

Metal speciation from stream to open ocean: modelling v. measurement
Edward Tipping A C , Stephen Lofts A and Anthony Stockdale BA Centre for Ecology and Hydrology, Lancaster Environment Centre, Lancaster, LA1 4AP, UK.
B School of Earth and Environment, University of Leeds, Leeds, LS2 9JT, UK.
C Corresponding author. Email: et@ceh.ac.uk
Environmental Chemistry 13(3) 464-477 https://doi.org/10.1071/EN15111
Submitted: 30 May 2015 Accepted: 16 July 2015 Published: 6 October 2015
Journal Compilation © CSIRO Publishing 2016 Open Access CC BY-NC-ND
Environmental context. The chemical speciation of metals strongly influences their transport, fate and bioavailability in natural waters. Analytical measurement and modelling both play important roles in understanding speciation, while modelling is also needed for prediction. Here, we analyse a large set of data for fresh waters, estuarine and coastal waters, and open ocean water, to examine how well measurements and modelling predictions agree.
Abstract. We compiled a data set of ~2000 published metal speciation measurements made on samples of fresh waters, estuarine and coastal waters, and open ocean waters. For each sample, we applied the chemical speciation model WHAM7 to calculate the equilibrium free metal ion concentrations, [M] (mol L–1), amounts of metal bound by dissolved organic matter (DOM), ν (mol g–1), and their ratio ν/[M] (L g–1), which is a kind of ‘local’ partition coefficient. Comparison of the measured and predicted speciation variables for the whole data set showed that agreements are best for fresh waters, followed by estuarine and coastal waters, then open-ocean waters. Predicted values of ν/[M], averaged over all results for each metal, closely follow the trend in average measured values, confirming that metal reactivity, and consequent complexation by DOM, in natural waters accord with the expectations of the speciation model. Comparison of model predictions with measurements by different analytical techniques suggests that competitive ligand–stripping voltammetry methods overestimate metal complexation by DOM, and therefore underestimate [M]. When measurements by other methods are compared with predictions, for all metals, reasonable agreement with little bias is obtained at values of ν > 10–6 mol g–1 DOM, but at lower values of ν, the model predictions of [M] are mostly higher than the measured values, and the predictions of ν and ν/[M] are mostly lower. Research is needed to establish whether this reflects analytical error or the failure of the model to represent natural high-affinity ligands.
Introduction
It has long been recognised that the bioavailability, transport and retention of metals in the natural environment depend very much on their chemical speciation, by which is meant the different chemical species that make up the total amount of metal in a given sample.[1] Bioavailability underpins the role of metals as essential nutrients and governs toxicity, and is considered to depend on the concentration of the free metal ion, combined with those of other competitors including protons.[2] The key process of metal partitioning between solutions and mobile or immobile solids depends on speciation in both phases.[3] A well-known example of the importance of metal speciation to ecosystem function is the case of freshwater acidification,[4] echoed in more recent concern about the acidifying effect of excess carbon dioxide on marine waters.[5,6]
Chemical speciation can be determined analytically and, in principle at least, this is the most reliable approach. However, direct measurements require considerable effort, either in terms of sampling and laboratory analysis[7] or in the development and maintenance of in situ techniques,[8] and of course cannot provide predictions of future conditions. To address these limitations, therefore, chemical speciation modelling is an essential complementary activity, offering the possibility to address ‘what if’ questions, and explain current conditions and forecast future ones, at different spatial scales. It is clearly desirable that measurements and model predictions should agree, and here we explore how well they do so. The analysis considers systems assumed to be at or near to chemical equilibrium, and thereby amenable to approximate but practical analysis using the internally consistent rules of equilibrium chemistry that follow from the laws of thermodynamics.
In aquatic systems, a dissolved cationic metal is present as the free metal ion, its complexes with inorganic ligands such as chloride and bicarbonate ions, and its complexes with organic compounds, either identifiable ones such as acetate or siderophores, or the complex mixture of partial breakdown products of living matter. How metals bind to the inorganic and identifiable organic ligands is, or can be, well understood, but interactions with the ‘natural organic matter’, ‘dissolved organic matter’ (DOM) or ‘humic substances’ are often quantitatively more important as repositories of the cations. Despite decades of research on this complex organic material, it remains poorly defined chemically. However, we know that it contains weak-acid functional groups, including carboxyl and phenolic groups, and entities containing N and S, and these provide a range of binding sites for cations.[9] Models have been developed that take into account this high degree of binding site heterogeneity, the most successful and widely used ones to date being the Windermere Humic Aqueous Model (WHAM)[10] and the Non-Ideal Competitive Adsorption–Donnan (NICA–Donnan) model,[11] both of which have been parameterised with laboratory data. These models take into account binding at different metal loadings and the effects of pH, competition by major cations and ionic strength, and so are suitable for field application. However, they have been parameterised with data for isolated natural organic matter (chiefly freshwater and soil humic and fulvic acids) obtained by experiments in which the analyst could impose the solution or suspension conditions and thereby optimise the measurement of the metal free ion or related variables. It is much more difficult to measure chemical speciation on natural samples, even more difficult to do so in situ, but this is what we really want to know.
Some efforts have been made to compare techniques applied to common samples,[12,13] and to compare results with model predictions, but progress has been limited, not least by the effort required and the time limitations of an individual study. A less direct but more widely applicable approach is the comparison of analytical results with outputs from the same model, and this also provides a test of the model. We have previously performed three such studies focussing on data from fresh waters,[14] estuarine and coastal waters[15] and the open ocean,[16] and comparing them with predictions obtained using WHAM6[17] or WHAM7.[18] We found that measurements and model predictions based only on metal binding by humic-type ligands (i.e. DOM) were in broad agreement, but in many cases differences were greater than could be explained by data uncertainty, and there was a tendency for the differences to be larger at low metal concentrations.
The work described here extends in two ways our previous efforts to compare observations and predictions of metal speciation in aquatic systems. First, by combining the results of the three previous studies, we obtained a large data set (~2000 samples) covering all types of surface water, including the deep ocean, and with some overlap of techniques applied to different types of water. Second, we improved the quantification of comparison between measurements and predictions. As before, we compared results in terms of the free-ion concentration [M] and DOM-bound metal (ν, mol g–1), but we added their ratio ν/[M] (L g–1), which is a kind of ‘local’ partition coefficient. We combined output statistics to create a single variable that quantifies the degree of agreement between measurement and prediction. Then we compared measured and modelled values by surface-water type, by metal and by measurement technique. This was an empirical comparison, with the simple aim of quantifying differences. We anticipated that analysis of the large combined data set would increase the possibility of identifying underlying trends and discrepancies, and therefore help to define future directions in efforts to understand and predict metal speciation in the field.
Methods
Speciation methods
The measured speciation data, taken from the literature, were obtained by eight different techniques, briefly described below. They are each designed to measure (quasi-) equilibrium speciation in terms of conventional chemistry, based on the concentration of the free metal ion.
Cation-exchange, dynamic mode (CED) was introduced by Driscoll[19] to determine Al speciation in acid to neutral fresh waters. It operates on the principle that most inorganic Al is cationic, and can be rapidly removed during passage through a column of cation-exchange resin (labile fraction), whereas Al complexed by organic matter remains unaffected (non-labile). A correction to account for the minor dissociation of the organic complex can be applied.[20] The free-ion (Al3+) concentration is not measured, but calculated by equilibrium speciation modelling of the inorganic forms only.
Competing ligand methods (CL-ASV, CL-CSV) involve the titration of multiple subsamples (to which a buffer and varying concentrations of a metal spike have been added) with a strongly complexing ligand (CL) as a means of converting the electrochemically inert fraction of metal into a single, well-characterised complex that is reducible and thus can be subsequently measured by adsorptive stripping voltammetry.[21] Anodic stripping voltammetry (ASV) uses a negative voltage for collection of the CL–metal complexes at the working electrode,[22] whereas cathodic stripping voltammetry (CSV) uses a zero or positive value.[23] Data from multiple experiments are then used to determine the conditional stability constant and complexation capacities of the organic ligand using non-linear curve fitting. Calculations require known side reaction coefficients for both the competing ligand and the inorganic metal.[24]
The Donnan membrane technique (DMT) involves the separation of the in situ solution being analysed from an acceptor solution by a cation-exchange membrane.[25] The acceptor solution contains Ca(NO3)2 and humic acid. The DMT cell is immersed into the river or lake for a set number of days, during which time free metal ions diffuse across the cation-exchange membrane and are complexed by the humic acid. After equilibration, metal concentrations in the acceptor solution are measured, and the free-ion concentration is computed by speciation modelling. If equilibrium is not achieved, the free-ion concentration is computed assuming diffusion-limited transport of the free metal ion across the cation-exchange membrane.
Differential pulse anodic stripping voltammetry (DPASV) uses the electrochemical technique used in CL-ASV but without the addition of a competing ligand. Samples are titrated with metal between voltammetric scans to estimate concentrations of labile metal, from which free-ion concentrations are calculated with inorganic side reaction coefficients, and then a linear transformation model is applied to determine conditional stability constants for one or two classes of ligands.[26]
In the ion-exchange column technique (IET),[27,28] a column of sulphonic acid-type resin is initially equilibrated with an electrolyte solution containing defined concentrations of Na, Mg, K and Ca. The resin is then calibrated by equilibrating it with solutions containing defined concentrations of the trace metal(s) of interest at a range of pH and a fixed ionic strength. A distribution coefficient, defining the ratio of resin-adsorbed metal to free metal ion, is calculated for the pH of the field water for which free-ion measurements are desired. The precalibrated column is then equilibrated with the field water of interest and the free metal ion concentrations computed using the distribution coefficient and the measured amounts of metal bound to the resin. Prior to equilibration with the column, the ionic strength of the field water is corrected to that of the calibration solutions using a multi-electrolyte stock containing Na, Mg, K and Ca nitrates.
In the ion-selective electrode (ISE) method, the electrode potential between an ion-selective electrode and a reference electrode is used for the direct determination of free copper ion concentration in a flow-through system.[29]
Permeable liquid membrane (PLM) techniques[30,31] are based on the measurement of metal flux, as generated by transport via a carrier molecule, through a hydrophobic membrane. The latter is sandwiched between a sample solution and a strip of receiving solution that contains a strong chelating agent to allow preconcentration. The metal of interest accumulates in the strip solution over time, the flux being directly related to either the concentration of free ion or to the free ion and labile metal complexes depending on the experimental setup.
The data set
The data set of measured speciation variables, together with the necessary additional information required as model inputs (pH, concentrations of major ions and DOM), was initially compiled from results for 2088 different samples. However, we restricted our analysis here to results that were realistic and complete, i.e. only accepting samples for which the values of ν were positive. This meant rejecting a total of 66 samples (3 %). The data set is summarised in Tables 1 and S1, with further details in references.[14–16]

|
Chemical speciation modelling
We used WHAM[10] incorporating Humic Ion-Binding Model VII[18] to perform the speciation calculations; previously, the open-ocean calculations[16] were done using Model VI.[17] Models VI and VII use structured formulations of discrete, chemically plausible binding sites for protons in humic and fulvic acids (HA, FA), in order to allow the creation of regular arrays of bidentate and tridentate binding sites for metals. Metal aquo ions (Al3+, Cu2+, Cd2+, etc.) and their first hydrolysis products (AlOH2+, CuOH+, CdOH+, etc.) compete with each other, and with protons, for binding. The same intrinsic equilibrium constant for binding to carboxyl or type A groups (KMA) is assumed to apply to the aquo ion and its first hydrolysis product. The constant for binding to weaker acid groups (KMB) is related to KMA, and the contributions of rarer ‘soft’ ligand atoms are factored in using a correlation with equilibrium constants for metal binding by NH3.[9,17] The intrinsic equilibrium constants are modified by empirical electrostatic terms that take into account the attractive or repulsive interactions between ions and the charged macromolecule. WHAM constants are derived from the results of numerous studies of proton and metal binding by isolated humic substances, together with linear free-energy relationships.[18] To make WHAM7, the humic ion-binding model is combined with an inorganic speciation model, the species list and constants for which were given by Tipping.[10] The inorganic reactions in this database are restricted to monomeric complexes of metals.
Temperature effects on reactions between inorganic species are taken into account using published or estimated enthalpy data, but in the absence of sufficient experimental information, reactions involving humic substances are assumed to be independent of temperature.
The effects of ionic strength on the inorganic reactions are taken into account using the extended Debye–Hückel equation, which relates the charge on an ion to an activity coefficient at a specified ionic strength. This approach is generally accepted for the low ionic strengths of fresh waters,[32] but in the modelling of high-ionic-strength systems, such as estuarine and marine systems, it is usually recommended to use the ‘mean salt method’ for calculating activity coefficients, rather than the methods normally applied to lower-ionic-strength fresh water systems. The ion-pairing model described by Millero and Schreiber[33] incorporates the mean salt method with Pitzer’s equations. We previously[15] compared the results from ion-pairing with the extended Debye–Hückel (EDH) equation, and found that for the great majority of the ions considered in the present work, differences in the free-ion activities calculated using the two methods were within ±16 %. An exception was nitrate, where the free ion activity was 24 % higher when the EDH method was employed. These differences are small in comparison with the variations when comparing modelled and measured values of trace metal free-ion concentrations, and so the use of the extended Debye–Hückel equation for all waters is justified, and consistent with several other studies that have used component-independent relationships for activities in marine systems.[34,35]
To run the model, we used measured total solute concentrations and pH. For all the freshwater samples, dissolved organic carbon (DOC) concentrations were available. For estuarine and coastal waters, some missing DOC concentrations had to be estimated from other studies carried out at the same location,[15] and for open ocean samples, [DOC] was taken to be the value at the nearest location for which data were available.[16]
The binding activity of DOM was estimated by assuming DOM to be 50 % carbon, and that 65 % of the DOM behaves like isolated FA whereas the rest is inert.[36,37] For example, a DOC concentration of 5 mg L–1 corresponds to [DOM] of 10 mg L–1, and so the concentration of FA for modelling is 6.5 mg L–1. All the predictions reported here were made using only humic-type DOM, i.e. no additional calculations were performed assuming the presence of anthropogenic ligands, e.g. ethylenediamine tetraacetic acid (EDTA).
For fresh waters from the field, we estimated truly dissolved FeIII concentrations with the empirical equation of Lofts et al.,[38] suitably modified for Humic Binding Model VII.[14] For samples where Fe was not the metal of interest with respect to speciation, the dissolved FeIII concentration was set to 10 nmol L–1 for estuarine and coastal sites and to 1 nmol L–1 for open-ocean sites. These procedures were adopted to take into account the significant competition by FeIII for binding at the stronger sites.[39]
Comparison of measured and model-predicted variables
We used three variables to compare measurements and model predictions. The ‘master’ variable is the free metal-ion concentration (Al3+, Cu2+, etc) denoted by [M] with units of moles per litre. The variable ν is used to quantify metal associated with DOM, with unit of moles per gram. A combination of these, ν/[M], with units of litres per gram, is useful because it permits comparisons of both strongly organically bound metals, for which the great majority of the metal is bound so that ν is well predicted, and the concern is about [M], and weakly bound ones, for which [M] is well predicted and the concern is about ν.
The expected behaviour of ν/[M] for the highly heterogeneous binding sites of DOM can be understood with reference to the theoretical calculations of Fig. 1. If DOM possessed only one type of binding site, with no electrostatic interactions, binding behaviour would be as shown in the upper three panels of Fig. 1. At low [M], ν increases such that the log–log plot is linear with a slope of unity, and log (ν/[M]) is constant, and equal to the equilibrium constant, when plotted against either log [M] or log ν. When the metal’s occupation of the sites passes ~10 % of the total (log ν ~ –3.3 in Fig. 1), log (ν/[M]) starts to decrease, and an increase in [M] causes a relatively smaller increase in ν. In other words, the affinity of the sites for metals effectively declines, owing to the fact that they are occupied already. We therefore can think of ν/[M] as a ‘local’ partition coefficient. The same behaviour is observed in the presence of competing metals (or the proton), which act to alter the effective equilibrium constant for binding.[9] For DOM with heterogeneous binding sites, the relationships are less straightforward (Fig. 1, lower panels). The slope of log ν against log [M] is no longer unity, because the system passes through sites with decreasing affinity, and the different metals display marked differences with respect to binding site heterogeneity, from the low heterogeneity of Zn to the high of Hg. Nonetheless, ν/[M] continues to operate as a ‘local’ partition coefficient, providing a measure of the propensity of the DOM to bind more metal.

|
We made comparisons of logarithmic values of the variables, (1) in order to be able to cover the large ranges, (2) because comparisons are better represented by ratios of observed to calculated values, rather than absolute differences, which are biased towards high values, and (3) because the data are noisy.
Measured and model-predicted values were compared by plotting log values and deriving regression slopes, forced through (0,0), and values of r2. Ideally the slopes and r2 values for all three variables would be close to unity. However, their values in practice inevitably depend on the ranges of the variables, and so they do not tell the full story. Therefore, we used as the main criteria of agreement (1) the root-mean-squared deviations (RMSD) between measured and predicted values, and (2) the average deviations (AD) between measured and predicted. Ideally, both of these should be zero. Because appreciation and discussion of these four measures (each applying to three variables) would be involved and complex, we used a single measure based on RMSD and AD. We refer to this as ΣRA, defined as:
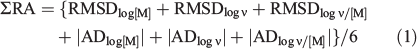
where the vertical lines indicate absolute values. Division by 6 simply reduces the total value so that it is more like the individual ones. Ideally, ΣRA should be zero. Because deviations are based on differences in log values, values of 0.5, 1 and 2 indicate overall agreement to approximately a factor of three, to one order of magnitude and to two orders of magnitude respectively.
Results and discussion
The WHAM model incorporating Humic Ion Binding Model VII provides a consistent means of calculating the speciation of dissolved metals in different surface waters, taking into account competition effects, including the effects of pH and ionic strength. The equilibrium and other constants used in the model are based on many laboratory measurements with isolated humic substances,[18] and therefore have not been calibrated with any field data. The factor of 65 % used to convert from DOM to FA (see above) is based on different field data from those considered here. Therefore, our speciation predictions are truly independent of the measured speciation variables, and the comparisons therefore provide an unbiased evaluation of the agreement or otherwise between measurements and predictions. In essence, we are taking information that many workers have obtained over many years from laboratory experiments, as encapsulated within WHAM, and comparing it with field observations.
The results cover the three major types of surface water, i.e. fresh, estuarine–coastal and the open ocean (Table 1). Therefore, they include wide ranges of ionic strength and of the concentrations of major metals (Mg, Ca) that compete significantly for binding to DOM. The DOM itself must vary in its composition and properties, given its different sources (terrestrial plants, freshwater and marine phytoplankton, microbes, etc). From Table S1, we see that whereas the estuarine–coastal and open-ocean samples refer to quite narrow ranges of pH, the range for fresh waters is wider, although acid systems are poorly represented, except in the Al data set. We have data for nine different metals, and these reflect research interest together with suitability for analysis. Some metals are better represented than others; for example, Al only appears in fresh waters, whereas most of the Fe measurements are for the open ocean. Copper is the most-analysed metal (35 % of all results) and covers all three of the surface-water types. To test model predictions and compare different analytical techniques against the model, the ideal data set would provide even coverage of conditions, a wide range of metals with similar amounts of data for each aquatic environment, and analysis by several methods on the same or similar samples common samples. Clearly, the present data set does not fully meet these characteristics, but it is large and wide-ranging and definitely useful for a meta-analysis.
Comparison of results by surface-water type
All data are plotted in Fig. 2, and statistical comparisons for the different types of surface water and the combined data set are given in Table 2. The results in Fig. 2 show strong correlations between predicted and measured values (high R2), which suggests that at the large scale, the general chemistry of metals in natural waters follows the expected trend from the laboratory-based model data. The regression depends considerably on data for Hg and Fe, both of which are very strongly bound (high ν/[M]), whereas the other metals show less variation at this scale.

|

|
Regression slope, RMSD and AD values show the same pattern for each surface-water type and for the combined data set (Table 2). The log [M] slopes are all fairly close to unity, but consistently too low, whereas the log ν slopes are consistently slightly greater than unity, and the log ν/[M] slopes are consistently less than unity. The AD values follow the pattern of the slope values, which means that predictions of log [M] tend to be greater, and those of log ν and log ν/[M] smaller, than the measured values.
Comparing the three types of surface water by the variable ΣRA, it is found that the best agreement between predictions and measurements is found for fresh waters (ΣRA = 0.52), followed by estuarine–coastal systems (ΣRA = 0.77) and then the open ocean (ΣRA = 1.24). The differences reflect increasing noise in the data rather than a systematic change in the relationship between measured and predicted values. This may simply reflect the greater analytical challenge of making speciation measurements in marine and marine-influenced waters.
For fresh waters, Lofts and Tipping[14] calculated how much error could be associated with the model predictions on the basis of uncertainties in input data, model parameters and variations in DOM binding properties. They then added uncertainty from the analytical precision of the speciation measurement. Only for Al, analysed by the CED method, could differences between measurements and predictions be accounted for by these factors, and it was concluded that there must be additional reasons for discrepancies in predicted and measured values. The same conclusion is likely to apply to results for estuarine and coastal systems and the open ocean, given that they show worse agreements than fresh waters. One possible reason for differences greater than those attributable to data uncertainty and DOM variations emerged in our analysis of data from estuarine and coastal waters[15] and is the presence of anthropogenic ligands, such as EDTA, which are found in the receiving waters of many industrial areas and can be extremely persistent in wastewater treatment plants and natural waters.[40] Better agreement with observed speciation was obtained when reasonable concentrations of EDTA were included in the model inputs (see also Baken et al.[41] and Ahmed et al.[42]). However, enhanced organic complexation due to anthropogenic ligands will apply only to a few of the samples considered here. Another possibility is that some natural waters contain high-affinity ligands that are not represented in humic-type DOM, as suggested in particular for marine waters.[43,44] Nonetheless, it is likely that in many or most cases, differences between predicted and measured speciation variables arise from faults in the measurement techniques, or in the model, or in both.
Comparison of results by metal
Fig. 3 compares results for the nine different metals for which we have data. The results for Al are good, helped by its relatively high concentrations, compared to those of the other metals, and the low pH values, which lead to readily measurable and predictable values of [M], ν and ν/[M]. In contrast, for FeIII total concentrations are rather low, pH values neutral or higher, and the samples are from estuarine and coastal and open ocean waters, and so [M] and ν values are much lower than those for Al, and measurement and prediction are harder. The predictions of [M] tend to be too high, more so at lower measured values. Values of ν for FeIII are well predicted because a very high proportion of truly dissolved Fe (i.e. not including oxide colloids) in natural waters is bound to organic matter, but because the [M] values are too high, values of ν/[M] are too low.

|
These trends for FeIII are also seen for Cu. For Ni, Zn, Cd and Pb, agreements of both log [M] and log ν are good at higher concentrations, but predicted log [M] is too high and predicted log ν and log ν/[M] too low at lower concentrations. Agreements for CoII are especially poor, perhaps because much of the metal was present as CoIII in marine samples.[16] The variable ν/[M] is poorly predicted for Cd in the open ocean.
The binding of Hg by natural organic matter is extremely strong[45] and consequently, there is almost perfect agreement between measured and predicted values of log ν (Fig. 3). This is the one case where predicted log [M] values are consistently lower than the measured ones, and consequently the model predicts higher values of log ν/[M]. Both measurements and predictions yield very high values of log ν/[M], of the order of 20, which is the highest of all the metals, ~5 log units greater than the values for the second most strongly bound metal, FeIII.
A plot of the predicted log ν/[M] averaged over all data for each metal v. the average of the measured values (Fig. 4) shows a strong relationship, which indicates that the order of reactivity of the metals, and their consequent binding to organic matter in natural waters follow the expectations of laboratory experiments, as encapsulated in WHAM7. (Only Co deviates strongly from the trend, as discussed above.) This reinforces the more general agreements shown in Fig. 2, confirming the basic assumption of our modelling approach, that the chemical speciation of metals in natural surface waters arises from equilibrium metal–ligand interactions involving humic-type organic matter.

|
Comparison of results by measurement technique
Fig. 5 summarises results obtained by different measurement methods. Table 3 compares results, including the variable ΣRA. The lowest ΣRA is produced by CED, because this method was used only for Al in fresh waters, providing favourable conditions for measurement, as discussed above. The next lowest ΣRA is given by PLM, but only on the basis of 19 points and 3 metals. Furthermore, the statistical variables for this method benefit from our removal of impossible results, i.e. negative values of ν.

|

|
The CL-ASV, DMT and IET methods give similar values of ΣRA (Table 3). DMT and IET both benefit statistically from the removal of impossible results, and neither produces any systematic deviations; the error terms are mainly due to noise. Predicted and measured results obtained by the CL-ASV method are in fair agreement, although there is a tendency for predicted free-ion values to be higher than measured ones, whereas the opposite applies to ν and ν/[M].
The DPASV method has been used widely in all three types of surface water; there is better agreement for fresh waters, but in estuarine and coastal samples, and more so in those from the open ocean, the model calculates [M] too high and ν and ν/[M] too low.
The most commonly applied technique (34 % of all analyses considered here) has been CL-CSV. The model predictions of [M] tend to be either in agreement or too high (Fig. 3), predictions of ν and ν/[M] either in agreement or too low. Consequently, the results overall are biased, leading to a high value of ΣRA.
The ISE results only apply to estuaries, and the results show very poor agreement between measurements and predictions. Because dissolved Cu is largely in the organically bound state, agreement is good for log ν but the predicted values of log [M] are much lower than the measured values and this leads to poor agreement in log ν/[M]. Reasons for this were discussed by Stockdale et al.[15] and the two most likely possibilities were first that the ‘dissolved’ Cu included significant amounts of colloidal material[46] and so the input concentration for modelling is too high, as well as the measured value of ν, and second that anthropogenic ligands were present in the samples, leading to more binding of the metal than would occur with only humic-type DOM.
The competitive ligand approach (i.e. the CL-ASV and CL-CSV methods) has been criticised on the grounds that equilibration is unlikely,[47] which would cause underestimation of [M], and overestimation of ν and ν/[M]; van Leeuwen and Town[47] showed that errors of several orders of magnitude in ν/[M] could result. Therefore, it is instructive to compare the results obtained by CL-ASV and CL-CSV with those obtained by the other six techniques (Table 4). Taken overall, the non-CL methods yield unbiased results, the average differences between measured and predicted values of log [M], log ν and log ν/[M] being close to zero. This applies whether or not the ISE data are included, but the standard deviations are appreciably reduced when the ISE results are omitted. In contrast, for the CL methods, the average difference in log [M] is –1.5, i.e. the measured values are lower than predicted ones by a factor of 30, the difference in log ν is 0.5 (factor of 3) and the difference in log ν/[M] is 2.0 (factor of 100). Only the few results for Hg (Fig. 3) show the opposite trend. On this basis, results from the CL methods differ from those obtained by other methods.

|
Deviations between measurements and predictions as a function of ν
A key aspect of DOM in its interactions with metals is binding site heterogeneity, and for many natural waters, especially seawater, where metal concentrations are low, the strongest binding sites are of primary importance. Therefore, it is worthwhile to consider how differences between measured and predicted values vary with the extent to which the strong and weaker sites are occupied, which can be most straightforwardly done by plotting the differences against log ν (Fig. 6). When all the data are considered, we find significant trends with log ν, so that as log ν becomes increasingly negative, measured log [M] values become more negative than the predicted ones, whereas the opposite is true for log ν and log ν/[M]. But if only data for log ν > –6 are considered (right-hand panels of Fig. 6), although statistically significant variations occur, they are much smaller than at low values of ν, a finding that fits with results of Ahmed et al.[48] For the data at log ν > –6 (838 samples), the value of ΣRA is quite small, at 0.34. Thus, there is little systematic bias and fair agreement between measured and predicted speciation variables at log ν > –6, whereas at lower loadings of the DOM, metal binding is stronger than predicted.

|
Model parameters for binding at the strong sites have been derived from laboratory studies in which the bias was towards higher loadings of the organic matter, i.e. high values of ν, largely because the determination of binding at high metal concentrations is easier and therefore within the capabilities of more researchers. Nonetheless, appreciable data at low values of ν were used by Tipping et al.[18] in parameterising Humic Ion-Binding Model VII. Of the 220 data sets employed, 41 contained values <–6 and 16 contained values <–8, the most negative value being –15. These cover the ranges found by measurement (Figs 3, 5, 6), and so it does not appear that the trends in Fig. 6 simply arise because data at low values of ν were not used in the model parameterisation. Indeed, the successful fitting of the laboratory data[18] means that the model’s formulation of strong binding sites present at low densities is adequate, i.e. there is presently no evidence for stronger binding sites in isolated humic substances that might explain the deviations between measurements and predictions in the field data analysed here. Therefore, if the measurements are accurate and unbiased, the trends in Fig. 6 would indicate the presence in field samples of ligands with stronger affinities than binding sites in isolated humic materials. These might include, for example, the metal-specific ligands thought to be produced by marine phytoplankton.[43,44]
The variable ν is central to the WHAM-FTox model of metal mixture toxicity,[49] in which the observed parallelism between WHAM-calculated binding of metals by HA and metal accumulation by aquatic organisms[50,51] is exploited to obtain a measure of toxic exposure in acute laboratory experiments. Toxicity of the accumulated metal is quantified by the variable FTox defined as Σαiνi where the αi are toxicity coefficients for the different metals. The highest value of αi derived so far is 2.1 × 106 g mol–1 (for cadmium effects on trout),[52] and toxic effects occur only when FTox exceeds ~2.0. Therefore, the minimum value of ν for toxicity is ~10–6 mol g–1. For less toxic metals, the required values of ν are much higher, of the order of 10–3 mol g–1 for Zn for example. Therefore, the range of ν over which measurements and model predictions are in reasonable agreement, i.e. >10–6 mol g–1 (Fig. 6), corresponds well to the range over which metal toxic effects are observed, implying that the WHAM7 model is appropriate for toxicity evaluation and prediction.
Future research
The comparisons of measured and predicted speciation variables presented here suggest that if we are to move closer to the reliable measurement and prediction of metal speciation in surface waters, three principal issues need to be resolved. The first is the apparent systematic difference between measurements made by the competitive ligand equilibrium techniques and those made by other methods. As noted above, there may theoretical reasons for this.[47] A way forward would be a concerted effort to compare techniques, building on previous work and ideas,[13,53–55] but covering a wider and more representative range of conditions and metals.
The second issue arises from our finding that model predictions differ most from measured values (obtained using techniques other than the CL-based ones) only at low values of ν (<10–6 mol g–1). The solution conditions under which such low loadings of the DOM occur are, of course, those that present the greatest analytical challenges, and therefore will produce the greatest uncertainties. Research is required to evaluate the analytical methodologies at low values of ν, for both natural water samples and laboratory-prepared solutions using isolated humic materials. The key requirement is to ascertain whether or not natural waters possess ligands with higher affinities for metals than humic-type DOM.
Third, the possible involvement in metal speciation of synthetic ligands such as EDTA in anthropogenically affected waters deserves more attention. By measuring their concentrations in surface-water samples for which speciation is determined analytically, and including them in the speciation calculations, the contributions of natural DOM and the synthetic ligands to metal speciation could be established.
The emphasis needs to be on analytical measurements, because ultimately, these are the basis of speciation knowledge, and further model developments based on existing data are unlikely to improve understanding or predictive capabilities. An alternative, more pragmatic, approach would be to accept the present field analytical data and use them to modify WHAM7, for example by creating additional strong binding sites, or altering competition effects, to make the results at low ν agree better. In our opinion, such modifications would be premature. We suggest that the best way forward is through coordinated analytical efforts and speciation method comparison, supported by speciation modelling.
Supplementary material
A summary of data used in the analysis is available in Table S1 and the source references are available in Table S2 (see http://www.publish.csiro.au/?act=view_file&file_id=EN15111_AC.pdf).
Acknowledgements
This work was supported by National Capability funding to the Centre for Ecology and Hydrology (E. Tipping, S. Lofts), and by the University of Leeds (A. Stockdale).
References
[1] T. M. Florence, G. E. Batley, P. Benes, Chemical speciation in natural waters. Crit. Rev. Anal. Chem. 1980, 9, 219.| Chemical speciation in natural waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXht1Kru7o%3D&md5=5452e43dc17f2c18ce4fc25b92aa3503CAS |
[2] P. G. C. Campbell, Interactions between trace elements and aquatic organisms: a critique of the free-ion activity model, in Metal Speciation and Bioavailability in Aquatic Systems (Eds A. Tessier and D.R. Turner) 1995, pp. 45–102 (Wiley: Chichester, UK).
[3] J. E. Groenenberg, S. Lofts, The use of assemblage models to describe trace element partitioning, speciation, and fate: a review. Environ. Toxicol. Chem. 2014, 33, 2181.
| The use of assemblage models to describe trace element partitioning, speciation, and fate: a review.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhsFKntrjP&md5=bb6ec4022067a322f9aeaa68b4a7ab10CAS | 24862928PubMed |
[4] P. G. C. Campbell, P. M. Stokes, Acidification and toxicity of metals to aquatic biota. Can. J. Fish. Aquat. Sci. 1985, 42, 2034.
| Acidification and toxicity of metals to aquatic biota.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL28Xotlelsw%3D%3D&md5=1f26c9c0ccd7d9e9a5f2431062a299cfCAS |
[5] M. Gledhill, E. P. Achterberg, K. Li, K. N. Mohamed, M. J. A. Rijkenberg, Influence of ocean acidification on the complexation of iron and copper by organic ligands in estuarine waters. Mar. Chem. 2015,
| Influence of ocean acidification on the complexation of iron and copper by organic ligands in estuarine waters.Crossref | GoogleScholarGoogle Scholar |
[6] F. J. Millero, R. Woosley, B. Ditrolio, J. Waters, Effect of ocean acidification on the speciation of metals in seawater. Oceanogr. 2009, 22, 72.
| Effect of ocean acidification on the speciation of metals in seawater.Crossref | GoogleScholarGoogle Scholar |
[7] M. Pesavento, G. Alberti, R. Biesuz, Analytical methods for determination of free metal ion concentration, labile species fraction and metal complexation capacity of environmental waters: a review. Anal. Chim. Acta 2009, 631, 129.
| Analytical methods for determination of free metal ion concentration, labile species fraction and metal complexation capacity of environmental waters: a review.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsFWis73L&md5=558d25cba093deffd9e9232673c8391cCAS | 19084618PubMed |
[8] J. Buffle, G. Horvai, In Situ Monitoring of Aquatic Systems: Chemical Analysis and Speciation 2000 (Wiley: New York).
[9] E. Tipping, Cation Binding by Humic Substances 2002 (Cambridge University Press: Cambridge, UK).
[10] E. Tipping, WHAM – a chemical equilibrium model and computer code for waters, sediments and soils incorporating a discrete-site electrostatic model of ion-binding by humic substances. Comput. Geosci. 1994, 20, 973.
| WHAM – a chemical equilibrium model and computer code for waters, sediments and soils incorporating a discrete-site electrostatic model of ion-binding by humic substances.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXhtlyhtrY%3D&md5=7ded37c9af50999f549148d9595f8de2CAS |
[11] C. J. Milne, D. G. Kinniburgh, W. H. Van Riemsdijk, E. Tipping, Generic NICA–Donnan model parameters for metal-ion binding by humic substances. Environ. Sci. Technol. 2003, 37, 958.
| Generic NICA–Donnan model parameters for metal-ion binding by humic substances.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXosVWgsQ%3D%3D&md5=2eda2310a7221ae42b73f399e3b9e8b5CAS | 12666927PubMed |
[12] J. W. Guthrie, N. M. Hassan, M. S. A. Salam, I. I. Fasfous, C. A. Murimboh, J. Murimboh, C. L. Chakrabarti, D. C. Grégoire, Complexation of Ni, Cu, Zn, and Cd by DOC in some metal-impacted freshwater lakes: a comparison of approaches using electrochemical determination of free-metal ion and labile complexes and a computer speciation model, WHAM V and VI. Anal. Chim. Acta 2005, 528, 205.
| Complexation of Ni, Cu, Zn, and Cd by DOC in some metal-impacted freshwater lakes: a comparison of approaches using electrochemical determination of free-metal ion and labile complexes and a computer speciation model, WHAM V and VI.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXkslyhsg%3D%3D&md5=4f6eabd66f3094be1b10367828dc9a03CAS |
[13] E. R. Unsworth, K. W. Warnken, H. Zhang, W. Davison, F. Black, J. Buffle, J. Cao, R. Cleven, J. Galceran, P. Gunkel, E. Kalis, D. Kistler, H. P. van Leeuwen, M. Martin, S. Noël, Y. Nur, N. Odzak, J. Puy, W. van Riemsdijk, L. Sigg, E. J. M. Temminghoff, M.-L. Tercier-Waeber, S. Topperwien, R. M. Town, L. Weng, H. B. Xue, Model predictions of metal speciation in freshwaters compared to measurements by in situ techniques. Environ. Sci. Technol. 2006, 40, 1942.
| Model predictions of metal speciation in freshwaters compared to measurements by in situ techniques.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtlSku7o%3D&md5=317db11fd744633a6ba8cae2f0cd8eaeCAS | 16570619PubMed |
[14] S. Lofts, E. Tipping, Assessing WHAM/Model VII against field measurements of free metal ion concentrations: model performance and the role of uncertainty in parameters and inputs. Environ. Chem. 2011, 8, 501.
| Assessing WHAM/Model VII against field measurements of free metal ion concentrations: model performance and the role of uncertainty in parameters and inputs.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlykt73J&md5=6d5203022a7c53649a5ef04da5785dcbCAS |
[15] A. Stockdale, E. Tipping, S. Lofts, Dissolved trace metal speciation in estuarine and coastal waters: comparison of WHAM/Model VII predictions with analytical results. Environ. Toxicol. Chem. 2015, 34, 53.
| Dissolved trace metal speciation in estuarine and coastal waters: comparison of WHAM/Model VII predictions with analytical results.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXitFaqtLfF&md5=57eef4367bc789fdb5ce2e81a0779c9fCAS | 25387688PubMed |
[16] A. Stockdale, E. Tipping, J. Hamilton-Taylor, S. Lofts, Trace metals in the open oceans: speciation modelling based on humic-type ligands. Environ. Chem. 2011, 8, 304.
| Trace metals in the open oceans: speciation modelling based on humic-type ligands.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptVWrsbc%3D&md5=79d2c4570666578baa92db699bb728f0CAS |
[17] E. Tipping, V. I. Humic Ion Binding Model, An improved description of the interactions of protons and metal ions with humic substances. Aquat. Geochem. 1998, 4, 3.
| An improved description of the interactions of protons and metal ions with humic substances.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXntlSjuro%3D&md5=cf0c6c40511e5000b173c62e5266b947CAS |
[18] E. Tipping, S. Lofts, J. E. Sonke, Humic Ion-Binding Model VII: a revised parameterisation of cation-binding by humic substances. Environ. Chem. 2011, 8, 225.
| Humic Ion-Binding Model VII: a revised parameterisation of cation-binding by humic substances.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptVWrsL0%3D&md5=bda1097b974d554ee7c343f673c6e24bCAS |
[19] C. T. Driscoll, A procedure for the fractionation of aqueous aluminium in dilute acid waters. Int. J. Environ. Anal. Chem. 1984, 16, 267.
| A procedure for the fractionation of aqueous aluminium in dilute acid waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXhvFOms78%3D&md5=425c8c68872e4b822a813f80c2a2ac1fCAS |
[20] C. A. Backes, E. Tipping, An evaluation of the use of cation-exchange resin for the determination of organically complexed Al in natural acid waters. Int. J. Environ. Anal. Chem. 1987, 30, 135.
| An evaluation of the use of cation-exchange resin for the determination of organically complexed Al in natural acid waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXls1WhsLo%3D&md5=7278d545f50bf3a0af175d3b0e1cd380CAS |
[21] G. Scarano, E. Bramanti, A. Zirino, Determination of copper complexation in seawater by a ligand competition technique with voltammetric measurement of the labile metal fraction. Anal. Chim. Acta 1992, 264, 153.
| Determination of copper complexation in seawater by a ligand competition technique with voltammetric measurement of the labile metal fraction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XltFyjtLw%3D&md5=2f117431becd1f65730bbc31b7c6a402CAS |
[22] F. L. L. Muller, Interactions of copper, lead and cadmium with the dissolved, colloidal and particulate components of estuarine and coastal waters. Mar. Chem. 1996, 52, 245.
| Interactions of copper, lead and cadmium with the dissolved, colloidal and particulate components of estuarine and coastal waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XjsFOntb8%3D&md5=f7484f59854ce2d0b41003cc5cad5b78CAS |
[23] D. Tang, K. W. Warnken, P. H. Santschi, Organic complexation of copper in surface waters of Galveston Bay. Limnol. Oceanogr. 2001, 46, 321.
| Organic complexation of copper in surface waters of Galveston Bay.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXjtVWgsLY%3D&md5=8e46e927edc5ba79861b98b3fa7a1eabCAS |
[24] C. M. G. van den Berg, Evidence for organic complexation of iron in seawater. Mar. Chem. 1995, 50, 139.
| Evidence for organic complexation of iron in seawater.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXotVaqu78%3D&md5=4101779cf873e2d3a2cbed4c5060f882CAS |
[25] E. J. J. Kalis, L. Weng, F. Dousma, E. J. M. Temminghoff, W. H. Van Riemsdijk, Measuring free metal ion concentrations in situ in natural waters using the Donnan membrane technique. Environ. Sci. Technol. 2006, 40, 955.
| Measuring free metal ion concentrations in situ in natural waters using the Donnan membrane technique.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtlars7rK&md5=05ff63516e243bb72f201b3e7303e1d9CAS |
[26] K. H. Coale, K. W. Bruland, Copper complexation in the north-east Pacific. Limnol. Oceanogr. 1988, 33, 1084.
| Copper complexation in the north-east Pacific.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXksVCluw%3D%3D&md5=7f7e91fb3bde3102334e068a20b07c56CAS |
[27] C. Fortin, P. G. C. Campbell, An ion-exchange technique for free-metal ion measurements (Cd2+, Zn2+): applications to complex aqueous media. Int. J. Environ. Anal. Chem. 1998, 72, 173.
| An ion-exchange technique for free-metal ion measurements (Cd2+, Zn2+): applications to complex aqueous media.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXis12gtLY%3D&md5=558cde496f597fdaaa6a6089bda8f0b1CAS |
[28] C. Fortin, Y. Couillard, B. Vigneault, P. G. C. Campbell, Determination of free Cd, Cu and Zn concentrations in lake waters by in situ diffusion followed by column equilibration ion-exchange. Aquat. Geochem. 2010, 16, 151.
| Determination of free Cd, Cu and Zn concentrations in lake waters by in situ diffusion followed by column equilibration ion-exchange.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFyjt7rN&md5=caf11fd6e783b47736715c86f6abf3e0CAS |
[29] A. Zirino, D. A. Van der Weele, S. L. Belli, R. DeMarco, D. J. Mackey, Direct measurement of CuIIaq in seawater at pH 8 with the jalpaite ion-selective electrode. Mar. Chem. 1998, 61, 173.
| Direct measurement of CuIIaq in seawater at pH 8 with the jalpaite ion-selective electrode.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXktVymsbY%3D&md5=f4943244e1870a69b224fdad712476e4CAS |
[30] S. Bayen, K. J. Wilkinson, J. Buffle, The permeation liquid membrane as a sensor for free nickel in aqueous samples. Analyst 2007, 132, 262.
| The permeation liquid membrane as a sensor for free nickel in aqueous samples.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXitVOksrk%3D&md5=d3264d33ab396acc4b2dbb2ff8640f3fCAS | 17325760PubMed |
[31] N. Parthasarathy, M. Pelletier, J. Buffle, Hollow fiber-based supported liquid membrane: a novel analytical system for trace metal analysis. Anal. Chim. Acta 1997, 350, 183.
| Hollow fiber-based supported liquid membrane: a novel analytical system for trace metal analysis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXlsVWgt70%3D&md5=169593e37fcf08a45d0b1800c9bb30c1CAS |
[32] W. Stumm, J. J. Morgan, Aquatic Chemistry, 3rd edn 1996 (Wiley: New York).
[33] F. J. Millero, D. R. Schreiber, Use of the ion-pairing model to estimate activity coefficients of the ionic components of natural waters. Am. J. Sci. 1982, 282, 1508.
| Use of the ion-pairing model to estimate activity coefficients of the ionic components of natural waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3sXksVejsL4%3D&md5=e7b3be7c14a9ef6699082c3a085ffd5dCAS |
[34] R. H. Byrne, L. R. Kump, K. J. Cantrell, The influence of temperature and pH on trace metal speciation in seawater. Mar. Chem. 1988, 25, 163.
| The influence of temperature and pH on trace metal speciation in seawater.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXhtlWqtg%3D%3D&md5=60df343f250dfb55f0754f635a47f964CAS |
[35] A. De Robertis, C. De Stefano, S. Sammartano, Equilibrium studies in natural fluids: a chemical speciation model for the major constituents of sea water. Chem. Spec. Bioavail. 1994, 6, 65.
| 1:CAS:528:DyaK2MXitVGjsr0%3D&md5=8d1272eb824ba8becb64e386d1ac5a78CAS |
[36] S. E. Bryan, E. Tipping, J. Hamilton-Taylor, Comparison of measured and modelled copper binding by natural organic matter in freshwaters. Comp. Biochem. Physiol. C – Toxicol. Pharmacol. 2002, 133, 37.
| Comparison of measured and modelled copper binding by natural organic matter in freshwaters.Crossref | GoogleScholarGoogle Scholar | 1:STN:280:DC%2BD38nmsFGhtA%3D%3D&md5=2dd391377220c31e2717c75fd746375cCAS | 12356515PubMed |
[37] E. Tipping, C. Woof, M. A. Hurley, Humic substances in acid surface waters – modeling aluminium binding, contribution to ionic charge-balance, and control of pH. Water Res. 1991, 25, 425.
| Humic substances in acid surface waters – modeling aluminium binding, contribution to ionic charge-balance, and control of pH.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXitlClsr4%3D&md5=a65a05259716c28016b79eba2658bd29CAS |
[38] S. Lofts, E. Tipping, J. Hamilton-Taylor, The chemical speciation of FeIII in freshwaters. Aquat. Geochem. 2008, 14, 337.
| The chemical speciation of FeIII in freshwaters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXht12gtLzN&md5=0c5f0efcdcd768cd55fdcd8bfc9f6428CAS |
[39] E. Tipping, C. Rey-Castro, S. E. Bryan, J. Hamilton-Taylor, AlIII and FeIII binding by humic substances in freshwaters, and implications for trace metal speciation. Geochim. Cosmochim. Acta 2002, 66, 3211.
| AlIII and FeIII binding by humic substances in freshwaters, and implications for trace metal speciation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XmslKktbk%3D&md5=c0d4c88b8a3edd2164fe3daa206e62b7CAS |
[40] M. Sillanpää, Environmental fate of EDTA and DTPA, in Reviews of Environmental Contamination and Toxicology (Ed. G. W. Ware) 1997, Vol. 152, pp. 85–111 (Springer: New York).
[41] S. Baken, F. Degryse, L. Verheyen, R. Merckx, E. Smolders, Metal complexation properties of freshwater dissolved organic matter are explained by its aromaticity and by anthropogenic ligands. Environ. Sci. Technol. 2011, 45, 2584.
| Metal complexation properties of freshwater dissolved organic matter are explained by its aromaticity and by anthropogenic ligands.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjtFKqtbo%3D&md5=1a4ba87011f61693f0b98519fda2c0b6CAS | 21405071PubMed |
[42] I. A. M. Ahmed, J. Hamilton-Taylor, M. Bieroza, H. Zhang, W. Davison, Improving and testing geochemical speciation predictions of metal ions in natural waters. Water Res. 2014, 67, 276.
| Improving and testing geochemical speciation predictions of metal ions in natural waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhs1WqsLfO&md5=c9ad572487aedf514c6a3cd08f42a5d4CAS |
[43] M. L. Wells, Marine colloids and trace metals, in Biogeochemistry of Marine Dissolved Organic Matter (Eds D. A. Hansell, C. A. Carlson) 2002, pp. 367–404 (Academic Press: London).
[44] F. M. M. Morel, N. M. Price, The biogeochemical cycles of trace metals in the oceans. Science 2003, 300, 944.
| The biogeochemical cycles of trace metals in the oceans.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjsVyjsLk%3D&md5=c9e39d3fdfa28c650219e4b0f457a654CAS |
[45] E. Tipping, Modelling the interactions of HgII and methylmercury with humic substances using WHAM/Model VI. Appl. Geochem. 2007, 22, 1624.
| Modelling the interactions of HgII and methylmercury with humic substances using WHAM/Model VI.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXpt1yls7w%3D&md5=404de1eb1931c9b177c74f82425e165bCAS |
[46] R. S. Eriksen, D. J. Mackey, R. van Dam, B. Nowak, Copper speciation and toxicity in Macquarie Harbour, Tasmania: an investigation using a copper ion-selective electrode. Mar. Chem. 2001, 74, 99.
| Copper speciation and toxicity in Macquarie Harbour, Tasmania: an investigation using a copper ion-selective electrode.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXhvFWls7k%3D&md5=a5d05cc35c9b6981ee203af6517c8b82CAS |
[47] H. P. van Leeuwen, R. M. Town, Kinetic limitations in measuring stabilities of metal complexes by competitive ligand exchange adsorptive stripping voltammetry (CLEAdSV). Environ. Sci. Technol. 2005, 39, 7217.
| Kinetic limitations in measuring stabilities of metal complexes by competitive ligand exchange adsorptive stripping voltammetry (CLEAdSV).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXns1aisLk%3D&md5=32716e96ac260ec7bd97e55bc9ce7c1cCAS | 16201651PubMed |
[48] I. A. M. Ahmed, J. Hamilton–Taylor, S. Lofts, J. C. L. Meeussen, C. Lin, H. Zhang, W. Davison, Testing copper-speciation predictions in freshwaters over a wide range of metal–organic matter ratios. Environ. Sci. Technol. 2013, 47, 1487.
| Testing copper-speciation predictions in freshwaters over a wide range of metal–organic matter ratios.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhslGhsw%3D%3D&md5=581330935373ead076e7eb0b0097e6b5CAS |
[49] A. Stockdale, E. Tipping, S. Lofts, S. J. Ormerod, W. H. Clements, R. Blust, Toxicity of proton–metal mixtures in the field: linking stream macroinvertebrate species diversity to chemical speciation and bioavailability. Aquat. Toxicol. 2010, 100, 112.
| Toxicity of proton–metal mixtures in the field: linking stream macroinvertebrate species diversity to chemical speciation and bioavailability.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFGjurbM&md5=6a9579e1eca139935498e8aeee3d20feCAS | 20701986PubMed |
[50] E. Tipping, C. D. Vincent, A. J. Lawlor, S. Lofts, Metal accumulation by stream bryophytes, related to chemical speciation. Environ. Pollut. 2008, 156, 936.
| Metal accumulation by stream bryophytes, related to chemical speciation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhsVSgtb3J&md5=50f02427f9ba4cb31d1e8d280e4874f1CAS | 18541353PubMed |
[51] E. Tipping, S. Lofts, Metal mixture toxicity to aquatic biota in laboratory experiments: application of the WHAM-FTOX model. Aquat. Toxicol. 2013, 142–143, 114.
| Metal mixture toxicity to aquatic biota in laboratory experiments: application of the WHAM-FTOX model.Crossref | GoogleScholarGoogle Scholar | 23994673PubMed |
[52] E. Tipping, S. Lofts, Testing WHAM-FTOX with laboratory toxicity data for mixtures of metals (Cu, Zn, Cd, Ag, Pb). Environ. Toxicol. Chem. 2015, 34, 788.
| Testing WHAM-FTOX with laboratory toxicity data for mixtures of metals (Cu, Zn, Cd, Ag, Pb).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXltFSksLs%3D&md5=a2856b734cc1fe90ff8cc8c60e923612CAS | 25318827PubMed |
[53] L. Sigg, F. Black, J. Buffle, J. Cao, R. Cleven, W. Davison, J. Galceran, P. Gunkel, E. Kalis, D. Kistler, M. Martin, S. Noel, Y. Nur, N. Odzak, J. Puy, W. Van Riemsdijk, E. Temminghoff, M. L. Tercier-Weber, S. Toepperwien, R. M. Town, E. Unsworth, K. W. Warnken, L. P. Weng, H. B. Xue, H. Zhang, Comparison of analytical techniques for dynamic trace metal speciation in natural freshwaters. Environ. Sci. Technol. 2006, 40, 1934.
| Comparison of analytical techniques for dynamic trace metal speciation in natural freshwaters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xht1yhsLk%3D&md5=d5016d472512f001bdd15dec070ccd7aCAS | 16570618PubMed |
[54] J. Feldmann, P. Salaün, E. Lombi, Critical review perspective: elemental speciation analysis methods in environmental chemistry – moving towards methodological integration. Environ. Chem. 2009, 6, 275.
| Critical review perspective: elemental speciation analysis methods in environmental chemistry – moving towards methodological integration.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVSlurfK&md5=3fede31ae41e217f2c5bc5bb8cb355ddCAS |
[55] A. M. Mota, J. P. Pinheiro, M. L. S. Goncalves, Electrochemical methods for speciation of trace elements in marine waters. Dynamic aspects. J. Phys. Chem. A 2012, 116, 6433.
| Electrochemical methods for speciation of trace elements in marine waters. Dynamic aspects.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xmt1ektb0%3D&md5=38f02b3dfdbc2401db343e8312b8bdf4CAS | 22540875PubMed |