
Direct sequencing technologies for bacterial sexually transmitted infections
Amy Jennison A * , Shivani Pasricha B and Francesca Azzato B CA
B
C

Assoc. Prof. Amy Jennison is the chief scientist of the Public Health Microbiology laboratory of the Queensland Department of Health, which is Queensland’s reference laboratory responsible for the molecular surveillance of notifiable bacterial pathogens and characterisation of public health related outbreaks. Dr Jennison has led the laboratory in the application of whole-genome sequencing for pathogen surveillance and has a particular interest in genomic analysis for understanding environmental threats from emerging pathogens and addressing antimicrobial resistance (AMR) issues in bacterial pathogens including Neisseria gonorrhoeae. |

Dr Shivani Pasricha is a microbiologist and laboratory head in the Department of Infectious Diseases of The University of Melbourne. Using molecular and genomic approaches, her research aims to improve the detection, prevention and surveillance of sexually transmitted infections (STIs). Her current research includes developing cutting-edge clustered regularly interspaced short palindromic repeats (CRISPR)-diagnostics for the point-of-care detection of STIs and AMR. |

Francesca Azzato is the section head of the Bacteriology, Victorian Mycology and Parasitology reference laboratories at the Victorian Infectious Diseases Reference Laboratory. She has expertise in the design, evaluation and implementation of novel molecular diagnostic assays. Currently, she is completing a PhD focusing on STI genomics and is involved in numerous projects, which focus on evaluating the use of new molecular methods to improve the diagnosis of bacterial and parasitic pathogens from clinical samples. |
Abstract
There is an important role for direct sequencing of patient samples to complement traditional culture-based methods for bacterial sexually transmitted infections (STIs), effectively overcoming limitations posed by fastidious or unculturable pathogens such as Neisseria gonorrhoeae, Treponema pallidum, Mycoplasma genitalium and Chlamydia trachomatis. Metagenomic techniques can enable profiling of antimicrobial resistance (AMR), strain typing and microbiome analysis in the absence of a cultured isolate, contributing critical information to understanding epidemiological trends and guiding targeted therapies. Despite significant advancements, challenges persist, such as cost, bioinformatics complexity and ethical considerations. The paper discusses current applications, technological innovations, and future prospects for integrating metagenomics into routine bacterial STI surveillance, emphasising the need to identify cost and time-effective workflows and enhanced accessibility of genomic data. By addressing these challenges, direct sequencing promises to fill critical gaps in AMR monitoring and pathogen typing, offering new avenues for enhancing public health strategies in combating bacterial STIs worldwide.
Keywords: AMR, antimicrobial resistance, direct sequencing, disease surveillance, metagenomics, public health, sexually transmitted infections, STI.
Background
Over the last 10 years, genomics, particularly next-generation sequencing (NGS), has become a crucial approach for the monitoring and diagnosing of infectious diseases. It plays a vital role in enabling high-resolution surveillance, assisting with outbreak identification and investigation, facilitating contact tracing through the identification of transmission pathways, and enabling antimicrobial resistance (AMR) target detection.1,2
However traditional microbial genomics is not a perfect fit for all bacterial pathogens, as it has typically been based on sequencing from a cultured isolate. In some cases, culturing of patient samples can prove time consuming and technically demanding, especially for sexually transmitted pathogens that might be fastidious or not readily culturable. Moreover, as diagnostic testing methods are increasingly shifting away from culture-based approaches to molecular testing direct on specimen and rapid point-of-care testing, there is a noticeable decline in the production of culture isolates.3
Although innovative culture-independent diagnostic approaches offer many advantages to patients and clinicians, dropping culture rates or an inability to culture can create gaps in disease surveillance including typing and AMR monitoring. Culturing for bacterial sexually transmitted infections (STIs) can be particularly challenging in remote areas meaning treatment surveillance data can be heavily skewed by urban cases. Problematically, some of the most overrepresented populations in Australian STI notifications are located in regional and remote areas.4 Therefore, it is increasingly important to develop surveillance approaches that are suitable for testing directly on patient specimens.
Metagenomic (direct sequencing) approaches have the potential to provide clinical and public health information directly from bacterial STI-positive patient samples. Although several challenges remain in integrating these methods into the routine testing of STIs in laboratories, particularly regarding cost, turnaround time and the need for suitable workflows and bioinformatic solutions, there are a number of promising approaches already developed for a range of pathogens.5
Neisseria gonorrhoeae
Neisseria gonorrhoeae is a major public health concern, with some lineages developing resistance to the last line of antibiotics used empirically to treat gonorrhoea.6–10 Despite global and Australian reports of N. gonorrhoeae isolates with reduced susceptibility to ceftriaxone and resistance to azithromycin, the standard treatment for gonorrhea in Australia remains a combination of injectable ceftriaxone and oral azithromycin.11 In response to increasing AMR infections, the absence of an effective vaccine and the lack of newly developed antibiotics classes, the World Health Organization (WHO) added N. gonorrhoeae to the list of high-priority pathogens.12
Phenotypic methods such as culture are still used for the diagnosis and AMR profiling of this pathogen, enabling genomics and population-level studies of N. gonorrhoeae diversity to occur. However, due to the high notification rate, most public health laboratories cannot afford to sequence all cultures, making routine comprehensive molecular surveillance uncommon. Molecular-based techniques on patient samples are the most widely used for diagnosing gonorrhoea (80% of notifications in Australia). However, these techniques require additional, often bespoke, molecular assays to detect AMR-associated mutations and associated genes or sequencing based typing schemes, which are costly and labour intensive.
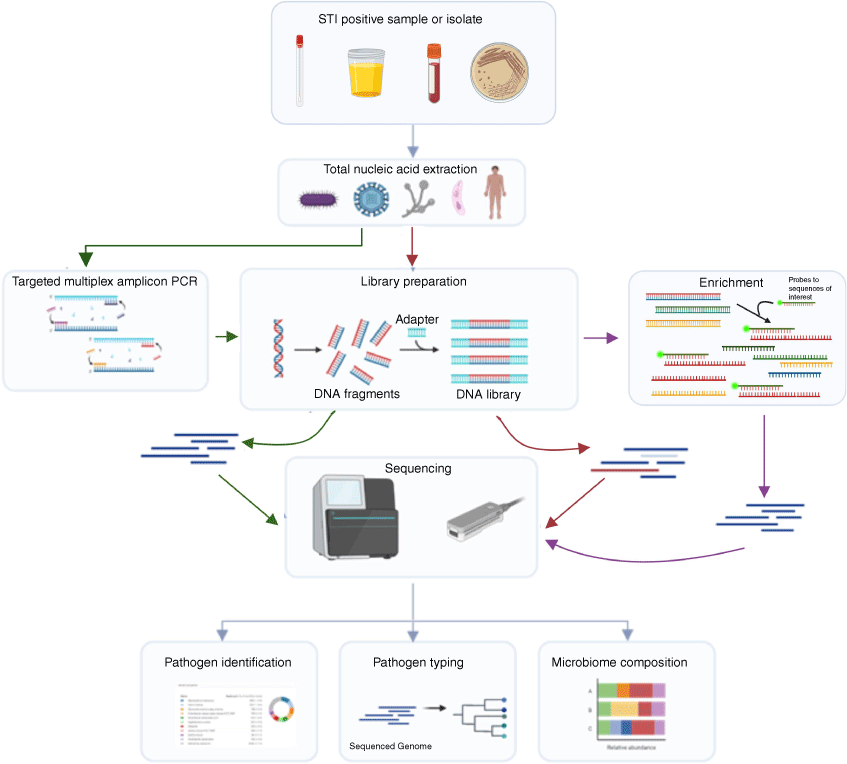
Advanced, culture-independent methods like metagenomic approaches offer significant advantages for simultaneous detection of AMR genes and mutations, virulence genes, and strain typing in real time. This approach has the potential to revolutionise the treatment of gonorrhoea by guiding targeted therapies more effectively even in regions where culture is too challenging.13 Several studies have successfully yielded AMR and typing information from N. gonorrhoeae-positive samples without the need for culture by using metagenomic approaches such as shotgun sequencing or capture hybridisation methods (Fig. 1).14,15 Additionally, amplicon-based techniques might also prove useful and has the potential to be particularly cost effective.16
Flowchart demonstrating the different workflows available for direct sequencing of STI-positive specimens. Common workflow is shown by grey arrows. Amplicon based sequencing steps have green arrows. Shotgun WGS has red arrows and enrichment-based sequencing is shown with purple arrows. This figure was created with Biorender.com.
Treponema pallidum
Syphilis, caused by Treponema pallidum, has re-emerged as a significant threat to public health. In 2020, there were 7.1 million new reported cases worldwide.17 Notably, in high-income regions such as Australia, heterosexual populations are now being affected after decades of low infection rates. Concerningly, congenital syphilis cases are rising across many countries, with a 937% rise in congenital syphilis cases in the USA and 358% increase in Australia over the past decade.17,18 Groups particularly vulnerable to syphilis include men who have sex with men (MSM) and Aboriginal and Torres Strait Islander communities.17,19
Syphilis offers significant diagnostic challenges as it is extremely difficult to culture. Diagnosis primarily relies on serological testing, with multiple tests needed to differentiate between active and past infections, a process that can be particularly challenging in remote areas. Serological testing for syphilis is negative in ~20% of primary syphilis cases. Nucleic-acid amplification tests (NAATs) such as quantitative polymerase chain reaction (qPCR) are uncommon but increasingly encouraged to improve the diagnosis of primary syphilis.20
In the absence of culture, genomic methods for T. pallidum have had to focus on metagenomic approaches. Enrichment-based whole-genome sequencing (WGS) has been successful in generating molecular-typing information able to inform on the emergence and evolution of T. pallidum, revealing the co-circulation of two major lineages (Nichols and SS14) across continents.21 This expansion aligns with the global resurgence of syphilis. In Australia, targeted whole-genome enrichment sequencing has also been applied to clinical T. pallidum samples, able to provide insights into the contemporary re-emergence of syphilis in Australia, revealing not only that multiple circulating strains linked to globally disseminated lineages are responsible for the epidemic, but that particular sub-lineages were associated with MSM and heterosexual groups.18
Mycoplasma genitalium
There has been a rise in Mycoplasma genitalium cases over the past decade, particularly among high-risk demographics, with reported incidence ranging from 5 to 20%.22 This surge coincides with a rise in infections resistant to treatment, leading to M. genitalium named as an emerging concern in STI treatment guidelines by the US Centers for Disease Control and Prevention (CDC).23 The incidence of macrolide resistance has surged to 50% and the efficacy of fluoroquinolones has declined across urban Australia.24 Understanding the AMR profile of M. genitalium is essential for delivering effective patient care and curbing the emergence of highly resistant strains.
Mycoplasma genitalium is highly fastidious, presenting a major hurdle in obtaining cultured isolates. Consequently, only a small number of isolates have been sequenced and made publicly available, leading to a limited understanding of the diversity and mechanisms of AMR in this organism.25 Presently, epidemiological studies mainly rely on individual molecular assays that target specific genes (such as resistance genes) or regions of the genome with high diversity to glean insights. Therefore, a method enabling genomic sequencing of M. genitalium directly from clinical samples would be ideal, offering a valuable tool to enhance our comprehension of the population structure of this organism.
Sequencing methods that incorporate enrichment strategies, where for example sequence specific baits are used to pull out M. genitalium-specific DNA over host and other DNA in the sample, present a promising approach to yield sufficient sequence information from M. genitalium-positive samples (Fig. 1). Preliminary data generated at the Doherty Institute, in collaboration with the Melbourne Sexual Health Centre, suggest that enrichment sequencing is a feasible approach to overcoming limited culturing capacity and provide information on prevalence and distribution of resistance mechanisms (F. Azzato, G. Taiaroa, J. Fernando, V. De Petra, M. Taouk, L. Vodstrcil, E. Plummer, S. Chea, S. Paricha, K. Raios, L. Caly, B. P. Howden, C. Bradshaw and D. A. Willamson, in prep.). Targeted amplicon-type approaches have also been shown to be successful in yielding sequences data on AMR-associated targets in M. genitalium-positive patient specimens.26,27
Chlamydia trachomatis
Chlamydia trachomatis is the most common bacterial sexually transmitted disease in females.28 Although classified as a bacterium, C. trachomatis will only grow intracellularly, similar to viruses. For this reason, culture of C. trachomatis is difficult, requiring time-consuming cell-culture methods. Instead, diagnosis of infection must focus on alternative methods including antigen tests, NAATs and occasionally serology. In the absence of high numbers of isolates, typing and phylogenetic studies using genomics have been performed using metagenomic shotgun WGS.29,30
Interestingly, shotgun metagenomic approaches offer additional benefits for C. trachomatis infections where sequencing all genetic material present in the patient sample has increased understanding of co-infections, whereby C. trachomatis may increase susceptibility to human papillomavirus (HPV) infection and play a role in viral persistence.31 Alternatively, studies utilising enrichment steps have been reported 10-fold higher sensitivity in generating C. trachomatis sequences, which, although more expensive, does increase the likelihood of yielding successful results.32 More recently, a primer-based amplification approach has been reported for C. trachomatis that has the potential to provide a cost-effective and sensitive approach to generating genomic sequences directly from clinical samples.33
Microbiome
Shotgun metagenomics offers the ability to generate a comprehensive analysis of all bacterial species present in a sample, which will allow for insights into mixed infections involving either multiple strains of a single pathogen or multiple pathogen species. Such studies show promise in assisting in identifying the role of different bacteria in STIs, and will potentially leading to more sophisticated diagnostic and treatment strategies, and ultimately a deeper understanding of the genital microbiome through metagenomics may contribute to unravelling complex conditions such as bacterial vaginosis.34,35
Summary
Direct sequencing presents a variety of exciting solutions to challenges in the STI public health space, including typing, AMR tracking and surveillance. However, several obstacles hinder routine adoption in public health and clinical microbiology laboratories. Cost remains a significant barrier, particularly for baiting approaches, necessitating ongoing efforts to reduce expenses. Additionally, bioinformatic tools must be optimised for accessibility and efficiency, and laboratory workflows need automation to handle large sample volumes efficiently, with reporting tailored to end-user needs for AMR and phylogenetic data. Ethical considerations must also be adequately addressed, especially regarding incidental findings and patient privacy and confidentiality raised by methods that sequence host DNA as well as pathogen DNA. Establishing robust protocols for patient de-identification, data security and handling incidental findings is imperative.
Despite these challenges to routine implementation, the future is promising for direct sequencing technologies addressing the current gaps in typing and AMR surveillance of under-cultured and unculturable STI pathogens.
Data availability
Data sharing is not applicable as no new data were generated or analysed during this study.
References
1 Baker KS et al. (2023) Genomics for public health and international surveillance of antimicrobial resistance. Lancet Microbe 4(12), e1047-e1055.
| Crossref | Google Scholar | PubMed |
2 Morens DM, Fauci AS (2020) Emerging pandemic diseases: how we got to COVID-19. Cell 182(5), 1077-1092.
| Crossref | Google Scholar | PubMed |
3 Mitchell SL, Simner PJ (2019) Next-generation sequencing in clinical microbiology: are we there yet? Clin Lab Med 39(3), 405-418.
| Crossref | Google Scholar | PubMed |
4 Ward J et al. (2013) STI in remote communities: improved and enhanced primary health care (STRIVE) study protocol: a cluster randomised controlled trial comparing ‘usual practice’ STI care to enhanced care in remote primary health care services in Australia. BMC Infect Dis 13, 425.
| Crossref | Google Scholar | PubMed |
5 Caruso G et al. (2021) Current and future trends in the laboratory diagnosis of sexually transmitted infections. Int J Environ Res Public Health 18(3), 1038.
| Crossref | Google Scholar | PubMed |
6 Jennison AV et al. (2019) Genetic relatedness of ceftriaxone-resistant and high-level azithromycin resistant Neisseria gonorrhoeae cases, United Kingdom and Australia, February to April 2018. Euro Surveill 24(8), 1900118.
| Crossref | Google Scholar | PubMed |
7 Martin I et al. (2019) Multidrug-resistant and extensively drug-resistant Neisseria gonorrhoeae in Canada, 2012–2016. Can Commun Dis Rep 45(2–3), 45-53.
| Crossref | Google Scholar | PubMed |
8 Maubaret C et al. (2023) Two cases of extensively drug-resistant (XDR) Neisseria gonorrhoeae infection combining ceftriaxone-resistance and high-level azithromycin resistance, France, November 2022 and May 2023. Euro Surveill 28(37), 2300456.
| Crossref | Google Scholar | PubMed |
9 Ouk V et al. (2024) High prevalence of ceftriaxone-resistant and XDR Neisseria gonorrhoeae in several cities of Cambodia, 2022–23: WHO Enhanced Gonococcal Antimicrobial Surveillance Programme (EGASP). JAC Antimicrob Resist 6(2), dlae053.
| Crossref | Google Scholar | PubMed |
10 Pleininger S et al. (2022) Extensively drug-resistant (XDR) Neisseria gonorrhoeae causing possible gonorrhoea treatment failure with ceftriaxone plus azithromycin in Austria, April 2022. Euro Surveill 27(24), 2200455.
| Crossref | Google Scholar | PubMed |
11 Australasian Society for HIV, Viral Hepatitis and Sexual Health Medicine (2021) Australian STI management guidelines for use in primary care – standard asymptomatic check-up. https://sti.guidelines.org.au/standard-asymptomatic-checkup/
12 World Health Organization (2021) WHO Bacterial Priority Pathogens List, 2024: bacterial pathogens of public health importance to guide research, development and strategies to prevent and control antimicrobial resistance. 17 May 2024, Report. WHO, Geneva, Switzerland. https://www.who.int/publications/i/item/9789240093461
13 Sánchez-Busó L et al. (2021) A community-driven resource for genomic epidemiology and antimicrobial resistance prediction of Neisseria gonorrhoeae at Pathogenwatch. Genome Med 13(1), 61.
| Crossref | Google Scholar |
14 Graham RM et al. (2017) Epidemiological typing of Neisseria gonorrhoeae and detection of markers associated with antimicrobial resistance directly from urine samples using next generation sequencing. Sex Transm Infect 93(1), 65-67.
| Crossref | Google Scholar | PubMed |
15 Street TL et al. (2024) Target enrichment improves culture-independent detection of Neisseria gonorrhoeae and antimicrobial resistance determinants direct from clinical samples with nanopore sequencing. Microb Genom 10(3), 001208.
| Crossref | Google Scholar | PubMed |
16 Zhang C et al. (2021) Multiplex PCR and nanopore sequencing of genes associated with antimicrobial resistance in Neisseria gonorrhoeae directly from clinical samples. Clin Chem 67(4), 610-620.
| Crossref | Google Scholar | PubMed |
17 Centers for Disease Control and Prevention (2022) Sexually transmitted infections surveillance, 2022. Last reviewed: 30 January 2024. CDC, US Department of Health & Human Services. https://www.cdc.gov/std/statistics/2022/default.htm
18 Taouk ML et al. (2022) Characterisation of Treponema pallidum lineages within the contemporary syphilis outbreak in Australia: a genomic epidemiological analysis. Lancet Microbe 3(6), e417-e426.
| Crossref | Google Scholar | PubMed |
19 Multijurisdictional Syphilis Outbreak Working Group (2019) Multijurisdictional Syphilis Outbreak (MJSO) Surveillance Report – consolidated reports May 2019 to January 2019. Australia Communicable Diseases Network of Australia, Australian Government Department of Health and Aged Care. https://www.health.gov.au/resources/publications/multijurisdictional-syphilis-outbreak-mjso-surveillance-report-consolidated-reports-may-2018-to-january-2019?language=en
20 World Health Organization (2023) The diagnostics landscape for sexually transmitted infections. WHO, Geneva, Switzerland. https://www.who.int/publications/i/item/9789240077126
21 Beale MA et al. (2021) Global phylogeny of Treponema pallidum lineages reveals recent expansion and spread of contemporary syphilis. Nat Microbiol 6(12), 1549-1560.
| Crossref | Google Scholar | PubMed |
22 Waites KB et al. (2023) Latest advances in laboratory detection of Mycoplasma genitalium. J Clin Microbiol 61(3), e0079021.
| Crossref | Google Scholar | PubMed |
23 Workowski KA et al. (2021) Sexually transmitted diseases treatment guidelines, 2021. MMWR Rercomm Rep 70(4), 1-187.
| Crossref | Google Scholar |
24 Trembizki E et al. (2017) High levels of macrolide-resistant Mycoplasma genitalium in Queensland, Australia. J Med Microbiol 66(10), 1451-1453.
| Crossref | Google Scholar | PubMed |
25 Fookes MC et al. (2017) Mycoplasma genitalium: whole genome sequence analysis, recombination and population structure. BMC Genomics 18(1), 993.
| Crossref | Google Scholar | PubMed |
26 Chiribau CB et al. (2024) Detection of resistance to macrolides and fluoroquinolones in Mycoplasma genitalium by targeted next-generation sequencing. Microbiol Spectr 12(3), e0384523.
| Crossref | Google Scholar | PubMed |
27 Plummer EL et al. (2020) A custom amplicon sequencing approach to detect resistance associated mutations and sequence types in Mycoplasma genitalium. J Microbiol Methods 179, 106089.
| Crossref | Google Scholar | PubMed |
28 Ojoo S (2003) Clinical practice in sexually transmissible infections. Sexually Transmitted Infect 79(5), 429.
| Crossref | Google Scholar |
29 Andersson P et al. (2013) Sequences of multiple bacterial genomes and a Chlamydia trachomatis genotype from direct sequencing of DNA derived from a vaginal swab diagnostic specimen. Clin Microbiol Infect 19(9), E405-E408.
| Crossref | Google Scholar | PubMed |
30 Suchland RJ et al. (2017) Demonstration of persistent infections and genome stability by whole-genome sequencing of repeat-positive, same-serovar Chlamydia trachomatis collected from the female genital tract. J Infect Dis 215(11), 1657-1665.
| Crossref | Google Scholar | PubMed |
31 Di Pietro M et al. (2018) HPV/Chlamydia trachomatis co-infection: metagenomic analysis of cervical microbiota in asymptomatic women. New Microbiol 41(1), 34-41.
| Google Scholar | PubMed |
32 Christiansen MT et al. (2014) Whole-genome enrichment and sequencing of Chlamydia trachomatis directly from clinical samples. BMC Infect Dis 14, 591.
| Crossref | Google Scholar | PubMed |
33 Chen W et al. (2024) Revealing the genetic diversity of Chinese Chlamydia trachomatis strains directly from clinical samples through selective whole-genome amplification. J Infect Dis jiae163.
| Crossref | Google Scholar | PubMed |
34 Bommana S et al. (2022) Metagenomic shotgun sequencing of endocervical, vaginal, and rectal samples among Fijian women with and without Chlamydia trachomatis reveals disparate microbial populations and function across anatomic sites: a pilot study. Microbiol Spectr 10(3), e0010522.
| Crossref | Google Scholar | PubMed |
35 Song J et al. (2024) Interpretation of vaginal metagenomic characteristics in different types of vaginitis. mSystems 9(3), e0137723.
| Crossref | Google Scholar | PubMed |
Assoc. Prof. Amy Jennison is the chief scientist of the Public Health Microbiology laboratory of the Queensland Department of Health, which is Queensland’s reference laboratory responsible for the molecular surveillance of notifiable bacterial pathogens and characterisation of public health related outbreaks. Dr Jennison has led the laboratory in the application of whole-genome sequencing for pathogen surveillance and has a particular interest in genomic analysis for understanding environmental threats from emerging pathogens and addressing antimicrobial resistance (AMR) issues in bacterial pathogens including Neisseria gonorrhoeae. |
Dr Shivani Pasricha is a microbiologist and laboratory head in the Department of Infectious Diseases of The University of Melbourne. Using molecular and genomic approaches, her research aims to improve the detection, prevention and surveillance of sexually transmitted infections (STIs). Her current research includes developing cutting-edge clustered regularly interspaced short palindromic repeats (CRISPR)-diagnostics for the point-of-care detection of STIs and AMR. |
Francesca Azzato is the section head of the Bacteriology, Victorian Mycology and Parasitology reference laboratories at the Victorian Infectious Diseases Reference Laboratory. She has expertise in the design, evaluation and implementation of novel molecular diagnostic assays. Currently, she is completing a PhD focusing on STI genomics and is involved in numerous projects, which focus on evaluating the use of new molecular methods to improve the diagnosis of bacterial and parasitic pathogens from clinical samples. |