

In Situ MOF-Templating of Rh Nanocatalysts under Reducing Conditions
Renata Lippi A B E , Campbell J. Coghlan A , Shaun C. Howard B , Christopher D. Easton B , Qinfen Gu C , Jim Patel D , Christopher J. Sumby A , Danielle F. Kennedy B and Christian J. Doonan A
A B E , Campbell J. Coghlan A , Shaun C. Howard B , Christopher D. Easton B , Qinfen Gu C , Jim Patel D , Christopher J. Sumby A , Danielle F. Kennedy B and Christian J. Doonan A
A Centre for Advanced Nanomaterials, Department of Chemistry, The University of Adelaide, Adelaide, SA 5005, Australia.
B CSIRO Manufacturing, Clayton, Vic. 3168, Australia.
C Australian Synchrotron (ANSTO), Clayton, Vic. 3168, Australia.
D CSIRO Energy, Clayton, Vic. 3168, Australia.
E Corresponding author. Email: renata.lippi@csiro.au
Australian Journal of Chemistry 73(12) 1271-1283 https://doi.org/10.1071/CH20193
Submitted: 12 June 2020 Accepted: 18 September 2020 Published: 3 November 2020
Journal Compilation © CSIRO 2020 Open Access CC BY-NC-ND
Abstract
Manganese-based metal–organic frameworks (MOFs) metalated with Rh were used as pre-catalysts for CO2 hydrogenation. Activated in situ (80 % H2, 20 % CO2, 350°C), the resulting templated catalysts displayed CO2 conversion of up to 20 %, with CH4 as the main product. Used catalysts were compared with samples templated in 5 % H2/Ar at 350°C using powder X-ray diffraction, electron microscopy, energy dispersive spectroscopy, and X-ray photoelectron spectroscopy. It was found that under reducing atmosphere Rh0 nanoparticles formed and organic MOF components decomposed, which allowed growth of MnO or MnCO3 and the formation of a mesh of catalytic Rh0 nanoparticles.
Introduction
Heterogeneous catalysts are widely applied in chemical industry, in part due to their facile separation and robust performance under severe conditions. However, understanding the catalytic reaction mechanisms can be difficult due to the challenges associated with the identification of catalytically active sites and reaction intermediates.[1] Active sites typically compose a small fraction of the overall surface area of a catalyst, thus catalyst optimisation aims to increase the surface density of these sites. Synthesis methods that provide control over chemical composition and structure may facilitate i) the identification of active sites and ii) an increase in the surface density of active sites. Metal nanocatalysts are synthesised via a variety of physicochemical methods designed to exert control over the cluster size, morphology, and available surface area.[2] However, there remains significant opportunity to develop new synthetic approaches for the preparation of catalytic materials with improved composition and morphology control to produce longer lasting and higher performing catalysts.[2a] Recently, metal–organic framework (MOF)-mediated synthesis has been demonstrated as a potential new route for the preparation of efficient catalysts.
MOFs are a class of porous materials comprised of inorganic nodes connected via organic linkers.[3] The large library of organic linkers and metal nodes available affords a vast number of possible structures.[4] Furthermore, the properties of MOFs can be tailored by the rational selection of these components. In addition, the functionality of synthesised MOF materials can be tuned by post synthetic modification techniques such as, covalent attachment, linker/cation exchange, or addition of a secondary metal.[5] A significant body of research has focussed on the gas capture,[6] separation,[7] and catalytic properties of MOFs.[8] Indeed, a recent development in the area of MOF chemistry has been to use their porous networks as self-sacrificing templates[9] to yield uniquely structured materials for electrochemical[9a] and catalytic[9c,9f] applications. Among the highly active MOF-templated catalysts reported are composites of metal, metal oxides, and porous carbon.[10] In these examples, the high activity of the MOF-derived catalysts has been attributed to factors, such as precise control of secondary metal dispersion in the derived catalyst,[10a] nanoparticle size control,[10b] and good nanoparticle dispersion on support.[10c] The control provided by MOF-templating is proposed to result from the uniform spacing of inorganic and organic subunits in the template.[9g] The underlying mechanism is not well understood, but insights into the mechanism may lead to catalyst synthesis optimisation.[11] Normally, the parent MOF is treated at high temperature in an inert atmosphere (pyrolysis) or in air (calcination) to yield the templated inorganic material.[9b,9d,9h] In general, pyrolysis of a MOF produces metal or metal oxide nanoparticles embedded in carbon, whereas calcination yields metal oxide nanoparticles alone. Of the few examples where MOF templating was carried out under a reducing atmosphere, H2, the synthesis of metallic nanoparticles has been reported.[12] Metallic nanoparticles are employed in several catalytic reactions, such as oxidations, hydrogenations, and C–C coupling, which can benefit from the control of nanoparticle dispersion, size, and morphology.[2a,2c,2d] This led us to investigate the effect of reducing conditions for the synthesis of metallic nanocatalysts via MOF templating.
Rhodium metal nanoparticles can be used as catalysts for both oxidation[13] and hydrogenation reactions,[14] including CO2 hydrogenation.[14d,15] Strategies to synthesise Rh0 nanoparticles of controlled size include the use of solid supports, surfactants or polymeric stabilisers.[14c,16] Previous reports of MOF-templated Rh materials are limited to two: Rh/CeO2 and Rh/C, obtained by laser processing[17] and by pyrolysis followed by acid etching[18] respectively.
Here, we explore the use of a Mn-based MOF, which can be post-synthetically metalated with Rh at precisely defined sites, as a template for a nanostructured Rh catalyst. The Rh-metalated MOF was thermally treated under CO2 hydrogenation conditions (80 % H2/CO2 at 350°C) to produce the MOF-templated Rh nanocatalyst ‘in situ’. We use X-ray diffraction, electron microscopy, energy dispersive spectroscopy, and X-ray photoelectron spectroscopy to provide insight into the templating mechanism of this novel material and compare the structure to control samples. Our data suggests that the presence of Rh in the MOF facilitated the decomposition of the organic linkers and, thus afforded the removal of carbon and nitrogen under reducing conditions. This resulted in crystals of MnO or MnCO3 distributed in a mesh of Rh0 nanoparticles of 6 to 9 nm in size.
Results and Discussion
The synthesis MnMOF (1) and its post synthetic metalation (PSM) with [Rh(CO)2Cl]2 to yield 1·[Rh(CO)2][RhCl2(CO)2] (1·Rh2) were performed as described in our previous work.[19] The inorganic nodes of 1 are composed of Mn trimers, which are coordinated by both the carboxylate and di-pyrazole moieties of L (Fig. 1). Due to the stoichiometry of the metal node one-third of all the organic linkers (L) in 1 presented free di-pyrazole moieties (Fig. 1). These free sites are available to chelate transition metals via PSM. In the case of PSM with [Rh(CO)2Cl]2 (1·Rh2), single crystal diffraction indicated occupancy of these sites greater than 90 %.[19a] In this report, the replacement of the cis-[RhCl2(CO)2]– anion by BF4– to yield 1·[Rh(CO)2]BF4 (1·Rh-BF4) was carried out to decrease the loading of RhI in the framework to provide a single RhI atom per available chelating site, as opposed to two found in to 1·Rh2. Evaluation by IR spectroscopy indicated the successful displacement of the counter-ion cis-[RhCl2(CO)2]– in 1·Rh-BF4 (Fig. S2, Supplementary Material). Fig. 1 provides a graphical representation of the structure of the MOFs tested in this work: 1, 1·Rh2, and 1·Rh-BF4. The resulting overall loading of Rh in the MOFs is ~11 and 6 wt-% for 1·Rh2 and 1·Rh-BF4, respectively.

|
We recently reported the MOF-templating of a Ru catalyst for CO2 methanation, which presented controlled nanoparticle size and distribution over the support.[20] Both Rh and Ru are known to promote high activity in CO2 hydrogenation reactions, and we hypothesised that MOF-templating could also afford the synthesis of Rh nanoparticles of controlled size and high activity.[2b,14d,15b,20] In this report, 1 is selected as the template due to the homogeneous distribution of Rh that can be achieved through metalation of the chelating sites. Accordingly, 1, 1·Rh2, and 1·Rh-BF4 were examined as precursors for producing CO2 hydrogenation catalysts. Each of the MOFs was transformed into an active catalyst under CO2 hydrogenation reaction conditions; 350°C and 4 bar under a flow of 80 % H2 and 20 % CO2 at 350°C and 4 bar (i.e. CO2 hydrogenation reaction conditions, Fig. S5, Supplementary Material). Therefore, activated samples of 1 (1-used), 1·Rh2 (1·Rh2-used), and 1·Rh-BF4 (1·Rh-BF4-used) were tested as in situ activated catalysts continuously over a 90 h period. For each sample, gradual changes in catalytic performance were observed over the first 25 h. The two Rh containing samples initially displayed a maximum production of CH4, 18 mmol min−1 gMOF−1 (4 h) and 16 mmol min−1 gMOF−1 (8 h) for 1·Rh2-used and 1·Rh-BF4-used, respectively, each of which decreased thereafter (Fig. 2). The CH4 production was accompanied by the consumption of H2 and CO2 (Fig. 2), which is strong evidence that methane was a product of CO2 hydrogenation and not a result of the decomposition of the organic linkers. Compound 1 and silicon carbide (SiC) were also tested as controls and displayed negligible activity. The significantly lower CH4 production, indicates that 1-used was inactive for the CO2 hydrogenation and that Rh is a necessary component of the active catalyst.

|
After activation and an initial deactivation period (Fig. 2, condition A), the catalysts displayed stable conversions for the duration of the experiment, 90 h (Fig. 2). The Rh-containing samples displayed higher CH4 production, however, 1·Rh2-used produced approximately double the amount of CH4 per gram of MOF than 1·Rh-BF4-used (Fig. S6, Supplementary Material), this difference can be correlated with Rh loading of the precatalysts, 11 and 6 wt-% respectively. Different product (CH4, CO, C3H8, C3H6) selectivity between 1·Rh2-used and 1·Rh-BF4-used (Fig. S7, Supplementary Material) suggest that the differences between the MOF-derived catalysts was not only dependent upon the amount of Rh but is also possibly related to other phases formed during the templating process.
The presence of different crystalline phases in each sample was determined by Rietveld-based quantitative phase analysis[21] on powder X-ray diffraction (PXRD) data of the used catalysts (Fig. 3 and Table 1). The remaining material was examined by X-ray photoelectron spectroscopy (XPS) and scanning electron microscopy/energy dispersive spectroscopy (SEM/EDS). The catalyst 1·Rh-BF4-used displayed a complex composition, containing cubic Rh0, trigonal MnCO3, Na salts, and at least one unidentified phase with a large unit cell as observed by the difference curve. The presence of Na salts is explained by the poor solubility of NaCl in acetonitrile. This results in the incomplete removal of NaCl, a by-product from the reaction of NaBF4 with the Rh dimer within the framework. A simpler composition was observed for 1·Rh2-used, with cubic Rh0 and cubic MnO in the used catalyst. SiC was also present in the diffractogram, as it was mixed with the catalyst as diluent for the performance testing and was a remnant in the isolated sample. It is evident that the composition of the precatalyst affects the final catalyst composition and, therefore, its catalytic properties.
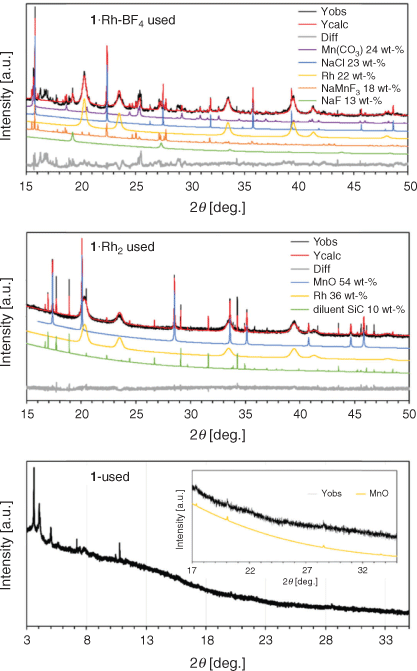
|

|
The diffractogram of the control sample, 1-used, showed low angle reflections suggestive of the planes (0 0 1) and (1 1 0) of the pristine MOF even after prolonged time on stream. However, these reflections in 1-used had broadened substantially and displayed decreased intensity consistent with a significant loss of crystallinity. In addition, low intensity reflections of MnO were identified in the wide-angle region (Fig. 3). This indicates that in the absence of Rh, the MOF undergoes partial decomposition under reaction conditions, forming a relatively small amount of MnO crystals, as observed from the low intensity peaks.
The difference in Rh loading of the two template MOFs was reflected in Rietveld-based quantitative phase analysis (QPA). Rh0 amounts for the derived catalysts 1·Rh2-used and 1·Rh-BF4-used were determined to be 40 and 22 wt-% respectively (excluding diluent SiC). The broad peaks for Rh0 reflections is a result of the small crystallite sizes in this phase, which are on average 10 nm, as measured via transmission electron microscopy (TEM). Volume weighted column height or apparent crystallite size (Lvol-IB) was also calculated via Rietveld refinement.[22] The MOF templating route yielded Rh nanoparticles of comparable size with an apparent size of 6 and 8 nm for 1·Rh2-used and 1·Rh-BF4-used, respectively. For both MOF-templated catalysts, Rh0 nanoparticles were substantially smaller than the other particles. For example, the apparent size of MnO in 1·Rh2-used, was 90 nm. For 1·Rh-BF4-used, the apparent size of MnCO3 was 32 nm and the Na salts were in the range of 20 to 200 nm (Table 1). The control 1-used presented MnO crystals with apparent size of 64 nm; these crystals were smaller than the crystals generated in the Rh containing sample, 1·Rh2.
Most MOF-templated materials are synthesised via calcination in air or under an inert atmosphere at temperatures ranging from 300 to 1000°C. The temperature of treatment is selected to exceed the thermal stability of the MOF, obtained via thermogravimetric analysis (TGA) in an inert atmosphere or in air.[9b–d,9h] The template MOFs 1, 1·Rh2, and 1·Rh-BF4 displayed thermogravimetric stability up to 400°C in N2 (Fig. S3, Supplementary Material). In this study the MOF-templating step occurs at a temperature below the thermal stability in N2 (350°C), however the presence of H2 (80 % H2 and 20 % CO2) can affect the MOF stability. Accordingly, to evaluate the effect of the atmosphere composition on the templating of the catalyst, samples of 1 and 1·Rh-BF4 were treated ex situ at 350°C under an inert (N2) and reducing atmosphere (5 % H2 in N2). The resulting samples (with added labels ‘N2’ or ‘5 %H2’) were characterised and are compared with the MOF-derived catalysts below.
Pyrolysis of MOFs 1 and 1·Rh-BF4 produced amorphous samples (Fig. 4); NaCl was present in the metalated MOF as noted above and was not formed during pyrolysis. In contrast, under a mildly reducing atmosphere (5 % H2), the resulting samples (1-5 %H2 and 1·Rh-BF4-5 %H2) displayed low angle peaks in agreement with the planes (0 0 1) and (1 1 0) of 1. This indicates that the samples heat-treated in 5 % H2 retained some structural features of the original MOF structure. No new phase was formed in 1-5 %H2, whereas Rh0 nanoparticles were formed in 1·Rh-BF4-5 %H2. Aside from Rh0 nanoparticles, no other phases previously observed for 1·Rh-BF4-used were present. These nanoparticles had the same phase, cubic-Rh0, as the ones formed during catalysis testing (1·Rh2-used and 1·Rh-BF4-used) but were smaller in apparent size (Lvol-IB = 5.3 nm). As Rh0 nanoparticles were not produced via pyrolysis, these samples were not further analysed.
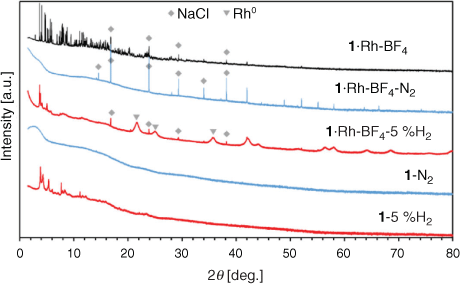
|
In situ MOF-templating of the catalyst was investigated in temperature and atmosphere controlled PXRD experiments. In these experiments, the phase transitions of 1 and 1·Rh-BF4 were observed while subjecting these samples to simulated CO2 hydrogenation reaction conditions: drying in 100 % Ar flow from room temperature to 200°C; followed by a flow of 75 % H2 and 25 % CO2 and temperature ramp to 350°C (Fig. 5). The crystalline structure of 1·Rh-BF4 was stable during the drying step, and abruptly collapsed on exposure to a H2-containing gas mixture at 200°C. At 350°C, increased intensity was observed in the region corresponding to Rh0 reflections revealing the formation of the Rh nanoparticles (Fig. S9, Supplementary Material). The sharp peak around 14° 2θ was identified as a reflection from NaCl, a by-product of the PSM step. Following the collapse of the MOF, this NaCl reflection displayed increasing intensity due to further growth of these crystals. Upon reaching 300°C, the intensity of NaCl reflections start to decrease, possibly due to formation of NaMnF3 and NaF, phases observed in 1·Rh-BF4-used. Other phases present in 1·Rh-BF4-used were not observed, thus indicating that they may only form after a prolonged exposure (10–25 h) to reactive gases (time restrictions when using the synchrotron source prevented further examination). The crystallinity of the control sample 1 was retained throughout the extended period of exposure to Ar and CO2 hydrogenation conditions (H2 + CO2), indicating that the metalation of the template MOF was required for the fast collapse of the structure under catalytic conditions.

|
The morphology of the MOF-derived samples produced after catalysis testing (used samples) and after tube furnace reduction (5 % H2) was studied by TEM (Fig. 6), SEM, and EDS (Fig. 7). The catalyst 1·Rh-BF4-used consisted of a mesh of Rh nanoparticles in the range of 6 to 15 nm (histogram in Fig. S10, Supplementary Material) among larger crystals of varying sizes ranging from 30 to 300 nm. Interestingly, at lower magnification it was possible to observe that these crystalline composite meshes retained the shape of the much larger original plate shaped monoclinic MOF crystals, another sign that the MOF served as a sacrificial template.[20] EDS mapping confirmed that the nanoparticles were composed of Rh and that the larger crystals were in agreement with the phases observed in the diffractogram, i.e. MnCO3 and Na salts.

|

|
The sample 1·Rh-BF4-5 %H2 had smaller Rh nanoparticles, from 3 to 12 nm (histogram in Fig. S10, Supplementary Material), embedded in larger lower electron density porous particles, which we hypothesise is a porous C doped with N amorphous matrix. EDS mapping confirmed that the high dispersion of Rh exists in the carbon-rich phase. This reduced sample, 1·Rh-BF4-5 %H2, displays characteristics of an intermediate material between the template MOF 1·Rh-BF4 and the activated catalyst 1·Rh-BF4-used, where the Rh nanoparticles have not reached their final form, the MOF linkers are not fully decomposed and Mn crystalline phases, e.g. MnO and MnCO3, are absent (Fig. 8).
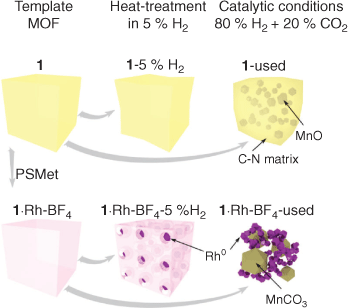
|
As for the non-metalated MOF (1), small medium electron density crystals embedded in N-doped carbon were present in 1-used (Figs 6 and 8). The crystals had been identified in PXRD as MnO derived from the aggregation of the Mn trimers in 1 (Fig. 3). Lastly, 1-5 %H2 consisted of low electron density micrometric particles, and no crystals or nanoparticles embedded within those particles were observed. This was in agreement with PXRD analysis that showed a partial loss of crystallinity, but no other crystalline phase formed.
The surface of the template MOFs (1 and 1·Rh-BF4), the reduced samples, and the used catalysts (Fig. 8) were analysed by XPS. First, the elements on the surface are quantified and discussed, followed by a detailed analysis of the assigned components to peak fitting of Mn 2p, Rh 3d, and N 1s high resolution spectra. The six samples are compared to investigate whether the reduced sample is a possible intermediate state of the MOF-templating process and to better understand the mechanism of this transformation.
Surface elemental quantification by XPS for the template MOFs and MOF-derived samples from reduction in 5 % H2 and used for CO2 hydrogenation catalysis testing are presented in Table 2. For the metalated MOF 1·Rh-BF4, a loss of 14 at.- % of carbon from its surface was observed after catalysis testing, indicating that the used catalyst still presents a significant amount of adventitious carbon. In contrast, N contained in the parent MOF linkers is completely absent in the used catalyst, evidence of the complete decomposition of the organic linkers. As a result of the linker decomposition, other elements which were in the interior of the MOF become exposed and display higher relative amounts, e.g. Mn.

|
The sample 1·Rh-BF4-5 %H2 derived from heat treatment of 1·Rh-BF4 in 5 % H2 displayed a lower amount of exposed Rh (1.2 at.- %) than 1·Rh-BF4-used (6.4 at.- %) but a higher amount than its precursor 1·Rh-BF4 (0.78 at.- %), suggesting an intermediate state between the MOF as-synthesised and the used catalyst. The compositions of C, N, and O remain relatively unchanged when comparing 1·Rh-BF4 and 1·Rh-BF4-5 %H2 suggesting that the synthesis of Rh nanoparticles occurs as a first step, which then assists in the catalytic decomposition of the organic linkers. Analysis of the bare MOF supports this theory as there is only a small difference in the element composition among 1, and the samples derived from reduction (1-5 %H2) and catalysis treatment (1-used). Nitrogen quantification was 9.9, 9.7, and 7.3 at.- % for 1, 1-5 %H2, and 1-used, respectively and even after 90 h on stream only 26 % of the N is lost in the absence of Rh. Mn quantification indicates an increase in surface Mn, possibly due to the formation of MnO crystals close to the surface.
Analysis of the high resolution XPS spectra clarified the state of the surface elements of the MOFs (1 and 1·Rh-BF4) at different stages of decomposition (as-synthesised, reduced in 5 % H2, and used). Starting with Mn, a strong satellite peak for Mn 2p relative to the main peak was observed for both 1 and 1·Rh-BF4 (Fig. 9). Resolved satellite peaks on the high binding energy (BE) side of the Mn 2p3/2 peak is consistent with MnO.[23] However, the overall peak shape and the intensity of the satellite peak relative to the main peak differs from reference Mn oxide spectra. The distinctive peak shape is attributed to the unique local environment of the Mn in the MOF framework, where the Mn trimer is coordinated by O and N from the organic linkers. Satellite peaks for Mn oxides have been reported to arise from shake-up processes[23a] that occur as a result of photoelectrons that lose energy through the promotion of valence electrons from an occupied state to a higher unoccupied level.[24] It is reasonable to assume that in the case of 1 and 1·Rh-BF4 this shake-up results in unusually intense satellite peaks due to the coordination of the Mn node by the chelating di-pyrazole moiety. Therefore, the presence of the intense shake-up for Mn 2p is indicative of the coordination of the inorganic node by the organic linkers and provides additional evidence for the presence of the MOF structure. After reduction of the MOFs, a minor increase in the intensity of the satellite peak for 1-5 %H2 and 1·Rh-BF4-5 %H2 was observed when comparing to the samples before reduction. The relative fraction of intensity for the satellite peak for each spectra was determined using component fitting, with the ratio of this peak for 1-5 %H2 (with respect to 1) at 1.04, and for 1·Rh-BF4-5 %H2 (with respect to 1·Rh-BF4) at 1.10. This minor increase can be attributed to the removal of adventitious carbon after the reduction treatment. Thus, despite the partial loss of crystallinity observed with PXRD, the inorganic nodes are still likely coordinated by the organic linkers in a disordered structure. In contrast, after catalysis testing, the satellite peak intensity for the Mn 2p peak is reduced for 1·Rh-BF4-used, due to the formation of MnCO3, with an intensity ratio of 0.71. Furthermore, the lack of a N signal for 1·Rh-BF4-used indicates the complete removal of the di-pyrazole moiety from the surface. The satellite peak is reduced further for 1-used, with a relative intensity ratio of 0.39 (with respect to 1), and whose spectroscopic envelope suggests contributions from MnII (MnO based on component Mn7) and MnIII (e.g. MnO(OH) based on relative fraction of the lower BE components).[23d] These results support the hypothesis that long-term testing of 1 for CO2 hydrogenation caused the formation of MnO on the surface and loss of the Mn trimer present in the original MOF.
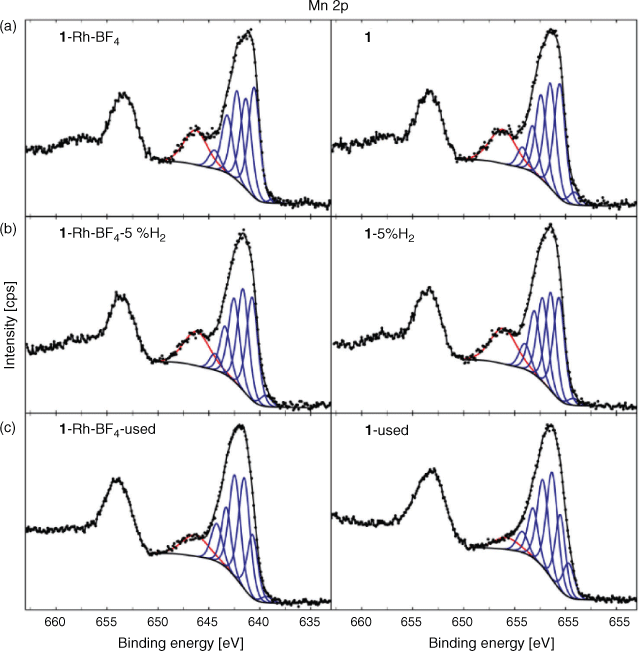
|
For the Rh containing samples, the high-resolution Rh 3d spectra of the parent sample 1·Rh-BF4 and its derivatives, 1·Rh-BF4-5 %H2 and 1·Rh-BF4-used, could be fitted using three doublets. The low BE doublet (red) is associated with Rh0, the middle (blue) doublet is assigned to both RhI and RhIII as Rh2O3, and the high BE doublet is assigned to RhIII. The spectrum for 1·Rh-BF4 is dominated by the high BE doublet, the presence of RhIII may be explained by the oxidation of Rh atoms bound to the linker after long-term storage in air, in agreement with previous observations for this MOF system.[19a] 1·Rh-BF4-used contains a significant fraction of Rh0 based on the component fitting of the Rh 3d peak (Fig. 10), whereas the sample reduced in a tube furnace, 1·Rh-BF4-5 %H2, can be fitted with three doublets of roughly equal proportions, suggesting the presence of Rh0, remnant linker-bound RhIII, as well as RhI and RhIII as Rh2O3 species, which are less evident in the parent sample. Furthermore, 1·Rh-BF4-5 %H2 may be interpreted as an intermediate material between the parent MOF and the used catalyst.
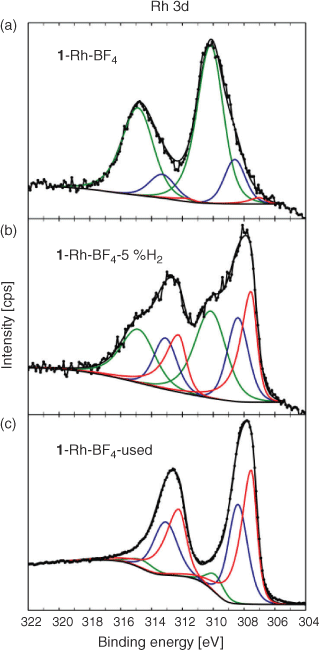
|
For the parent samples 1 and 1·Rh-BF4 and reduced samples, 1-5 %H2 and 1·Rh-BF4-5 %H2, the high-resolution N 1s spectra could be fitted with a model spectrum of the ligand as described in the Supplementary Material at ~399.5 and 401.1 eV (Fig. 11). The blue component at lower BE (MC1) is assigned to the linker bridging two Mn nodes in the MOF while the higher BE component (MC2) in red represents the ligand coordinated with Mn. Comparing the MOFs with the reduced samples, the higher BE component MC2 has increased in intensity relative to MC1. In addition, MC1 has shifted to a lower BE position consistent with the original di-pyrazole moiety. This is still present with considerable intensity, in agreement with the surface decomposition of a fraction of the organic ligands and with the preferential decomposition of the ligands bridging the Mn nodes. The templated catalyst 1-used required a modification of the fitting components, where the spectrum is dominated by a peak at 398.9 eV. The higher BE contributions have significantly reduced, but still comprise a considerable amount of overall N in the sample, indicating that the ligand has likely decomposed, and N has now formed a different phase, for example pyridinic N.[25] Lastly, N is not detected for 1·Rh-BF4-used indicating that not only has the ligand decomposed, but N has been completely removed from the structure during the catalysis tests.

|
Conclusions
In this study we describe the use of Rh-metalated Mn-MOFs as templates for Rh0 nanoparticles. The MOF-templating occurred under highly reducing conditions (80 % H2 and 20 % CO2 at 350°C), as opposed to commonly used pyrolysis and calcination methods. The MOF-derived nanoparticles were applied as catalysts for CO2 hydrogenation. The chemical composition of the template MOF influenced the composition of the active catalyst and the chemical selectivity obtained. The catalyst derived from 1·Rh2 had the simplest composition, Rh0 nanoparticles and MnO microcrystals. In comparison, the addition of the counter-ion BF4– in the template 1·Rh-BF4 resulted in a catalyst with Rh0 nanoparticles, MnCO3, and other Na-phases.
The thorough characterisation of the samples by PXRD, electron microscopy, and XPS allowed us to understand the MOF-templating mechanism for this MOF-derived catalyst. As illustrated in Fig. 8, Rh0 is formed by reduction of the Rh atoms within the metalated MOF in the presence of H2 gas. These Rh0 nanoparticles dispersed in a porous C-matrix may assist with the decomposition of the organic ligands, through processes such as Rh0 catalysed hydrogenolysis,[26] until the final catalyst is composed of a 3D mesh of Rh nanoparticles, MnCO3 or MnO, and other salts depending on the counter-ion. In the absence of Rh, the reducing treatment does not affect the MOF significantly; loss of crystallinity is observed after reduction, but the organic ligands and the Mn atoms are not substantially reacted and display similar properties to the as-synthesised MOF. However, long-term exposure to CO2 hydrogenation reaction conditions caused the full decomposition of the framework, as the final material has substantially lost both crystallinity and the binding state of the elements. Interestingly, the catalyst derived from 1 was composed of MnO crystals embedded in an amorphous phase containing both C and N. As such a phase is not observed in the Rh-containing catalyst, this result provides further evidence that the Rh hydrogenolysis properties facilitate the removal of these elements. Understanding the mechanism of MOF-templating in H2 may permit the synthesis of different metallic nanoparticles of controlled size for numerous applications, including catalysis.
Experimental
Synthetic Procedures
MnMOF (1)
The organic ligand bis(4-(4′-carboxyphenyl)-3,5-dimethylpyrazolyl)methane (H2L) was synthesised according to Bloch et al.[27] Compound 1 was synthesised by solvothermal synthesis reported by Bloch et al.[19a] In a typical synthesis, the organic ligand (63.2 mg H2L) and the Mn salt (49.9 mg MnCl2·4H2O) were dissolved in a mixture of DMF (8 mL) and water (4 mL). The solution was kept static at 100°C for 24 h. The white precipitate formed was washed with DMF (5 × 20 mL). The MOF structure was verified by PXRD.
PSM of 1
This was achieved by soaking 1 in anhydrous acetonitrile with excess of di-μ-chloro-tetracarbonyldirhodium(i) (1·Rh2) or with both di-μ-chloro-tetracarbonyldirhodium(i) and sodium tetrafluoroborate (1·Rh-BF4) (Fig. 1). After 48 h, the solution was decanted and the PSM MOFs were washed with anhydrous acetonitrile (7 × 20 mL CH3CN) and dried under a stream of Ar.
Catalysis Testing
Catalysis testing was performed in a multi-channel testing rig with parallel fixed-bed microreactors using a method previously described in detail in Lippi et al.[20] Each MOF (15 mg of 1, 18.8 mg of 1·Rh-BF4 and 18.7 mg of 1·Rh2) was mixed with SiC (50 mg) and loaded into a different microreactor. In addition, a reactor filled with SiC (100 mg) was used as a negative control. The catalysis experiments started with a 30 min drying step, under an Ar flow (1 mL min−1 per reactor) at 220°C and 1 bar, to remove any solvent guest molecules within the MOF pores. Subsequently, a reactive gas mixture (2.9 mL min−1 per reactor, 71.4 vol.- % H2, 17.9 vol.- % CO2, and 10.7 vol.- % Ar) H2/CO2 = 4 : 1 was flowed through the reactors and activation was performed at 4 bar and 350°C (Condition A). To evaluate the effect of space velocity, the flow of the reactive gas mix was increased to 4.7 mL min−1 per reactor (74.6 vol.- % H2, 18.7 vol.- % CO2, and 6.7 vol.- % Ar) H2/CO2 = 4 : 1, while keeping the temperature and pressure constant (Condition B). Flow was set back to match activation flow (2.9 mL min−1 per reactor, 71.4 vol.- % H2, 17.9 vol.- % CO2, and 10.7 vol.- % Ar) and catalysis testing was then performed at a lower temperature, 250°C (Condition C). Lastly, the first reaction conditions (Condition A) were re-established in order to determine the stability of the catalyst and identify any hysteresis (Condition D).
Effluent gas composition for each individual reactor was analysed using an online gas chromatograph with He as carrier gas.
Controlled Atmosphere Thermal Treatment
The samples 1, 1·Rh2, and 1·Rh-BF4 were loaded into quartz crucibles. The crucibles were placed inside the quartz tube of a tube furnace with three hot zones, each zone independently controlled by a 1/16 DIN temperature controller (Watlow, USA). The temperature of each zone was calibrated before the experiment. The ends of the quartz tube were connected to in-house built gas connections. The upstream end was connected to the feed gas line and the downstream end was connected to an oil bubbler. One set of samples was treated in pure N2 and another set in 5 vol- % H2 in Ar. All samples were treated at 350°C for 4 h under continuous gas flow.
Powder X-Ray Diffraction
PXRD experiments were carried out at the Powder Diffraction Beamline at the Australian Synchrotron. A Mythen microstrip detector[28] was used for data collection.
Ex Situ Analysis
The samples were loaded into special glass 0.7 mm wide capillaries (The Charles Supper Co., USA). The capillary was kept rotating during acquisition for better averaging of reflections. The beam energy during data acquisition was 16 keV (Fig. 3) and 15 keV (Fig. 4) with a current of 200 mA.
In Situ Analysis
For the experiments performed under a controlled atmosphere and variable temperature, the samples (1 and 1·Rh-BF4) were loaded into open-ended special glass 0.7 mm wide capillaries (The Charles Supper Co., USA) glass wool was used to contain the powder within the capillary while allowing gas to flow. The capillary was kept oscillating during acquisition for better average of reflections. A hot-air blower positioned below the capillary was used for temperature control. A capillary holder adapted for controlled atmosphere experiments was connected to a gas manifold allowing the selection of gas. The beam energy during data acquisition was 17.9 keV with a current of 200 mA. The sample was first heated at 5°C min−1 to 200°C under Ar flow, then the gas flow was switched to a mixture of CO2/H2 = 1 : 3. The temperature profile for each sample is indicated in Fig. 5.
Rietveld Refinement
Phase identification of the collected patterns were obtained via the use of search and match algorithm in X’pert Highscore Plus (PANalytical, the Netherlands). The obtained phases were quantified via Rietveld refinement based quantitative phase analysis[21] using Topas V5 software.[29]
Transmission Electron Microscopy (TEM)
Samples were suspended in ethanol and deposited on carbon-coated copper TEM grids and allowed to dry. Grids were examined in a Tecnai 12 G2 TEM (FEI, The Netherlands), operating at 120 kV, and images were recorded with a MegaView III CCD (Olympus, Tokyo).
Scanning Electron Microscopy (SEM) and Energy Dispersive Spectroscopy (EDS)
The samples loaded on TEM grids, as previously described, were placed on a SEM stage and examined using a Zeiss Merlin field emission scanning electron microscope operated in the secondary electron (SE) mode and back-scattered mode (BSE). EDS was used to identify elements present within the samples. The EDS system used was AZTEC, manufactured by Oxford Instruments Pty Ltd. An accelerating voltage of 15 kV was used for EDS mapping. The scale bars in the images are indicative of the magnification used.
X-Ray Photoelectron Spectroscopy (XPS)
XPS analysis was performed using an AXIS Nova spectrometer (Kratos Analytical Inc., Manchester, UK) using our standard protocol detailed elsewhere.[30] The following parameters were employed during analysis: X-ray source and power – monochromated Al Kα source at 180 W; system pressure – between 10–9 and 10–8 mbar; pass energy – 160 eV (survey) and 40 eV; step size – 0.5 eV (survey) and 0.1 eV (high resolution); emission angle – 0° as measured from the surface normal; charge neutraliser – on.
Data processing was performed using CasaXPS processing software version 2.3.15 (Casa Software Ltd, Teignmouth, UK). The atomic concentrations of the detected elements were calculated using integral peak intensities and the sensitivity factors supplied by the manufacturer. Binding energies (BE) were referenced to the C 1s peak at 284.8 eV. Peak fitting of high resolution spectra was performed using a ‘U 3 Tougaard’ background and a generalised Voigt line shape detailed in the Supplementary Material.
Supplementary Material
Additional experimental details and supporting catalytic and characterisation results (XRD, IR, TGA, N2 sorption, SEM, and XPS) are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
Aspects of this work were undertaken at the powder diffraction beamline of the Australian Synchrotron. The authors acknowledge funding from the Science and Industry Endowment Fund (SIEF), the AIM FSP funding from Commonwealth Scientific and Industrial Research Organisation (CSIRO) and the University of Adelaide. RL acknowledges the University of Adelaide for the Beacon of Enlightenment PhD Scholarship.
References
[1] (a) F. Zaera, Chem. Rec. 2005, 5, 133.| Crossref | GoogleScholarGoogle Scholar | 15889409PubMed |
(b) E. Roduner, Chem. Soc. Rev. 2014, 43, 8226.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. S. Taylor, Proc. R. Soc. Lond., Ser. A 1925, 108, 105.
| Crossref | GoogleScholarGoogle Scholar |
(d) S. Zhang, L. Nguyen, Y. Zhu, S. Zhan, C.-K. Tsung, F. Tao, Acc. Chem. Res. 2013, 46, 1731.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) B. R. Cuenya, Thin Solid Films 2010, 518, 3127.
| Crossref | GoogleScholarGoogle Scholar |
(b) P. Frontera, A. Macario, M. Ferraro, P. Antonucci, Catalysts 2017, 7, 59.
| Crossref | GoogleScholarGoogle Scholar |
(c) R. J. White, R. Luque, V. L. Budarin, J. H. Clark, D. J. Macquarrie, Chem. Soc. Rev. 2009, 38, 481.
| Crossref | GoogleScholarGoogle Scholar |
(d) J. M. Campelo, D. Luna, R. Luque, J. M. Marinas, A. a. Romero, ChemSusChem 2009, 2, 18.
| Crossref | GoogleScholarGoogle Scholar |
[3] S. R. Batten, N. R. Champness, X. M. Chen, J. Garcia-Martinez, S. Kitagawa, L. Ohrstrom, et al. Pure Appl. Chem. 2013, 85, 1715.
| Crossref | GoogleScholarGoogle Scholar |
[4] (a) H. Furukawa, K. E. Cordova, M. O’Keeffe, O. M. Yaghi, Science 2013, 341, 1230444.
| Crossref | GoogleScholarGoogle Scholar | 23990564PubMed |
(b) Y. G. Chung, J. Camp, M. Haranczyk, B. J. Sikora, W. Bury, V. Krungleviciute, et al. Chem. Mater. 2014, 26, 6185.
| Crossref | GoogleScholarGoogle Scholar |
[5] (a) S. M. Cohen, Chem. Rev. 2012, 112, 970.
| Crossref | GoogleScholarGoogle Scholar | 21916418PubMed |
(b) J. D. Evans, C. J. Sumby, C. J. Doonan, Chem. Soc. Rev. 2014, 43, 5933.
| Crossref | GoogleScholarGoogle Scholar |
[6] (a) Y. He, W. Zhou, G. Qian, B. Chen, Chem. Soc. Rev. 2014, 43, 5657.
| Crossref | GoogleScholarGoogle Scholar | 24658531PubMed |
(b) H. Furukawa, K. E. Cordova, M. O’Keeffe, O. M. Yaghi, Science 2013, 341, 1230444.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. P. Suh, H. J. Park, T. K. Prasad, D.-W. Lim, Chem. Rev. 2012, 112, 782.
| Crossref | GoogleScholarGoogle Scholar |
[7] J.-R. Li, J. Sculley, H.-C. Zhou, Chem. Rev. 2012, 112, 869.
| Crossref | GoogleScholarGoogle Scholar | 21978134PubMed |
[8] A. Corma, H. Garcia, F. X. L. I. Xamena, Chem. Rev. 2010, 110, 4606.
| Crossref | GoogleScholarGoogle Scholar | 20359232PubMed |
[9] (a) W. Xia, A. Mahmood, R. Zou, Q. Xu, Energy Environ. Sci. 2015, 8, 1837.
| Crossref | GoogleScholarGoogle Scholar |
(b) Y. Song, X. Li, L. Sun, L. Wang, RSC Adv. 2015, 5, 7267.
| Crossref | GoogleScholarGoogle Scholar |
(c) K. Shen, X. Chen, J. Chen, Y. Li, ACS Catal. 2016, 6, 5887.
| Crossref | GoogleScholarGoogle Scholar |
(d) X. Cao, C. Tan, M. Sindoro, H. Zhang, Chem. Soc. Rev. 2017, 46, 2660.
| Crossref | GoogleScholarGoogle Scholar |
(e) Z. Xie, W. Xu, X. Cui, Y. Wang, ChemSusChem 2017, 10, 1645.
| Crossref | GoogleScholarGoogle Scholar |
(f) S.-N. Zhao, X.-Z. Song, S.-Y. Song, H.-j. Zhang, Coord. Chem. Rev. 2017, 337, 80.
| Crossref | GoogleScholarGoogle Scholar |
(g) L. Oar-Arteta, T. Wezendonk, X. Sun, F. Kapteijn, J. Gascon, Mater. Chem. Front. 2017, 1, 1709.
| Crossref | GoogleScholarGoogle Scholar |
(h) Y. V. Kaneti, J. Tang, R. R. Salunkhe, X. C. Jiang, A. B. Yu, K. C. W. Wu, et al. Adv. Mater. 2017, 29, 1604898.
| Crossref | GoogleScholarGoogle Scholar |
[10] (a) H. Wang, M. Liu, S. Guo, Y. Wang, X. Han, Y. Bai, Mol. Catal. 2017, 436, 120.
| Crossref | GoogleScholarGoogle Scholar |
(b) V. P. Santos, T. A. Wezendonk, J. J. D. Jaen, A. I. Dugulan, M. A. Nasalevich, H. U. Islam, et al. Nat. Commun. 2015, 6, 6451.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. Y. Niu, S. L. Liu, Y. Q. Cai, F. C. Wu, X. L. Zhao, Microporous Mesoporous Mater. 2016, 219, 48.
| Crossref | GoogleScholarGoogle Scholar |
[11] D. Xu, Y. Pan, M. Chen, Q. Pan, L. Zhu, M. Xue, et al. RSC Adv. 2017, 7, 26377.
| Crossref | GoogleScholarGoogle Scholar |
[12] (a) D. Chen, M. Huang, S. He, S. He, L. Ding, Q. Wang, et al. Appl. Clay Sci. 2016, 119, 109.
| Crossref | GoogleScholarGoogle Scholar |
(b) Z. Wang, X. Li, Y. Yang, Y. Cui, H. Pan, Z. Wang, et al. J. Mater. Chem. A Mater. Energy Sustain. 2014, 2, 7912.
| Crossref | GoogleScholarGoogle Scholar |
[13] S. Senkan, M. Kahn, S. Duan, A. Ly, C. Leidholm, Catal. Today 2006, 117, 291.
| Crossref | GoogleScholarGoogle Scholar |
[14] (a) M. J. Jacinto, P. K. Kiyohara, S. H. Masunaga, R. F. Jardim, L. M. Rossi, Appl. Catal. A 2008, 338, 52.
| Crossref | GoogleScholarGoogle Scholar |
(b) X.-R. Ye, Y. Lin, C. Wang, M. H. Engelhard, Y. Wang, C. M. Wai, J. Mater. Chem. 2004, 14, 908.
| Crossref | GoogleScholarGoogle Scholar |
(c) N. Yan, Y. Yuan, P. J. Dyson, Chem. Commun. 2011, 47, 2529.
| Crossref | GoogleScholarGoogle Scholar |
(d) A. Karelovic, P. Ruiz, Appl. Catal. B 2012, 113–114, 237.
| Crossref | GoogleScholarGoogle Scholar |
[15] (a) R. Buchel, A. Baiker, S. E. Pratsinis, Appl Catal. A. 2014, 477, 93.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. Karelovic, P. Ruiz, J. Catal. 2013, 301, 141.
| Crossref | GoogleScholarGoogle Scholar |
[16] (a) Y. Yuan, N. Yan, P. J. Dyson, ACS Catal. 2012, 2, 1057.
| Crossref | GoogleScholarGoogle Scholar |
(b) B. Jiang, C. Li, Ö. Dag, H. Abe, T. Takei, T. Imai, et al. Nat. Commun. 2017, 8, 15581.
| Crossref | GoogleScholarGoogle Scholar |
[17] S. Guo, Y. Zhao, H. Yuan, C. Wang, H. Jiang, G. J. Cheng, Small 2020, 16, 2000749.
| Crossref | GoogleScholarGoogle Scholar | 32285619PubMed |
[18] J. Li, H. Huang, Y. Li, Y. Tang, D. Mei, C. Zhong, J. Mater. Chem. A Mater. Energy Sustain. 2019, 7, 20239.
| Crossref | GoogleScholarGoogle Scholar |
[19] (a) W. M. Bloch, A. Burgun, C. J. Coghlan, R. Lee, M. L. Coote, C. J. Doonan, et al. Nat. Chem. 2014, 6, 906.
| Crossref | GoogleScholarGoogle Scholar | 25242486PubMed |
(b) A. Burgun, C. J. Coghlan, D. M. Huang, W. Chen, S. Horike, S. Kitagawa, et al. Angew. Chem. Int. Ed. 2017, 56, 8412.
| Crossref | GoogleScholarGoogle Scholar |
[20] R. Lippi, S. C. Howard, H. Barron, C. D. Easton, I. C. Madsen, L. J. Waddington, et al. J. Mater. Chem. A Mater. Energy Sustain. 2017, 5, 12990.
| Crossref | GoogleScholarGoogle Scholar |
[21] I. C. Madsen, N. V. Y. Scarlett, D. P. Riley, M. D. Raven, in Modern Diffraction Methods (Eds E. J. Mittemeijer, U. Welzel) 2012, pp. 283–320 (Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim).
[22] I. C. Madsen, N. V. Y. Scarlett, in Powder Diffraction: Theory and Practice (Eds R. E. Dinnebier, S. J. L. Billinge) 2008, pp. 298–331 (The Royal Society of Chemistry: Cambridge, UK).
[23] (a) M. Oku, K. Hirokawa, S. Ikeda, J. Electron Spectrosc. Relat. Phenom. 1975, 7, 465.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson, R. S. C. Smart, Appl. Surf. Sci. 2011, 257, 2717.
| Crossref | GoogleScholarGoogle Scholar |
(c) V. Di Castro, G. Polzonetti, J. Electron Spectrosc. Relat. Phenom. 1989, 48, 117.
| Crossref | GoogleScholarGoogle Scholar |
(d) E. S. Ilton, J. E. Post, P. J. Heaney, F. T. Ling, S. N. Kerisit, Appl. Surf. Sci. 2016, 366, 475.
| Crossref | GoogleScholarGoogle Scholar |
[24] P. Casey, A. P. McCoy, J. Bogan, C. Byrne, L. Walsh, R. O’Connor, et al. J. Phys. Chem. C 2013, 117, 16136.
| Crossref | GoogleScholarGoogle Scholar |
[25] J. R. Pels, F. Kapteijn, J. A. Moulijn, Q. Zhu, K. M. Thomas, Carbon 1995, 33, 1641.
| Crossref | GoogleScholarGoogle Scholar |
[26] (a) M. J. Holgado, V. Rives, React. Kinet. Catal. Lett. 1986, 32, 215.
| Crossref | GoogleScholarGoogle Scholar |
(b) J. L. Carter, J. A. Cusumano, J. H. Sinfelt, J. Catal. 1971, 20, 223.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. H. Sinfelt, Catal. Rev. 1970, 3, 175.
| Crossref | GoogleScholarGoogle Scholar |
[27] W. M. Bloch, C. J. Doonan, C. J. Sumby, CrystEngComm 2013, 15, 9663.
| Crossref | GoogleScholarGoogle Scholar |
[28] B. Schmitt, C. Bronnimann, E. F. Eikenberry, F. Gozzo, C. Hormann, R. Horisberger, et al. Nucl. Instrum. Methods Phys. Res. Sect A. 2003, 501, 267.
| Crossref | GoogleScholarGoogle Scholar |
[29] Topas V5: General Profile and Structure Analysis Software for Powder Diffraction Data 2012 (Bruker AXS GmbH: Karlsruhe, Germany).
[30] C. D. Easton, C. Kinnear, S. L. McArthur, T. R. Gengenbach, J. Vac. Sci. Technol. A 2020, 38, 023207.
| Crossref | GoogleScholarGoogle Scholar |