-H), 7.84 (4H, d, Ar-H)
Proligand 2: Pentane-2,4-diylidene)bis(azaneylylidene))bis(ethan-1-ol)
Benzoylacetone (1 g, 6.16 × 10−3 moles) and ethanolamine (0.7420 mL (0.7532 g), 12.33 × 10−3 moles) were combined in ethanol (15 mL) and stirred for 16 h at room temperature. An immediate yellow solution was produced which developed to orange. Evaporation of the solvent under vacuum produced an oily orange solid. Yield: 1.31 g, 86 %. δH (CDCl3) 2.07 (3H, s, CH3), 2.10 (2H, s, OH), 2.83 (2H, t, CH2), 3.48 (2H, t, CH2), 3.61 (2H, t, CH2), 3.81 (2H, t, CH2), 5.68 (1H, s, CH), 7.39 (3H, m, Ar-H), 7.82 (2H, d, Ar-H).
H3banme
Proligand 1 (0.6995 g, 2.0 × 10−3 moles) was dissolved in anhydrous methanol (10 mL) and transferred to a dropping funnel with 1 mL of acetic acid. 4-Methyl-3-thiosemicarbazide (0.8440 g, 8.029 × 10−3 moles, a 2-fold excess) was dissolved in anhydrous methanol (10 mL) in a three necked round bottom flask with 0.5 mL of acetic acid. The solution containing proligand 1 was added dropwise over three days to the stirred solution at 70°C, with the reaction left for an additional day. The reaction mixture was evaporated to dryness to produce a yellow powder. The powder was suspended in diethyl ether (10 mL) and the insoluble precipitate was filtered off and washed with diethyl ether (5 mL). The diethyl ether extracts contained the mono-thiosemicarbazone byproduct, which was discarded. The solid was suspended in chloroform (10 mL) and the insoluble precipitate of 4-methyl-3-thiosemicarbazide washed with a further 5 mL of chloroform. The chloroform extracts were combined and evaporated producing an oil, which upon addition of ethanol (10 mL) resulted in precipitation of the desired product. Yield: 0.3502 g, 52 %. Anal. Calc. for C14H20N6S2: C 50.0, N 25.0, H 6.00, S 19.1. Found: C 49.38, N 24.69, H 5.89, S 20.34 %. δH (DMSO-d6) 1.89 (3H, s, CH3), 3.17(6H, dd, CH3), 3.20 (2H, AB quartet, CH2), 6.75 (1H, s, NH), 6.96 (1H, s, NH), 7.34 (1H, quartet, NH), 7.42 (3H, m, Ar-H), 7.62 (1H, quartet, NH), 7.65 (2H, dd, Ar-H).
H3banet
Proligand 2 (0.5293 g, 2.13 × 10−3 moles) was dissolved in anhydrous methanol (15 mL) with 1.5 mL of acetic acid in a dropping funnel. Dry 4-ethyl-3-thiosemicarbazide (0.7496 g, 6.29 × 10−3 moles) was added to the three-necked round bottom flask with anhydrous methanol (15 mL) and 0.325 mL of acetic acid. The solution containing proligand 2 was added dropwise over three days at 70°C. The solution was evaporated to dryness to produce a yellow/orange oil. Ethanol (10 mL) was added and the mono-thiosemicarbazone byproduct precipitated overnight, which was filtered, washed with cold ethanol (5 mL), and discarded. Evaporation of the filtrate produced an oil, which upon the addition of chloroform (10 mL) precipitated unreacted 4-ethyl-3-thiosemicarbazide, which was filtered and washed with 5 mL of chloroform. The chloroform extracts were evaporated to dryness to obtain an oil, which was taken up in ethanol (15 mL) and this was left to slowly evaporate at room temperature. The resulting solid containing the desired product was filtered off and washed with diethyl ether. Yield: 0.0573 g, 7.4 %. Anal. Calc. for C16H24N6S2: C 52.7, N 23.0, H 6.6, S 17.6. Found: C 52.66, N 23.09, H 6.61, S 18.18 %. δH (DMSO-d6) 1.29 (6H, m, CH3), 1.89 (3H, s, CH3), 3.19 (2H, AB quartet, CH2), 3.69 (4H, m, CH2), 6.75 (1H, s, NH), 6.92 (1H, s, NH), 7.39 (3H, m, Ar-H), 7.43 (1H, t, NH), 7.62 (2H, d, Ar-H). Crystals suitable for X-ray work were obtained from slow evaporation of an EtOH solution of the compound.
H3banphe
Proligand 2 (0.2511 g, 1.01 × 10−3 moles) with 0.35 mL of acetic acid and 4-phenyl-3-thiosemicarbazide (0.4800 g, 2.87 × 10−3 moles) with 0.175 mL acetic acid were reacted using the same method as above for H3banet. The work-up was conducted in a different order, whereupon evaporation of the reaction mixture was followed by addition of chloroform (10 mL) to precipitate unreacted 4-phenyl-3-thiosemicarbazide, which was filtered off and discarded. Evaporation of the chloroform filtrate and addition of ethanol (15 mL) saw the mono-thiosemicarbazone precipitate as a crystalline solid overnight, which was filtered off and discarded. The ethanolic filtrate was then left to evaporate before diethyl ether (10 mL) was added, precipitating the di-substituted product overnight. The product was then filtered off and washed with cold ethanol. Yield: 0.0398 g, 8.5 %. Anal. Calc. for C28H34N6OS2 (diethyl ether solvate): C 62.89, N 15.72, H 6.41, S 11.99. Found: C 63.9, N 15.9, H 5.2, S 11.9 %. δH (DMSO-d6) 2.36 (3H, s, CH3), 3.15 (2H, AB quartet, CH2), 7.28 (11H, m, Ar-H), 7.41 (2H, d, Ar-H), 7.55 (1H, d, NH), 7.98 (2H, m, Ar-H), 9.07 (1H, s, NH), 10.01 (1H, s, NH), 10.55 (1H, s, NH).
Complexation with Copper
All measurements were undertaken on complexes prepared in situ by combining stoichiometric amounts of copper acetate monohydrate and free ligand (each dissolved in DMSO at approximately millimolar concentrations) under a dinitrogen atmosphere within a Belle Technology anaerobic glovebox (O2 < 20 ppm). Due to their reactivity, no attempt was made to isolate the complexes in pure form and characterisation was carried out in DMSO by UV-vis and EPR spectroscopy as well as cyclic voltammetry and UV-vis spectroelectrochemistry as described for each experiment.
Physical Methods
Electrochemistry
Cyclic voltammetry measurements were carried out with a BAS100B/W potentiostat employing a glassy carbon working electrode, platinum counter electrode, and Ag wire (in 0.1 M Et4NClO4, DMSO) reference electrode. DMSO (containing 0.1 M Et4NClO4 as supporting electrolyte) was used in all experiments. Complex solutions were made up in the glovebox as 8.856 × 10−3 M [Cu(banme)], 8.175 × 10−3 M [Cu(banet)], and 6.513 × 10−3 M [Cu(banphe)] with 3 equivalents of triethylamine in 5 mL and left to complex under anaerobic conditions for 2 days, before collecting electrochemical data. Upon completion of the sample measurements under anaerobic conditions, the solutions were removed from the glovebox and left to spontaneously oxidise in air for 2 days before final measurements of the aerobically oxidised samples being taken. All electrochemical potentials were referenced externally against the ferrocene/ferrocenium couple in DMSO.
UV-Vis Spectroelectrochemistry
Spectroelectrochemistry measurements were undertaken using a 1.7 mm pathlength spectroelectrochemical cell (Pine Instruments), in combination with an integrated platinum ‘honeycomb’ working and counter electrode, and a Ag wire in DMSO with 0.1 M Et4NClO4 as the reference electrode. Stock solutions of 1 × 10−2 M complex (with base) were diluted 12-fold in DMSO (0.1 M Et4NClO4) before measurement. Data were modelled by global analysis of the entire potential dependent UV-vis spectrum with the program Reactlab Redox.[5]
Time Resolved UV-Vis Spectroscopy
Approximately 1 mM ligand solutions and a stock solution of Cu(OAc)2·H2O (~166 mM) in DMSO were deoxygenated in the glovebox overnight. An aliquot of 0.8 mL of H3banme or H3banet was added to a cuvette with 1.2 mL of deoxygenated DMSO or 0.5 mL of H3banphe with 1.5 mL of deoxygenated DMSO; the smaller amount being due to the greater molar absorptivity of the phenyl-substituted ligand and complex. Three equivalents of Et3N were added. An equimolar amount of the copper acetate solution (a droplet) was added into the top corner of the cuvette and held there by surface tension to delay mixing with the ligand solution. Silicon grease coated lids sealed the cuvettes before them being removed from the glovebox. The cuvette contents were mixed and then time-dependent spectroscopic measurements were taken every 30 s for the first 3 h, thereafter every 300 s up to 48 h. The same solutions were used to monitor the reactivity of the complexes aerobically by removing the lids on the cuvettes. The aerobic oxidation experiment was run for 24 h with measurements taken every 5 min. Global analysis of the time-dependent spectroscopic data was carried out with Reactlab Kinetics.[6]
EPR Spectroscopy
Q-band (~34 GHz) EPR spectra were measured on an Bruker ElexE580 spectrometer equipped with a 150 W TWT Amplifier (Model 187Ka, Applied Systems Engineering, Inc.), a Bruker Q-band probe head (taking 3 mm EPR tubes, ER5106QT-2), and a cryogen-free variable temperature cryostat (No. 3679A, Cryogenics, Inc.). Field-sweep data were acquired by integration of the signal (free induction decay) from a single microwave pulse of 600 ns in length using a repetition time of 500 μs and 400 shots per point. The derivative of this absorption spectrum was calculated numerically with MatLab using a second order polynomial fitted ± 5 points either side of the intensity point being processed. Samples were prepared by combining ligand solutions (1 mM in DMSO) with an equimolar amount of Cu(OAc)2·H2O solution (in DMSO) plus three equivalents of Et3N in a glovebox and the mixture was allowed to equilibrate over a week to generate the anaerobic complexes. The solutions were then exposed to the atmosphere for 2 days before measurement of the aerobic products. Simulation of all EPR spectra were carried out with the program EasySpin.[7]
X-Ray Crystallography
Crystallographic data were collected on an Oxford Diffraction Gemini CCD X-ray diffractometer using Cu-Kα (1.54184 Å) radiation. Data reduction was carried out with the CrysAlisPro software (Rigaku Oxford Diffraction). The samples were cooled to 190 K with an Oxford Cryosystems Desktop Cooler. The structures were solved with SHELXS and refined with SHELXL[8] within the WinGX graphical user interface.[9] The crystal of [(CuIII(banphe))2]·6DMF was found to be a non-merohedral twin and data reduction and integration was achieved within the CryAlisPro program and refinement was carried out using both components (HKLF 5 option) in SHELXL. The thermal ellipsoid diagrams were produced with Mercury.[10] Crystallographic data in CIF format have been deposited with the Cambridge Crystallographic Data Centre (CCDC 2013101–2013102).
Results and Discussion
Ligand Synthesis
Synthesis of the benzoylacetone bis(thiosemicarbazone) ligands was first attempted using the method previously published by Akbar Ali et al.[11] where the β-diketone is added to two equivalents of thiosemicarbazide with acid and refluxed in methanol. While this methodology works well with acetylacetone, with benzoylacetone poor yields of the bis(thiosemicarbazone) target compounds resulted. Large amounts of the unwanted mono-thiosemicarbazone (monosubstituted) ligand were found, always in its cyclised pyrazoline form (Scheme 1). Attempts to ring open this monosubstituted ligand using acid or base to enable condensation with another equivalent of thiosemicarbazide were unsuccessful.

|
Alternatively, the method used by Cowley et al.[12] was employed to synthesise H3banme, whereby two equivalents of benzoylacetone were condensed with ethylenediamine in water to give proligand 1. The 1H NMR spectrum identified a single C–H resonance at 5.70 ppm (Fig. S1, Supplementary Material) indicative of the enol tautomer and a symmetrical diimine. In principle, either symmetrical isomer could be present (depending on which carbonyl/enol group condenses with the primary amine), but the acetyl group is evidently more reactive and the isomer shown in Scheme 1 forms exclusively as illustrated by the many structural reports of its complexes.[13–19] The transamination reaction with N-methylthiosemicarbazide still leads to a mixture including the target compound, mono-thiosemicarbazone, and excess thiosemicarbazide but these can be removed by selective precipitation leading to pure bis-thiosemicarbazone H3banme in reasonable yield. Again the pyrazoline form of H3banme was isolated due to spontaneous cyclisation through nucleophilic attack of one ‘arm’ of the linear bis-thiosemicarbazone on the adjacent C=N bond. The 1H NMR spectrum of H3banme was conducted in both DMSO-d6 and CDCl3 (Fig. S3, Supplementary Material), as the pyrazoline methylene signal is obscured by the water peak in DMSO, but clearly seen as an AB quartet in CDCl3. The combination of both solvents enabled structural confirmation of the disubstituted ligand. A single isomer was found and cyclisation apparently occurs by nucleophilic attack at the more reactive acetyl imine (as opposed to the conjugated benzoyl imine) as the C-methyl resonance is shifted upfield relative to proligand 1 (2.07 → 1.87 ppm) indicating it is now attached to a saturated C-atom.
However, the same synthetic approach was unsuccessful for the synthesis of H3banet and H3banphe so a new method was devised utilising proligand 2 (Scheme 2). Proligand 2 was characterised as one tautomer in solution by 1H NMR spectroscopy, preferentially forming the conjugated imine/ene-amine over the β-diimine (Fig. S2, Supplementary Material). The olefinic C–H peak at 5.68 ppm dictates that the two ethanolamine arms are distinctly different and four well separated CH2 resonances are seen. Upon reaction with excess thiosemicarbazide, transamination led to the desired bis-thiosemicarbazone but the cyclised mono-thiosemicarbazone was still formed.
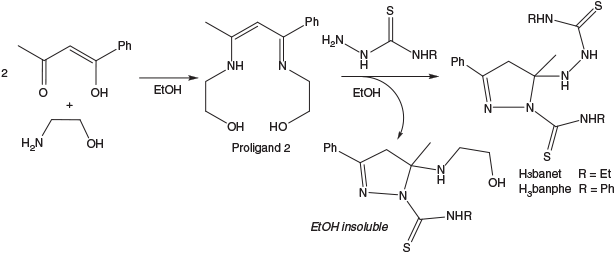
|
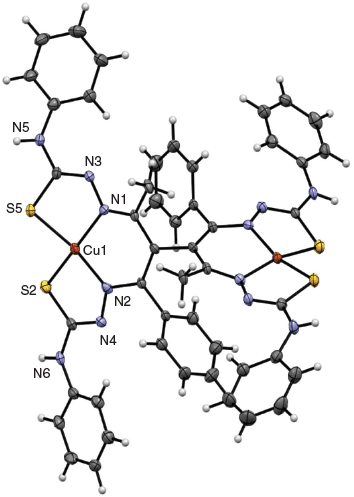
Therefore, regardless of which proligand was used, the reactions invariably led to mixtures of products. It was not uncommon for multiple recrystallisations with ethanol and chloroform to be required before all of the mono-thiosemicarbazone byproduct was removed and the disubstituted product was sufficiently pure. The pyrazoline form of the ligand was confirmed by the crystal structure of H3banet (Fig. 3). The crystal structure also confirms that cyclisation occurs by nucleophilic attack of the hydrazine derived N-atom (N5A) on the acetyl imine and not the benzoyl imine as suggested by NMR analysis.
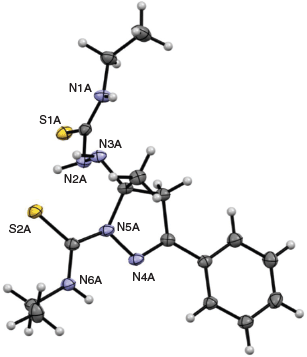
|
Copper Complexation
Previous work with the H3acacR ligands[4] has shown that their complexation reactions with CuII are complicated and involve several steps. They are also affected by the presence of oxygen. As will be seen, several species are typically present during the course of each complexation reaction, so isolation of the complexes as pure solids was not pursued. Instead spectroscopic and electrochemical characterisation was carried out on complexes prepared in situ by combining equimolar amounts of Cu(OAc)2 and bis(thiosemicarbazone) initially under anaerobic conditions and then subsequently monitoring the oxidation of these complexes by atmospheric oxygen. Investigations were carried out in DMSO solution, as preliminary studies in DMF resulted in precipitation of some compounds upon oxidation. To eliminate complications from different protonated and tautomeric forms, all data shown were acquired with three equivalents of Et3N added in line with the studies reported previously[4] generating a trianionic ligand initially coordinated to CuII (Fig. 2). Anaerobic solutions of [CuII(banme)]– and [CuII(banet)]– were olive green, while [CuII(banphe)]– was emerald green. After exposure to air all solutions turned brown. Assignment of the structures of each intermediate was assisted by comparisons with the [CuII(acacR)]– series as a point of reference.[4]
Time Resolved UV-Vis Spectroscopy
UV-Vis spectroscopy is well suited to studying copper thiosemicarbazone complex formation as the complexes exhibit some quite intense ligand-to-metal charge transfer absorption bands (ϵ > 1000 M−1 cm−1) that are characteristic of the metal in different oxidation states. To limit the number of species being generated at one time the complexation study was partitioned into two phases; an anaerobic phase and an aerobic phase. Also to avoid complications from different protonated and tautomeric forms, all data shown were acquired with three equivalents of Et3N added so that all labile protons on the complexes were removed leading to the monoanion [CuII(banR)]– at the completion of this phase (Scheme 3).
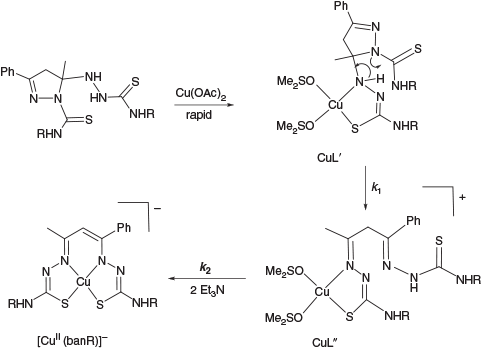
|
The time-dependent anaerobic complexation data are shown in Fig. 4a–c for the three ligand systems over a period of 48 h at 25°C. Global analysis of the spectroscopic data followed a mechanism comprising two first order reactions. The initial (nominally second order) complexation step, indicated by a rapid colour change on mixing, was too fast to measure i.e. CuII + H3L → CuIIL′. This meant the first observable species was CuIIL′. Scheme 3 illustrates the proposed intermediates in this phase. The complex CuIIL′ is partially coordinated to the ring-closed ligand. There is a crystallographic precedent for this coordination mode in the TcV complex of a related dithiocarbazate Schiff base.[20] This pyrazoline complex converts slowly into CuIIL′′ (second intermediate, rate constant k1) before conversion into the fully formed N2S2-coordinated complex CuIIL (rate constant k2) where charges have been omitted for simplicity. The rate constants obtained for the two first order reactions are given in Table 1 and the spectroscopic data obtained by analysis of the time-dependent spectroscopic data with Reactlab Kinetics appear in Fig. 5a–c.

|

|

|
The spectroscopic changes leading to the CuN2S2 complexes [CuII(banme)]– and [CuII(banet)]– (Fig. 4a, b and Fig. 5a, b) are very similar and the most prominent features to emerge in the final products are a very broad maximum around 780 nm and a shoulder around 440 nm, which was also seen in the [Cu(acacR)]– series.[4] For [CuII(banphe)]– (Figs 4c and 5c) a much more intense maximum appears at 440 nm with a shoulder at 460 nm. This transition must be linked with the N-phenyl chromophore.
The rate constants k1 and k2 (Table 1) are of the order of 10−5 s−1 (t1/2 ~18 h) and 1–2 orders of magnitude slower than seen for the [CuII(acacR)]– series (measured in DMF), but the spectroscopic changes are similar. In this case the solvent DMSO appears to be responsible for the rate decrease (not the change from [Cu(acacR)]– to [Cu(banR)]–) as preliminary experiments in DMF led to much faster complexation rates. As mentioned above the three complexes were not all soluble in DMF under both anaerobic and aerobic conditions so all kinetic studies were restricted to DMSO.
Upon exposure to oxygen (the aerobic phase), two consecutive first order reactions were found; CuL + O2 → CuL′′′ → CuLO, and k3 (strictly a pseudo-first order rate constant with excess O2) and k4 were determined to differ by an order of magnitude (Table 1). The time dependent spectroscopic changes are seen in Fig. 4d–f. The initial spectra of all three complexes are consistent with the [CuII(banR)]– compounds formed under anaerobic conditions. For [CuII(banme)]– and [CuII(banet)]– the bands at 440 and 790 nm decrease to be replaced by an intermediate and final spectrum with a single maximum around 500 nm (Fig. 5d, e). For [CuII(banphe)]– on the other hand, the initial intense peak at 440 nm is replaced by an intermediate with spectroscopic peaks at 620 and 780 nm before converting into a final species with a shoulder at 518 nm (Fig. 5f). The nature of these intermediates and final products were revealed by electrochemical and EPR spectroscopy measurements.
Cyclic Voltammetry
Electrochemical measurements were carried out on the Cu complexes (under the same conditions) at the end of the anaerobic complexation and the aerobic oxidation phases. At the end of the anaerobic phase, the dominant species exhibited a reversible redox couple in the range −700 to −500 mV versus Fc+/0 (Fig. 6a–c). This is in the same region as the [CuII(acacR)]– complexes.[4] Taken with the results from the dithiocarbazate Schiff base relatives[11,21] and spectroelectrochemistry results (below), this corresponds to the CuIII/II couple. Additional irreversible responses were found around +200 mV (oxidation) and –1600 mV (reduction). These were not examined in further detail due to their poor reversibility. The high potential irreversible responses (~100–200 mV) are most likely ligand centred oxidations.

|
Aerobic oxidation of the solutions of [CuII(banme)]–, [CuII(banet)]‐, and [CuII(banphe)]‐ for 2 days gave the cyclic voltammograms shown in Fig. 7a–c. The [CuII(banme)]– + O2 and [CuII(banet)]– + O2 CVs are essentially the same and show a low potential (~ –1000 mV) quasi-reversible single electron response in addition to a high potential (~200 mV) response. By analogy with the studies of the [CuII(acacR)]– system,[4] the redox couples at –925 and –861 mV respectively are assigned as CuII/I couples of the ketone complexes [CuII(banmeO)] and [CuII(banetO)] where a carbonyl group has been installed at the apical C-atom (Scheme 4). The analogous [CuII(acacRO)] (R = Et, Ph) compounds were crystallographically characterised to support this assignment.[4] The higher potential couples are most likely ligand centred processes according to spectroelectrochemical analysis (see below).

|
|
However, the CV from the [CuII(banphe)]– oxidation reaction is obviously different, and comprises a pair of closely spaced quasi-reversible single electron couples at –589 and –834 mV, in addition to a multi-electron oxidation wave at high potential. Two closely spaced couples of similar magnitude suggest a symmetrical dimeric complex and the potentials are in the same region as the CuIII/II couples seen in Fig. 6 for the mononuclear precursor. Similar but less well defined peaks around –500 mV appear for the oxidation products of [Cu(banme)]– and [Cu(banet)]– (Fig. 7a, b). Crystallographic characterisation of the [CuII(banphe)]– oxidation product established the structure of this putative dimer.
A solution of [CuII(banphe)]– in 0.1 M Et4NClO4 DMF that had undergone aerobic oxidation produced a precipitate on standing. After suspension in EtOH and filtration, the powder was redissolved in 0.1 M Et4NClO4 DMF solution. Slow evaporation over about one month gave fine black needles. The crystal structure of this compound confirmed the above assignment as the unusual dimer [(CuIII(banphe))2] (Fig. 8). The dimer is located on a two-fold axis so both Cu centres are identical and joined through a new C–C bond between the apical (α) carbon atoms of the benzoylacetone moiety. The N2S2 chelating units are trianionic (deprotonated at the α-carbon and both hydrazide N-atoms) and the neutral charge of the complex indicates that both metals are formally in the CuIII oxidation state.
|
This is the first crystal structure of a CuIII complex of a bis(thiosemicarbazone) ligand. The Cu–N and Cu–S bond lengths of [(CuIII(banphe))2] are also consistent with trivalent copper in related dithiocarbazate Schiff base copper complexes.[11,21] In contrast, there are numerous examples of structurally comparable CuII thiosemicarbazone complexes in the Cambridge Structural Database[22] and the average coordinate bonds in those compounds (from a Mogul search) are significantly longer: 2.00 Å (Cu–N, n = 55) and 2.24 Å (Cu–S, n = 186).
The two CuN2S2 planes are twisted by ~78° to one another. The C–C and C–N bond lengths within the six-membered chelate ring are intermediate of double and single bonds. The mean length for a localised C=N (imine) group coordinated to a transition metal is 1.287 Å (n = 40), while a C–C (single) bond within a protonated acac-diimine configuration bearing an sp3 hybridised apical C-atom is on average 1.480 Å (n = 17). Of those complexes recorded as delocalised across the six-membered chelate ring (all sp2 hybridised) the mean values were 1.321 Å (C–N, n = 21) and 1.385 Å (C–C, n = 98), which agree with the bond lengths in this structure. There is also an appreciable tetrahedral distortion of the CuN2S2 plane. This is apparent in Fig. 8 and from inspection of the trans-S–Cu–N bond angles which are both much less than 180°. This distortion may be quantified with the parameter τ4 = (360 – (α + β))/141), where α and β are the two largest coordinate angles.[23] The parameter τ4 is 0.0 for ideal square planar and 1.0 for tetrahedral. In this case, τ4 = 0.25. By analogy with the isoelectronic low spin d8 NiII analogues, the coordination geometry for this electronic configuration should be square planar. Therefore, the out of plane distortion is driven by the ligand and can be attributed to the short CuIII–N and CuIII–S bond lengths which enforce a twisting of the ligand while the bonds to the larger CuII ion in related CuIIN2S2 complexes are better suited to a planar ligand conformation.
Spectroelectrochemistry
UV-Vis spectroelectrochemistry was employed to determine the spectroscopic features of the various oxidised and reduced forms observed in the CV experiments. This links changes in oxidation state with the UV-vis spectra seen in the time-resolved kinetic experiments measured during the anaerobic complexation and aerobic oxidation reactions. The potential dependent spectroscopic data (Fig. 9) were analysed with Reactlab Redox[5] to determine the spectra of the fully oxidised and reduced forms for each reversible couple identified by CV (Fig. 10). The calculated (midpoint) redox potentials from this analysis were consistent with the CV results.

|

|
Scanning the CuIII/II redox couple at –678 mV for [Cu(banme)]0/– (Fig. 9a, purple) revealed that the oxidised CuIII form exhibits peaks at 430 (ϵ 9200 M−1 cm−1), 569 (ϵ 850 M−1 cm−1), and 801 nm (ϵ 1650 M−1 cm−1). Upon reduction to CuII, only one band remains; 441 nm (ϵ 7370 M−1 cm−1). The molar extinction coefficients are very informative and warrant analysis. In Figs 4 and 5 it is apparent that at the end of the anaerobic complexation reaction, Cu complexes are formed that exhibit weak absorption maxima around 800 and 580 nm. The spectroelectrochemistry results (Fig. 9) show that the [CuII(banR)]– complexes have no absorption in this region, but their CuIII relatives do and in their fully oxidised form the bands at 800 nm have large extinction coefficients (ϵ > 1500 M−1 cm−1). This shows that the final products of the ‘anaerobic’ complexation reaction (Figs 4 and 5) had in fact been partially oxidised over the course of the 2 days of the kinetic measurement presumably by adventitious oxygen.
The spectra produced by [Cu(banet)]0/– when probed at the corresponding couple gave almost identical results (Fig. 9b). The oxidised spectrum exhibits maxima at 435 (ϵ 10700 M−1 cm−1), 575 (ϵ 1130 M−1 cm−1), and 795 nm (ϵ 1890 M−1 cm−1), while the reduced spectrum shows one band at 441 nm (ϵ 8620 M−1 cm−1). The results for [Cu(banphe)]0/– are again similar, although the intense absorption peaks around 440 nm for both oxidation states do not change upon oxidation or reduction (Fig. 9c). The corresponding peaks for the oxidised CuIII species were observed at 413 (ϵ 24500 M−1 cm−1), 611 (ϵ 1070 M−1 cm−1), and 786 nm (ϵ 920 M−1 cm−1), while reduction generates the CuII complex with a single band at 421 nm (ϵ 24200 M−1 cm−1).
The three Cu compounds were oxidised in air for three days and spectroelectrochemistry was carried out over the region of the reversible lower potential couples (see Fig. 7). The [Cu(banmeO)]0/– and [Cu(banetO)]0/– systems were very similar as expected. In the oxidised CuII form a single peak is present at 500 nm (Fig. 10), along with a transition that extends past the 400 nm window. Reduction to (formally) CuI gave three transitions around 407, 520, and 800 nm. This broad maximum around 800 nm was also seen in the related [CuI(acacRO)]– complexes.
The CuIII dimer [(CuIII(banphe))2] was analysed by spectroelectrochemistry (Fig. 11) in the potential window of the consecutive single electron CuIII/II couples (see Fig. 7c). The data were modelled with a two-electron transfer model within Reactlab Redox[5] yielding three spectra (CuIII–CuIII, CuIII–CuII, and CuII–CuII) and two redox potentials.[5] The fully reduced CuII–CuII species [(CuII(banphe))2]2– presents peaks at 470 and 489 nm (shoulder) and the spectrum resembles that of the monomeric [CuII(banphe)]– (Fig. 9c), although both peaks of the dimer are shifted bathochromically. The fully oxidised CuIII–CuIII species exhibits a large peak at 431 nm, in addition to shoulders at 500, 544, and 585 nm and a broad maximum at 785 nm. The spectrum is again very similar to the monomeric CuIII complexes in (Fig. 9). The intermediate mixed valent CuIII–CuII spectrum is essentially an average of the fully oxidised and fully reduced spectra. No extra peaks that might be due to a metal-to-metal (intervalence) charge transfer transitions are apparent in the experimental wavelength range (400–1000 nm).

|
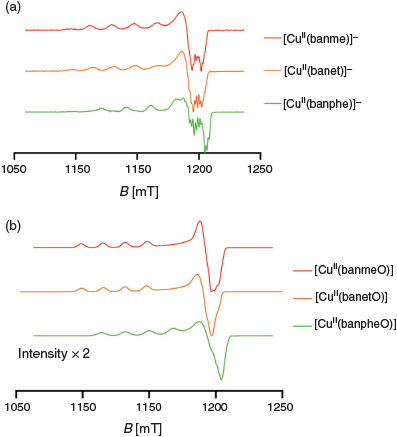
EPR Spectroscopy
EPR spectra at the Q-band frequency (33.95 GHz) were measured on all complexes prepared under anaerobic conditions (Fig. 12a) in DMSO after equilibration for 1 week (to give [CuII(banR)]–). In each case approximately axial spectra were found (gz >> gx,y, Az >> Ax,y) with the data summarised in Table 2. Not surprisingly given the variety of products and intermediates seen in the UV-vis spectroscopy and electrochemical experiments, mixtures of complexes were present in some cases. The samples of [CuII(banme)]– and [CuII(banet)]– prepared anaerobically gave EPR spectra comprising two components (Figs S6 and S7, Supplementary Material). The major component was assigned to [CuII(banR)]– while the minor component exhibited a spectrum that matched that of the sample that had been exposed to oxygen ([CuII(banRO)], see below) indicating that partial oxidation had occurred over the week that the compounds were left to equilibrate within the glovebox. The corresponding spectrum of [CuII(banphe)]– prepared anaerobically gave a single species (Fig. S8, Supplementary Material).
|

|
The spectra of [CuII(banme)]– and [CuII(banet)]– are essentially the same, while the spectrum of [CuII(banphe)]– exhibits a significantly larger Az and smaller gz value, which indicates a more planar CuIIN2S2 configuration and stronger Cu–ligand bonding in [CuII(banphe)]–.[24] The g values of CuII complexes also are very sensitive to changes in the donor atoms and the values found here lie in the region expected for CuN2S2 complexes.[25] The near axial symmetry strongly supports a symmetric coordination mode i.e. 5–6–5-membered chelate rings which is also consistent with the crystal structure of dimeric [(CuIII(banphe))2].
Upon oxidation all solutions yielded a single EPR active species (Figs S9–S11, Supplementary Material). The gz values of [CuII(banmeO)] and [CuII(banetO)] (Fig. 12b, Table 2) are similar to their [CuII(banR)]– precursors and the Az values only increase slightly, which is indicative of very similar CuN2S2 coordination geometries. Interestingly, the spectrum of [CuII(banpheO)] exhibits higher gz and smaller Az values than [CuII(banphe)]– which indicates some out of plane distortion in the CuN2S2 of [CuII(banpheO)]. It is also notable that the intensity of the [CuII(banpheO)] signal is about half of the other two analogues. This seems to be a consequence of a greater formation of the (diamagnetic and EPR silent) dimer [(CuIII(banphe))2].
Conclusions
As seen with the [Cu(acacR)]– analogues, complexation of CuII with the asymmetric H3banR (R = Me, Et, Ph) bis-thiosemicarbazones led to a variety of products depending on the reaction conditions and to some extent the R-substituent. In DMSO, the anaerobic complexation was very slow compared with previous investigations in DMF, but all spectroscopic data (UV-vis and EPR) point to a symmetrical 5–6–5 chelate ring binding mode for all Cu complexes reported here.
The aerobic oxidation reactions of the [CuII(banR)]– complexes comprised two quite slow first order processes enabling the observation of an intermediate before the final product was identified. Spectroscopic similarity with the CuIII complexes formed during the spectroelectrochemistry experiments indicate initial oxidation to CuIII occurs and it is this form that is the reactive species, triggering ligand centred oxidation reactions. The final spectra in the [CuII(banme)]– and [CuII(banet)]– oxidation reactions are predominantly of [CuII(banRO)] on the basis of the UV-vis spectra, which match the corresponding structurally characterised [CuII(acacRO)] complexes.[4] The time-resolved UV-vis data for [CuII(banphe)]– are similar, indicating formation of [CuIII(banphe)] as an intermediate but the final spectrum appears to be a mixture of [(Cu(banphe))2] and [Cu(banpheO)], which was supported by electrochemical and crystallographic data. In fact electrochemical and spectroelectrochemical analysis of this system found that dimeric [(Cu(banphe))2] was the major species present.
Actually all three aerobic oxidation reactions lead to mixtures of products comprising the monomeric ketone complex [CuII(banRO)] and dimeric [(CuIII(banR))2] with different ratios. Although the [CuII(banphe)]– system seems to produce the largest amount of dimer, corresponding peaks in the CVs of [CuII(banmeO)] and [CuII(banetO)] are apparent in the region –700 to –500 mV (Fig. 7a, b) that can be attributed to the dimers [(CuIII(banme))2] and [(CuIII(banet))2]. It seems likely that the two oxidation products (ketone and dimer) share a common precursor and the time-resolved spectra from the oxidation reaction indicates formation of the reactive [CuIII(banR)] intermediate that can either undergo O-atom transfer or dimerisation.
The C–C bond formation reaction leading to the dimer must result from a radical homocoupling reaction and this can be rationalised by considering the resonance structures in Scheme 5 comprising a mixture of singlet (CuIII low spin d8) and triplet (CuII d9: radical cation) electronic states. After C–C coupling, the acidic α-CH groups are immediately deprotonated by the excess Et3N present, restoring electron delocalisation, before reoxidation to the CuIII dimer product. The alternative pathway leading to [CuII(banRO)] is more speculative. It is clear that O2 is the source of the O-atom in the product and an organic peroxo or superoxo intermediate is possible but none were observed during the time-resolved UV-vis experiments.

|
The observation that [CuII(banphe)]– is seemingly more susceptible to dimer formation is interesting. The CV results show that the CuIII oxidation state of this complex is destabilised by around 200 mV relative to [CuII(banme)]– and [CuII(banet)]– and this can be attributed to the electron withdrawing effect of the N-phenyl groups. The EPR results for [CuII(banphe)]– also indicate a more planar coordination geometry and stronger CuII–N and CuII–S bonding than in [CuII(banme)]– and [CuII(banet)]–. These effects seem to combine to make [CuIII(banphe)] more reactive, particularly suggesting that its CuII-radical resonance structure is more relevant in this compound and that redox non-innocence of the ligand in [CuIII(banphe)] is a feature.
This is the first example of a bis-thiosemicarbazone dimer. So far, the library of dimers of this sort, connected through a C–C bond between the α-carbons of the β-diketone (typically acetylacetone) moiety, are of isothiocarbazones,[26] and salicyl[27] or benzoyl hydrazones[28] complexed with nickel or copper. The rarity of observing a dimer and one that supports both metal centres in the trivalent oxidation state is intriguing. As has been seen above, the complexes [CuII(banme)]– and [CuII(banet)]– are less inclined to dimerise, although small amounts of both are visible in the CV analysis. Finally, the phenyl perched on the β-diketone moiety may play a part in electron delocalisation leading to radical stabilisation and greater ligand non-innocence as dimeric complexes were not observed in the analogous [Cu(acacR)]– system.
Supplementary Material
All NMR spectra and EPR spectroscopy analysis including simulations are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
Financial support from the Australian Research Council (DP190103158) is gratefully acknowledged. The authors are sincerely grateful to the Inorganic Chemistry Division of the Royal Australian Chemical Institute for the honours of a 2019 Don Stranks Award (to JKB) and the 2019 Burrows Award (to PVB).
References
[1] B. M. Paterson, J. A. Karas, D. B. Scanlon, J. M. White, P. S. Donnelly, Inorg. Chem. 2010, 49, 1884.| Crossref | GoogleScholarGoogle Scholar | 20055473PubMed |
[2] B. M. Paterson, C. Cullinane, P. J. Crouch, A. R. White, K. J. Barnham, P. D. Roselt, W. Noonan, D. Binns, R. J. Hicks, P. S. Donnelly, Inorg. Chem. 2019, 58, 4540.
| Crossref | GoogleScholarGoogle Scholar | 30869878PubMed |
[3] L. E. McInnes, A. Noor, K. Kysenius, C. Cullinane, P. Roselt, C. A. McLean, F. C. K. Chiu, A. K. Powell, P. J. Crouch, J. M. White, P. S. Donnelly, Inorg. Chem. 2019, 58, 3382.
| Crossref | GoogleScholarGoogle Scholar | 30785268PubMed |
[4] J. K. Bilyj, J. R. Harmer, P. V. Bernhardt, Eur. J. Inorg. Chem. 2018, 4731.
| Crossref | GoogleScholarGoogle Scholar |
[5] P. King, M. Maeder, Reactlab Redox, Ver. 1.1 2014 (J Plus Consulting Pty Ltd: Karawarra, Western Australia).
[6] P. King, M. Maeder, Reactlab Kinetics, Ver. 1.1 2009 (J Plus Consulting Pty Ltd: Karawarra, Western Australia).
[7] S. Stoll, A. Schweiger, J. Magn. Reson. 2006, 178, 42.
| Crossref | GoogleScholarGoogle Scholar | 16188474PubMed |
[8] G. M. Sheldrick, Acta Crystallogr. Sect. A 2008, 64, 112.
| Crossref | GoogleScholarGoogle Scholar |
[9] L. J. Farrugia, J. Appl. Cryst. 1999, 32, 837.
| Crossref | GoogleScholarGoogle Scholar |
[10] C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek, P. A. Wood, J. Appl. Cryst. 2008, 41, 466.
| Crossref | GoogleScholarGoogle Scholar |
[11] M. Akbar Ali, P. V. Bernhardt, M. A. H. Brax, J. England, A. J. Farlow, G. R. Hanson, L. L. Yeng, A. H. Mirza, K. Wieghardt, Inorg. Chem. 2013, 52, 1650.
| Crossref | GoogleScholarGoogle Scholar | 23324063PubMed |
[12] A. R. Cowley, J. R. Dilworth, P. S. Donnelly, A. D. Gee, J. M. Heslop, Dalton Trans. 2004, 2404.
| Crossref | GoogleScholarGoogle Scholar | 15303151PubMed |
[13] M. Salehi, R. Kia, A. Khaleghian, J. Coord. Chem. 2012, 65, 3007.
| Crossref | GoogleScholarGoogle Scholar |
[14] R. K. Sherwood, C. L. Kent, B. O. Patrick, W. S. McNeil, Chem. Commun. 2010, 46, 2456.
| Crossref | GoogleScholarGoogle Scholar |
[15] M. Amirnasr, R. Vafazadeh, A. H. Mahmoudkhani, Can. J. Chem. 2002, 80, 1196.
| Crossref | GoogleScholarGoogle Scholar |
[16] S.-X. Liu, Y.-L. Feng, Polyhedron 1996, 15, 4195.
| Crossref | GoogleScholarGoogle Scholar |
[17] X. Pang, H. Du, X. Chen, X. Wang, X. Jing, Chem. – Eur. J. 2008, 14, 3126.
| Crossref | GoogleScholarGoogle Scholar | 18232044PubMed |
[18] D. Xu, B. Chen, K. Chen, C. Chen, K. Miki, N. Kasai, Bull. Chem. Soc. Jpn. 1989, 62, 2384.
| Crossref | GoogleScholarGoogle Scholar |
[19] M. Salehi, F. Rahimifar, M. Kubicki, A. Asadi, Inorg. Chim. Acta 2016, 443, 28.
| Crossref | GoogleScholarGoogle Scholar |
[20] T. I. A. Gerber, E. Hosten, O. Knoesen, P. Mayer, J. Coord. Chem. 2007, 60, 2369.
| Crossref | GoogleScholarGoogle Scholar |
[21] J. K. Bilyj, N. V. Silajew, G. R. Hanson, J. R. Harmer, P. V. Bernhardt, Dalton Trans. 2019, 48, 15501.
| Crossref | GoogleScholarGoogle Scholar | 31304485PubMed |
[22] F. H. Allen, Acta Crystallogr. Sect. B 2002, 58, 380.
| Crossref | GoogleScholarGoogle Scholar |
[23] L. Yang, D. R. Powell, R. P. Houser, Dalton Trans. 2007, 955.
| Crossref | GoogleScholarGoogle Scholar | 17308676PubMed |
[24] B. J. Hathaway, D. E. Billing, Coord. Chem. Rev. 1970, 5, 143.
| Crossref | GoogleScholarGoogle Scholar |
[25] J. Peisach, W. E. Blumberg, Arch. Biochem. Biophys. 1974, 165, 691.
| Crossref | GoogleScholarGoogle Scholar | 4374138PubMed |
[26] V. B. Arion, N. V. Gerbeleu, V. G. Levitsky, Y. A. Simonov, A. A. Dvorkin, P. N. Bourosh, J. Chem. Soc., Dalton Trans. 1994, 1913.
| Crossref | GoogleScholarGoogle Scholar |
[27] D. Laziz, C. Beghidja, N. Baali, B. Zouchoune, A. Beghidja, Inorg. Chim. Acta 2019, 497, 119085.
| Crossref | GoogleScholarGoogle Scholar |
[28] A. Mukhopadhyay, S. Pal, Eur. J. Inorg. Chem. 2009, 4141.
| Crossref | GoogleScholarGoogle Scholar |
* Jessica K. Bilyj is the recipient of a 2019 RACI Inorganic Chemistry Division Don Stranks Award and Paul V. Bernhardt is the recipient of the 2019 RACI Inorganic Chemistry Division Burrows Award.