

Evaluation of a Continuous-Flow Photo-Bromination Using N-Bromosuccinimide for Use in Chemical Manufacture
Matthew Waterford A B , Simon Saubern A and Christian H. Hornung A
A and Christian H. Hornung A
A CSIRO Manufacturing, Bag 10, Clayton South, Vic. 3169, Australia.
B Corresponding author. Email: matthew.waterford@csiro.au
Australian Journal of Chemistry 74(8) 569-573 https://doi.org/10.1071/CH20372
Submitted: 21 December 2020 Accepted: 22 March 2021 Published: 9 April 2021
Journal Compilation © CSIRO 2021 Open Access CC BY
Abstract
A continuous-flow photo-bromination reaction on benzyl and phenyl groups was conducted using N-bromosuccinimide as the bromine source inside a preparatory-scale glass plate reactor. This flow reactor system was capable of independently controlling light intensity, wavelength, and reaction temperature, hence exerting an exceptional level of control over the reaction. A short optimisation study for the synthesis of 2-bromomethyl-4-trifluoromethoxyphenylboronic acid pinacol ester resulted in best conditions of 20°C and 10 min residence time using an LED (light-emitting diode) array at 405 nm and acetonitrile as the solvent. The present study evaluates the potential for this easy-to-handle bromination system to be scaled up for chemical manufacture inside a continuous-flow glass plate reactor. The combination with an in-line continuous flow liquid–liquid extraction and separation system, using a membrane separator, demonstrates the potential for continuous flow reaction with purification in an integrated multi-stage operation with minimal manual handling in between.
Keywords: continuous flow chemistry, photochemistry, photo-bromination, microreactor, in-line separation, photoactivation.
Introduction
The scale-up of photochemical processes for industry is an underdeveloped field that offers promising opportunities to perform chemical reactions in a green and efficient manner. Traditionally, batch processes have been utilised for photochemistry using mercury or xenon lamps, which are very inefficient, consume high amounts of energy, and can cause undesired side reactions occurring from broad-wavelength irradiation. Batch processes also suffer from inefficient photo-harvesting as governed by the Beer–Lambert Law, which states that the transmission of light decreases with the path length through the vessel, leading to lower reaction conversions.[1] These shortcomings have not been looked on favourably in the past when considering the scale-up of photochemical processes.[2]
Aryl bromides have a very broad and widespread use in the synthesis of fine chemicals. They have applications as intermediates in cross-couplings[3] and halogen displacement reactions.[4] In addition, they are used in the synthesis of antibacterial, antifungal, and antiviral agents.[5]
Recent interest in brominations under continuous-flow conditions including Cantillo et al.[6] has prompted us to investigate one of the earliest bromination reactions. The Wohl–Ziegler bromination was the first reported general method for the direct introduction of bromine using N-bromosuccinimide (NBS) in 1919.[7] This reagent has found widespread use as a brominating agent owing to it being much less hazardous to use than molecular bromine, stable, and practical.[8]
A recent paper by Cantillo et al. investigated this reaction using a compact fluorescent lamp (CFL) as the light source.[6] However, improvements on efficiencies can be made, as a paper from Bonfield et al. notes that the maximum absorption of NBS is 400 nm,[9] which should allow the use of a lower-energy LED (light-emitting diode) light with a more selective wavelength. Also, the paper notes that only 40 % maximum LED light intensity was required for optimal results.
Another limitation of the Wohl–Ziegler bromination was the use of CCl4 as the solvent. This solvent has since been banned for industrial scale under the Montreal Protocol and acetonitrile has since been widely adopted as the solvent of choice for this reaction owing to the high solubility and stability of NBS and absence of a significant exotherm.[10]
The bromination of electron-rich arenes was chosen as a model to demonstrate the scale-up of a photochemical process utilising flow chemistry on a meso-scale, without the need to use harsh reaction conditions necessary for electron-poor systems.[11]
The Corning G1 photoreactor[12] is a versatile glass-plate-type continuous-flow reactor that overcomes many of the limitations when scaling up photochemistry and herein we aimed to test its capabilities, as well as the development of a continuous in-line work-up of the reaction mixture via the use of a Zaiput PTFE membrane liquid–liquid separator.[13]
Results and Discussion
The Corning G1 photoreactor is a continuous-flow photoreactor utilising an array of LEDs of different wavelengths that can be matched to the UV/VIS absorbance of the substrate for maximum efficiency. Previous analysis of NBS by UV-vis absorption spectroscopy found λmax is ~400 nm[6] and therefore a wavelength of 405 nm was chosen to be most suitable for this reaction. The temperature, residence time, solvent, and wavelength conditions were screened to determine the best reaction conditions and the benzylic bromination of the boronic acid 1 was selected as a model substrate. The results of these conditions can be found in Table 1. Benzylic brominations usually require an initiator such as AIBN (azobisisobutyronitrile), which forms a toxic by-product (tetramethylsuccinonitrile) so a photo-initiated approach that eliminates this reagent is desirable in an industrial setting.

|
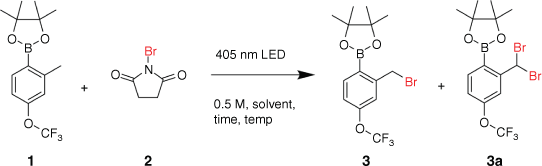
The initial screening of the reaction conditions in acetonitrile found promising results when a residence time of 30 min was chosen (entry 1) and 93 % conversion to 3 was observed. In subsequent experiments, the reaction time was decreased to 15 min (entry 2), which gave the same result; however with a 5 min residence time (entry 3), a decrease in conversion was then found at 67 %. For this set of conditions, the optimal residence time was found to be 10 min (entry 4) as conversion plateaued at 93 %, similarly to entries 1 and 2.
Changing the wavelength by switching to either the 365- or 470-nm LEDs (entries 5 and 6) produced a decrease in conversion compared with the same conditions at 405 nm. This result aligns with the expectation that 405 nm would be the most efficient wavelength due to the λmax of NBS being ~400 nm.[9] An increase in the amount of NBS in the feed (1.5 equiv., entry 7) showed that the excess bromine source was causing an increase of dibromination as selectivity for 3 decreased to 92 %.
The solvent was then switched to acetone (entries 8–11) and this had the effect of slowing reaction kinetics, possibly owing to the somewhat lower solubility of NBS in acetone; however, selectivity was improved slightly to 99 %.
Next, we wanted to try and develop a continuous in-line workup procedure to complement the flow bromination reaction, and for this purpose a Zaiput membrane-based liquid–liquid separator was used. This separator contains a PTFE hydrophobic membrane and exploits the difference in surface tension between organic and aqueous phases to separate them into separate streams.[7]
The outlet stream of the Corning G1 photoreactor was connected to a four-way junction where two separate pumps of toluene and water were pumped simultaneously with the reaction mixture into a 10-mL Vapourtec static mixer coil,[14] then fed into the Zaiput membrane separator. As the reaction mixture is in acetonitrile, which is miscible with water, an immiscible solvent is required to facilitate liquid–liquid separation. Toluene was chosen for this purpose; this reaction stream now becomes a ternary mixture of three solvents, where the product and starting material migrate to the toluene layer, while the by-product of the reaction being succinimide migrates into the aqueous layer (Fig. 1).

|
After processing through the Zaiput membrane, the organic process stream was then collected and analysed. As this is a complex ternary mixture, it was found that a single pass through the Zaiput membrane separator was not enough for all of the succinimide to migrate to the aqueous stream and a further two passes were necessary to ensure all the succinimide was removed (Fig. 2). This result aligns with the results of ternary mixtures used by another group who performed extraction efficiency experiments on toluene/acetone/water and ethyl acetate/acetic acid/water systems.[8]
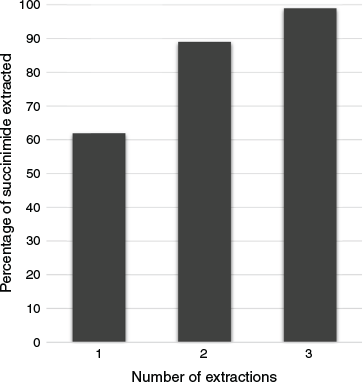
|
This is a limitation of using a ternary solvent system for workup, and a fully continuous process would require a complex multi-stage separator set-up. Indeed, recognising the limitations of a single-pass membrane separation, Zaiput have made available pre-assembled trains of separators for more challenging purification needs.[15] Ideally, a binary solvent system would solve this issue as doing so has resulted in much-increased extraction efficiency with only a single pass.[16] However, as acetonitrile is the most suitable solvent for this reaction and it is miscible with water, the use of multi-stage extraction can only be avoided by a solvent swap, i.e. acetonitrile would be removed before the product mixture was redissolved in an appropriate ratio of toluene and water and then fed into the liquid–liquid separation system. As this is difficult to achieve in a fully continuous fashion, we have opted for the less efficient ternary system for this limited flow study; for scale-up, a different approach would be advisable to increase overall process efficiency, taking the considerations outlined above into account.
Such methods have been presented in the literature and applied to flow bromination. For example, the bromination of a precursor to Rosuvastatin when performed on an industrial scale utilised the much simpler method of addition of water to the reaction mixture, causing the precipitation of the product, which was subsequently filtered and recrystallised to obtain the pure compound.[9]
Another alternative in-line workup purification approach presented in the literature is the use of a gravity-based computer-vision controlled system, which has also been successfully used in the removal of succinimide from a bromination reaction.[10]
The space–time yield, STY, can be derived as follows:
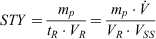
where mp is product mass (kg); 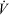 , total volumetric flow rate (L h−1); VR, reactor volume (L); VSS, total stock solution volume processed (L); and tR, total processing time (h). Here, the total processing time tR can be expressed as:
, total volumetric flow rate (L h−1); VR, reactor volume (L); VSS, total stock solution volume processed (L); and tR, total processing time (h). Here, the total processing time tR can be expressed as: 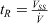 .
.
The reaction scope was expanded to demonstrate the general applicability of the bromination procedure and several electron-rich substrates were successfully brominated (see Table 2). All of the reactions were carried out on a 10 mmol scale in acetonitrile and very good conversions and selectivities were obtained. Examples 4, 6, 7, and 8 could be synthesised in quantitative yields at very short residence times of only 3 or 4 min, resulting in extremely high space–time yields of up to 2.3 kg L−1 h−1, especially when compared with classical batch processes. Recent work has shown that in the dark and the absence of any other catalyst, these reactions proceed ‘sluggishly’ (24 h).[17] When a substrate with multiple electron-rich sites is used, site selectivity can be decreased as each site competes for the bromine atom; hence, the substrate needs to be chosen carefully as this is difficult to overcome and site selectivity cannot be controlled.

|
The large parameter space that can be scanned with this reactor system provides a series of options to optimise reaction conditions for the desired outcome. With a separate heating and cooling cycle regulating the temperature of the reaction chamber, which was not affected by the heat output of the light sources, it was possible to decouple and independently control light intensity and reaction temperature. In combination with choosing the wavelength of the light source from a set of six different LED arrays, this set-up was capable of finely controlling the main reaction parameters, which increased the operational space accessible to optimise the reaction systems shown in Table 2. Compared with the majority of batch photo reactors, this is an enormous improvement, allowing access to control over parameter space not previously feasible. While small-scale laboratory batch systems may also be reasonably efficient in controlling temperature and light flux, this very quickly changes when scaling up to batch reaction vessels. The limited light penetration depth renders illumination of larger batch vessels highly inefficient, and precise temperature control at large batch scale also becomes a major concern and is often not achievable. In contrast, both light harvesting and temperature control are extremely efficient in flow reactor channels at small and large scale, as demonstrated with the reactor system presented herein.
Conclusions
We have demonstrated a simple continuous-flow photo-bromination inside a highly efficient preparatory-scale glass plate reactor. Using NBS as the bromine source avoids handling of highly hazardous bromine species used in many other direct brominations; acetonitrile and acetone proved to be useful solvents for this system, avoiding the formation of precipitates inside the continuous flow reactor. Using a membrane separator, it was possible to combine the photo-bromination reactor with in-line liquid–liquid extraction in an integrated multi-stage operation, with minimal manual handling between steps. However, the ternary solvent system used here required at least three membrane separation units for optimal recovery of the product. The use of a binary toluene/water system would be more productive, but in the absence of an efficient solvent swap system that could remove the acetonitrile before separation, a multiple-membrane separator train is necessary for a fully continuous flow system.
While for most batch reactions photoinitiation is not an option for production scale, and often a thermal initiation route needs to be chosen, we believe that flow photochemistry will provide the chemical manufacturing industry with the option to consider light as an alternative.
Experimental
Reagents and solvents were used as purchased. NMR spectra were recorded on a Bruker Avance 400 spectrometer operating at 400 MHz. Chemical shifts are expressed on the δ scale (ppm). Multiplicities are indicated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad.
The reactions were conducted in a standard Corning Advanced-Flow G1 photoreactor equipped with five glass fluidic modules connected in series (8.2 mL volume each). Each module was illuminated from both sides by a multiple-wavelength LED panel (>100 mW cm−2). The chosen wavelength was 405 nm, and the intensity was adjusted to 100 %. The temperature of the fluidic modules was regulated by a Lauda Integral XT 150 thermostat using silicone oil as the thermofluidic fluid (transparent at 405 nm), and the LED panels were cooled by a Lauda Microcool MC 250 thermostat using ethylene glycol as the thermofluidic fluid.
General Procedure for Continuous-Flow Bromination
NBS (1.87 g, 10.50 mmol, 1.05 equiv.), HPLC-grade acetonitrile, and the corresponding substrate (10 mmol) were placed into a flask and stirred until completely homogeneous. The solution was then pumped through the Corning G1 photoreactor at the desired flow rate. The reaction mixture was collected from the reactor and combined with the two separate streams of toluene and water into a 10-mL Vapourtec static mixer coil; then, the mixture was pumped into a Zaiput liquid–liquid membrane separator, the organic phase was collected and passed twice more through the Zaiput membrane separator. The organic phase was evaporated under reduced pressure, providing the corresponding brominated product.
2-Bromomethyl-4-trifluoromethoxyphenylboronic Acid, Pinacol Ester (3)
δH (400 MHz, CDCl3) 1.38 (s, 12H), 4.89 (s, 2H), 7.12 (d, 1H), 7.24 (s, 1H), 7.88 (d, 1H).
1-Bromo-4-methoxybenzene (4)[8]
δH (400 MHz, CDCl3) 3.77 (s, 3H), 6.79 (dd, 2H), 7.38 (dd, 2H). δC (100 MHz, CDCl3) 55.4, 112.8, 115.7, 132.2, 158.7.
5-Bromo-1,3-benzodioxole (5)[18]
δH (400 MHz, CDCl3) 5.95 (s, 2H), 6.67 (d, 1H), 6.94 (d, 2H). δC (100 MHz, CDCl3) 101.6, 109.6, 112.3, 113.0, 124.3, 147.0, 148.6.
4-Bromoacetanilide (6)[19]
δH (400 MHz, [D6]DMSO) 2.05 (s, 3H), 7.47 (d, 2H), 7.55 (d, 2H), 10.06 (s, 1H). δC (100 MHz, DMSO-d6) 24.5, 114.9, 121.3, 131.9, 139.12, 168.9.
1-Bromo-2-naphthol (7)[20]
δH (400 MHz, CDCl3) 5.94 (s, 1H), 7.28 (d, 1H), 7.40 (t, 1H), 7.58 (t, 1H), 7.76 (q, 2H), 8.04 (d, 1H). δC (100 MHz, CDCl3) 106.15, 117.16, 124.15, 125.33, 127.85, 128.22, 129.34, 129.69, 132.29, 150.58.
1-Bromo-4-methoxynaphthalene (8)[8]
δH (400 MHz, CDCl3) 3.98 (s, 3H), 6.66 (d, 1H), 7.56 – 7.60 (m, 1H), 7.64 – 7.70 (m, 1H), 8.24 (dd, 1H), 8.34 (dd, 1H). δC (100 MHz, CDCl3): δ 55.69, 104.54, 113.28, 122.51, 126.00, 126.89, 127.82, 129.53, 132.45, 155.26.
Supplementary Material
Supplementary material including 1H and 13C NMR spectra is available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Declaration of Funding
This research did not receive any specific funding.
References
[1] D. F. Swinehart, J. Chem. Educ. 1962, 39, 333.| Crossref | GoogleScholarGoogle Scholar |
[2] J. P. Knowles, L. D. Elliott, K. I. Booker-Milburn, Beilstein J. Org. Chem. 2012, 8, 2025.
| Crossref | GoogleScholarGoogle Scholar | 23209538PubMed |
[3] N Miyaura, A Suzuki, Chem. Rev. 1995, 95, 2457.
| Crossref | GoogleScholarGoogle Scholar |
[4] R. Cramer, D. R. Coulson, J. Org. Chem. 1975, 40, 2267.
| Crossref | GoogleScholarGoogle Scholar |
[5] R. Hosseinzadeh, M. Tajbakhsh, M. Mohadjerani, Z. Lasemi, Synth. Commun. 2010, 40, 868.
| Crossref | GoogleScholarGoogle Scholar |
[6] D. Cantillo, O. de Frutos, J. A. Rincon, C. Mateos, C. O. Kappe, J. Org. Chem. 2014, 79, 223.
| Crossref | GoogleScholarGoogle Scholar | 24261546PubMed |
[7] A. Wohl, Ber. Dtsch. Chem. Ges. B 1919, 52, 51.
| Crossref | GoogleScholarGoogle Scholar |
[8] M. C. Carreno, J. L. Garcia Ruano, G. Sanz, M. A. Toledo, A. Urbano, J. Org. Chem. 1995, 60, 5328.
| Crossref | GoogleScholarGoogle Scholar |
[9] H. E. Bonfield, J. D. Williams, W. X. Ooi, S. G. Leach, W. J. Kerr, L. J. Edwards, ChemPhotoChem 2018, 2, 938.
| Crossref | GoogleScholarGoogle Scholar |
[10] S. Shimizu, Y. Imamura, T. Ueki, Org. Process Res. Dev. 2014, 18, 354.
| Crossref | GoogleScholarGoogle Scholar |
[11] K. Rajesh, M. Somasundaram, R. Saiganesh, K. K. Balasubramanian, J. Org. Chem. 2007, 72, 5867.
| Crossref | GoogleScholarGoogle Scholar | 17590047PubMed |
[12] Corning, Advanced-FlowTM Reactors (AFR). Available at: https://www.corning.com/au/en/innovation/corning-emerging-innovations/advanced-flow-reactors.html (accessed 7 July 2020).
[13] A. Adamo, P. L. Heider, N. Weeranoppanant, K. F. Jensen, Ind. Eng. Chem. Res. 2013, 52, 10802.
| Crossref | GoogleScholarGoogle Scholar |
[14] Vapourtec, Large-Diameter Tubular Reactor for Rapid Mixing. Available at: https://www.vapourtec.com/products/flow-reactors/large-diameter-tubular-reactor-for-rapid-mixing-features/ (accessed 14 July 2020)
[15] Zaiput Flow Technologies, Multistage Extraction. Available at: https://www.zaiput.com/product/multistage-extraction/ (accessed 27 July 2020)
[16] J. Britton, T. F. Jamison, Nat. Protoc. 2017, 12, 2423.
| Crossref | GoogleScholarGoogle Scholar | 29072707PubMed |
[17] X. He, X. Wang, Y.-L. S. Tse, Z. Ke, Y.-Y. Yeung, Angew. Chem. Int. Ed. 2018, 57, 12869.
| Crossref | GoogleScholarGoogle Scholar |
[18] D. C. Braddock, G. Cansell, S. A. Hermitage, Synlett 2004, 461.
| Crossref | GoogleScholarGoogle Scholar |
[19] M. B. Smith, L. Guo, S Okeyo, J Stenzel, J Yanella, E LaChapelle, Org. Lett. 2002, 4, 2321.
| Crossref | GoogleScholarGoogle Scholar | 12098237PubMed |
[20] S. Adimurthy, G. Ramachandraiah, A. V. Bedekar, S. Ghosh, B. C. Ranu, P. K. Ghosh, Green Chem. 2006, 8, 916.
| Crossref | GoogleScholarGoogle Scholar |