

Unlocking therapeutic potential: the role of adamantane in drug discovery
Chianna Dane A , Grace A. Cumbers A , Beau Allen A , Andrew P. Montgomery A , Jonathan J. Danon A and Michael Kassiou A *
A , Beau Allen A , Andrew P. Montgomery A , Jonathan J. Danon A and Michael Kassiou A *
A
Abstract
The unique structural and physicochemical properties of adamantane and its derivatives have attracted considerable attention in the field of medicinal chemistry. Substituting phenyl rings for adamantane or its derivatives has provided a promising strategy to introduce lipophilicity and escape the ‘flat land’ of modern drug discovery. Additionally, the unique three-dimensional structure of adamantane facilitates the precise positioning of substituents allowing for a more effective exploration of drug targets. Evidently, we have seen an increased use of adamantane in pharmaceutically relevant molecules. The following Account highlights our group’s research in five drug discovery programs over the past 15 years showcasing the use of adamantane and its analogues in these studies.
Keywords: adamantane, cannabinoid receptors, CNS, medicinal chemistry, NMDA receptor, P2X7 receptor, sigma-2 receptor, structure–activity relationships, tau aggregation.
Introduction
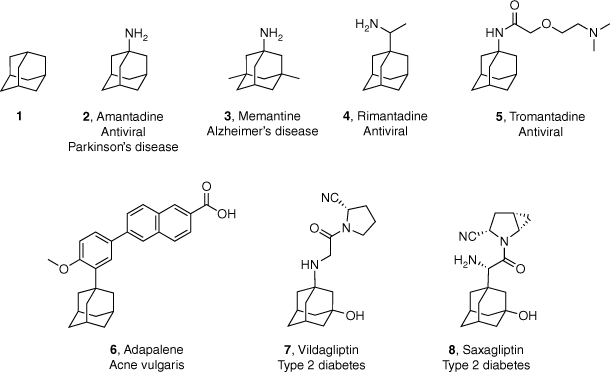
Adamantane (tricyclo[3.3.1.11,7]decane; 1, Fig. 1), the smallest of the diamonoids, was first elucidated in 1933 following its isolation from crude oil.1,2 Despite being synthesised chemically for the first time in 1941, it was not until Schleyer reported the synthesis in 1957 that adamantane and its derivatives became readily available.3,4 Subsequently this moiety began to be incorporated into drug discovery programs, with the first promising pharmaceutical breakthrough occurring with the identification of the antiviral activity of amantadine (2, Fig. 1) in 1963.5,6 In the 60 years following this seminal report, adamantyl-based compounds have seen numerous applications in medicinal chemistry and drug discovery, with seven adamantyl-based drugs (3–8, Fig. 1) currently in clinical use. These compounds are used to treat a range of diseases including viral infections, neurodegenerative disorders, acne vulgaris and type 2 diabetes mellitus.7
Adamantane (1) and the structure and target indication of adamantyl-based drugs 2–8 that are currently available for clinical use.
Incorporation of adamantane or its derivatives into drug-like molecules has been adopted by medicinal chemists for a variety of reasons. For instance, drug discovery programs targeting ion channels have exploited the three-dimensional space occupied by the adamantyl group. The rigid, highly symmetric (Td point group) hydrocarbon scaffold can be used to control the orientation of functional groups to facilitate the optimisation of both potency and selectivity at a particular target.2,5,8 Additionally, inclusion of the sterically bulky adamantyl group can increase stability through the contribution of both London dispersion forces and electrostatic interactions.9,10 Adamantane has also shown utility as a suitable polycyclic replacement of phenyl rings to escape the ‘flat land’ of modern drug discovery.11 Finally, the ADMET (absorption, distribution, metabolism, excretion and toxicity) properties of a drug candidate can be modulated by the incorporation of an adamantyl group. For example, the increased lipophilicity of an adamantyl-derivative can enhance central nervous system (CNS) drug exposure by improving blood–brain barrier (BBB) permeability and membrane solubility.5,7 Inclusion of hydrophobic substituents such as adamantane has been estimated to increase the calculated partition coefficients (c logP) value of a given drug by ~3.1 log units and therefore may be clinically useful for highly water soluble compounds.7 In terms of metabolism, the rigid hydrocarbon scaffold of adamantane has been demonstrated to protect functional groups in its proximity from metabolic cleavage, thereby increasing drug stability and plasma half-life.7 Further insight into the benefits of using adamantyl-based small molecules in medicinal chemistry campaigns can be found in a number of detailed reviews.5,7,8,12
Over the past 15 years, our research group has attempted to harness the beneficial properties of the adamantyl group and its derivatives in our CNS drug discovery projects. The following section highlights five of these projects.
Adamantane in CNS drug discovery
The sigma-2 receptor
The sigma (σ) receptors are a unique class of proteins located throughout the CNS and peripheral organs.13 Categorised into two subtypes, the sigma-1 and sigma-2 receptors (σ1R and σ2R respectively) acknowledge similar ligands, suggesting significant homology within the ligand-binding domain. Structurally and genetically, however, the relationship between the two remains unclear as the σ2R has yet to be cloned. The σ1R is a chaperone protein that resides in the endoplasmic reticulum, although it can still be regulated by small molecule antagonists and agonists. Within the CNS both the σ1R and σ2R play a notable role in synaptic signalling pathways by modulating ion channels and regulating calcium homeostasis.14 Evidently, both σ-receptors have been implicated in a multitude of neurological disorders, prompting the development of selective and potent ligands.15
The incorporation of polycyclic moieties into medicinal chemistry campaigns has been an area of growing interest over the past several decades. Amantadine (2, Fig. 2), the first FDA-approved drug containing the adamantane scaffold, was clinically approved for the treatment of Parkinson’s disease (PD) in 1968. Despite the mechanism of action being poorly understood there is considerable evidence that it acts as an antagonist of the N-methyl-D-aspartate (NMDA) glutamate receptor thereby increasing dopamine release.16 Following this, studies showed that amantadine was found to behave as a σ1R agonist at therapeutically relevant concentrations.16 This initiated the inclusion of polycyclic frameworks, such as trishomocubane and adamantane, in the development of novel σ-receptor agonists.
Structure–activity relationship studies conducted on the trishomocubane scaffold led to the exploration of polycycles such as azaadamantane.17,19–21,23,24 Data extracted from Liu et al.19,20 and Banister et al.21,23,24 Competitive binding affinity measurements were performed using [3H](+)-pentazocine and [3H]DTG as radioligands and either guinea pig or rat as the source of σ1 and σ2 receptors.
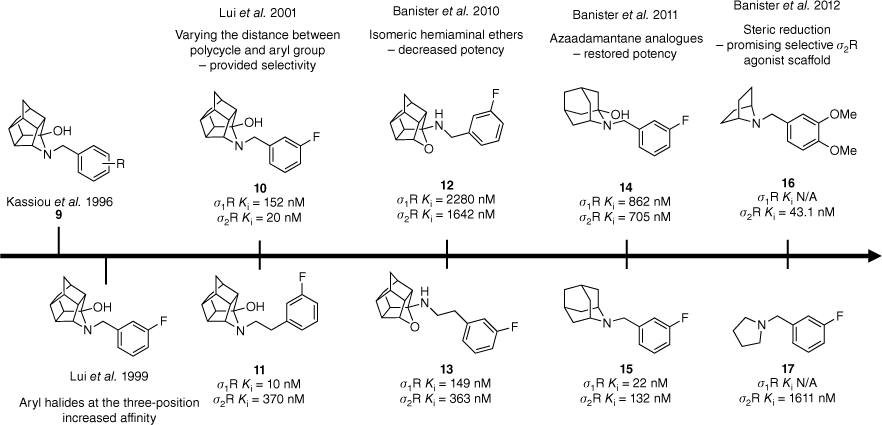
Initially, the trishomocubane framework 9 was explored resulting in a novel series of compounds exhibiting selectivity and high affinity for the σ-binding site.17 Subtype selectivity was achieved by varying the distance between the polycyclic hemiaminal and the aryl group (Fig. 2). Benzyl derivatives 10 displayed a preference for the σ1R, whereas phenethyl derivatives 11 had better activity at the σ2R (Fig. 2).18–20 Affinity and selectivity were shown to both be improved when aryl halides were utilised, especially when substituted at the 3-position (10 and 11).18–20 The isomeric hemiaminal ethers 12 and 13 did not retain activity as expected despite their structural similarity to previously reported agonists (Fig. 2).21,22
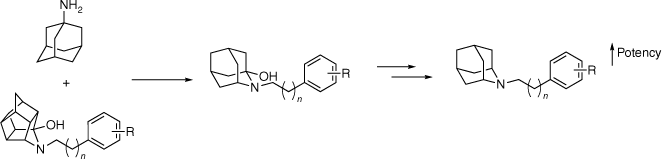
Considering the affinity of amantadine (2) at σ-receptors, and the promising results observed within the trishomocubane scaffold 9, a structure–activity relationship (SAR) study of polycarbocyclic amines was explored. N-Arylalkyl-2-azaadamantan-1-ols 14 and 19 and their deoxygenated derivatives 15 and 20 were synthesised to gain information about the steric and electronic tolerance of the hydrophobic region of the σ-receptor binding site. The hydroxy functional group was found to be detrimental to binding affinity with the achiral azaadamantane derivatives 15 and 20, increasing affinity for both receptors by an order of magnitude (Fig. 3).23 The azaadamantane analogues displayed excellent selectivity over 42 other CNS targets screened; however, only minimal selectivity between the σ-receptor subtypes was observed. A similar deoxygenation method was attempted for trishomocubane derivatives; however, it was deemed to be not synthetically viable.23
Hybrid scaffold campaign between amantadine (2) and the trishomocubane σ-agonists. Deoxygenation of the N-arylalkyl-2-azaadamantan-1-ols was found to increase affinity at both σ-receptors.
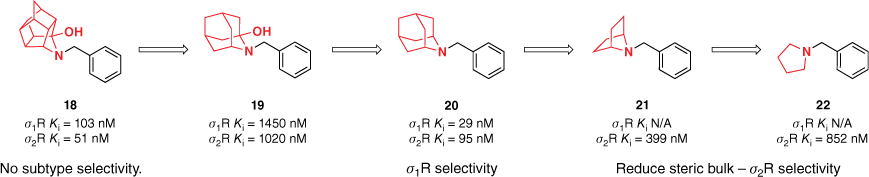
The effects of the steric contribution of polycarbocyclic amines on σ-receptor binding and selectivity were expanded upon through a series of azabicyclo[2.2.1]heptanes, representing the simplest aza-bridged bicyclic subunit, and its analogous N-substituted pyrrolidines (Fig. 2).24 Aryl and cubyl moieties were connected to the polycarbocycle by alkyl chains of varying length. Reducing the steric bulk of the polycarbocycle in the azabicyclo[2.2.1]heptane derivatives 16 and 21 diminished binding affinity at both σ-receptors and was further illustrated by the significant decrease in σ2R activity for the pyrrolidine derivatives 17 and 22. Among the azabicyclo[2.2.1]heptane library, subtype discrimination was achieved with analogues incorporating benzylic substituents. This was further improved through electron-rich functional groups, such as a methoxy group (16, Fig. 2). The electronic argument was supported by the high σ1R affinity of cyclohexyl-derived azabicyclo[2.2.1]heptane.24
Substitution of the trishomocubane moiety for other polycycles provided valuable insight into the steric and electronic tolerance of the σ-receptor binding sites (Fig. 4). This information can then be utilised for the development of selective agonists and improve our general understanding of these two receptors. Reducing steric bulk through the introduction of an azabicyclo[2.2.1]heptane polycycle reduced binding affinity, however, highlighted a promising scaffold for the development of selective σ2R agonists. Furthermore, the azaadamantane framework was highlighted as a novel lead for σ1R selective agonists.
Exploration of the effects of steric contribution on polycarbocyclic amines on σ-receptor binding. Azaadamantane and azabicyclo[2.2.1]heptane frameworks were highlighted as promising scaffolds for selective σ1R and σ2R agonists respectively. Data extracted from Liu et al.20 and Banister et al.23,24 Competitive binding affinity measurements were performed using [3H](+)-pentazocine and [3H]DTG as radioligands and either guinea pig or rat as the source of σ1 and σ2 receptors.
The N-methyl-D-aspartate receptor
NMDA receptors (NMDARs) are distributed widely throughout the CNS and play an essential role in memory function and information processing.25,26 Under physiological conditions, activation by their endogenous ligand L-glutamate in the presence of glycine results in the influx of calcium ions. Defects within the functioning of NMDAR signalling pathways have been implicated in a multitude of CNS pathologies including PD, neuropathic pain and stroke, thus highlighting this family of receptors as a therapeutic target for symptomatic treatment of such diseases.27,28 NMDAR antagonists have previously been reported, however, have failed preclinical trials due to non-selective subunit inhibition.28
Structurally, NMDARs are hetero-oligomeric ligand-gated ion channels (LGIC) that exist as either tetrameric or pentameric complexes of two glycine-binding GluN1 subunits and two glutamate-binding GluN2 (A–D) subunits.29 Subunit selectivity of potential therapeutics is crucial as broad-range inhibition of NMDARs can lead to serious adverse effects such as hallucinations and hypertension.29 Inhibiting the GluN2B subunit selectively has been proposed to reduce the adverse effect profiles due to its expression being confined to regions of the forebrain, unlike GluN2A which is distributed throughout the brain.30
In 2003 BZAD01 (23), a benzamidine reported by Claiborne et al. was shown to be an NR2B-subtype selective NMDAR antagonist (hNr2B Ki = 47 nM, hNR2B FLIPR half maximal inhibitory concentration (IC50) = 47 nM, Fig. 5).27,31 When co-administered with Levodopa, BZAD01 demonstrated positive results in a rat model of PD. To investigate this scaffold further a library of N-benzyl benzamidines (24–28, Fig. 5) was prepared to determine the steric and electronic requirements of the GluN2B binding site. The synthesised library included a range of substituted benzene (24–26) and polycycle moieties, such as adamantane 28 (Fig. 5).26 The library was evaluated for affinity at the ifenprodil binding site of GluN2B-containing NMDARs. The binding affinity (Ki) was determined by the compound’s ability to outcompete the binding of [3H]ifenprodil (5 nM) at various concentrations.26
The introduction of polycycles reduced activity at the hNR2B receptor by several orders of magnitude.26,31 Data extracted from Moir et al.26 Competitive binding affinity measurements were performed using [3H]ifenprodil as the radioligand and rat as the source of GluN2B-containing NMDA receptors.
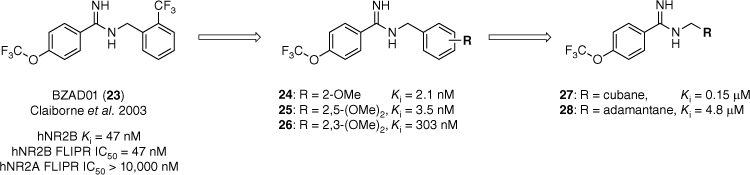
Replacement of the benzyl ring with polycycles identified that steric bulk was not tolerated in this part of the molecule, with the adamantyl analogue 27 reducing the activity by several orders of magnitude (Ki = 4.8 µM).26 Our findings also suggested a subtle electronic requirement for affinity at the GluN2B-containing NMDA receptors. This was evident by the replacement of the electron-withdrawing trifluoromethyl group of BZAD01 with an electron-donating methoxy moiety. Direct substitution saw an increase in binding affinity (24), and di-substitution was generally well tolerated around the benzene ring except at the 3-position (26).26 The synthesised library provided valuable SAR data that increased our understanding of GluN2B pharmacology.
Within this case study, substitution of the phenyl ring for adamantane was detrimental to activity. Therefore it was proposed that the GluN2B binding pocket does not favour sterically bulky functional groups and aromatic interactions may confer activity.
Tau aggregation inhibitors
Alzheimer’s disease (AD) is one of the most prevalent neurodegenerative diseases, accounting for 60–70% of all cases of dementia.32 In 2019, 55 million people worldwide were reported to be living with dementia, with this number projected to increase to 139 million by 2050.32,33 However, strategies that slow or prevent its clinical progression have largely remained elusive. Within AD and other neurodegenerative disorders, a common characteristic of the disease is the accumulation of aggregates comprised of post-translationally modified tau proteins in the cytoplasm of neurons.34,35 Many tau-centric approaches to treating AD have been proposed, including the direct inhibition of tau aggregation.36 Small molecule tau aggregation inhibitors have been the subject of many drug candidate campaigns. A high throughput screening (HTS) campaign identified aminothienopyridazines (ATPZs) as potent inhibitors of tau aggregation with favourable physiochemical properties.35,37 However, the mechanism of action employed by ATPZs and similar compounds is unknown.36
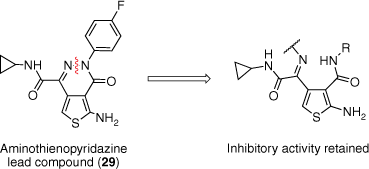
Preliminary SAR of the thienopyridazine substituents by Ballatore et al. modified the ATPZ scaffold for optimal activity and pharmacokinetic (PK) properties.37 ATPZ 29 was thereby proposed as a suitable lead compound for further structural modifications and in vivo evaluation. A compound library was thus proposed of a series of ring-opened analogues of ATPZs (Fig. 6).38 Disconnection of the nitrogen–nitrogen bond of the pyridazine ring was anticipated to produce analogues with a similar arrangement of functional groups in space and therefore have similar or improved binding to tau monomers or aggregates. These compounds would also allow for the evaluation of oxidation mechanisms in tau aggregation inhibition due to their differing redox properties to that of the lead ATPZs.37,38 Through simple modifications, this also provided us with the opportunity to improve the drug-like properties such as solubility. A range of oxime and oxime ether derivatives were also synthesised to account for the potential hydrolytic instability of the imine motif.38
Structural modifications to the lead ATPZ compound 29 were explored through the disconnection of the pyridazine nitrogen–nitrogen bond to result in the respective imine. Reprinted (adapted) with permission from Moir et al.38 Copyright 2017 The Royal Society of Chemistry.
Our group reported a novel synthetic approach that enabled the synthesis of ring-opened ATPZ analogues and easy functionalisation of the C3 position of the thiophene with aliphatic amines. This methodology provided an efficient divergent synthesis to further explore the chemical space. The library was assessed for in vitro activity by both a fluorescence thioflavin T assay and an aggregate trap assay. Disconnection of the nitrogen–nitrogen bond of the pyridazine ring was shown to not affect activity with the ring-opened oxime allyl ether analogue 30 achieving 33% inhibition in the thioflavin T assay (29: 51% inhibition).38 Aromatic amides at the 3-position of the thiophene were shown to not be necessary for inhibition of tau aggregation with the adamantane derivate 32 reporting 30% inhibition. The aggregate trap assay demonstrated that the oxime analogues 31 and 32 had a higher tendency to reduce higher-order tau aggregates compared to 29 and 30 (Fig. 7).38
Biological assessment of tau aggregation and redox properties. (a) Analogues assessed with percentage inhibition values determined from a thioflavin T assay. (b) A decrease in spot intensity is consistent with a decreased amount of fibrillar tau aggregates. Quantified from seven independent experiments (*, P < 0.05; ****, P < 0.0001). (c) Lower band intensities of IAA-OF Oregon green indicate increased oxidation. Quantified from five independent experiments and normalised to total recombinant tau proteins, stained in Coomassie blue ( *, P < 0.01; ***, P < 0.001). Reprinted (adapted) with permission from Moir et al.38 Copyright 2017 The Royal Society of Chemistry.
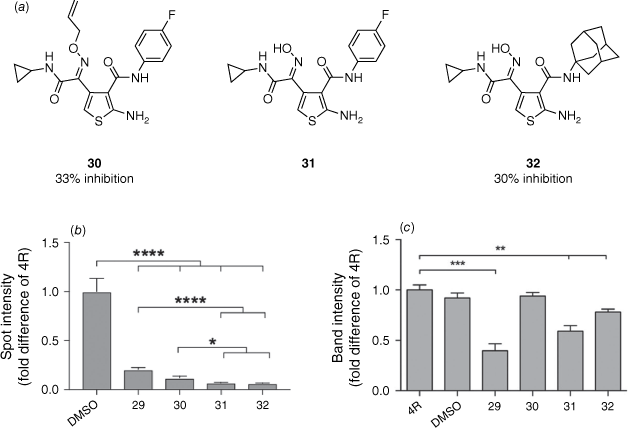
Oxidation of the cysteine residues in tau has been highlighted as a potential mechanism by which ATPZs exhibit their anti-aggregation effects.35 Therefore, each compound’s ability to oxidise tau was also assessed using Oregon green binding to non-oxidised cysteine. The oxime analogues 31 and 32 demonstrated oxidising activity, whereas 30 did not (Fig. 7). This suggested that the mechanism by which 31 and 32 inhibit tau aggregation is independent of oxidising effects.38
Therefore, we have demonstrated that the pyridazine ring of the ATPZ scaffold is not imperative for activity. Aliphatic amides were demonstrated to be well tolerated at the 3-position of the thiophene providing an excellent handle for further SAR exploration. Aromatic interactions were similarly shown not to be crucial for inhibition of tau aggregation, with the adamantane analogue demonstrating similar results to that of the 4-fluorophenyl oxime allyl ether analogue 30.38
Synthetic cannabinoids
Cannabinoid (CB) receptors are G-protein coupled receptors (GPCRs) that are categorised into two subtypes: CB1 and CB2. Responsible for the mediation of the endocannabinoid system, CB receptors play a key role in synaptic plasticity and homeostatic processes in the brain.39 Endogenously, CB receptors are activated by endocannabinoids such as anandamide and 2-arachidonoylglycerol. However, numerous exogenous ligands, both natural and synthetic, have been shown to act as CB receptor agonists.39 Activation of the CB1 receptor is responsible for the psychotropic effects associated with recreational cannabis, whereas CB2 receptor agonists have been highlighted as potential therapeutic targets in a multitude of preclinical models. Expressed throughout the CNS on immune cells such as microglia and astrocytes, activation of the CB2 receptor has been demonstrated to result in the resolution of inflammation following an assault or injury.40
Synthetic cannabinoids (SCs) are the most rapidly emerging class of designer drugs and notably have been for the past 20 years.41 In 2022–23, synthetic cannabinoid receptor agonists (SCRAs) made up 40% of new substances reported to the United Nations Office on Drugs and Crime.41 SCRAs intentionally target the endocannabinoid system, activating the CB1 receptor to mimic the intoxicating effects of known recreational drugs, namely cannabis.42 Owing to the rapid emergence of new SCs, the implementation of appropriate legal restrictions is significantly delayed because of a lack of reference materials and analytical data. The lack of data surrounding key synthetic intermediates and major metabolites also prompts significant health dangers and issues within the public health sector.
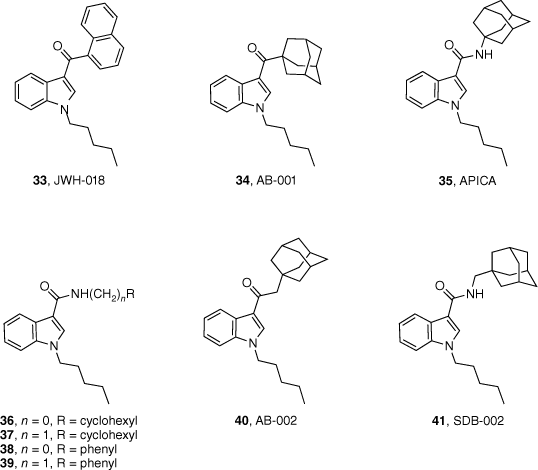
Over the past 24 years, adamantane-derived SCRAs have been consistently identified in illicit material. First detected by the European Monitoring Centre for Drugs and Drug Addiction in 2010, adamantan-1-yl(1-pentyl-1H-indol-3-yl)methanone (AB-001, 34) had no synthetic details or pharmacological evaluation in scientific literature.43 Soon after, N-(adamantan-1-yl)-1-pentyl-1H-indole-3-carboxamide (APICA, 35) was also detected. Despite their novel structures, it was suspected that AB-001 and APICA were products of clandestine rational drug design as they both exploited known SAR data for previously reported cannabimimetic indoles.
Our study in 2013 represented the first attempt to characterise and evaluate the pharmacology of two novel synthetic cannabimimetics of abuse, AB-001 (34) and APICA (35).43 The synthetic pathway proposed allowed for an efficient method to substituted analogues of the adamantyl-indole scaffold, and their corresponding intermediates for the development of reference standards of potentially emerging SCRAs. Preliminary SAR into the AB-001 and APICA chemotypes was also explored. All compounds (36–41, Fig. 8) were identified to be full agonists at CB1 and CB2 receptors in vitro (with the exception of SDB-002 41), demonstrating that structural heterogeneity is well tolerated at the 3-indole position.43 Interestingly, these results did not correlate to in vivo assays, with only APICA (35) demonstrating activity. It was therefore inferred that the nature of the functional group joining the adamantane and indole units determined whether a compound had cannabimimetic activity or not, whereas distance between the two groups was found to have no effect.
Synthetic cannabimimetics of abuse identified in seizures and preliminary SAR of the indole chemotype.
The pharmacology and toxicity of fluorinated SCRA scaffolds have been highlighted as an area of concern. Fluorinated N-pentyl indoles are susceptible to thermolytic defluorination due to the smoking route of administration and metabolic oxidative defluorination in vivo.44 Consequently, pharmacological evaluation of such compounds is imperative to assess any potential toxicity. A series of 5-fluoropentyl analogues (42, 44–46, Fig. 9) were synthesised from a series of different cannabimimetic chemotypes including APICA (35) which incorporates the adamantane motif.44 In vitro potency at the CB1 receptor was found to increase for fluorinated analogues, except for 46. Although 46 did not improve potency v. its non-fluorinated analogue 41 it did improve efficacy at both the CB1 and CB2 receptors (hCB1 maximum: 87 v. 53% respectively). Compounds 42–46 were found to dose-dependently decrease body temperature and heart rate in rats. However, this did not correlate to in vivo potency in which no obvious trends were observed among the fluorinated and non-fluorinated analogues. It was suggested that the former discrepancy may be a correlation between the compound’s PK properties, species-related differences in ligand affinity and efficacy or an error in measuring parameters such as body temperature to identify small differences.44
A sample set of the fluorinated analogues was analysed for functional activity. Fluorination generally resulted in increased potency at the hCB1 receptor. Data extracted from Shi et al.44 EC50 determined by a FLIPR membrane potential assay in AtT-20 cells expressing human CB1 or CB2 receptors.
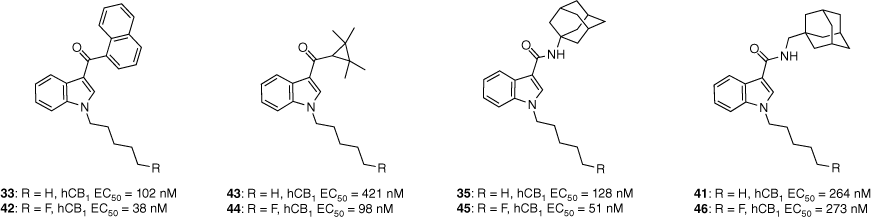
Major metabolites of the adamantane-containing APICA (35) were synthesised and pharmacologically characterised by our group in 2017.45 The adamantane motif is known to undergo oxidation during Phase 1 metabolism. Other major sites of metabolism include the N-alkyl group and carboxamide, with 1-adamantylamine identified as an APICA biomarker. All metabolites were shown to have comparable cannabimimetic activity to APICA at both CB receptors.45 This suggested that they could be contributing to the physiological effects of the leads. Selectivity for the CB2 receptor was observed for metabolites exploring oxidation on the N-pentyl chain regardless of the heterocyclic core. Oxidation of the adamantane bridgehead carbon demonstrated an increased potency at both cannabinoid receptors. To confirm whether the metabolites included in this study act synergistically with their parent compound requires further investigation into their biodistribution.
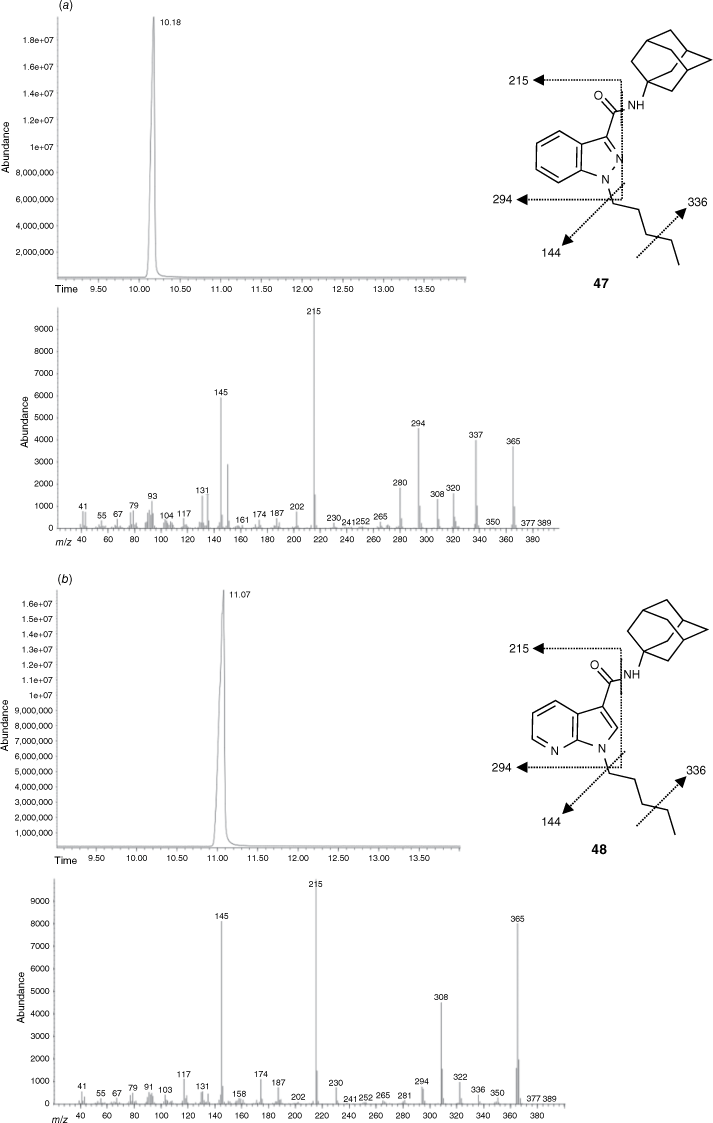
Forensic samples of street seizures have confirmed that new modifications to the SC scaffold are consistently occurring. SC scaffolds with an ester linker and azaindole motif were reported in 2013 and 2015 respectively. Owing to the isomeric relationship between azaindole and indazoles, routine methods of characterisation such as mass spectrometry were unable to distinguish between the two.46 The indazole (47) and azaindole (48) derivatives of APICA were synthesised through previously reported methods (Fig. 10). Total ion chromatograms (TICs) and electron ionisation (EI) mass spectra were reported for all compounds and intermediates synthesised. Through gas chromatography–mass spectrometry (GC-MS) analysis we were able to reliably differentiate between each compound of the carboxamide series exclusively on retention times, despite each derivative producing similar EI fragmentation patterns (Fig. 10).46 Fragmentation of the C–N amide bond and cleavage of the pentyl chain was commonly observed. Similarly, carboxylates incorporating the azaindole scaffold could be distinguished based on retention times and fragmentation ratios.46 Substantial SAR data was also elucidated within this study with several compounds having no activity at either CB receptor, demonstrating the importance of the bicyclic ring system. However, no consistent trends were observed for the change of linker within each scaffold.
GC-MS analyses showing the total ion chromatograms (TICs; time recorded in minutes, top) and electron ionisation mass spectra (EIMS, bottom) for indazole 47 (a) and azaindole 48 (b). Reprinted (adapted) from an Open-Access article distributed under the terms of the CC BY license.46
Selective agonists for the CB2 receptor are crucial to avoid the psychotropic effects associated with the activation of the CB1 receptor.47 Multiple efforts have been made to synthesise CB2 selective analogues; however, most have failed in the clinic due to species receptor differences and lack of selectivity. A study in 2017 by Shi et al. identified that for the APICA SC scaffold, CB2 selectivity could be achieved by shifting the amide at the 3-position to the 2-position of the indole.48 By incorporating an adamantyl amide at the 2-position (49) the lipophilicity was increased and only resulted in low to moderate potency. We had previously shown that the substitution of the adamantane for bulky amino acid derived amides and cumyl amides at the 3-position restores the potency and decreases the lipophilicity of the compounds.43 Within our development of an EI mass spectrometry method for the distinguishment between azaindole and indazole derivatives we also observed that the azaindole scaffold reported some CB2 selectivity.46 Combining the former SAR knowledge, a hybrid strategy (Fig. 11) was established for the development of selective CB2 agonists from non-selective synthetic cannabinoid designer drugs.47
Strategies to develop CB2 selective agonists from non-selective synthetic cannabinoids such as APICA.45,47,48 Data extracted from Moir et al.47 Half maximal effective concentration (EC50) determined by a FLIPR membrane potential assay in AtT-20 cells expressing human CB1 or CB2 receptors.
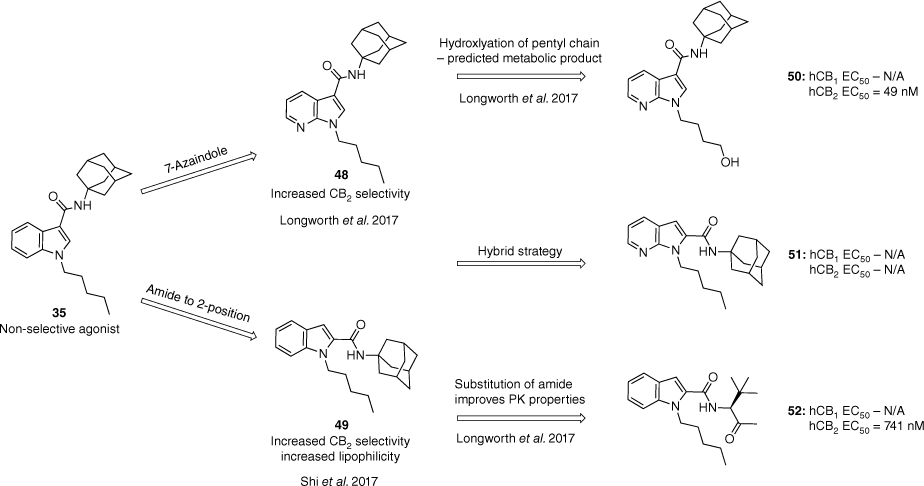
Building off the 2-amide strategy reported by Shi et al., CB1 activity was found to be suppressed within analogues incorporating the 7-azaindole core, and the addition of a polar hydroxy group at the terminal position of the pentyl chain (50).47,48 By contrast, the hybrid structure 51 lost all activity at both subtypes (Fig. 11). Substitution of the 2-amide adamantane for amino acid (52) and cumyl amides mirrored that at the 3-position with analogues retaining potency and selectivity while improving the PK properties and solubility. Polar amino acid substituents were demonstrated to disrupt CB2 activity due to unfavourable intramolecular H-bonding interactions. The following strategies were all supported through molecular modelling data.47
Adamantane-derived SCRAs are consistently identified in illicit material and thereby remain a key interest within academia. Characterising their synthetic pathway and pharmacology is imperative for both law enforcement and public safety. As demonstrated above, our group has greatly contributed to this knowledge by deducing novel synthetic pathways and analytical methods to distinguish between structural isomers of both SCs and their metabolites. The lipophilicity issues corresponding to the adamantyl group were addressed through substitution with cumyl amides. Retaining the adamantyl motif, effective modifications were also reported to confer CB2 selectivity, providing invaluable data for the development of novel CB2 agonists for the treatment of chronic pain.49
The P2X7 receptor
The P2X7 receptor (P2X7R) is a non-selective, LGIC that is upregulated on immune cells following a peripheral or central insult.50 Activated by ATP, the P2X7R mediates the processing and release of pro-inflammatory cytokines, interleukin 1β (IL-1β), and thus plays a role in a variety of cellular actions. Therapeutic intervention through targeting the P2X7R has demonstrated success in eliminating or reducing symptoms of a multitude of neuroinflammatory diseases such as AD and multiple sclerosis.50 However, despite multiple pursuits, only a few have proceeded to clinical trials with many failing due to unfavourable ADMET properties.
In 2014, our group synthesised the first molecule to possess CNS-modifying activity with a carborane motif (Fig. 12).51 Similar to that of adamantane, carboranes are bioisosteres of phenyl rings, however, their unique structure and composition present an exciting avenue for drug discovery.52 Structurally, carboranes are pseudoaromatic polyhedral clusters consisting of boron, carbon and hydrogen atoms.52 They were originally utilised within boron neutron capture therapy and can exist as a neutral, lipophilic closo cluster or the anionic, hydrophilic nido species. Within our study, the carborane motif was utilised as a bioisosteric replacement of the adamantane within the AstraZeneca benzamide P2X7 antagonist 53.51–53 Previous SAR studies of the benzamide chemotype identified that removing the three-dimensionality of the polycycle for simpler cycloalkane functional groups was detrimental to activity.53 Therefore, to explore the SAR further, a series of polycyclic cages such as cubane and trishomocubane were included in the library. In vitro activity was assessed in a functional dye uptake assay in THP-1 cells utilising BzATP (100 µM) to induce pore formation.
Exploring the effect of polycyclic cages with varying lipophilic and steric properties on P2X7R antagonism. Reprinted (adapted) with permission from Banister et al.43 Copyright 2014 American Chemical Society.
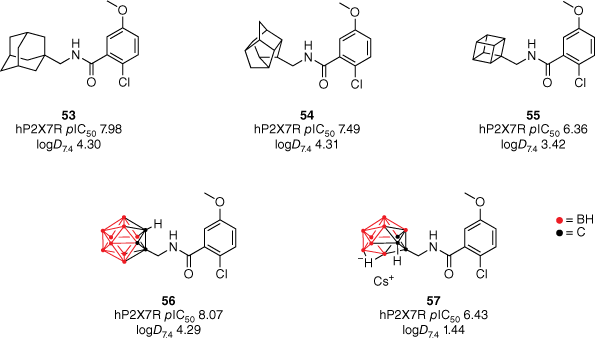
Reducing the size of the polycyclic cage reduced potency at the hP2X7R accordingly.51 The trishomocubane analogue 54 was found to reduce potency by approximately one-third (hP2X7R pIC50 7.49), whereas the cubane analogue 55 saw a decrease in over one order of magnitude (hP2X7R pIC50 6.36). Although equivalent in size, the closo-1,2-carborane cage is more lipophilic compared to adamantane due to the low polarity of the B–H bonds, whereas the nido-7,8-carboranyl 57 has significantly less lipophilic properties due to its anionic charge.54 Both analogues were shown to maintain potency, with the closo derivative 56 displaying a slight improvement compared to the adamantane lead (Fig. 12).
The analogues’ CNS activity was validated through animal behavioural tests to measure their in vivo efficacy and BBB permeability.51 A forced-swim test (FST) was implemented as a method of evaluating the antidepressant potential of the benzamide analogues. Compounds 55 and 57 were shown to have significant antidepressant activity (16 and 13% more mobile than wild-type respectively), despite being the least potent analogues in vivo.51 Compounds 53, 54 and 56 demonstrated no activity within the FST, therefore suggesting insufficient BBB permeability. This was rationalised by their experimental logD7.4 values being much greater than the range reported for successful CNS drugs (c logP < 3). Furthermore, the compounds ‘drug-likeness’ was further evaluated by determining their ligand-lipophilicity efficiency (LLE; LLE = pIC50 − logD) value.51 The nido-7,8-carborane 57 reported a LLE of 4.99, making it the most promising drug candidate of the reported benzamides (LLE ≥ 5). Despite reporting comparable in vivo and in vitro results to 57, the cubanyl 55 derivative is suspected to be less bioavailable due to its high lipophilicity and thereby is less likely to be a good drug candidate (LLE = 2.94).51
Expanding on the AstraZeneca benzamide scaffold 53, a series of hybrid structures with the reported cyanoguanidine chemotype 58 by Abbott Laboratories was synthesised and evaluated at the hP2X7R.53,55,56 This study investigated whether combining favourable motifs from two distinct compound classes would retain activity at the hP2X7R. Choosing to keep the adamantyl moiety from the AstraZeneca scaffold as a hydrophobic portion, the hybrid molecules include various substituted aryl groups connected by a cyanoguanidine linker (e.g. 59, Fig. 13).55 SAR studies on this hybrid skeleton structure have spanned multiple years and remain under investigation in our group.
Investigational SAR study of adamantyl-cyanoguanidine hybrid compounds53,55,56 first explores the linker length between the cyanoguanidine linker and the adamantyl (L1) and aryl (L2) groups.55
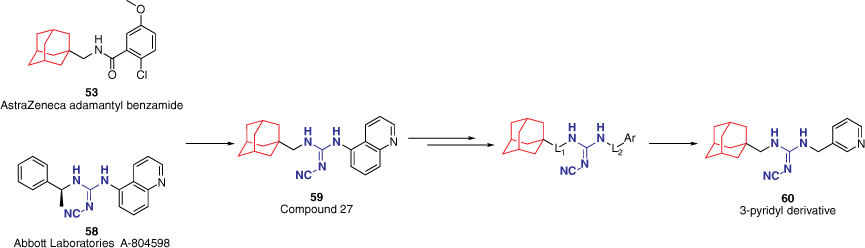
The optimal distance of the cyanoguanidine linker to the adamantyl (L1 = 0–2) and aryl (L2 = 0–2) groups was initially explored.55 Variations to L1 were not tolerated with only L1 = 1 maintaining activity, whereas L2 allowed for more variance. The effect of L2 was found to vary in conjunction with the electronics of the aryl moiety. Benzene derivatives were found to be more potent when L2 = 0, although a methylene linker was favoured for heteroaromatic analogues. This was thought to be due to the electron-deficient pyridinyl analogues being unable to effectively participate in cation–π interactions in the active site when directly attached (L2 = 0).55 This notion was further supported through the comparison of the 5-quinolinyl analogue 59 (Fig. 13) and the 3-pyridyl analogue 60 (Fig. 13). Compound 59 features the quinoline directly attached (L2 = 0), however, sufficient cation–π interactions can still occur by the phenyl portion. The drug-likeness of the analogues were assessed through a lipophilic efficiency (LiPE; LiPE = pIC50 − c logP) score. Despite having better in vitro potency, the 5-quinolinyl derivative 59 yielded a lower LiPE value to that of the 3-pyridyl derivative 60 due to its inherent higher calculated logP.55 As a direct result, 60 was deemed the more ‘drug-like’ candidate based on in silico physicochemical and PK properties and thereby utilised within in vivo studies. Compound 60 demonstrated unfavourable in vivo PK properties, however, was able to demonstrate the ability of the hybrid analogues to act centrally and produce antidepressant activity in the FST, thereby validating the hybrid approach and prompting further SAR studies on the adamantyl-cyanoguanidine core structure for the development of potent CNS-permeable P2X7R antagonists.55
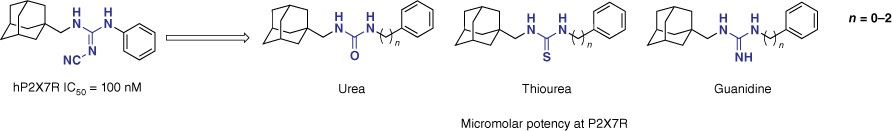
To optimise lipophilicity and thereby increase the potential for clinical translation, we next explored the cyanoguanidine linker.57 Chemical modification to the cyanoguanidine moiety was made to explore its significance. Such substitutions included the introduction of a urea, thiourea and guanidine motif (Fig. 14). A methylene group between the linker motif and the adamantyl remained constant, whereas the spacing of the aryl analogue (n) varied between 0 and 2. Of the 10 analogues synthesised, no derivative improved potency v. the lead, highlighting the importance of the cyanoguanidine moiety for potent P2X7R inhibition within this chemotype.57
Substitution of the cyanoguanidine linker to optimise lipophilicity resulted in a significant decrease in hP2X7R antagonism.
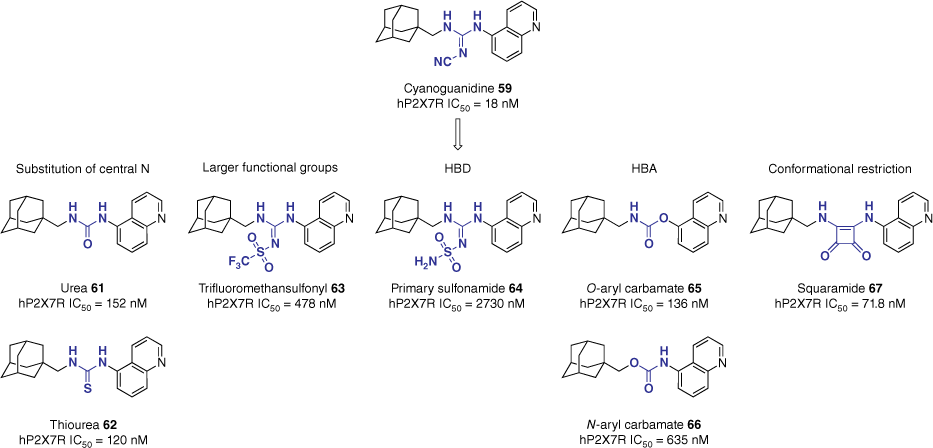
Expanding on this, key pharmacophoric features of the linker were further explored through bioisosteric replacement and systematic deconstruction of the cyanoguanidine motif.58 Retaining the methyl adamantane and the superior 5-quinoline heterocycle, 12 additional analogues were synthesised. The library explored the effects of hydrogen bond donors (HBDs) and acceptors (HBAs), tolerance of larger functional groups and conformational restriction (Fig. 15). In vitro evaluation of the compound library reported all compounds to be less potent than the lead, however, key structural information was extrapolated from the study.58 Mirroring the results of the formerly described study, the urea (61, 152 nM) and thiourea (62, 120 nM) analogues resulted in a seven-fold reduction in potency compared to the lead (59, 18 nM). Larger functional groups, such as trifluoromethanesulfonyl (63, 478 nM) demonstrated moderate potency, whereas the primary sulfonamide HBD 64 (2730 nM) was not tolerated. Interestingly, the introduction of a HBA to form the carbamate analogues 65 and 66 yielded very different results based on orientation. The O-aryl carbamate 65 (136 nM) retained potency, whereas the N-aryl carbamate 66 (635 nM) saw a significant reduction. Conformational restriction of the linker motif was explored through the non-linear squaramide functionality. The squaramide analogue 67 (71.8 nM) demonstrated a moderate increase in potency relative to the urea analogue 61, however, it still displayed a four-fold reduction compared to the lead (59).58
SAR evaluation of the linker motif. Data extracted from Garrett et al.58 IC50 assessed by inhibition of hP2X7R-mediated pore formation using the YO-PRO-1 dye uptake assay in human THP-1.
Despite the rigorous efforts made to discover potent and selective P2X7R antagonists, many fail to proceed to clinical translation due to poor ADMET properties. Extensive SAR studies exploring the chemical space of the AstraZeneca benzamide scaffold have continuously highlighted the importance of the adamantyl moiety on activity.53,55 However, the compounds have high lipophilicity and poor metabolic stability as a result. Through a systematic approach, our group explored two common bioisosteric strategies. Firstly, we replaced the benzene ring with heteroaromatic motifs. Substituting for azabenzenes and diazenes successfully reduced the lipophilicity, however, also significantly reduced the potency at the hP2X7R.59 The bridgehead carbons of the adamantane cage are known to be prone to oxidation. Introducing carbon–fluorine bonds is a common approach to overcome this in drug discovery. The highly electronegative fluorine atom forms a strong, polar covalent bond with carbon that reduces lipophilicity and blocks metabolically labile sites. The trifluoroadamantane derivative 68 was well tolerated with only a small loss of activity, yet significantly improved physicochemical and PK properties compared to the lead benzamide (Table 1).59 For example, it was identified to have a half-life six times longer than that of the lead (53) in rat liver microsomes and was effective across several isoforms of the hP2X7R. These data identified 68 as a potential preclinical ligand to validate models of CNS disorders and as a suitable PET radiotracer.
| Pharmacokinetic properties |  | 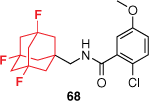 | |
|---|---|---|---|
| logD7.4 | 4.23 | 2.83 | |
| hP2X7R IC50 (nM) | 10.5 | 33.9 | |
| LLE | 3.75 | 4.64 | |
| rLiver t1/2 (min) | 7.8 | 46.8 | |
| rBrain t1/2 (min) | 9.6 | 75.6 | |
| rPlasma t1/2 (min) | 28.2 | 103 | |
| CI (mL min−1 kg−1) | 86.90 | 41.81 | |
| CYP1A2 (%) | 25.7 | 5.48 | |
| CYP2C9 (%) | 75.0 | 9.30 | |
| CYP2C19 (%) | 84.4 | 12.9 | |
| CYP2D6 (%) | −47.3 | −29.1 | |
| CYP3A4 (%) | 84.4 | 89.9 |
logD7.4 data were determined experimentally by HPLC. hP2X7R IC50 values were the mean values (n > 4) ± standard deviation derived from a dye uptake assay with THP-1 cells. LLE = pIC50 – logD, where pIC50 was from dye uptake and logD was from HPLC. Reprinted (adapted) with permission from Wilkinson et al.59 Copyright 2017 American Chemical Society.
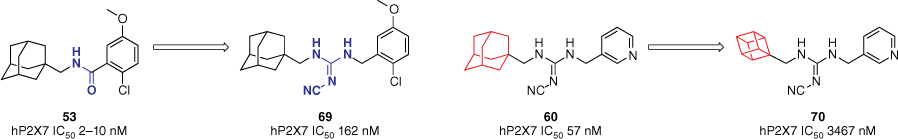
Combining the formerly mentioned SAR data of the hybrid scaffold and AstraZeneca’s benzamide, the role of polycycles and the cyanoguanidine linker was explored by incorporating them with other known P2X7R antagonists.60 For example, the original AstraZeneca adamantyl benzamide 53 was modified to include a cyanoguanidine linker. Aiming to improve the lipophilicity of these compounds, different polycycles were introduced. Substituting the amide for a cyanoguanidine linker in the benzamide scaffold to give 69 resulted in a decrease in potency (162 v. 2–10 nM for the 2-chloro-5-methoxy analogue, Fig. 16) across the board.60 Reducing the polycycle size significantly reduced activity (3467 v. 57 nM, Fig. 16), mirroring observations from our previous study.60 This further confirms the important role adamantane plays in the functional activity of P2X7R inhibitors.
Series of hybrid structures to confirm key SAR data. Adamantane was confirmed to be an important motif within P2X7R antagonists. Data extracted from Callis et al.60 Values refer to pore-formation inhibition and were obtained from a fluorescent dye uptake assay in THP-1 cells.
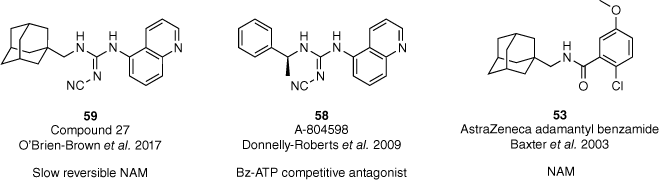
Despite the extensive research into P2X7R antagonists, a severe lack of mechanistic pharmacological characterisation has been noted.61 Therefore, the suitability of previously discussed antagonists as clinical tools or drug candidates has been restricted. In 2022, our group determined the mechanism of action of our lead compound 59, and other previously reported antagonists (53 and 58, Fig. 17).53,55,56,62 The adamantyl-cyanoguanidine 59 was determined to be a slowly reversible negative allosteric modulator (NAM) of the hP2X7R highlighting its potential for use in in vitro and in vivo studies.62 Furthermore, our characterisation also identified that the adamantane moiety promotes binding in the allosteric site of the hP2X7R.
Pharmacological evaluation of our lead compound and reported P2X7 antagonists to determine the mechanism of action.53,55,56
The adamantyl-cyanoguanidine scaffold 59, initiated from our exploratory hybrid approach, resulted in a largely successful novel P2X7R antagonist chemotype.55 Since its conceptualisation our group has conducted extensive SAR studies on the aryl and linker motifs.55,57,58 The adamantane functional group has been a consistent feature in new analogues and has thus been identified as a privileged structure for the P2X7R. Metabolic soft spots, such as the bridgehead carbons, have been shown to be easily resolved through a hydrogen-to-fluorine bioisosteric replacement.59 Our studies have also shown adamantane’s superiority over other polycycles such as cubane, within multiple P2X7R antagonist chemotypes.60
Summary and future directions
The adamantane motif and its analogues continue to be useful tools in drug discovery campaigns to alter PK properties and exploit precise substitution orientation to explore three-dimensionality. Since 2011, our group has published and patented work surrounding the synthesis and validation of adamantyl-containing compounds. We have demonstrated the effects of adamantane within multiple SAR studies on unique targets and validated these in vivo. Most notably, we have confirmed adamantane to be a privileged scaffold of the P2X7R and supplied a multitude of reference materials and data to assist in SC identification by law enforcement. Current work within the group continues to include adamantane as a key benchmark of steric allowance and tolerance of hydrophobic motifs. However, a key gap in our current utility of adamantane is exploiting the three-dimensionality of the scaffold beyond the bridgehead carbons. Utilising synthetic strategies, that are becoming readily available, to access more diverse substitution patterns will provide us with further avenues to explore the potential of this moiety in CNS drug discovery.
Data availability
Data sharing is not applicable as no new data were generated or analysed during this study.
Declaration of funding
The work presented herein was supported by the Australian Research Council and the National Health and Medical Research Council.
References
1 Landa S, Macháček V. Sur l’adamantane, nouvel hydrocarbure extrait du naphte. Collect Czechoslov Chem Commun 1933; 5: 1-5 [In French].
| Crossref | Google Scholar |
3 Prelog V, Seiwerth R. Über die Synthese des Adamantans. Ber Dtsch Chem Ges 1941; 74(10): 1644-1648 [In German].
| Crossref | Google Scholar |
4 von R, Schleyer P. A simple preparation of adamantane. J Am Chem Soc 1957; 79(12): 3292.
| Crossref | Google Scholar |
5 Wanka L, Iqbal K, Schreiner PR. The lipophilic bullet hits the targets: medicinal chemistry of adamantane derivatives. Chem Rev 2013; 113(5): 3516-3604.
| Crossref | Google Scholar | PubMed |
6 Jackson GG, Muldoon RL, Akers LW. Serological evidence for prevention of influenzal infection in volunteers by an anti-influenzal drug adamantanamine hydrochloride. Antimicrob Agents Chemother 1963; 161: 703-707.
| Google Scholar | PubMed |
7 Liu J, Obando D, Liao V, Lifa T, Codd R. The many faces of the adamantyl group in drug design. Eur J Med Chem 2011; 46(6): 1949-1963.
| Crossref | Google Scholar | PubMed |
8 Lamoureux G, Artavia G. Use of the adamantane structure in medicinal chemistry. Curr Med Chem 2010; 17(26): 2967-2978.
| Crossref | Google Scholar | PubMed |
9 Gryn’ova G, Corminboeuf C. Steric “attraction”: not be dispersion alone. Beilstein J Org Chem 2018; 14: 1482-1490.
| Crossref | Google Scholar | PubMed |
10 Rummel L, Schreiner PR. Advances and prospects in understanding london dispersion interactions in molecular chemistry. Angew Chem Int Ed 2024; 63(12): e202316364.
| Crossref | Google Scholar | PubMed |
11 Tse EG, Houston SD, Williams CM, Savage GP, Rendina LM, Hallyburton I, et al. Nonclassical phenyl bioisosteres as effective replacements in a series of novel open-source antimalarials. J Med Chem 2020; 63(20): 11585-11601.
| Crossref | Google Scholar | PubMed |
12 Ragshaniya A, Kumar V, Tittal RK, Lal K. Nascent pharmacological advancement in adamantane derivatives. Arch Pharm 2024; 357(3): 2300595.
| Crossref | Google Scholar | PubMed |
13 Pergolizzi J, Varrassi G, Coleman M, Breve F, Christo DK, Christo PJ, et al. The sigma enigma: a narrative review of sigma receptors. Cureus 2023; 15(3): e35756.
| Crossref | Google Scholar | PubMed |
14 Pergolizzi Jr J, Varrassi G. The emerging role of sigma receptors in pain medicine. Cureus 2023; 15(7): e42626.
| Crossref | Google Scholar | PubMed |
15 Piechal A, Jakimiuk A, Mirowska-Guzel D. Sigma receptors and neurological disorders. Pharmacol Rep 2021; 73(6): 1582-1594.
| Crossref | Google Scholar | PubMed |
16 Peeters M, Romieu P, Maurice T, Su TP, Maloteaux JM, Hermans E. Involvement of the sigma 1 receptor in the modulation of dopaminergic transmission by amantadine. Eur J Neurosci 2004; 19(8): 2212-2220.
| Crossref | Google Scholar | PubMed |
17 Kassiou M, Nguyen VH, Knott R, Christie MJ, Hambley TW. Trishomocubanes, a new class of selective and high affinity ligands for the sigma binding site. Bioorg Med Chem Lett 1996; 6(6): 595-600.
| Crossref | Google Scholar |
18 Nguyen VH, Kassiou M, Johnston GAR, Christie MJ. Comparison of binding parameters of σ1 and σ2 binding sites in rat and guinea pig brain membranes: novel subtype-selective trishomocubanes. Eur J Pharmacol 1996; 311(2): 233-240.
| Crossref | Google Scholar |
19 Liu X, Kassiou M, Christie MJ. Synthesis and binding studies of trishomocubanes: novel ligands for σ binding sites. Aust J Chem 1999; 52(7): 653-656.
| Crossref | Google Scholar |
20 Liu X, Kassiou M, Christie MJ, Hambley TW. Trishomocubanes: requirements for σ receptor binding and subtype selectivity. Aust J Chem 2001; 54(1): 31-36.
| Crossref | Google Scholar |
21 Banister SD, Moussa IA, Jordan MJT, Coster MJ, Kassiou M. Oxo-bridged isomers of aza-trishomocubane sigma (σ) receptor ligands: synthesis, in vitro binding, and molecular modeling. Bioorg Med Chem Lett 2010; 20(1): 145-148.
| Crossref | Google Scholar | PubMed |
22 Glennon RA, Ablordeppey SY, Ismaiel AM, El-Ashmawy MB, Fischer JB, Howie KB. Structural features important for sigma1 receptor binding. J Med Chem 1994; 37(8): 1214-1219.
| Crossref | Google Scholar | PubMed |
23 Banister SD, Yoo DT, Chua SW, Cui J, Mach RH, Kassiou M. N-Arylalkyl-2-azaadamantanes as cage-expanded polycarbocyclic sigma (σ) receptor ligands. Bioorg Med Chem Lett 2011; 21(18): 5289-5292.
| Crossref | Google Scholar | PubMed |
24 Banister SD, Rendina LM, Kassiou M. 7-Azabicyclo[2.2.1]heptane as a scaffold for the development of selective sigma-2 (σ2) receptor ligands. Bioorg Med Chem Lett 2012; 22(12): 4059-4063.
| Crossref | Google Scholar | PubMed |
25 Beinat C, Banister S, Moussa I, Reynolds AJ, McErlean CSP, Kassiou M. Insights into structure–activity relationships and CNS therapeutic applications of NR2B selective antagonists. Curr Med Chem 2010; 17(34): 4166-4190.
| Crossref | Google Scholar | PubMed |
26 Beinat C, Banister SD, Hoban J, Tsanaktsidis J, Metaxas A, Windhorst AD, et al. Structure–activity relationships of N-substituted 4-(trifluoromethoxy)benzamidines with affinity for GluN2B-containing NMDA receptors. Bioorg Med Chem Lett 2014; 24(3): 828-830.
| Crossref | Google Scholar | PubMed |
27 Warraich ST, Allbutt HN, Billing R, Radford J, Coster MJ, Kassiou M, et al. Evaluation of behavioural effects of a selective NMDA NR1A/2B receptor antagonist in the unilateral 6-OHDA lesion rat model. Brain Res Bull 2009; 78(2): 85-90.
| Crossref | Google Scholar | PubMed |
28 Truong L, Allbutt HN, Coster MJ, Kassiou M, Henderson JM. Behavioural effects of a selective NMDA NR1A/2B receptor antagonist in rats with unilateral 6-OHDA + parafascicular lesions. Brain Res Bull 2009; 78(2): 91-96.
| Crossref | Google Scholar | PubMed |
29 Liu W, Jiang X, Zu Y, Yang Y, Liu Y, Sun X, et al. A comprehensive description of GluN2B-selective N-methyl-D-aspartate (NMDA) receptor antagonists. Eur J Med Chem 2020; 200: 112447.
| Crossref | Google Scholar |
30 Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 1994; 12(3): 529-540.
| Crossref | Google Scholar | PubMed |
31 Claiborne CF, McCauley JA, Libby BE, Curtis NR, Diggle HJ, Kulagowski JJ, et al. Orally efficacious NR2B-selective NMDA receptor antagonists. Bioorg Med Chem Lett 2003; 13(4): 697-700.
| Crossref | Google Scholar | PubMed |
32 2024 Alzheimer’s disease facts and figures. Alzheimers Dement 2024; 20(5): 3708-3821.
| Crossref | Google Scholar | PubMed |
34 Burns S, Selman A, Sehar U, Rawat P, Reddy AP, Reddy PH. Therapeutics of Alzheimer’s disease: recent developments. Antioxidants 2022; 11(12): 2402.
| Crossref | Google Scholar | PubMed |
35 Crowe A, Huang W, Ballatore C, Johnson RL, Hogan AML, Huang R, et al. Identification of aminothienopyridazine inhibitors of tau assembly by quantitative high-throughput screening. Biochemistry 2009; 48(32): 7732-7745.
| Crossref | Google Scholar | PubMed |
36 Self WK, Holtzman DM. Emerging diagnostics and therapeutics for Alzheimer disease. Nat Med 2023; 29(9): 2187-2199.
| Crossref | Google Scholar | PubMed |
37 Ballatore C, Crowe A, Piscitelli F, James M, Lou K, Rossidivito G, et al. Aminothienopyridazine inhibitors of tau aggregation: evaluation of structure–activity relationship leads to selection of candidates with desirable in vivo properties. Bioorg Med Chem 2012; 20(14): 4451-4461.
| Crossref | Google Scholar | PubMed |
38 Moir M, Chua SW, Reekie T, Martin AD, Ittner A, Ittner LM, et al. Ring-opened aminothienopyridazines as novel tau aggregation inhibitors. MedChemComm 2017; 8(6): 1275-1282.
| Crossref | Google Scholar | PubMed |
39 Kendall DA, Yudowski GA. Cannabinoid receptors in the central nervous system: their signaling and roles in disease. Front Cell Neurosci 2017; 10: 294.
| Crossref | Google Scholar | PubMed |
40 Mackie K. Cannabinoid receptors: where they are and what they do. J Neuroendocrinol 2008; 20: 10-14.
| Crossref | Google Scholar | PubMed |
41 United Nations Office on Drugs and Crime. Current NPS Threats Volume VII July 2024. Vienna, Austria: United Nations; 2024. Available at https://www.unodc.org/documents/scientific/Current_NPS_threats_VII.pdf
42 Banister SD, Connor M The chemistry and pharmacology of synthetic cannabinoid receptor agonist new psychoactive substances: evolution. In: Maurer HH, Brandt SD, editors. New Psychoactive Substances: Pharmacology, Clinical, Forensic and Analytical Toxicology. Cham, Switzerland: Springer International Publishing; 2018. pp. 191–226. 10.1007/164_2018_144
43 Banister SD, Wilkinson SM, Longworth M, Stuart J, Apetz N, English K, et al. The synthesis and pharmacological evaluation of adamantane-derived indoles: cannabimimetic drugs of abuse. ACS Chem Neurosci 2013; 4(7): 1081-1092.
| Crossref | Google Scholar | PubMed |
44 Banister SD, Stuart J, Kevin RC, Edington A, Longworth M, Wilkinson SM, et al. Effects of bioisosteric fluorine in synthetic cannabinoid designer drugs JWH-018, AM-2201, UR-144, XLR-11, PB-22, 5F-PB-22, APICA, and STS-135. ACS Chem Neurosci 2015; 6(8): 1445-1458.
| Crossref | Google Scholar | PubMed |
45 Longworth M, Connor M, Banister SD, Kassiou M. Synthesis and pharmacological profiling of the metabolites of synthetic cannabinoid drugs APICA, STS-135, ADB-PINACA, and 5F-ADB-PINACA. ACS Chem Neurosci 2017; 8(8): 1673-1680.
| Crossref | Google Scholar | PubMed |
46 Longworth M, Reekie TA, Blakey K, Boyd R, Connor M, Kassiou M. New-generation azaindole-adamantyl-derived synthetic cannabinoids. Forensic Toxicol 2019; 37(2): 350-365.
| Crossref | Google Scholar |
47 Moir M, Lane S, Lai F, Connor M, Hibbs DE, Kassiou M. Strategies to develop selective CB2 receptor agonists from indole carboxamide synthetic cannabinoids. Eur J Med Chem 2019; 180: 291-309.
| Crossref | Google Scholar | PubMed |
48 Shi Y, Duan YH, Ji YY, Wang ZL, Wu YR, Gunosewoyo H, et al. Amidoalkylindoles as potent and selective cannabinoid type 2 receptor agonists with in vivo efficacy in a mouse model of multiple sclerosis. J Med Chem 2017; 60(16): 7067-7083.
| Crossref | Google Scholar | PubMed |
49 Han S, Thatte J, Buzard DJ, Jones RM. Therapeutic utility of cannabinoid receptor type 2 (CB2) selective agonists. J Med Chem 2013; 56(21): 8224-8256.
| Crossref | Google Scholar | PubMed |
50 Bartlett R, Stokes L, Sluyter R. The P2X7 receptor channel: recent developments and the use of P2X7 antagonists in models of disease. Pharmacol Rev 2014; 66(3): 638-675.
| Crossref | Google Scholar | PubMed |
51 Wilkinson SM, Gunosewoyo H, Barron ML, Boucher A, McDonnell M, Turner P, et al. The first CNS-active carborane: a novel P2X7 receptor antagonist with antidepressant activity. ACS Chem Neurosci 2014; 5(5): 335-339.
| Crossref | Google Scholar | PubMed |
52 Issa F, Kassiou M, Rendina LM. Boron in drug discovery: carboranes as unique pharmacophores in biologically active compounds. Chem Rev 2011; 111(9): 5701-5722.
| Crossref | Google Scholar | PubMed |
53 Baxter A, Bent J, Bowers K, Braddock M, Brough S, Fagura M, et al. Hit-to-lead studies: the discovery of potent adamantane amide P2X7 receptor antagonists. Bioorg Med Chem Lett 2003; 13(22): 4047-4050.
| Crossref | Google Scholar | PubMed |
54 Lesnikowski ZJ. Boron units as pharmacophores – new applications and opportunities of boron cluster chemistry. Collect Czech Chem Commun 2007; 72(12): 1646-1658.
| Crossref | Google Scholar |
55 O’Brien-Brown J, Jackson A, Reekie TA, Barron ML, Werry EL, Schiavini P, et al. Discovery and pharmacological evaluation of a novel series of adamantyl cyanoguanidines as P2X7 receptor antagonists. Eur J Med Chem 2017; 130: 433-439.
| Crossref | Google Scholar | PubMed |
56 Donnelly-Roberts DL, Namovic MT, Surber B, Vaidyanathan SX, Perez-Medrano A, Wang Y, et al. [3H]A-804598 ([3H]2-cyano-1-[(1S)-1-phenylethyl]-3-quinolin-5-ylguanidine) is a novel, potent, and selective antagonist radioligand for P2X7 receptors. Neuropharmacol 2009; 56(1): 223-229.
| Crossref | Google Scholar | PubMed |
57 Wong ECN, Reekie TA, Werry EL, O’Brien-Brown J, Bowyer SL, Kassiou M. Pharmacological evaluation of a novel series of urea, thiourea and guanidine derivatives as P2X7 receptor antagonists. Bioorg Med Chem Lett 2017; 27(11): 2439-2442.
| Crossref | Google Scholar | PubMed |
58 Garrett T, Gilchrist J, McKenzie A, Larik FA, Danon J, Werry E, et al. An investigation on linker modifications of cyanoguanidine-based P2X7 receptor antagonists. ChemMedChem 2024; e202400163.
| Crossref | Google Scholar | PubMed |
59 Wilkinson SM, Barron ML, O’Brien-Brown J, Janssen B, Stokes L, Werry EL, et al. Pharmacological evaluation of novel bioisosteres of an adamantanyl benzamide P2X7 receptor antagonist. ACS Chem Neurosci 2017; 8(11): 2374-2380.
| Crossref | Google Scholar | PubMed |
60 Callis TB, Reekie TA, O’Brien-Brown J, Wong ECN, Werry EL, Elias N, et al. The role of polycyclic frameworks in modulating P2X7 receptor function. Tetrahedron 2018; 74(12): 1207-1219.
| Crossref | Google Scholar |
61 Michel AD, Chambers LJ, Walter DS. Negative and positive allosteric modulators of the P2X7 receptor. Br J Pharmacol 2008; 153(4): 737-750.
| Crossref | Google Scholar | PubMed |
62 Jackson A, Werry EL, O’Brien-Brown J, Schiavini P, Wilkinson S, Wong ECN, et al. Pharmacological characterization of a structural hybrid P2X7R antagonist using ATP and LL-37. Eur J Pharmacol 2022; 914: 174667.
| Crossref | Google Scholar | PubMed |