
Hacking nature: genetic tools for reprograming enzymes
Carol J Hartley A , Matthew Wilding A and Colin Scott A BA CSIRO Land and Water
Black Mountain Research and Innovation Park
Canberra, ACT, Australia
B Tel: +61 2 6246 4090
Fax: +61 2 6246 4176
Email: colin.scott@csiro.au
Microbiology Australia 38(2) 73-75 https://doi.org/10.1071/MA17032
Published: 22 March 2017
Enzymes have many modern industrial applications, from biomass decomposition in the production of biofuels to highly stereospecific biotransformations in pharmaceutical manufacture. The capacity to find or engineer enzymes with activities pertinent to specific applications has been essential for the growth of a multibillion dollar enzyme industry. Over the course of the past 50–60 years our capacity to address this issue has become increasingly sophisticated, supported by innumerable advances, from early discoveries such as the co-linearity of DNA and protein sequence1 to modern computational technologies for enzyme design. The design of enzyme function is an exciting nexus of fundamental biochemical understanding and applied engineering. Herein, we will cover some of the methods used in discovery and design, including some ‘next generation’ tools.
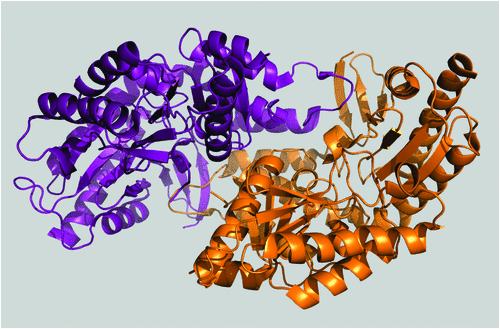
Traditionally, enzymes with useful biochemical properties have been sourced from nature, tapping into the natural diversity generated by evolution. Where known physiological functions are useful in an industrial setting, it is relatively simple to match an enzyme to an application (e.g. amylase-mediated glucose production from starch). Where novel functions are required, enrichment culturing of microbes can be used: for example, the recent isolation of bacteria capable of using nylon intermediates as a nitrogen source with potential utility in nylon manufacture (Figure 1)3. Non-culture based methods can also be applied to enzyme discovery, for example by exploiting the explosion of genetic information that followed the ‘omics’ revolution. Driven by technological advances in DNA sequencing, and computational power, this has provided an enormous resource, accessible by bioinformatic analysis, and leading to the discovery of enzymes with industrial applications, such as novel imine reductases for asymmetric organic synthesis4.
|
Methods have also been developed to move beyond the repertoire of enzymes currently known in nature. One of the most prevalent approaches has been to use atomic information about structure and function to rationally redesign enzymes, often to expand or alter substrate range, change stereospecificity or alter physical properties, such as thermostability5. Enzyme engineers have also exploited the power of evolution, by applying artificial selection pressures or selective screens to probe libraries of enzyme variants, often generated by low frequency amino acid randomisation6,7. This method has been especially effective in accessing the cryptic biochemical diversity available through promiscuous enzyme activities, i.e. physiologically irrelevant ‘side-reactions’ that do not confer a fitness cost or benefit8,9. Combining rational engineering principles with an evolutionary approach has proven particularly powerful: small focused libraries are synthesised, guided by structure-function information, and then screened/selected for properties of interest10. Such strategies have been used to alter properties such as stereospecificity, expression level11 and Michaelis constants (KM)12 and to overcome functional constraints, such as inhibition by substrates and/or products leading to improved reaction yields13.
Impressively, it has also been possible to engineer wholly new catalytic functions for enzymes, such as direct amination of unactivated carbon atoms14, cyclopropanation15 and Diels-Alder cycloaddition16 (albeit, naturally-occurring enzymes for the latter reaction have since been discovered17). Several stratagems have been used to introduce ‘unnatural’ functionality into enzymes, with examples of mechanism-based re-engineering of extant natural enzymes6,14,15 and computer-aided de novo design of new active sites18–20. Often, enzymes constructed using the current iteration of such techniques have limited catalytic functionality; however, the catalytic properties of such synthetic and semisynthetic enzymes can be improved by direction evolution and related methodologies21,22.
In the approaches considered above, enzyme engineers have been content to explore the chemical space provided by the 20 canonical proteinogenic amino acids. However, there have been considerable efforts to expand the chemical repertoire of amino acids and add functionality to enzymes by introducing non-natural amino acids. The synthesis of hundreds of non-natural amino acids have already been reported, with functionalities including halides, cyanides, azides and alkenyl/alkynyl groups for ‘click’ chemistry, as well as fluorophores and a range of others23.
Incorporating these amino acids into proteins and living organisms has proven challenging; however, an approach in which components of the protein synthesis apparatus have been re-engineered is proving successful. By reprograming aminoacyl tRNA synthetases to accept non-natural amino acids, rather than their native ones, it is now possible to repurpose codons for the incorporation of non-natural amino acids23,24. In some recently reported methods, this approach has been used to incorporate dehydroalanine via dephosphorylation of O-phosphoserine that had been incorporated using a repurposed tRNA synthase25–27. The highly reactive dehydroalanine allows post-translational installation of non-natural side-chains. The potential of such methods is enormous, nonetheless, there is another problem that needs to be addressed. The genetic code is already fully utilised encoding the naturally occurring proteinogenic amino acids. Repurposing a codon for a non-natural amino acid will affect every gene that uses that codon, and modify every protein transcribed by those genes. How, then do we make space for an expanded repertoire of amino acids?
Recent advances in synthetic biology may hold the key. One option, which has recently been reported to be successful, is to recode entire genomes, removing codons from use28. In principle, the redundancy of the genetic code can be eliminated so that each amino acid is encoded by a single codon, making available coding space for novel amino acids. More ambitiously, it is possible to expand the genetic code by introducing non-natural nucleotides29, and in doing so introduce an array of new codons with no natural function. Albeit this additional information is currently ‘inaccessible’ to the cell, pending reengineering of the cellular machinery to recognise and translate this new coding space.
The rate of advancement in our ability to design and repurpose enzymes has been enormous, with much of our fundamental understanding of protein biochemistry and laboratory and computational tools developed over the course of the past 50 years. In that time, we have progressed from randomly surveying the natural diversity of enzymes in easily cultured organisms, to de novo enzyme design and overcoming the limitations of nature’s chemical toolbox. As new tools from disciplines such as synthetic biology will support the expanding utility of enzymes, we can expect this field to continue to evolve and play a role in the next industrial revolution.
References
[1] Kresge, N. et al. (2005) Using tryptophan synthase to prove gene-protein colinearity: the work of Charles Yanofsky. J. Biol. Chem. 280, e43.| 1:CAS:528:DC%2BD2MXht1SktLbI&md5=88f29bcca5033595080f0bd804a31615CAS |
[2] Wilding, M. et al. (2017) Crystal structure of a putrescine aminotransferase from Pseudomonas sp. strain AAC. Acta Crystallogr. F Struct. Biol. Cryst. Commun. 73, 29–35.
| Crystal structure of a putrescine aminotransferase from Pseudomonas sp. strain AAC.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2sXhtFCnug%3D%3D&md5=dcbe0c322e7fd687bec28bc34f7ce36aCAS |
[3] Wilding, M. et al. (2015) Identification of novel transaminases from a 12-aminododecanoic acid-metabolizing Pseudomonas strain. Microb. Biotechnol. 8, 665–672.
| Identification of novel transaminases from a 12-aminododecanoic acid-metabolizing Pseudomonas strain.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhtVGnu7jN&md5=3110f4561eab72ed52dc95db9da8724cCAS |
[4] Mangas-Sanchez, J. et al. (2016) Imine reductases (IREDs). Curr. Opin. Chem. Biol. 37, 19–25.
| Imine reductases (IREDs).Crossref | GoogleScholarGoogle Scholar |
[5] Böettcher, D. and Bornscheuer, U.T. (2010) Protein engineering of microbial enzymes. Curr. Opin. Microbiol. 13, 274–282.
| Protein engineering of microbial enzymes.Crossref | GoogleScholarGoogle Scholar |
[6] Arnold, F.H. (2015) The nature of chemical innovation: new enzymes by evolution. Q. Rev. Biophys. 48, 404–410.
| The nature of chemical innovation: new enzymes by evolution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXhvVWru73O&md5=9b30be4754f26736b31c449011233c18CAS |
[7] Renata, H. et al. (2015) Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution. Angew. Chem. Int. Ed. Engl. 54, 3351–3367.
| Expanding the enzyme universe: accessing non-natural reactions by mechanism-guided directed evolution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXjslCiur4%3D&md5=797dd31627db2a119b30715d8301bf1cCAS |
[8] Copley, S.D. (2015) An evolutionary biochemist’s perspective on promiscuity. Trends Biochem. Sci. 40, 72–78.
| An evolutionary biochemist’s perspective on promiscuity.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXltVynug%3D%3D&md5=697e76ea8f73971c389c0c9197949ce3CAS |
[9] Khersonsky, O. and Tawfik, D.S. (2010) Enzyme promiscuity: a mechanistic and evolutionary perspective. In Annual Review of Biochemistry, Vol 79 (Kornberg, R.D., et al., eds), pp. 471–505
[10] Li, G. and Reetz, M.T. (2016) Learning lessons from directed evolution of stereoselective enzymes. Org. Chem. Front. 3, 1350–1358.
| Learning lessons from directed evolution of stereoselective enzymes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhtVOgu7zK&md5=8a10286843da3254b2bae3047f8642c5CAS |
[11] Reetz, M.T. and Zheng, H. (2011) Manipulating the expression rate and enantioselectivity of an epoxide hydrolase by using directed evolution. ChemBioChem 12, 1529–1535.
| Manipulating the expression rate and enantioselectivity of an epoxide hydrolase by using directed evolution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXotVCmt7Y%3D&md5=c66cf03b07cb771e1ec636f859b9ece3CAS |
[12] Scott, C. et al. (2009) Catalytic improvement and evolution of atrazine chlorohydrolase. Appl. Environ. Microbiol. 75, 2184–2191.
| Catalytic improvement and evolution of atrazine chlorohydrolase.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXksFWltb4%3D&md5=c7d8dcbda38ae053b9b9149124c904cbCAS |
[13] Kim, J.H. et al. (2004) Enhanced thermostability and tolerance of high substrate concentration of an esterase by directed evolution. J. Mol. Catal., B Enzym. 27, 169–175.
| Enhanced thermostability and tolerance of high substrate concentration of an esterase by directed evolution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhtFWnurk%3D&md5=95c481eb016cda758ec33beb777b6da2CAS |
[14] Farwell, C.C. et al. (2015) Enantioselective enzyme-catalyzed aziridination enabled by active-site evolution of a cytochrome P450. ACS Cent. Sci. 1, 89–93.
| Enantioselective enzyme-catalyzed aziridination enabled by active-site evolution of a cytochrome P450.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXntVCntbg%3D&md5=76220fc20c5b902394e396ace96ed9b0CAS |
[15] Renata, H. et al. (2016) Identification of mechanism-based inactivation in P450-catalyzed cyclopropanation facilitates engineering of improved enzymes. J. Am. Chem. Soc. 138, 12527–12533.
| Identification of mechanism-based inactivation in P450-catalyzed cyclopropanation facilitates engineering of improved enzymes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhsVagu7rP&md5=f1967d77f37c9bd853f9be86e98349e9CAS |
[16] Cottet, K. et al. (2016) Artificial enzyme-catalyzed Diels-Alder cycloadditions. Curr. Org. Chem. 20, 2254–2281.
| Artificial enzyme-catalyzed Diels-Alder cycloadditions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhtlCru7nM&md5=9a21c8e27821037db65b5accc4d66ba8CAS |
[17] Ose, T. et al. (2003) Insight into a natural Diels-Alder reaction from the structure of macrophomate synthase. Nature 422, 185–189.
| Insight into a natural Diels-Alder reaction from the structure of macrophomate synthase.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXhvFKgsrs%3D&md5=f47349c4d3d8843e65f2302d031a1341CAS |
[18] Kries, H. et al. (2013) De novo enzymes by computational design. Curr. Opin. Chem. Biol. 17, 221–228.
| De novo enzymes by computational design.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXjvFCnsrY%3D&md5=5484d5a5492dee10d96efebde286ab26CAS |
[19] Garrabou, X. et al. (2016) Fast Knoevenagel condensations catalyzed by an artificial Schiff base-forming enzyme. J. Am. Chem. Soc. 138, 6972–6974.
| Fast Knoevenagel condensations catalyzed by an artificial Schiff base-forming enzyme.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28Xot1ertL8%3D&md5=8e98917f9aba7c5689346fe69f363d21CAS |
[20] Huang, P.S. et al. (2016) The coming of age of de novo protein design. Nature 537, 320–327.
| The coming of age of de novo protein design.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhsFajsrzL&md5=98987fd51882cb259f3d5ff5f5a5b46fCAS |
[21] Kiss, G. et al. (2013) Computational enzyme design. Angew. Chem. Int. Ed. Engl. 52, 5700–5725.
| Computational enzyme design.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXksVOjsb0%3D&md5=73783606bf12ed6fd047cdd26f9ebbfcCAS |
[22] Obexer, R. et al. (2016) Efficient laboratory evolution of computationally designed enzymes with low starting activities using fluorescence-activated droplet sorting. Protein Eng. Des. Sel. 29, 355–365.
| Efficient laboratory evolution of computationally designed enzymes with low starting activities using fluorescence-activated droplet sorting.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhslWksr7I&md5=d8814b84c84a810468ed836ae73b5f98CAS |
[23] Xiao, H. and Schultz, P.G. (2016) At the interface of chemical and biological synthesis: an expanded genetic code. Cold Spring Harb. Perspect. Biol. 8, .
[24] Chatterjee, A. et al. (2013) A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli. Biochemistry 52, 1828–1837.
| A versatile platform for single- and multiple-unnatural amino acid mutagenesis in Escherichia coli.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvVynt7s%3D&md5=b2aa1394b6a8b06c1a4cab1a64445cd6CAS |
[25] Hofmann, R. and Bode, J.W. (2016) A radical approach to posttranslational mutagenesis. Science 354, 553–554.
| A radical approach to posttranslational mutagenesis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhvVCksb3P&md5=dcdc7ee6adc38c035e8755e55a7c609eCAS |
[26] Yang, A. et al. (2016) A chemical biology route to site-specific authentic protein modifications. Science 354, 623–626.
| A chemical biology route to site-specific authentic protein modifications.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhslKgt7vL&md5=54071f2031129b0920629f9daf8998fbCAS |
[27] Wright, T.H. et al. (2016) Posttranslational mutagenesis: a chemical strategy for exploring protein side-chain diversity. Science 354, 597–605.
| Posttranslational mutagenesis: a chemical strategy for exploring protein side-chain diversity.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XhslKgt7jP&md5=7601678f876b40cf58914b7f1db98381CAS |
[28] Hutchison, C.A. et al. (2016) Design and synthesis of a minimal bacterial genome. Science 351, 1414–1473.
| Design and synthesis of a minimal bacterial genome.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC28XksFCht7k%3D&md5=78f3ac25d91fc803b8c77f93d57dd834CAS |
[29] Zhang, Y. et al. (2017) A semisynthetic organism engineered for the stable expansion of the genetic alphabet. Proc. Natl. Acad. Sci. USA 114, .
| A semisynthetic organism engineered for the stable expansion of the genetic alphabet.Crossref | GoogleScholarGoogle Scholar |
Biographies
Dr Carol Hartley is a research scientist and leader of the Biocatalysis and Synthetic Biology research team within CSIRO in Canberra, Australia. She obtained a PhD in microbiology from Rhodes University, South Africa, before joining the CSIRO and has a strong interest in biocatalysis and the use of enzymes to advance biotechnology and synthetic biology.
Matt Wilding was awarded his PhD in Biological Chemistry in 2012 from the University of Manchester, UK. He moved to Australia later that year to take up a prestigious OCE Post-Doctoral Fellowship with the Biocatalysis Team at CSIRO in Canberra, and in 2015 became a Research Scientist in the team. His research interests include synthetic biology, protein evolution and biocatalyst engineering.
Dr Colin Scott obtained his PhD in molecular microbiology from the University of Sheffield in the UK in 2000 before taking up a post-doctoral fellowship with the CSIRO. He currently leads the Biotechnology and Synthetic Biology Group at the CSIRO. He has strong interests in enzyme evolution, biocatalysis, microbial physiology and synthetic biology.