

Hierarchical population genetic structure and signatures of adaptation in Lates calcarifer
Matthew A. Campbell A * and Joy A. Becker A
A * and Joy A. Becker A
A
Abstract
Lates calcarifer is a widespread Indo-Pacific fish that is important in aquaculture, recreational and commercial fisheries. Genetic divergences from different data sources and sampling schemes have been reported.
To conduct phylogenetic and population genetic analyses from a geographically and phylogenetically representative data set to identify hierarchical divisions within L. calcarifer. We further test the evolutionary significance of genetic units in terms of signatures of adaptation.
Using a whole-genome sequence data set of 61 fish, including an outgroup, we conducted phylogenetic and population genetic analyses. We also generated measures of fixation index (Fst), nucleotide diversity (π) and Tajima’s D (D).
We identified three main lineages of L. calcarifer, corresponding to the Indian subcontinent, South-east Asia and Australasia. Subdivision within each of the three main lineages was also identified and characterised. Adaptively significant differences are indicated within and among the three main lineages.
L. calcarifer exhibits genetic divergences at different levels that originate before and during the Pleistocene. These divergences are associated with adaptive divergence but unclear phenotypic changes.
This study has highlighted the need for comprehensive sampling and integrative study of genotypes and phenotypes across the range of L. calcarifer.
Keywords: adaptive genetic variation, aquaculture, Asian sea bass, barramundi, conservation genetics, Latidae, phylogenetics, population genetics, recreational fisheries.
Introduction
Essential processes necessary for life on Earth are sustained by biodiversity, and biodiversity provides for human needs in various ways. Regardless of its limits and challenges, the basic unit for understanding biodiversity is the concept of species (e.g. Agapow et al. 2004). Although the recognition of species is indeed important, so is the recognition of units below the species level that contribute to biodiversity. The ecological effects of intraspecific variation can meet or exceed species effects and, therefore, are important to document and recognise (Des Roches et al. 2018). Conservation of both species-level and intraspecific variation serves to promote ecological, cultural and economic functions of organisms, resilience and genetic variation for future adaptive potential (e.g. Schindler et al. 2010; Layton et al. 2021; Lehnert et al. 2023). Specific and intraspecific variation can be considered through both isolation and adaptation, without the reliance on formal species taxonomy (Waples 1991).
We apply these basic ideas of isolation and adaptation to the Indo-Pacific fish genus Lates. Most Indo-Pacific Lates are classified under Lates calcarifer (Bloch, 1790), and it is a valuable commercial taxon of cultural and recreational significance e.g. (Cluney 2004; Edwards-Vandenhoek 2015; Shamsi et al. 2020). The common names ‘barramundi’ and ‘Asian sea bass’ are widely used. A broad-ranging taxon, L. calcarifer is often considered to be distributed from the Persian Gulf eastward to Taiwan and Australia, and into Oceania (Fig. 1). Abed et al. (2023) noted that there are no reliable reports from the Persian Gulf of native Lates and indicated a natural distribution through western India. Within this broader range, two regional endemics may be recognised, in Sri Lanka L. lakdiva Pethiyagoda & Gill, 2012 and Myanmar L. uwisara Pethiyagoda & Gill, 2012 (Pethiyagoda and Gill 2012). We refer to all these fishes together as Indo-Pacific Lates.
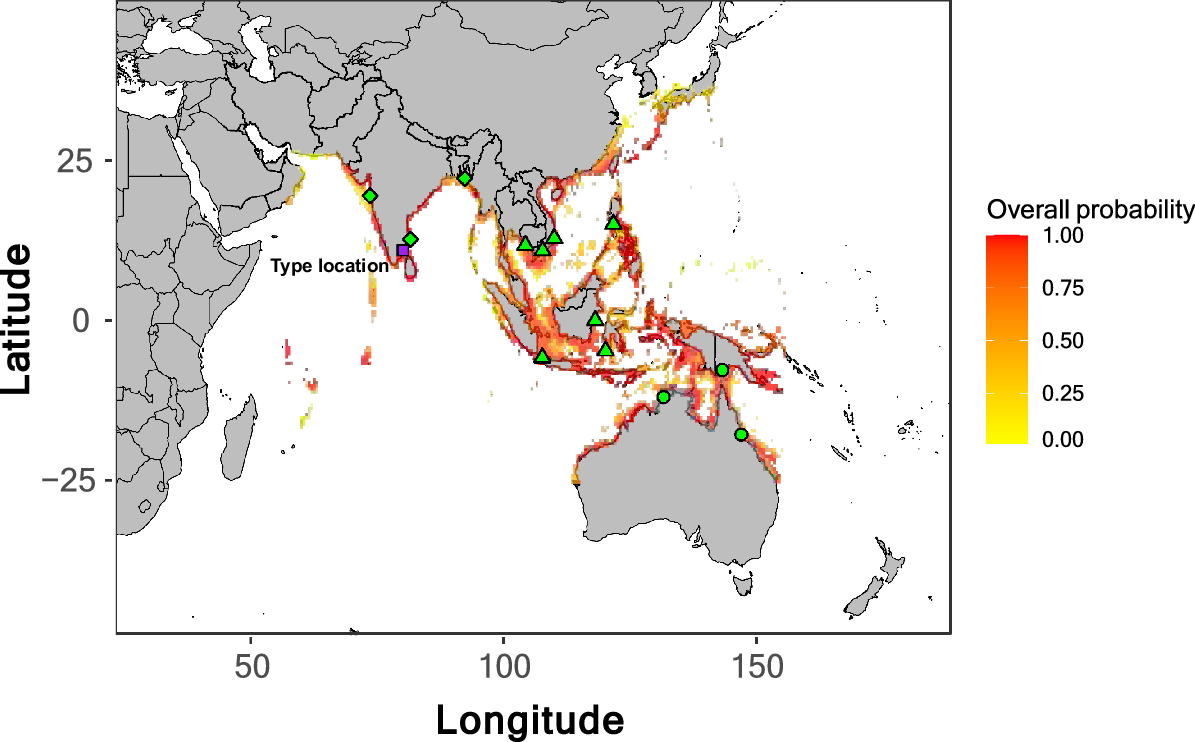
Distribution of Lates calcarifer sensu lato from AquaMaps (ver. 10/2019, K. Kaschner, K. Kesner-Reyes, C. Garilao, J. Segschneider, J. Rius-Barile, T. Rees and R. Froes, see https://www.aquamaps.org, accessed 8 August 2022), with probability of occurrence (overall probability) coloured from yellow (low) to red (high). The type location of L. calcarifer, Tamil Nadu, India, is indicated by a purple square. The main genetic lineages sampled and included in this paper are indicated by shape: AUS + NG (circles), SEA (triangles) and IND (diamonds). Sampling locations are approximate because specific coordinates are not available. Cambodia is represented by four separate collection locations (shown as one).
Aquaculture of Indo-Pacific Lates, beginning in Thailand in the 1970s, has increased greatly and Indo-Pacific Lates species are now widely propagated across their range, with some aquaculture being located outside the native range. Indo-Pacific Lates is now grown in diverse locations such as the United States of America (Arizona, Hawaii and Massachusetts), Israel, Taiwan and the Persian Gulf (Iraq) (Mathew 2009; Abed et al. 2023; Chen et al. 2024). Propagation is also undertaken for stocking programs in lakes across northern Australia (e.g. Manton Dam, Northern Territory; Lake Kununurra, Western Australia) to support recreational fisheries and tourism in remote communities. Generally, Indo-Pacific Lates fishes are considered to be protandrous haemaphrodites, catadromous, and tolerant of a wide range of salinity (Moore 1979; Moore and Reynold 1982). The sequential hermaphrodism of the taxon requires the importation of young males into aquaculture facilities because older fish become females, a potential source of hybridisation and mixing of stocks of different origin. Movement for aquaculture, for example, from Thailand to Taiwan, also can result in the mixing of fish of different backgrounds and opportunities for release or escape (Chen et al. 2024). Other qualities of Indo-Pacific Lates such as high fecundity, ease of culture and rapid growth (i.e. harvestable size of 3 kg in less than 2 years) encourage their production (Rimmer 2024).
The widespread culture and commercial fishing for Indo-Pacific Lates has led to genetic studies, so as to understand population genetic structure. Initial mitochondrial barcoding showed a surprisingly large divergence between Australian and Myanmarese fish classified under L. calcarifer (K2P distance of ~9.50%) (Ward et al. 2008). Broader sampling geographically of mitochondrial lineages has substantiated this divergence, showing a distinctive clade of Indian subcontinent and Bay of Bengal fish (IND, including the Andaman Islands and Myanmar), separate from South-east Asia (SEA, e.g. Singapore, Malaysia, Thailand and Indonesia) and northern Australia (Vij et al. 2014). Vij et al. (2014) included the sequences from Ward et al. (2008) and demonstrated that they are similar to a more broadly distributed IND mitochondrial lineage. Restriction site-associated DNA sequencing (RADseq) applied to population-level samples of Indo-Pacific Lates (n = 131) from six localities (Thailand, Malaysia, Indonesia, western Australia, eastern Australia and Papua New Guinea) showed a high degree of differentiation of SEA fish from Australia + New Guinea (AUS + NG). Pairwise fixation index (Fst) within SEA was 0.005–0.009 but 0.360–0.443 between SEA and AUS + NG, without evidence of contemporary gene flow (Wang et al. 2016). Some investigation of population genomic structure was applied to a range-wide data set at a high level as part of a genome assembly study, but did not investigate concordance between mitochondrial DNA (mtDNA) and genomic DNA or population genetic metrics (Vij et al. 2016). Broadly, Vij et al. (2016) identified a separation of Indian Ocean fish, South-east Asian fish and Australasian fish into genetic clusters. Population genetic structure range-wide examined with microsatellites (K = 4 genetic clusters presented) produced rather unclear distinctions of Indian Ocean fish from parts of South-east Asia, and indicated a central Indonesian phylogroup and Australasian phylogroup (Jerry and Smith-Keune 2014). No study has analysed both mitochondrial data and genomic data explicitly from the same individuals to evaluate concordance between data sources and to resolve possible discordance between mitochondrial and genomic signals.
Although there is evidence of genetic differentiation within Indo-Pacific Lates at a high phylogenetic level (Vij et al. 2014), this has been evaluated thoroughly only through molecular phylogenetic approaches using mtDNA. Within major genetic lineages, there is also genetic differentiation evident (e.g. Shaklee and Salini 1985; Chenoweth et al. 1998; Yue et al. 2009), and the evolutionary significance of these differences has not been substantiated. We apply a hierarchical approach to identify isolation (discreteness) of Indo-Pacific Lates genetic lineages by using molecular phylogenetic and population genetic approaches, and test for adaptive significance among these lineages. Within lineages, we test for additional isolation and adaptive significance of these differences by using population genetic approaches. (Lehnert et al. 2023). We further evaluate evidence of hybridisation among main genetic lineages to determine whether the hybridisation is recent (F1, F2) and, presumably, a result of human activities, or represents older admixture events.
Methods
We approached the question of isolation within Indo-Pacific Lates by first constructing phylogenies by using mtDNA and genome-wide single-nucleotide polymorphism (SNPs). Subsequently, population genetic techniques were used, and divisions were examined at a high level (genetic lineages) and then within those lineages. We tested for evolutionary significance through identifying possible adaptive loci among and within main lineages of Indo-Pacific Lates. An outstanding question is whether hybridisation is recent, as a result of human-mediated translocations, or a result of natural admixture among lineages. We applied statistical testing to this question to determine wheter evidence of recent introgression in mtDNA was corroborated at the genetic level. Finally, we developed a timeline for diversification of Indo-Pacific Lates by using a time-calibrated phylogenetic approach.
Whole-genome sequence data set
Whole genome sequencing (WGS) data from Indo-Pacific Lates were downloaded from the NCBI Sequence Read Archive (see https://www.ncbi.nlm.nih.gov/sra) under the BioProject accession PRJNA311498. The 61 samples in PRJNA311498 covered a broad distribution, from the Indian subcontinent, parts of South-east Asia, northern Australia and Papua New Guinea. Two additional sequences of Indo-Pacific Lates were obtained from Bangladesh (PRJNA1021005) and, for phylogenetic analyses, an outgroup was obtained (L. japonicus, n = 1, PRJDB13763). The paired-end data were aligned to the L. calcarifer reference genome GCF001640805.2 (TLL_Latcal_v3) by using the Burrows–Wheeler aligner (BWA, ver. 0.7.17, see https://github.com/lh3/bwa) with the mem algorithm (Li and Durbin 2009, 2010). Alignments were sorted, properly paired reads kept and polymerase chain reaction (PCR) duplicates removed by using SAMtools (ver. 1.13, see http://samtools.sourceforge.net; Li et al. 2009; Danecek et al. 2021). Total read counts, filtered read counts and depth of coverage were calculated from alignments with SAMtools. Those samples with coverage of ≤3× were excluded from further analyses.
Mitochondrial phylogeny
A mitochondrial alignment from the WGS data was created by creating a binary alignment map (BAM) file for each individual with more than 3× coverage, including only reads aligned to the reference mitochondrial genome (NC_007439.1, SAMtools view). These BAM files were converted to consensus FASTQ files with the bam2cns subprogram of proovread (ver. 2.14.0, see https://github.com/BioInf-Wuerzburg/proovread; Hackl et al. 2014) then to FASTA files turning all bases with quality scores of <20 to N with seqtk (ver. 1.3-r117, seq -a -q20 -n N, see https://github.com/lh3/seqtk). The mitochondrial cytochrome oxidase I (mtCOI) gene was extracted from the complete mitochondrial FASTA-formatted sequences with SAMtools (faidx NC_007439.1:6329-7879). For rooting, the corresponding region of the L. japonicus mitochondrial genome was extracted from the NCBI reference sequence (NC_034339.1) and sequences were aligned with MAFFT (ver. 7.520, see https://mafft.cbrc.jp/alignment/software/; Katoh et al. 2002; Katoh and Toh 2008). A maximum-likelihood (ML) phylogeny of sequences was generated with IQ-TREE (ver. 2.0.3, see http://www.iqtree.org/) specifying a Hasegawa–Kishino–Yano model of nucleotide substitution with gamma-distributed rate variation (-m HKY + G), and support for nodes was evaluated with rapid bootstrapping (-bb 10000) (Nguyen et al. 2015; Hoang et al. 2018). The tree was imported into R and visualised by using the ggtree package (ver. 3.4.4, see https://doi.org/10.18129/B9.bioc.ggtree; Yu et al. 2017), collapsing nodes to polytomies receiving less than 50% bootstrap support (BS) and rooting by the longest branch.
Genome-wide phylogenetic and network analyses
We generated a genome-wide set of called SNPs with analysis of next-generation sequencing data (ANGSD, ver. 0.930, see https://github.com/ANGSD/angsd) from the 24 main linkage groups in the L. calcarifer assembly (Korneliussen et al. 2014). Quality of called SNPs was enforced by ensuring that a SNP was present in at least 90% of individuals, had a minimum minor allele frequency (MAF) of 0.05, a minimum mapping quality of 20, minimum base quality of 20, a SNP P-value of 1.00 × 10−6 and a posterior cutoff of 0.90. A SAMtools model was specified and a PLINK-formatted file created as the output. We converted the PLINK-formatted file to a VCF-formatted file with PLINK (ver. 1.90b6.16, see https://cran.r-project.org/package=plink; Purcell et al. 2007). Linked SNPs were removed by BCFtools (ver. 1.10.1, +prune, -l 0.20 -w 10000, see https://github.com/samtools/bcftools).
The multispecies coalescence was applied to model gene-tree and species-tree discordance resulting from incomplete lineage sorting (ILS) (Maddison 1997) on a set of called SNPs that included the outgroup L. japonicus. A species tree was generated using SVDQuartets in PAUP* (ver. 4.0a, see http://phylosolutions.com/paup-test/; Swofford 2003; Chifman and Kubatko 2014, 2015). Samples were pooled by sampling region for the species tree. For a random set of quartets (evalQuartets = random), partially ambiguous SNPs were distributed over all compatible site patterns (ambigs = distribute), and 1000 bootstrap replicates (bootstrap = standard nreps = 1000) were option-specified.
For ML phylogenetic analysis and network analysis we called SNPs and a phylogeny of SNPs was inferred with IQ-TREE by identifying a model of nucleotide evolution with ModelFinder Plus (see http://www.iqtree.org/ModelFinder/) and indicating that the ascertainment bias correction should be applied (-m MFP + ASC) (Nguyen et al. 2011; Kalyaanamoorthy et al. 2017). IQ-TREE was used to further filter SNPs for suitability for ascertainment bias correction. Support for nodes was evaluated with 10,000 rapid boostraps (Hoang et al. 2018). Because concatenated phylogenetic analyses do not explicitly model hybridisation or ILS, we created an implicit network to identify potential hybridisation or ILS in our data set with SplitsTree (ver. 4.15.1, see https://github.com/husonlab/splitstree4; Huson and Bryant 2006). The same alignment as provided to IQ-TREE was analysed with SplitsTree.
Population genetic structure and statistics
Total signal in the data set was assessed by the generation of covariance matrix by using ANGSD (ver. 0.930) and a principal component (PC) analysis. The covariance matrix was generated by with a single-read sampling approach because of a wide range of individual coverage (minimum 4.73, maximum 34.81, mean 9.38, and median 6.41) with the options -doIBs 1 and -doCov 1 in ANGSD. The 24 main linkage groups of the reference assembly were sampled for this analysis. A variable site was required to be present in >90% of individuals with a minimum MAF of 0.05. A minimum mapping quality of 10 was specified with a minimum base quality of 20 and a GATK-genotype likelihood model was used (-GL 2). The resulting covariance matrix was imported into R for PC analysis.
The genetic differentiation in values of Fst was calculated by generating site-frequency spectra (SFS) for each sampling region by using ANGSD and the realSFS subprogram (ver. 0.930), splitting Indonesia into two locations on the basis of metadata available with the sequences (‘K’, n = 5; and and ‘SJ’, n = 5). Vietnam (n = 2), Indonesia ‘SU’ (n = 1) and Bangladesh (n = 2) were excluded because of small sample sizes. Two-dimensional SFS were calculated between each possible pair of sampling regions and Fst was calculated. Nucleotide diversity (π) and Tajima’s D (D) estimates were calculated for each chromosome by using realSFS, using the saf2theta option followed by the subprogram of ANGSD, thetaStat, and the do_stat option.
Admixture analysis was conducted by using NGSadmix (ver. 0.930, see https://www.popgen.dk/software/index.php/NgsAdmix; Skotte et al. 2013) on genotype likelihoods generated by ANGSD, as for the PC analysis using a single-read sampling approach. The resulting BEAGLE-formatted file was used with NGSadmix to produce an admixture plot of K = 2–5 genetic clusters.
Tests for adaptative genetic variation
Genotype calls produced for phylogenetic network analysis after pruning were imported into R for analysis by using PCAdapt (ver. 4.4.0, see https://cran.r-project.org/package=pcadapt; Luu et al. 2017; Privé et al. 2020). The pcadapt function was then used to generate a scree plot and identify an objective number of PCs, with the first 20 PCs examined. From the resulting object from the pcadapt function, outlier loci were identified by filtering for Benjamini and Hochberg (1995) and Bonferroni-corrected P-values with significance values of <0.10. We repeated this analysis for individuals in each main genetic lineage by filtering for variants with a minimum minor allele frequency of 0.05 and examining the first five PCs.
Evaluation of hybridisation levels
It is possible that South-east Asian Lates exhibit a mixture of hybridisation levels between IND and SEA lineages, with possible F1s, F2s and advanced backcrosses. Two wild-caught samples in the data set from Cambodia (Sequence-read accessions SRR3165998, SRR3165999) had IND lineage mtDNA sequences (see Results) (Fig. 2). We applied NewHybrids (ver. 1.1 beta, see https://github.com/eriqande/newhybrids; Anderson and Thompson 2002) to test whether these two Cambodian fish were recent (F1, F2) or older hybrids (advanced backcrosses). By using the called and pruned SNP data sets, we excluded recently reported potential chromosomal inversions (Campbell and Hale 2024) and imported the data into R and analysed with functions of the dartR package (ver. 2.97, see https://cran.r-project.org/package=dartR; Gruber et al. 2018). We restricted the analysis to fish from the SEA and IND lineages because NewHybrids functions with two populations and mixtures of those populations then filtered out low MAF SNPs (threshold of 0.25). The gl.nhybrids function of dartR was used to drive a NewHybrids analysis with India western coast and Bangladesh sampling locations designated as one parental lineage (P0) and Indonesia-K and Indonesia-SU as another (P1). We allowed SNPs to not be entirely fixed between parental populations (threshold = 0.01) and chose a subset of 200 SNPs for analysis with the ‘AvgPIC’ criterion. Analysis included a 100,000-step burnin and 50,000 sweeps.
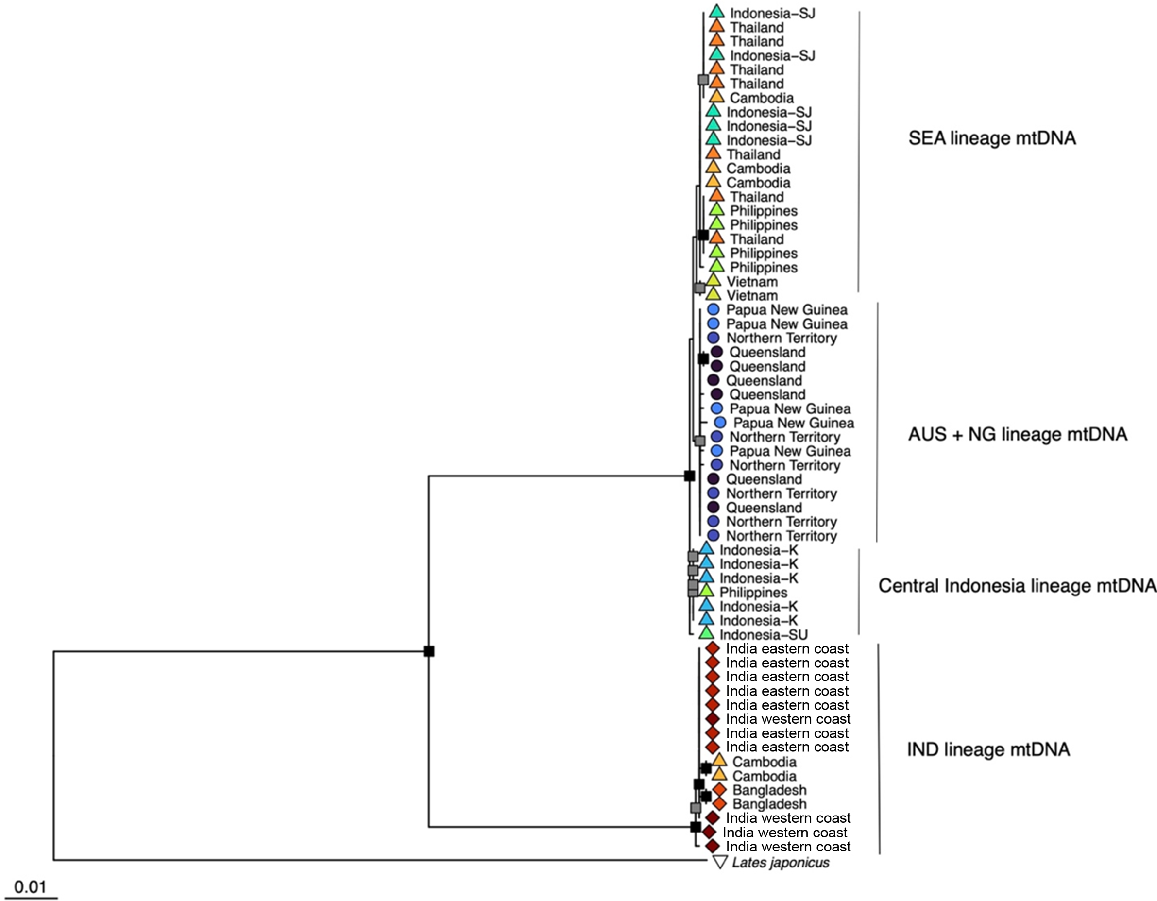
Maximum-likelihood phylogeny of mitochondrial cytochrome oxidase I from 60 Indo-Pacific Lates individuals derived from whole-genome sequence data. The tree is rooted by L. japonicus. Node shape indicates the putative lineage of origin and collection information is provided as tip labels. Nodes with <50% bootstrap support are collapsed to polytomies. Nodes with ≥75% bootstrap support but <90% bootstrap support are indicated as grey squares and nodes of ≥90% bootstrap support as black squares.
Divergence time estimation
Existing efforts have covered a broad geographic and lineage sampling of Indo-Pacific Lates with barcoding (mitochondrial cox1) (Ward et al. 2008; Vij et al. 2014). The effort of this study was then to calculate divergence times of the three main lineages (IND, SEA and AUS + NG). We downloaded barcode sequences from the Barcode of Life Database and Genbank for Latidae. By using BLAST (blastn, ver. 2.9.0, see https://blast.ncbi.nlm.nih.gov/), we confirmed the major lineages of Indo-Pacific Lates in barcode data (Altschul et al. 1997), then selected representative sequences from these three lineages to conduct a divergence-time analysis (Table 1). We also searched for representation across Latidae for broad taxonomic representation of this family, including sequences from L. japonicus, L. microlepis and L. niloticus. We included representatives of the two main mitochondrial lineages present in Psammoperca corresponding to P. waigiensis and P. datnioides. Centropomidae has been confirmed to be the sister lineage of Latidae in molecular phylogenetic study e.g. (Chen et al. 2007; Li et al. 2011; Campbell et al. 2013) and inclusion of Centropomidae allows dating the time to most recent common ancestor (TMRCA) of Centropomidae and Latidae to 48.6 million years ago (Ma) (Otero 2004; Campbell et al. 2013). We also included sequences from the outgroup lineages Channidae and Nandidae as an additional calibration point dating the TMRCA of those taxa to be 48 Ma (Santini et al. 2009).
| Region | Additional geographic information | Source | Sample size | Average coverage | |
|---|---|---|---|---|---|
| Northern Territory, Australia | Darwin Harbour | Wild-caught | 6 | 5.80 | |
| Queensland, Australia | Hinchinbrook channel | Wild-caught | 6 | 5.34 | |
| Papua New Guinea | Fly River | Wild-caught | 5 | 5.83 | |
| Indonesia | Kalimantan (K) | Unknown | 5 | 5.73 | |
| Indonesia | South of Jakarta (SJ) | Wild-caught | 5 | 17.8 | |
| Indonesia | Sulawesi (SU) | Unknown | 1 | 6.13 | |
| Philippines | Hatchery broodstock | 5 | 6.32 | ||
| Vietnam | Hatchery Broodstock | 2 | 6.21 | ||
| Cambodia | Three locations | Wild-caught | 5 | 5.90 | |
| Thailand | Eastern coast | Wild-caught | 7 | 15.60 | |
| Bangladesh | Bakkhali River | Unknown | 2 | 10.04 | |
| India eastern coast | Tamil Nadu | Wild-caught | 7 | 8.50 | |
| India western coast | Mumbai | Wild-caught | 4 | 20.22 |
The sample size and average coverage are presented for each sampling region.
Sequences were aligned using MAFFT (Katoh et al. 2002; Katoh and Toh 2008), in-frame coding was confirmed by translation in the NCBI ORF finder and a NEXUS file made for a time-calibrated phylogenetic analysis in MrBayes (ver. 3.2.2, see https://github.com/NBISweden/MrBayes/; Ronquist et al. 2012). The third codon position was excluded owing to the age of the data set and expected saturation occurring in mitochondrial sequences from carangimorph taxa at these timescales e.g. (Campbell et al. 2014). A nucleotide substitution model of general time reversible (GTR) with a proportion of invariant sites (I) and gamma-distributed rate variation (Γ) was specified. Constraints were applied to Centropomidae + Latidae to date, with an exponential distribution used, offsetexp (48.60, 63.18), and the outgroup, offsetxp (48.00, 62.40). Initially, three runs and three chains were specified with a run length of 1 million generations sampled ever 1000 generations and a 25% burnin applied. Evaluation of parameters and convergence with Tracer (ver. 1.7.2, https://github.com/beast-dev/tracer/releases/tag/v1.7.2; Rambaut et al. 2018) was assessed, and run length increased until sufficient effective sample sizes (ESS) for all parameters (>200) was reached. Tree summarisation was performed in MrBayes with the sumt command and an all compatible consensus tree was made.
Results
Whole-genome sequence data set
From the original 63 Indo-Pacific Lates sequences, 60 had coverage over 3× and were included in additional analyses (Table 1). Sample sizes for regions were 13 for putative members of the IND lineage, 30 for putative members of the SEA lineage, and 17 for putative members of the AUS + NG lineage from ten sampling regions (Fig. 1). The L. japonicus sequence aligned to the L. calcarifer reference genome with high success with 110× coverage and was down-sampled to 20× coverage with SAMTools to be more consistent with genome coverage measures of Indo-Pacific Lates.
Mitochondrial phylogeny
The 61 samples (60 Indo-Pacific Lates and 1 L. japonicus) produced an alignment of 1576 base pairs in length with 71 distinct patterns and 128 parsimony-informative sites. In the alignment, 1441 sites were invariant. The ML phylogeny indicates a clear division of L. calcarifer IND individuals with two Cambodian-origin fish from all other fish (Fig. 2). Support for monophyly of this IND mtDNA lineage is maximal. Support for monophyly of SEA + AUS + NG is maximal, and support for a clade of L. calcarifer AUS + NG is moderate (BS = 80%). Individuals sourced from the putative L. calcarifer SEA clade do not form clear geographic clusters, with the exception of a Central Indonesia mtDNA lineage that receives low support for monophyly (BS = 57%) and includes a single sequence from the Philippines. Support for the arrangement of major mtDNA lineages SEA and AUS + NG lineages is low (BS = 55%). The ‘assemble species by automatic partition’ algorithm indicated the strongest support for two species, with a threshold distance of 4.10%. This division separates the IND mitochondrial lineage from the combined SEA and AUS + NG lineage.
Genome-wide phylogenetic and network analyses
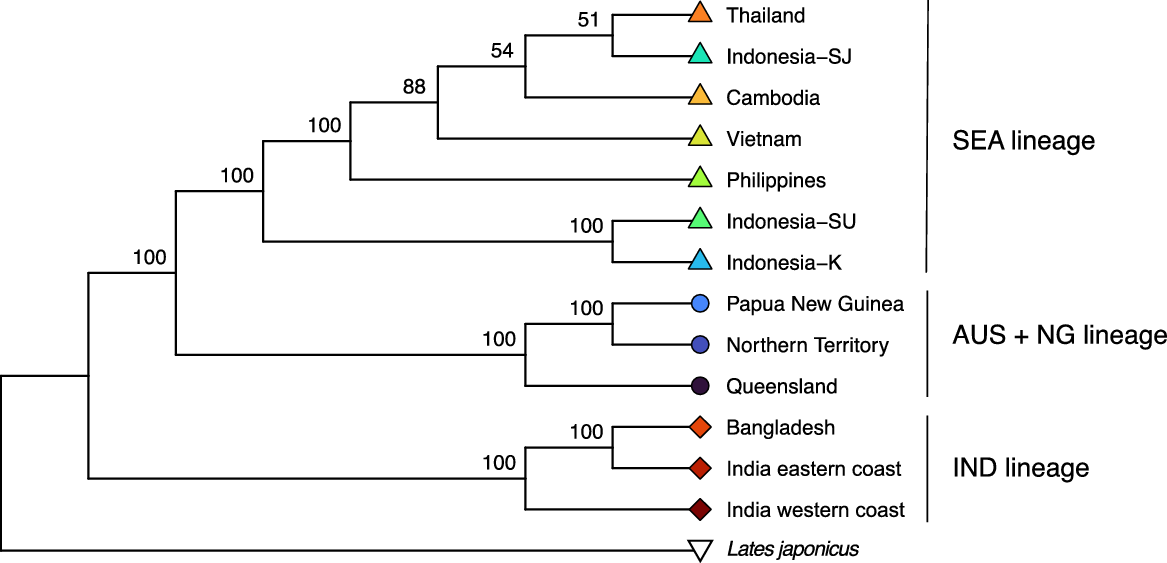
For analysis with SVDQuartets to generate a rooted species tree, 8529 variants that all have unique alignment patterns were analysed and the species tree is presented in Fig. 3. The SNP alignment consisted of 6456 parsimony-informative and 2073 singleton sites. Three lineages were apparent in the species tree, with maximal support for monophyly corresponding to the L. calcarifer IND, L. calcarifer SEA and L. calcarifer AUS + NG. The SEA and AUS + NG lineages were highly supported (BS = 100%) as sister lineages. Within the IND lineage, the India eastern coast and Bangladesh sampling locations were indicated to be sister lineages (BS = 100%). The AUS + NG lineage individuals from the Northern Territory and Papua New Guinea were indicated to be the most closely related (BS = 100%). The first division within the SEA lineage is between central Indonesia sampling locations (Indonesia-SU + Indonesia-K) and receives maximal support for monophyly and branching patterns. The Philippines sampling location separates with the next successive branching within the SEA lineage (BS = 100%). Relationships among Cambodia, Indonesia-SJ, Thailand and Vietnam sampling locations were not clearly resolved.
Species tree from SVDQuartets of Indo-Pacific Lates sampling locations in this study, based on 8529 variants rooted by Lates japonicus. Values at nodes are bootstrap support values.
For ML phylogenetic analysis and phylogenetic network analysis, an alignment of 60 individuals with 7661 SNPs having 7661 distinct unique alignment patterns and 5571 parsimony-informative sites was created. The alignment contains 2090 singleton sites. Support for monophyly of IND lineage fish is maximal, as well as geographic divisions into eastern and western Indian subcontinent (Fig. 4a). Indo-Pacific Lates from SEA as a grouping received low support (BS = 76%), with concordance between sampling locations and placement not being complete. Indonesia-K and the Philippines sampling locations were highly supported to be monophyletic, but Cambodia, Thailand and Indonesia-SJ were not. Support for a group of AUS + NG sampling locations is maximal, with maximal support for division between Queensland and Northern Territory + Papua New Guinea fish. Monophyly of samples from the Northern Territory is poorly supported (BS = 68%).
(a) Maximum-likelihood phylogeny of genome-wide SNPs of 60 individuals of Indo-Pacific Lates. Nodes with <75% bootstrap support are collapsed to polytomies. Nodes with ≥75% bootstrap support but <90% bootstrap support are indicated as grey squares and nodes with ≥90% bootstrap support as black squares. (b) Implicit phylogenetic network of genome-wide SNPs from 60 individuals of Indo-Pacific Lates. Node that shape indicates the putative lineage of origin and colour indicates collection location.
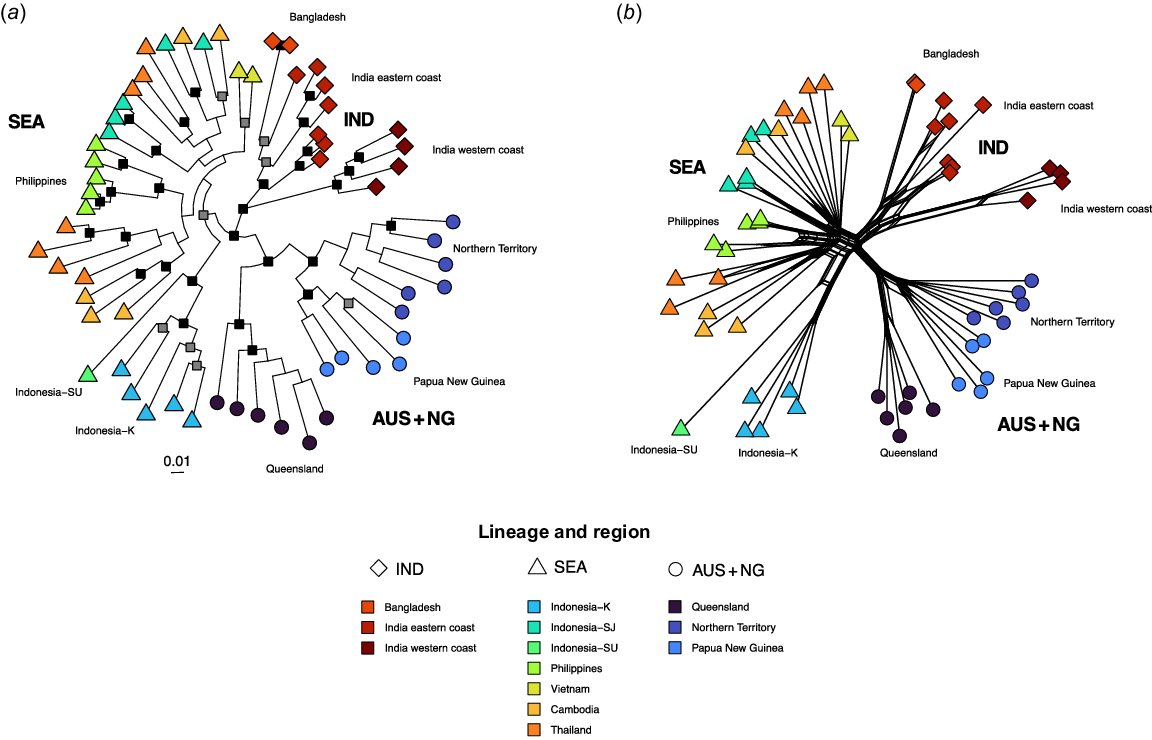
The phylogenetic network resolves an IND lineage containing two sublineages (India eastern coast + Bangladesh, India western coast), with separation of all three sampling locations (Fig. 4b). The SEA lineage is present, separated between central Indonesia and all other locations as two main sublineages, with reticulations with the IND lineage occurring with a subgroup of 10 individuals, disrupting monophyly of some sampling locations. The Indonesia-K and Philippines locations were monophyletic, but the Cambodia, Thailand and Indonesia-SJ sampling locations were not. The AUS + NG grouping is composed of three subclades, namely, Queensland (Australia), Northern Territory (Australia) and Papua New Guinea. In the network, Northern Territory and Papua New Guinea fish were most-closely related to each other.
Population genetic structure and statistics
The PC analysis included 7,321,852 variable sites in the genome and indicated a separation into three larger genetic clusters corresponding to IND, SEA and AUS + NG individuals (Supplementary material Fig. S1). The first PC separated AUS + NG, SEA and IND individuals (PC 1 12.57% of variation) with the second PC showing variation within SEA and IND clusters (PC2 4.71%). Clear separation of India eastern coast + Bangladesh and India western coast individuals is present on both PC2 and PC3 (PC 3 3.51%).
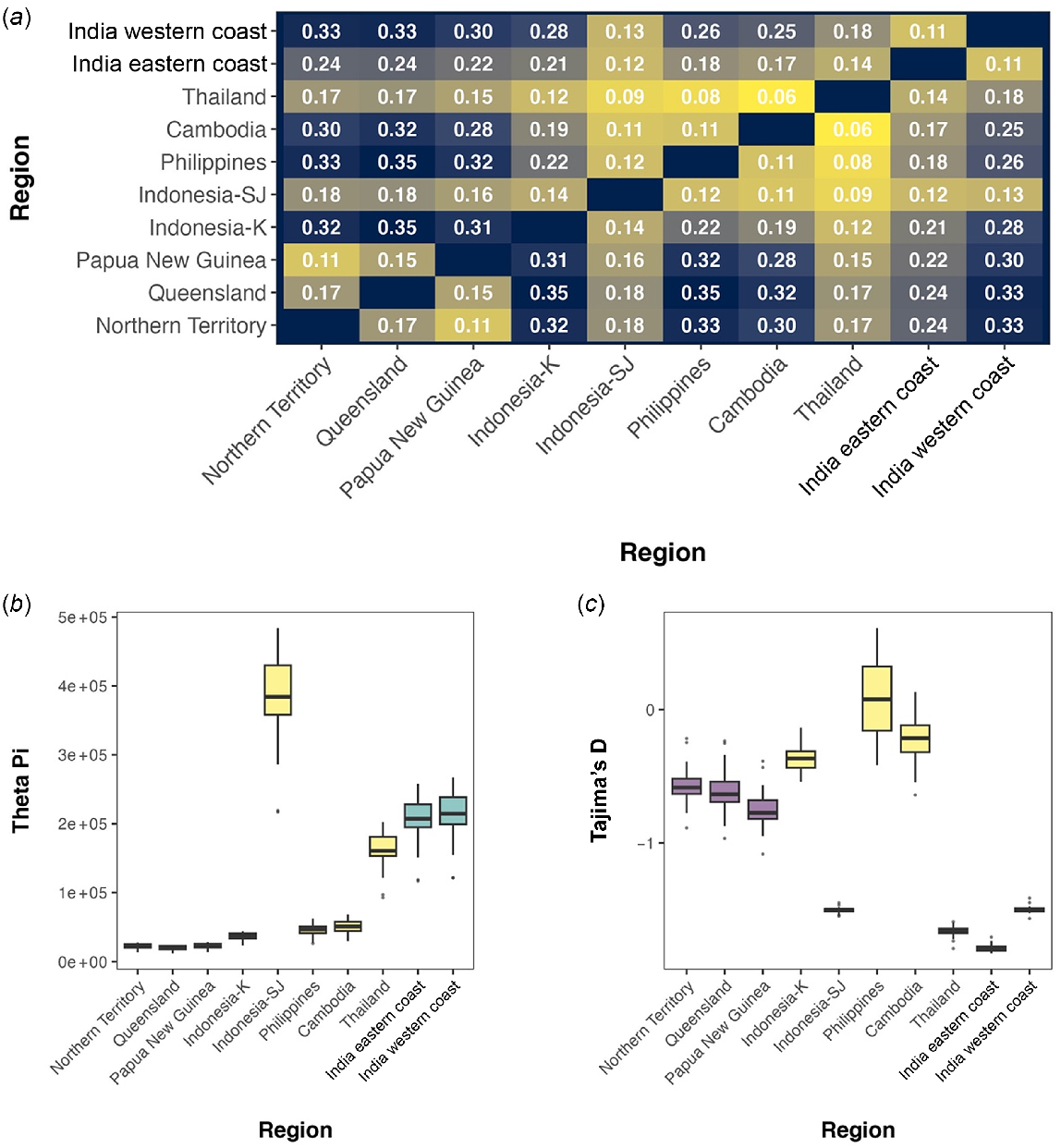
Genetic differentiation (Fst) was lowest within main genetic lineages, such as Thailand and Cambodia (Fst = 0.06) within the L. calcarifer SEA lineage and between the India eastern coast and India western coast sampling locations (Fst = 0.11) within the L. calcarifer IND lineage (Fig. 5a). The highest levels of genetic differentiation were found between main genetic lineages (between AUS + NG and SEA, Fst = 0.15–0.35; between AUS + NG and IND, Fst = 0.22–0.33; between SEA and IND Fst = 0.12–0.26). Nucleotide diversity is the lowest in the AUS + NG lineage (π = 20,067–22,775) and highest in the IND lineage (π = 207,299–214,485) (Fig. 5b). Tajima’s D was strongly negative (<−1) in the Indonesia-SJ, Thailand, India eastern Coast and India western coast sampling locations (Fig. 5c). The AUS + NG sampling locations had D values between −0.58 and −0.77.
Population genetic statistics of Indo-Pacific Lates from WGS data. (a) Pairwise Fst values between regions. (b) Nucleotide diversity of sampling locations in the WGS data set. (c) Tajima’s D of sampling locations in the WGS data set. Indonesia is represented by two sampling locations. Indonesia-SU, Vietnam and Bangladesh were excluded because of low sample sizes (n < 2).
Admixture analysis of 7,321,852 genotype likelihoods from 60 Indo-Pacific Lates individuals for K = 2–5 is presented as Fig. S2. At K = 2, the Bangladesh, India eastern coast and India western coast have high ancestry coefficients (Q > 0.99) for a single genetic cluster, and the AUS + NG lineage and Indonesia-K sampling location have high ancestry coefficients for the other. The remaining SEA sampling locations have a mixture of contributions from both genetic clusters. At K = 3, the AUS + NG individuals formed a clearly genetic cluster with Q > 0.99 for ancestry coefficients, the SEA sampling locations were heterogenous mixture proportions from sampling locations largely composed of the third genetic cluster, with the Indonesia-K and Indonesia-SU sampling locations exhibiting a near uniform ~0.27% admixture proportion with the genetic cluster typical of AUS + NG sampling locations. Higher values of K lead to separating of genetic units within the SEA lineage that may or may not correspond to sampling locations.
Tests for adaptation
The variation within the pruned genotype calls of 22,088 SNPs from the n = 60 WGS sequences with >3× coverage supported an optimal K = 3 PCs (Fig. S3). These PCs separated the three main lineages of L. calcarifer, with the third PC separating the Indonesia-K sampling location and to a lesser degree the Indonesia-SU individual from other SEA fish. Benjamini and Hochberg (1995) adjusted P-values (BH) identified 134 candidate adaptive SNPs and Bonferroni-corrected P-values (BF) indicated 34 candidate adaptive SNPs across the three PC axes.
Filtering to IND individuals (n = 13) resulted in 12,075 SNPs with a minimum MAF of 0.05, with the first PC separating the Bangladesh, India eastern coast and India western coast with 738 BH and 379 BF candidate adaptive SNPs identified on this axis (Fig. S4). The second PC axis separated India eastern coast with 285 BH and 59 BF candidate adaptive SNPS. From the SEA lineage, 30 individuals with 17,556 SNPs with a minimum MAF of 0.05 were analysed. The first three PCs were considered, separating Indonesia-K and Indonesia-SU sampling locations on PC1, the Philippines along PC2 and some Thailand and Cambodia individuals along PC3, with 190 BH and 29 BF candidate adaptive SNPs being identified in total (Fig. S5). The data set of AUS + NG individuals (n = 17) for loci with a minimum MAF of 0.05 resulted in 14,642 SNPs. We found strong support for K = 2 PCs, the first PC separating Queensland from the Northern Territory and Papua New Guinea (Fig. S6). The second PC separated Papua New Guinea from the other locations with 234 BH candidate adaptive SNPs and 57 BF candidate adaptive SNPs across the two PC axes.
Evaluation of hybridisation levels
The NewHybrid analysis of 43 individuals from the SEA and IND lineages indicated that all IND lineage fish were most likely a parent population (P0) (Fig. S7). This includes India eastern coast, which was not designated a priori as a potential parent population. Indonesia-K and Indonesia-SU were resolved as P1 fish with high probability. The two wild-caught fish from Cambodia with IND lineage mtDNA were indicated to be F2s, with other fish from Cambodia P1 (n = 2) and a backcrossed P1 fish.
Divergence-time estimation
We compiled a barcode alignment of 13 sequences (Table 2) with 660 characters, resulting in 440 characters (399 invariant) when including the first and second codon positions only. Sufficient effective sample sizes (ESS > 200, range of 712–3002) were reached with 20 million generations and two runs of three chains each, as evaluated after a 25% burnin. The resulting consensus tree presented is in Fig. 6. The median divergence time of the IND lineage from the other two lineages is estimated to be 6.76 Ma, with a 95% highest posterior density (HPD) of 1.31–14.24 Ma. The divergence of AUS + NG from SEA is estimated to be 1.02 Ma, with a 95% HPD range of 0.00–5.03 Ma. The relationships of Indo-Pacific Lates mitochondrial lineages were highly supported, with the placement having a posterior probability (PP) of 0.99, and the sister relationship between AUS + NG and SEA having a PP of 0.97.
| Family | Taxon | Accession | Origin | |
|---|---|---|---|---|
| Latidae | Lates calcarifer AUS + NG | EU189379.1 | Western Australia | |
| Lates calcarifer SEA | KJ573921.1 | Indonesia | ||
| Lates calcarifer IND | KJ573895.1 | Andaman and Nicobar Islands | ||
| Lates japonicus | LC269832.1 | Japan | ||
| Lates microlepis | ISZA082–21 | Democratic Republic of the Congo | ||
| Lates niloticus | LT853908.1 | Not Specified | ||
| Hypopterus macropterus | LC269834.1 | Australia | ||
| Psammoperca waigiensis | OQ387793.1 | Philippines, Luzon | ||
| Psammoperca datnioides | NC035737.1 | Australia, Darwin | ||
| Centropomidae | Centropomus ensiferus | MW183492.1 | Brazil, Para, Braganca | |
| Centropomus undecimalis | JQ841102.1 | Belize, Belize City | ||
| Channidae | Channa maculata | KP112195.1 | China, Hubei, Wuhan | |
| Nandidae | Nandus nandus | KX245097.1 | India |
Lates calcarifer is represented by three lineages, namely, Australia + New Guinea (AUS + NG), South-east Asia (SEA) and Indian subcontinent (IND), as described in the text. The family is given, with the taxon, corresponding BOLD or NCBI accession and the geographic origin of the sample if known.

Discussion
We identified from mitochondrial and nuclear genomic data three main genetic lineages of Indo-Pacific Lates. These are an Indian Ocean lineage, and a Pacific Ocean lineage split between South-east Asia and Australasia. A central Indonesian genetic cluster, representing Borneo and Sulawesi, appears as a distinctive group, with placement poorly supported in our mtDNA analysis (Fig. 2). Phylogenetic and population genetic analyses using data from the nuclear genome place the central Indonesian samples as more closely related to South-east Asian fish, and we treat them as part of that lineage (SEA).
The three main genetic lineages of Indo-Pacific Lates are present with substantial differences quantified in terms of genome-wide estimates of Fst, candidate adaptive loci and deep divergence times. We observed Fst values up to 0.35 and, in terms of divergence times, we estimated 6.76 million years for the divergence of IND from other Indo-Pacific Lates and a divergence-time estimate of 1.02 million years for AUS + NG from SEA. The previously published estimate based on RADseq data for divergence times by Wang et al. (2016) is restricted to AUS + NG from the SEA lineage and is an approximate of 644,100–878,900 years before present. Both of these estimates, the estimate presented here from fossil-calibrated phylogeny and that of Wang et al. (2016), support a divergence during the glacial cycling and changes in sea level and conditions during the Quaternary. The three main lineages of Indo-Pacific Lates exhibit discreteness (genetic isolation) and evolutionary significance (i.e. adaptive differentiation).
Within each of the three main lineages, there was additional evidence for discreteness and evolutionary significance. The IND sampling locations were divided phylogenetically (Fig. 3, 4), and in PC analysis (Fig. S4), with candidate adaptive SNPs being identified. We found that all three sampling locations within IND (Bangladesh, India eastern coast and India western coast) can be separated clearly with genomic data, but exhibit some mixing of mtDNA haplotypes. The SEA lineage exhibited notable separation of the Indonesia-K and Indonesia-SU sampling locations, the Philippines sampling location and some Thailand fish (Fig. 4, S5). Clear differentiation of all Philippines and Thailand fish may be affected by hybridisation (Fig. S2). The RADseq data set of Wang et al. (2016) did not show clear evidence of genetic structure, with three sampling locations within SEA, but broader sampling with microsatellites did appear to support genetic structuring (Yue et al. 2009). The AUS + NG sampling exhibited a clear division between Queensland and other sampling locations (Fig. 3, 4) and all three sampling locations could be clearly distinguished on two PC axes, with candidate adaptive loci being apparent (Fig. S6). Within the AUS + NG lineage, several studies have demonstrated genetic differentiation within that lineage (e.g. Shaklee and Salini 1985; Chenoweth et al. 1998; Wang et al. 2016; Loughnan et al. 2019), with 16 discrete genetic populations having been identified in Australia only (Keenan 1994).
What is Lates calcarifer?
Our fundamental understanding of nature is based on the species level and the lack of recognition of species is a major knowledge shortfall in conservation and management (Brown and Lomolino 1998). Molecular genetic techniques have continued to identify candidate species of fishes that may or may not have diagnostic morphological characters typical of species descriptions (e.g. Moyle and Campbell 2022; Campbell et al. 2023). Instead of being a historical aspect of biology, the need for continued taxonomic work is very important (e.g. Godfray et al. 2004; Delić et al. 2017). Taxonomic work often involves the detailed anatomical investigation of organisms that can show phenotypic differences related not only to speciation, but also life-history variation. What remains outstanding is whether Indo-Pacific Lates can be accurately represented within Linnean taxonomy or whether an alternative approach is better suited to the biological realities of Indo-Pacific Lates.
The most recent taxonomic effort relevant to Indo-Pacific Lates led to the description of L. lakdiva Pethiyagoda & Gill, 2012 from western Sri Lanka and L. usiwara Pethiyagoda & Gill, 2012 from eastern Myanmar (Pethiyagoda and Gill 2012). The specimens of L. usiwara were the same as those featured in the barcoding study of Ward et al. (2008) highlighting deep mitochondrial divergences; however, in a wider context, the barcode sequences of L. usiwara are identical to samples from the Indian subcontinent (Vij et al. 2014). In comparison to L. calcarifer, a differential diagnosis was possible and the distribution of L. usiwara is perhaps indicative of a recently derived species or ecotype. The anatomical study undertaken by Vij et al. (2014) along with barcoding did not clearly separate all these newly described species of Lates from L. calcarifer, noting small sample sizes used in the description of L. usiwara and L. lakdiva, and concluded that there is substantial mismatch between anatomical descriptions of new species of Lates (within the Indian Ocean) and candidate species of Lates apparent genetically (between the Indian and Pacific oceans at the broadest scale). Barcode sequences, it should be noted, will not reflect proportion of ancestry as do genomic data and ascertaining the ancestry proportions of fish used in taxonomic studies is critical going forward because of human translocations of Lates. Furthermore, it is possible for a high degree of genome-wide differentiation or substantial reproductive isolation to be present that would not be apparent in barcode data. Pethiyagoda and Gill (2012) themselves observed that because of the large body size and sequential hermaphodism of L. calcarifer sensu lato, they were restricted to examining many juveniles for their study.
The three genetic lineages of Indo-Pacific Lates remain unclear in distribution and have unclear anatomical differentiation (Vij et al. 2014). Portions of the range, such as Taiwan, remain unsampled for genomic data and genetic structuring is apparent across all three lineages (Fig. 3, 4, S1–S6). Differentiation between and within the main lineages can be clarified with additional sampling and representation of nominal taxa in genomic data sets (i.e. L. usiwara and L. lakdiva). Given the strong genetic differentiation and deep inferred time scales (Fig. 6), one possible resolution is to recognise at least two species-level taxa that correspond to the IND lineage and a combined SEA and AUS + NG lineage. At a potential species level, the taxonomy of Indo-Pacific Lates is complex and the recognition of types and their origins is also challenging (reviewed by Pethiyagoda and Gill 2012). The type locality of L. calcarifer (Bloch, 1790) may be Tamil Nadu, India (10.4°N, 79.3°E) (Pethiyagoda and Gill 2012). Thus, the IND lineage may be represented by this name. There are numerous available names that may be applicable to the SEA lineage, but this requires substantial time and expertise to resolve. However, there is an available name, L. cavifrons (Alleyne & Macleay, 1877), that could be applied to AUS + NG fish. The collection locality is either the Torres Strait or the coast of New Guinea with a lost holotype, with the drawing providing the current synonymy (Pethiyagoda and Gill 2012).
Without formal taxonomic recognition, the three main genetic lineages of Indo-Pacific Lates can be recognised for management purposes such as mixed-stock fisheries. Vij et al. (2014) noted that the assumption of single species of Indo-Pacific Lates has undoubtedly influenced previous studies, and microsatellite data from Thailand showing the highest allelic richness may reflect genotyping of two divergent genetic lineages along the western coast of Thailand (Yue et al. 2009). If different species of Indo-Pacific Lates are recognised, then questions of biology can be researched, such as the causes of speciation and what the mechanisms for reproductive isolation of IND may be. Development of an anatomical key usable in the field that can distinguish these species would be of great practical utility and could be used to resolve the distributions and overlap of IND and SEA lineages. A high-throughput sequencing assay that can discriminate the main lineages could provide a rapid assessment of genetic lineage, hybridisation levels and possible provenance in studies of food adulteration.
Admixture
Overall, analyses that can quantify hybridisation in this study are challenging because of the high innate diversity of the SEA lineage and limited sample sizes, notably that of the IND lineage (n = 13). The sequences were also obtained opportunistically from the NCBI Sequence Read Archive and not collected by the authors. With these cautions in mind, we found evidence of admixture among major genetic lineages within our data set, as evidenced by the placement of Cambodia-origin samples, with a largely SEA genomic background, within the mitochondrial IND lineage (Fig. 2–4). When examining all 60 individuals at K = 3, the two Cambodian individuals with IND mtDNA had ancestry coefficients (Q) of 0.29 and 0.43 with the IND genetic cluster; however, there is such a degree of variation in the SEA group with respect to admixture proportions and inconsistency across sampling locations that these measurements should not be accepted at face value. The Cambodian group has two individuals with Q = 0.00 for the IND genetic cluster and one with 0.17, for example, when examining all individuals. Separate analysis of Cambodian and L. calcarifer IND individuals showed only Q = 0.23 and 0.33 for the two Cambodian individuals with IND mtDNA and no evidence for nuclear genomic admixture of other Cambodian individuals with IND. This finding is congruent with the NewHybrids analysis, indicating the presence of two F2 hybrids in the Cambodian fish, in line with the ancestry proportions of these fish analysed with IND fish separately (Fig. S7).
Nucleotide diversity in the SEA Indonesia-SJ location is very high (Fig. 5b) and is a likely contributor to the reduced Fst of this lineage to other sampling regions (Fig. 5a). Combined with the large negative D value, which arises from admixture (Hahn et al. 2013), this sampling location may contain admixed individuals. A similar pattern of high nucleotide diversity, low Fst, and strongly negative D is apparent with the Thailand sampling location (Fig. 5). Both the Indonesia-SJ and Thailand sampling locations show substantial contributions of the IND genetic cluster in their ancestry (Fig. S2); however, it is not uniformly distributed across sampling locations. If the distribution of ancestry is uniform across sampling locations, it would suggest an older age of hybridisation. In NewHybrid analyses, Indonesia-SJ and Thailand contained numerous recent (F2) hybrids (Fig. S7). Furthermore, the admixture analyses suggested that Thailand at K = 4 is represented by more than one distinct genetic unit from within the SEA lineage, with additional contributions from the IND lineage. The dearth of F1 hybrids found by NewHybrids was surprising but may have to do with a high degree of diversity within the SEA lineage as a result of (1) retained ancestral variation and (2) historical admixture. Furthermore, the selection of a random set of 200 SNPs because of computational limitations may not capture informative loci for hybridisation assessment.
Conclusions
We have provided several advances in the study of Indo-Pacific Lates, presenting molecular phylogenetic analyses of a geographically and genomically representative data set and testing for adaptive differentiation and recent or more advanced hybridisation. The analysis of genome-wide nuclear variants also provided evidence of hierarchical divergences; that is, within the IND, SEA and AUS + NG lineages, there is genetic structuring. The SEA lineage demonstrated a complex history with a combination of natural genetic differentiation and what is likely to be human-mediated admixture events. Clearly, samples of known provenance, the use of informative loci that are diagnostic to lineages, and boosting of sampling locations and sample sizes focused across the possible intergrade zone IND and SEA would help resolve these ambiguities. Future studies making use of archive and museum samples to provide genetic baselines prior to use of Indo-Pacific Lates in aquaculture can be used to create a genetic baseline to compare modern genetics against, so as to identify potential translocations and adaptation to changing environments (Turbek et al. 2023).
Phenotypic and life-history variation within Indo-Pacific Lates remain poorly characterised and these are other aspects of variation that can substantially contribute to ecosystem productivity and commercial applications. Research has indicated that within Australia, for instance, there is strong local adaptation related to temperature (Newton et al. 2013) that could be applied to various goals. The wide and varied use of Indo-Pacific Lates in commercial, recreation and subsistence fisheries as well as aquaculture creates differing priorities across user groups. Further exploration of genotypic and phenotypic variation can support conservation aquaculture and food production as well as the preservation of productivity and adaptive potential of Indo-Pacific Lates stocks.
Data availability
Genomic sequence data are available from the NCBI Sequence Read Archive under BioProject accessions PRJNA311498, PRJNA1021005 and PRJDB13763. Alignments for phylogenetic analyses, the control block for SVDQuartets and the time-calibrated phylogeny tree file are available in File S1 of the Supplementary material. Sequence data examined in this study for time-calibrated phylogenetic analysis are publicly available with mitochondrial sequence accessions provided in Table 2.
Declaration of funding
Support for M. A. Campbell was received from the School of Life and Environmental Sciences at The University of Sydney and the Charles Gilbert Heydon Traveling Fellowship.
Acknowledgements
The authors recognise the two anonymous reviewers that provided valuable feedback, which improved the paper.
References
Abed JM, Ali AH, Yaseen AT, Al-Faisal A, Mutlak F, Jassim FK, Jerry DR, Jawad LA (2023) Barramundi (Lates calcarifer) from Iraq: a new record for the Arabian Gulf, with a highlight on it genetic origins and description of two skeletal deformities. New Zealand Journal of Zoology [Published online 20 September 2023].
| Crossref | Google Scholar |
Agapow P, Bininda-Emonds OR, Crandall KA, Gittleman JL, Mace GM, Marshall JC, Purvis A (2004) The impact of species concept on biodiversity studies. The Quarterly Review of Biology 79(2), 161-179.
| Crossref | Google Scholar | PubMed |
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 25(17), 3389-3402.
| Crossref | Google Scholar | PubMed |
Anderson EC, Thompson EA (2002) A model-based method for identifying species hybrids using multilocus genetic data. Genetics 160(3), 1217-1229.
| Crossref | Google Scholar | PubMed |
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society – B. Statistical Methodology 57(1), 289-300.
| Crossref | Google Scholar |
Campbell MA, Hale MC (2024) Genomic structural variation in Barramundi Perch Lates calcarifer and potential roles in speciation and adaptation. G3: Genes|Genomes|Genetics 14(8), jkae141.
| Crossref | Google Scholar |
Campbell MA, Chen W-J, López JA (2013) Are flatfishes (Pleuronectiformes) monophyletic? Molecular Phylogenetics and Evolution 69(3), 664-673.
| Crossref | Google Scholar | PubMed |
Campbell MA, López JA, Satoh TP, Chen W-J, Miya M (2014) Mitochondrial genomic investigation of flatfish monophyly. Gene 551(2), 176-182.
| Crossref | Google Scholar | PubMed |
Campbell MA, Badger ME, Buckmaster N, Starostka AB, Hawks T, Finger AJ (2023) Molecular phylogenetic and population genetic relationships of a putative species of sucker (Catostomus sp.) from Surprise Valley in the Great Basin, USA. Transactions of the American Fisheries Society 152(3), 273-286.
| Crossref | Google Scholar |
Chen W-J, Ruiz-Carus R, Ortí G (2007) Relationships among four genera of mojarras (Teleostei: Perciformes: Gerreidae) from the western Atlantic and their tentative placement among percomorph fishes. Journal of Fish Biology 70(sb), 202-218.
| Crossref | Google Scholar |
Chen TY, Nazir A, Lim HC, Shiao JC (2024) Genetic diversity and population structure of cultured and wild barramundi Lates calcarifer (Latidae) in Taiwan. Journal of Ichthyology 64(5), 842-853.
| Crossref | Google Scholar |
Chenoweth SF, Hughes JM, Keenan CP, Lavery S (1998) Concordance between dispersal and mitochondrial gene flow: isolation by distance in a tropical teleost, Lates calcarifer (Australian barramundi). Heredity 80, 187-197.
| Crossref | Google Scholar |
Chifman J, Kubatko L (2014) Quartet inference from SNP data under the coalescent model. Bioinformatics 30(23), 3317-3324.
| Crossref | Google Scholar | PubMed |
Chifman J, Kubatko L (2015) Identifiability of the unrooted species tree topology under the coalescent model with time-reversible substitution processes, site-specific rate variation, and invariable sites. Journal of Theoretical Biology 374, 35-47.
| Crossref | Google Scholar | PubMed |
Danecek P, Bonfield JK, Liddle J, Marshall J, Ohan V, Pollard MO, Whitwham A, Keane T, McCarthy SA, Davies RM, Li H (2021) Twelve years of SAMtools and BCFtools. GigaScience 10(2), giab008.
| Crossref | Google Scholar |
Delić T, Trontelj P, Rendoš M, Fišer C (2017) The importance of naming cryptic species and the conservation of endemic subterranean amphipods. Scientific Reports 7, 3391.
| Crossref | Google Scholar | PubMed |
Des Roches S, Post DM, Turley NE, Bailey JK, Hendry AP, Kinnison MT, Schweitzer JA, Palkovacs EP (2018) The ecological importance of intraspecific variation. Nature Ecology & Evolution 2, 57-64.
| Crossref | Google Scholar | PubMed |
Edwards-Vandenhoek S (2015) This my country, I’m painting here: the role of social practice art and participatory design in building resilience and place-making in the Warmun Aboriginal Community, Kimberley, Western Australia. In ‘Australia Council of University Art and Design Schools Conference Proceedings: ACUADS Conference 2015: Art and Design Education in the Global 24/7’, 24–25 September 2015, Adelaide, SA, Australia. (ACUADS) Available at https://acuads.com.au/wp-content/uploads/2016/01/Edwards-Vandenhoek.pdf
Godfray HCJ, Knapp S, Mace GM (2004) The role of taxonomy in species conservation. Philosophical transactions of the Royal Society of London – B. Biological sciences 359(1444), 711-719.
| Crossref | Google Scholar | PubMed |
Gruber B, Unmack PJ, Berry OF, Georges A (2018) dartR: an R package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Molecular Ecology Resources 18(3), 691-699.
| Crossref | Google Scholar | PubMed |
Hackl T, Hedrich R, Schultz J, Förster F (2014) Proovread : large-scale high-accuracy PacBio correction through iterative short read consensus. Bioinformatics 30(21), 3004-3011.
| Crossref | Google Scholar | PubMed |
Hahn C, Bachmann L, Chevreux B (2013) Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads – a baiting and iterative mapping approach. Nucleic Acids Research 41(13), e129.
| Crossref | Google Scholar |
Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS (2018) UFBoot2: improving the ultrafast bootstrap approximation. Molecular Biology and Evolution 35(2), 518-522.
| Crossref | Google Scholar | PubMed |
Huson DH, Bryant D (2006) Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution 23(2), 254-267.
| Crossref | Google Scholar | PubMed |
Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods 14, 587-589.
| Crossref | Google Scholar | PubMed |
Katoh K, Toh H (2008) Recent developments in the MAFFT multiple sequence alignment program. Briefings in Bioinformatics 9(4), 286-298.
| Crossref | Google Scholar | PubMed |
Katoh K, Misawa K, Kuma K-I, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Research 30(14), 3059-3066.
| Crossref | Google Scholar | PubMed |
Keenan CP (1994) Recent evolution of population structure in Australian barramundi, Lates calcarifer (Bloch): an example of isolation by distance in one dimension. Australian Journal of Marine and Freshwater Research 45(7), 1123-1148.
| Crossref | Google Scholar |
Korneliussen TS, Albrechtsen A, Nielsen R (2014) ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15, 356.
| Crossref | Google Scholar |
Layton KKS, Snelgrove PVR, Dempson JB, Kess T, Lehnert SJ, Bentzen P, Duffy SJ, Messmer AM, Stanley RRE, DiBacco C, Salisbury SJ, Ruzzante DE, Nugent CM, Ferguson MM, Leong JS, Koop BF, Bradbury IR (2021) Genomic evidence of past and future climate-linked loss in a migratory Arctic fish. Nature Climate Change 11, 158-165.
| Crossref | Google Scholar |
Lehnert SJ, Bradbury IR, Wringe BF, Van Wyngaarden M, Bentzen P (2023) Multifaceted framework for defining conservation units: an example from Atlantic salmon (Salmo salar) in Canada. Evolutionary Applications 16(9), 1568-1585.
| Crossref | Google Scholar | PubMed |
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25(14), 1754-1760.
| Crossref | Google Scholar | PubMed |
Li H, Durbin R (2010) Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26(5), 589-595.
| Crossref | Google Scholar | PubMed |
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25(16), 2078-2079.
| Crossref | Google Scholar | PubMed |
Li C, Ricardo B-R, Leo Smith W, Ortí G (2011) Monophyly and interrelationships of Snook and Barramundi (Centropomidae sensu Greenwood) and five new markers for fish phylogenetics. Molecular Phylogenetics and Evolution 60(3), 463-471.
| Crossref | Google Scholar | PubMed |
Loughnan SR, Smith-Keune C, Beheregaray LB, Robinson NA, Jerry DR (2019) Population genetic structure of barramundi (Lates calcarifer) across the natural distribution range in Australia informs fishery management and aquaculture practices. Marine and Freshwater Research 70(11), 1533-1542.
| Crossref | Google Scholar |
Luu K, Bazin E, Blum MGB (2017) Pcadapt: an R package to perform genome scans for selection based on principal component analysis. Molecular Ecology Resources 17(1), 67-77.
| Crossref | Google Scholar | PubMed |
Maddison WP (1997) Gene trees in species trees. Systematic Biology 46(3), 523-536.
| Crossref | Google Scholar |
Mathew G (2009) Taxonomy, identification and biology of Seabass (Lates calcarifer). In ‘National Training on “Cage culture of seabass”’, 14–23 December 2009, Kochi, India. pp. 38–43. (Central Marine Fisheries Research: India) Available at https://eprints.cmfri.org.in/6062/1/7._Gra.pdf
Moore R (1979) Natural sex inversion in the giant perch (Lates calcarifer). Australian Journal of Marine and Freshwater Research 30(6), 803-813.
| Crossref | Google Scholar |
Moore R, Reynold LF (1982) Migration patterns of barramundi, Lates calcarifer (Bloch), in Papua New Guinea. Australian Journal of Marine and Freshwater Research 33(4), 671-682.
| Crossref | Google Scholar |
Moyle PB, Campbell MA (2022) Cryptic species of freshwater sculpin (Cottidae: Cottus) in California, USA. Zootaxa 5154(5), 501-527.
| Crossref | Google Scholar | PubMed |
Newton JR, Zenger KR, Jerry DR (2013) Next-generation transcriptome profiling reveals insights into genetic factors contributing to growth differences and temperature adaptation in Australian populations of barramundi (Lates calcarifer). Marine Genomics 11, 45-52.
| Crossref | Google Scholar | PubMed |
Nguyen H-V, Legras J-L, Neuvéglise C, Gaillardin C (2011) Deciphering the hybridisation history leading to the lager lineage based on the mosaic genomes of Saccharomyces bayanus strains NBRC1948 and CBS380T. PLoS ONE 6(10), e25821.
| Crossref | Google Scholar | PubMed |
Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Molecular Biology and Evolution 32(1), 268-274.
| Crossref | Google Scholar | PubMed |
Otero O (2004) Anatomy, systematics and phylogeny of both recent and fossil latid fishes (Teleostei, Perciformes, Latidae). Zoological Journal of the Linnean Society 141(1), 81-133.
| Crossref | Google Scholar |
Pethiyagoda R, Gill AC (2012) Description of two new species of sea bass (Teleostei: Latidae: Lates) from Myanmar and Sri Lanka. Zootaxa 3314(1), 1-16.
| Crossref | Google Scholar |
Privé F, Luu K, Vilhjálmsson BJ, Blum MGB (2020) Performing highly efficient genome scans for local adaptation with R package pcadapt version 4. Molecular Biology and Evolution 37(7), 2153-2154.
| Crossref | Google Scholar | PubMed |
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de de Bakker PIW, Daly MJ, Sham PC (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics 81(3), 559-575.
| Crossref | Google Scholar | PubMed |
Rambaut A, Drummond AJ, Xie D, Baele G, Suchard MA (2018) Posterior summarization in Bayesian phylogenetics using Tracer 1.7. Systematic Biology 67(5), 901-904.
| Crossref | Google Scholar | PubMed |
Rimmer MA (2024) Cultured aquatic species information programme: Lates calcarifer (Bloch 1790). In ‘Fisheries and Aquaculture’, updated 2006-07-19. (Food and Agriculture Organization of the United Nations: Rome, Italy) Available at https://www.fao.org/fishery/en/culturedspecies/lates_calcarifer/en
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology 61(3), 539-542.
| Crossref | Google Scholar | PubMed |
Santini F, Harmon L, Carnevale G, Alfaro M (2009) Did genome duplication drive the origin of teleosts? A comparative study of diversification in ray-finned fishes. BMC Evolutionary Biology 9, 194.
| Crossref | Google Scholar |
Schindler DE, Hilborn R, Chasco B, Boatright CP, Quinn TP, Rogers LA, Webster MS (2010) Population diversity and the portfolio effect in an exploited species. Nature 465, 609-612.
| Crossref | Google Scholar | PubMed |
Shaklee JB, Salini JP (1985) Genetic variation and population subdivision in Australian barramundi, Lates calcarifer (Bloch). Australian Journal of Marine and Freshwater Research 36(2), 203-218.
| Crossref | Google Scholar |
Shamsi S, Williams M, Mansourian Y (2020) An introduction to Aboriginal fishing cultures and legacies in seafood sustainability. Sustainability 12(22), 9724.
| Crossref | Google Scholar |
Skotte L, Korneliussen TS, Albrechtsen A (2013) Estimating individual admixture proportions from Next Generation Sequencing data. Genetics 195(3), 693-702.
| Crossref | Google Scholar | PubMed |
Turbek SP, Bossu C, Rayne C, Gruppi C, Kus BE, Whitfield M, Smith TB, Paxton EH, Bay RA, Ruegg KC (2023) Historical DNA reveals climate adaptation in an endangered songbird. Nature Climate Change 13, 735-741.
| Crossref | Google Scholar |
Vij S, Purushothaman K, Gopikrishna G, Lau D, Saju JM, Shamsudheen KV, Kumar KV, Basheer VS, Gopalakrishnan A, Hossain MS, Sivasubbu S, Scaria V, Jena JK, Ponniah AG, Orbán L (2014) Barcoding of Asian seabass across its geographic range provides evidence for its bifurcation into two distinct species. Frontiers in Marine Science 1, 30.
| Crossref | Google Scholar |
Vij S, Kuhl H, Kuznetsova IS, Komissarov A, Yurchenko AA, Van Heusden P, Singh S, Thevasagayam NM, Prakki SRS, Purushothaman K, Saju JM, Jiang J, Mbandi SK, Jonas M, Hin Yan Tong A, Mwangi S, Lau D, Ngoh SY, Liew WC, Shen X, Hon LS, Drake JP, Boitano M, Hall R, Chin C-S, Lachumanan R, Korlach J, Trifonov V, Kabilov M, Tupikin A, Green D, Moxon S, Garvin T, Sedlazeck FJ, Vurture GW, Gopalapillai G, Kumar Katneni V, Noble TH, Scaria V, Sivasubbu S, Jerry DR, O’Brien SJ, Schatz MC, Dalmay T, Turner SW, Lok S, Christoffels A, Orbán L (2016) Chromosomal-level assembly of the Asian Seabass genome using long sequence reads and multi-layered scaffolding. PLoS Genetics 12, e1005954.
| Crossref | Google Scholar | PubMed |
Wang L, Wan ZY, Lim HS, Yue GH (2016) Genetic variability, local selection and demographic history: genomic evidence of evolving towards allopatric speciation in Asian seabass. Molecular Ecology 25(15), 3605-3621.
| Crossref | Google Scholar | PubMed |
Ward RD, Holmes BH, Yearsley GK (2008) DNA barcoding reveals a likely second species of Asian sea bass (barramundi) (Lates calcarifer). Journal of Fish Biology 72(2), 458-463.
| Crossref | Google Scholar |
Yu G, Smith DK, Zhu H, Guan Y, Lam TT-Y (2017) ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods in Ecology and Evolution 8(1), 28-36.
| Crossref | Google Scholar |
Yue GH, Zhu ZY, Lo LC, Wang CM, Lin G, Feng F, Pang HY, Li J, Gong P, Liu HM, Tan J, Chou R, Lim H, Orban L (2009) Genetic variation and population structure of Asian seabass (Lates calcarifer) in the Asia–Pacific region. Aquaculture 293(1–2), 22-28.
| Crossref | Google Scholar |