

Glyoxal secondary organic aerosol chemistry: effects of dilute nitrate and ammonium and support for organic radical–radical oligomer formation
Jeffrey R. Kirkland A , Yong B. Lim A , Yi Tan A C , Katye E. Altieri B and Barbara J. Turpin A DA Department of Environmental Sciences, Rutgers University, New Brunswick, NJ 08901, USA.
B Department of Geosciences, Princeton University, Princeton, NJ 08540, USA.
C Present address: The Center for Atmospheric Particle Studies, Carnegie Mellon University, Pittsburgh, PA 15213, USA.
D Corresponding author. Email: turpin@envsci.rutgers.edu
Environmental Chemistry 10(3) 158-166 https://doi.org/10.1071/EN13074
Submitted: 30 March 2013 Accepted: 25 May 2013 Published: 28 June 2013
Environmental context. Atmospheric waters (clouds, fogs and wet aerosols) are media in which gases can be converted into particulate matter. This work explores aqueous transformations of glyoxal, a water-soluble gas with anthropogenic and biogenic sources. Results provide new evidence in support of previously proposed chemical mechanisms. These mechanisms are beginning to be incorporated into transport models that link emissions to air pollution concentrations and behaviour.
Abstract. Glyoxal (GLY) is ubiquitous in the atmosphere and an important aqueous secondary organic aerosol (SOA) precursor. At dilute (cloud-relevant) organic concentrations, OH• radical oxidation of GLY has been shown to produce oxalate. GLY has also been used as a surrogate species to gain insight into radical and non-radical reactions in wet aerosols, where organic and inorganic concentrations are very high (in the molar region). The work herein demonstrates, for the first time, that tartarate forms from GLY + OH•. Tartarate is a key product in a previously proposed organic radical–radical reaction mechanism for oligomer formation from GLY oxidation. Previously published model predictions that include this GLY oxidation pathway suggest that oligomers are major products of OH• radical oxidation at the high organic concentrations found in wet aerosols. The tartarate measurements herein provide support for this proposed oligomer formation mechanism. This paper also demonstrates, for the first time, that dilute (cloud or fog-relevant) concentrations of inorganic nitrogen (i.e. ammonium and nitrate) have little effect on the GLY + OH• chemistry leading to oxalate formation in clouds. This, and results from previous experiments conducted with acidic sulfate, increase confidence that the currently understood dilute GLY + OH• chemistry can be used to predict GLY SOA formation in clouds and fogs. It should be recognised that organic–inorganic interactions can play an important role in droplet evaporation chemistry and in wet aerosols. The chemistry leading to SOA formation in these environments is complex and remains poorly understood.
Introduction
Atmospheric waters (e.g. clouds, fogs and wet aerosols) serve as a medium in which water-soluble organic compounds react. These water-soluble organics are abundant in the atmosphere due to the gas-phase fragmentation and oxidation of primary emissions. Reactions in atmospheric waters can produce low volatility products that remain in the particle phase upon droplet evaporation (e.g. oxalate, glycolate, oligomers, epoxide sulfates and imidazoles).[1–11] As a result, gas-phase followed by aqueous-phase chemistry contributes to secondary organic aerosol (SOA).[12,13] SOA is also formed when semi- and low-volatility products of gas-phase chemistry sorb to particulate organic matter[14,15] (here SOAgas is used to denote SOA formed by this latter pathway and SOAaq to denote SOA formed with a contribution from aqueous-phase chemistry).
Although the magnitude of SOAaq remains uncertain, the inclusion of aqueous organic chemistry in global models has led to a substantial enhancement of aerosol burden,[16–19] improved correlations between models and measurements in the North-eastern United States[20] and helped to explain the highly oxidised nature of atmospheric organic aerosols.[13,21] (SOAgas is not as oxidised as measured atmospheric aerosol.) SOAaq and SOAgas form from different precursors under different atmospheric conditions and have different properties and behaviour. For example, because the precursors of SOAaq are water-soluble gases with high O/C ratios, SOAaq is expected to have high O/C ratios, be more hygroscopic and be better able to serve as cloud condensation nuclei than other organics.[22] For these reasons, an improved understanding of SOAaq is needed to link emissions to ambient aerosol concentrations, composition and effects through predictive models that enable the development of effective air quality management strategies. This ultimate goal motivates the work reported herein.
Glyoxal (GLY) is ubiquitous in the atmosphere and an important SOAaq precursor because GLY is highly water soluble, reactive to OH• radicals and forms oligomers at high concentrations (millimolar and above). GLY is the smallest dicarbonyl and forms through the gas-phase photooxidation of alkene and aromatic compounds.[23] The largest global source of GLY is the gas-phase oxidation of isoprene (biogenic).[24] A major anthropogenic source of GLY is the oxidation of acetylene.[25] Although its marine sources are uncertain, satellite measurements reveal substantial concentrations of GLY in marine air.[26] Acetylene, with an atmospheric lifetime near 18 days,[16] is one possible GLY source in marine air.[27] GLY (HGLY: 3 × 105 M atm–1)[28] readily partitions into atmospheric waters where it is present at concentrations from one to several hundred micromolar and can react further.[29–31] GLY can react in atmospheric waters to form lower volatility products (e.g. oxalate, oligomers, organosulfate and imidazole).[6,8,32–34] Because these compounds are found predominantly in the condensed phase in the atmosphere, aqueous reaction of GLY contributes to SOAaq.[32,33]
Organic nitrogen and sulfur compounds have been observed to form in highly concentrated aqueous solutions relevant to wet aerosols.[6,34–37] Evidence for the photochemical formation of organosulfates from GLY and other aldehydes is provided through studies of bulk sulfate solutions and through smog chamber experiments with ammonium sulfate seed particles and UV irradiation.[6,35,36] Aerosol mimic solutions containing GLY and inorganic ammonium salts have yielded products with carbon–nitrogen bonds in the absence of major atmospheric oxidants (i.e. O3, OH• and NO3•).[37] In addition, imidazole formation has been detected in (dark) concentrated aldehyde–ammonium sulfate solutions and chamber experiments.[6,34] Inorganic and organic concentrations are orders of magnitude smaller in clouds and fogs. Although experiments have been performed to examine the effect of dilute (cloud-relevant) concentrations of acidic sulfate on GLY chemistry,[33] to our knowledge the degree to which GLY + OH• chemistry is altered by the presence of cloud-relevant concentrations of nitrate or ammonium has not been examined. Inorganic nitrate and ammonium are found in atmospheric cloud and fog waters (e.g. ~1 µM–3 mM for NO3– and NH4+ in fogs),[38–41] and organic nitrogen species have been measured in California fogs[42] and New Jersey rainwater.[41] Although organic nitrogen is clearly formed through gas-phase chemistry and there is evidence for its formation in aerosols, it could plausibly also form through cloud and fog chemistry. In this work, we conducted chemical modelling and aqueous GLY + OH• experiments with cloud and fog-relevant concentrations of inorganic nitrogen (i.e. ammonium sulfate or nitric acid) to determine whether the commonly understood dilute GLY + OH• chemistry leading to oxalate (and therefore SOA) is altered by the presence of cloud-relevant concentrations of ammonium sulfate or nitric acid.
Tan et al.[33] studied the aqueous OH• radical oxidation of GLY at cloud-relevant and higher concentrations (30– 3000 µM). At cloud-relevant (dilute) concentrations, oxalate was the major product and the addition of cloud and fog-relevant concentrations of H2SO4 had little effect on the chemistry. At higher (3000 µM) concentrations Tan et al.[33] observed the formation of products with higher carbon numbers than the precursor, GLY. These higher carbon number products only formed in the presence of OH• radicals. By accounting for organic radical–radical reactions in the GLY oxidation mechanism, Lim et al.[21] were able to reproduce the concentration dynamics of the major products in the experiments of Tan et al.[33] Using GLY as a surrogate for the behaviour of dissolved organics at the high (1–10 M) concentrations found in wet aerosols, Lim et al.[21] predicted that oligomers would be the main products of organic OH• radical oxidation in wet aerosols. Tartarate is a key product in the organic radical–radical mechanism proposed by Lim et al.[21] However, tartarate was not definitively measured in the Tan et al.[33] experiments because tartarate and malonate co-eluted and thus could not be distinguished. In model runs that simulated the experimental conditions and included organic radical–radical chemistry,[21] tartarate forms early in the experiment, peaking after ~25 min. Malonate formation is minor and slower, because it is formed through radical–radical reactions followed by acid catalysed dehydration. The experimental results reported herein provide definitive evidence for the formation of tartarate from GLY + OH•.
This paper examines for the first time the effect of cloud and fog relevant concentrations of inorganic nitrogen on the aqueous oxidation of GLY. The finding that oxalate is not affected by cloud-relevant concentrations of ammonium sulfate and nitric acid increases confidence that the currently understood dilute GLY + OH• chemistry can be used to predict GLY SOA formation in clouds and fogs. This paper also provides critical laboratory evidence for oligomer formation through organic radical–radical reactions; this chemistry is evident in 1 mM GLY experiments and is the dominant OH• radical oxidation product at the much higher (1–10 M) organic concentrations present in wet aerosols.[21]
Experimental
Batch reactions
Bulk aqueous phase reactions of GLY (1 mM) and the hydroxyl radical (~10–12 M, modelled OH•) with nitric acid (1.68 mM) or ammonium sulfate (840 μM) and control experiments were conducted in a 1-L glass reaction vessel, as described in detail elsewhere.[1] OH• radicals were continuously produced in situ by direct photolysis of hydrogen peroxide (5 mM) using a 254-nm mercury lamp (Heraeus Noblelight, Inc. Duluth, GA) placed in a quartz immersion well in the centre of the reaction vessel. Surrounding the reaction vessel was a water jacket that maintained a temperature of 26 ± 2 °C. The reaction vessel was wrapped in aluminium foil to eliminate exposure to ambient light. The following control experiments were conducted: GLY + OH• without nitric acid or ammonium sulfate, GLY + HNO3 + UV, GLY + (NH4)2SO4 + UV, GLY + HNO3 + H2O2, GLY + (NH4)2SO4 + H2O2, GLY + HNO3, GLY + (NH4)2SO4 and H2O2 + UV.
OH• radical concentrations
Aqueous hydroxyl radical concentrations in clouds and fogs are predicted to be 10–12 to 10–13 M.[43] Hydroxyl radical concentrations in these GLY experiments were as follows: [OH•]average = (1 ± 2) × 10–12 M, [OH•]initial = 7.8 × 10–13 M and [OH•]final = 6.0 × 10–12 M. These concentrations were estimated using the GLY photooxidation model of Lim et al.[21] and a H2O2 photolysis rate constant (k1) equal to 1.0 × 10–4 s–1. The value of k1, which depends on the photon flux of the lamp, was determined by fitting modelled to measured H2O2 concentrations in H2O2 + UV control experiments (Fig. S1). Hydrogen peroxide was quantified by the triiodide method and analysed with a UV-Vis spectrometer.[44]
Product analysis and analytical methods
Discrete samples from aqueous experiments were analysed by ion chromatography (IC, Dionex ICS 3000; IonPac AS11-HC column; 30 °C, AG11-HC guard column; conductivity detector; 35 °C), IC coupled to electrospray ionisation mass spectrometry (ESI-MS) (HP–Agilent 1100; negative ion mode) and Fourier transform ion cyclotron resonance mass spectrometry (FT-ICR-MS) (Thermo-Finnigan LTQ-XL: positive and negative ion modes, Woods Hole Oceanographic Institution Mass Spectrometry Facility) as described in detail previously and briefly below.[41,45] Discrete samples for IC and IC/ESI-MS (~1.2 mL) were analysed immediately (same day), whereas discrete samples for FT-ICR-MS (~5 mL; 40-min time points only) were frozen at –20 °C for later analysis as recommended by Seitzinger et al.[46] for preservation of stored atmospheric water samples for mass spectrometric analysis.
Carboxylic acids and inorganic anions were quantified by IC with five point calibration curves (accept R2 > 0.98) developed from authentic standards, including: glycolate (5.8 min), formate (6.7 min), glyoxylate (9.5 min), succinate (20.2 min), tartarate (20.8 min) and oxalate (24.2 min). Note that glycolate co-elutes with acetate, succinate with malate, and tartarate with malonate. These peaks were quantified as glycolate, succinate and tartarate respectively. Nitrate (16.7 min) and sulfate (22.8 min) are also detected within the 40-min IC program.
Negative ionisation mode mass spectrometric analysis (50–1000 amu) of each peak in the sample chromatograms was performed by IC/ESI-MS as described by Tan et al.[45] Briefly, the IC conductivity detector effluent (0.4 mL min–1) was directed into the ESI-MS with 40 % of 0.05 % formic acid in water and 60 % methanol mobile phase (0.22 mL min–1). After IC separation, mass spectrometric analysis was performed with a fragmentor voltage of 40 V and a capillary voltage of 3000 V (nitrogen drying gas; 10 L min–1; 350 °C). Mass assignments were verified with a mixed standard analysed at the beginning and end of each sequence.
A discrete sample (40 min into the reaction) from each experiment, control experiment and a 1 mM GLY standard was also analysed by FT-ICR-MS in positive and negative ionisation modes (mass resolution 100 000) to determine elemental formulae of reaction products from 50 to 500 amu. A syringe pump delivered analyte into the spray chamber at 4 µL min–1 with a capillary temperature of 260 °C and a capillary voltage of –17.50 V. The FT-ICR-MS was calibrated using a Thermo Scientific LTQ-FT external calibration mix. For both positive and negative ion modes, at least 200 scans were collected using the parameters described in Kido Soule et al.[47] The transients were processed using SimStich[48] and aligned with MATLAB code provided by Mantini et al.[49] as described in Bhatia et al.[50] Exact masses were processed with Midas Molecular Formula Calculator (v1.1) as described by Altieri et al.[41] to provide elemental formulae of detected species.
Quality control
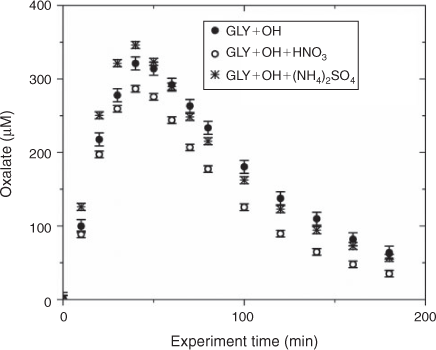
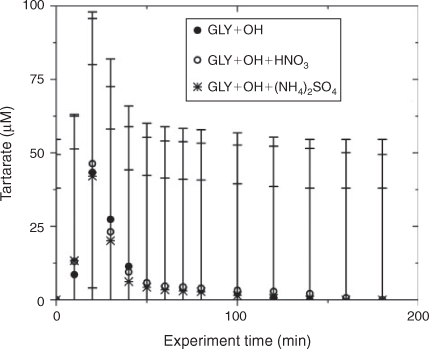
Water blanks and dynamic blanks were respectively sampled from the water source (17 MΩ cm Barnstead E-Pure MilliQ water) and reaction vessel, and analysed in the same manner as samples, described above. Blanks had negligible (signal <0.5 µS) IC peaks, thus no subtraction was performed. IC/ESI-MS analysis of the blanks revealed that m/z –113 had a strong signal (abundance >5000) throughout all spectra and thus was subtracted from the spectra. The remaining peaks were small (abundance <2000) and did not interfere with products (abundance of major products >10 000; e.g. tartarate, oxalate, nitrate and sulfate). Mixed standards were analysed with samples to assess the accuracy of quantified organic acids and to check for shifts in retention times. The retention times varied less than 10 %. Greater than 20 % of samples were run in duplicate. Experiments were run in triplicate. Error bars for oxalate and tartarate concentrations (Figs 1, 2) are the coefficients of variation across experiments.
|
|
Accounting for nitrate photolysis
The effect of OH• production by nitrate photolysis on the batch experiments was examined by adding Reactions 1–7 to the aqueous GLY + OH• chemistry documented in Lim et al.[21]:

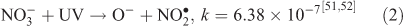
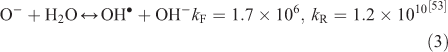

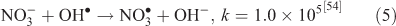

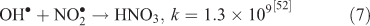
where kF is the rate of the forward reaction and kR is the rate of the reverse reaction. Nitric acid dissociates to H+ and nitrate (Reaction 1). The photolysis of nitrate produces OH• radicals (Reactions 2 and 3). Nitric acid and nitrate react with OH• radicals to form nitrate radicals (Reactions 4 and 5). GLY reacts with nitrate radicals by H-atom abstraction to give a GLY• radical and the rate constant is assumed to be 1 × 104 M–1 s–1 based on Neta and Huie[54] (Reaction 6). Finally, nitric acid can be regenerated by the reaction of OH• radicals with nitrite radicals (Reaction 7). This chemistry was used to simulate batch experiments (Figs S2–S5).
Results and discussion
Dilute glyoxal chemistry in the presence of inorganic nitrogen
The addition of ammonium sulfate (840 μM) had no effect on the production of oxalate (Fig. 1), the major product of aqueous GLY photooxidation. The addition of nitric acid (1.68 mM) decreased oxalate production slightly (~10 % at peak) when compared with oxalate production in the absence of nitric acid. Mean oxalate concentrations during GLY + OH• experiments in the presence of nitric acid are significantly less than (95 % confidence) mean oxalate concentrations in GLY + OH• experiments at every time point. This finding can be understood by examining model simulations. The addition of HNO3 decreased oxalate formation (Fig. S3) because HNO3 lowered the pH (Fig. S5) affecting glyoxylic acid dissociation and subsequently oxalate formation. Simulated OH• radical concentrations were unchanged by the addition of HNO3 (Fig. S2) because OH• produced by nitrate photolysis reacts with nitrite radicals to regenerate nitric acid.[52] The production of oxalate was much slower in GLY + HNO3 + UV control experiments (Fig. S6) compared with OH• radical experiments, also suggesting that OH• radical production by HNO3 photochemistry was modest (Fig. S7). Note that simulated and measured nitrate in the GLY + OH• + HNO3 system remained constant over the course of the reaction (Figs. S4, S8) and no substantial change in other quantified GLY oxidation products (i.e. tartarate plus malonate) was observed either with the addition of ammonium sulfate or nitric acid (Fig. 2). This suggests that the presence of ammonium and inorganic nitrate in clouds and fogs has little effect on the formation of cloud SOAaq from GLY, and supports the use of dilute GLY + OH• chemistry (e.g. Lim et al.[21]) to predict GLY cloud SOA.
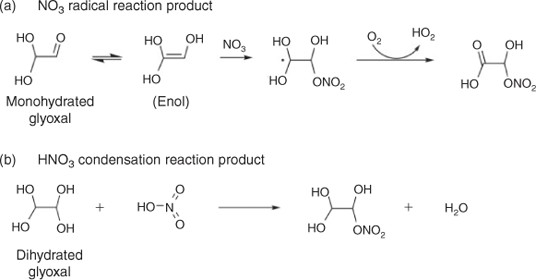
When beginning this work, we hypothesised that the addition of cloud-relevant concentrations of nitric acid might lead to the formation of organonitrates by radical–radical reactions or condensation reactions shown in Fig. 3. It is plausible that aqueous nitrate radical chemistry might be a source of oxygenated organic nitrogen compounds measured in fog and rain water,[41,42] although these compounds could also have been formed through gas-phase chemistry. If the nitrate radicals were formed by photolysis in the aqueous phase and if these reactions occur at dilute concentrations, we should see organonitrate products in experimental samples. Note that uptake from the gas phase provides a source of night-time nitrate radicals in atmospheric waters; this source is not accounted for in our experimental samples. Below we look for evidence of the formation of these and other organic nitrogen products in experimental samples.
|
Organic nitrogen
Organonitrates are known to form by gas-phase OH• radical reactions at high NOx during the daytime and gas-phase NO3• radical reactions at night.[55,56] In the aqueous phase, organonitrates are not expected to form photochemically with NO or NO2 due to low NO and NO2 water solubilities (i.e. the Henry’s Law constants for the species are HNO = 1.9 × 10–3 M atm–1 and HNO2 = 7.0 × 10–3 M atm–1).[52] However, organonitrate formation by NO3• radical reactions could occur because NO3• radicals are water soluble (HNO3 = 2 M atm–1)[52] and reactive with respect to organics with rate constants of ~103 to ~104 M–1 s–1.[54] In the night-time atmosphere, we expect that some NO3• radicals will dissolve in cloud, fog and aerosol liquid water. Photolysis of HNO3 in the aqueous phase can also form NO3• radicals. In the presence of NO2, NO3• radicals form N2O5, which is highly water soluble (HN2O5 = 1.4 × 1010 M atm–1).[52] HNO3 forms by hydrolysis when N2O5 dissolves in the aqueous phase. Thus, GLY + OH• + HNO3 is an appropriate system to examine to look for the formation of organonitrates in clouds and fogs.
Experiments conducted with nitric acid versus without nitric acid provide no evidence for organonitrate formation. Modelled (Fig. S4) and measured nitrate (Fig. S8) concentrations show no decrease with time across the experiments. IC/ESI-MS results provide no evidence of unique IC peaks or unique masses during GLY photooxidation in the presence of nitrate or ammonium (Fig. 4c, d), when compared with GLY photooxidation without nitrate or ammonium (Fig. 4b). Hypothesised organonitrate products shown in Fig. 3 were not detected by FT-ICR-MS analysis.

|
IC/ESI-MS results are shown in Fig. 4. Fig. 4a is a mixed standard analysed by IC/ESI-MS. The lettered peaks in the IC chromatogram at the left of the figure correspond to the mass spectrum labelled with the same letter at the right. Fig. 4b–d show the IC/ESI-MS analysis of 40-min samples. The IC chromatogram of the 1 mM GLY + OH• sample (Fig. 4b) is consistent with the IC chromatogram of similar (3 mM) GLY + OH• experiments conducted by Tan et al.,[33] showing a large peak with the retention time of oxalate (F) and small peaks with the retention time of malonate + tartarate (E) and with the retention time of mesoxalate (Y). In the 3-mM experiments of Tan et al.,[33] an even smaller peak with the retention time of succinate + malate (D) was detectable; this peak was not detected in the current (1 mM) experiments. The mass spectra of Fig. 4b verify that peak F is in fact oxalate (m/z –89) and peak Y is mesoxalate (m/z –117) as proposed by Tan et al.[33] Both malonate and tartarate are detectable in the 40-min GLY + OH• sample (peak E, m/z –103 and –149 respectively). The IC/ESI-MS of GLY + OH• + HNO3 (Fig. 4c) and GLY + OH• + (NH4)2SO4 (Fig. 4d) were identical to that of GLY + OH• with the exception of the nitrate peak (Fig. 4c; peak N; m/z –62) and the sulfate peak (Fig. 4d; peak S; m/z –97). Thus, the IC/ESI-MS provides no evidence for the formation of organic nitrogen species when GLY is oxidised in the presence of cloud-relevant concentrations of HNO3 or (NH4)2SO4. It is possible that the nitrate peak quantified by the IC includes organic nitrates that formed and elute at the same retention time. However, the IC/ESI mass spectrum for peak N has a single signal at m/z –62, where we expect nitrate ion detection.
Hypothesised organonitrate products shown in Fig. 3 were not detected by FT-ICR-MS analysis. Some nitrogen-containing elemental formulae were identified in samples from experiments conducted both in the presence and in the absence of nitrogen, suggesting the presence of trace nitrogen-containing contaminants. Ultra-high resolution FT-ICR MS has sub-parts per million mass accuracy and a resolution >100 000, enabling the determination of elemental formulae for thousands of compounds in a complex organic mixture. However, ion abundance depends both on compound concentration and on the matrix of other compounds present. Thus quantification is challenging and the ion abundance above a signal-to-noise threshold is used to determine the presence or absence of that compound. In this case, positive and negative mode FT-ICR-MS analyses did not provide definitive evidence for the formation of organic nitrogen species, including those proposed in Fig. 3, in the presence of cloud-relevant concentrations of HNO3 or (NH4)2SO4 when compared with samples from GLY + OH• alone.
This finding is, perhaps, not surprising. In order to form organic nitrogen, NO3• radical reactions (Fig. 3a) require a C=C double bond. The enol form of GLY has this C=C double bond. But because the GLY enol forms from monohydrated or unhydrated GLY, not dihydrated GLY, the reaction in Fig. 3a is unlikely to occur in dilute aqueous solution. Although the nitration of alcohols with HNO3 is common, there is little evidence of GLY nitration by HNO3. A recent study suggests that HNO3 acts as an oxidising agent that oxidises GLY to glyoxylic acid,[57] although we did not see evidence of this using cloud-relevant HNO3 as a source of NO3• radicals and in the presence of OH• radicals.
In the daytime atmosphere, NO3• radical reactions are slow relative to OH radical reactions. The rate constant for GLY with NO3• is 1 × 104 M–1 s–1[54] and the rate constant with OH radicals is 1 × 109 M–1 s–1. Plus, daytime gas phase nitrate radicals are removed by photolysis in seconds. For these reasons, nitrate radical reactions are unlikely to be important to daytime cloud chemistry. At night, NO3• radicals are taken up into the aqueous phase from the gas phase. They can slowly oxidise organics by hydrogen atom abstraction and peroxy radical formation (e.g. forming glyoxylic acid from GLY) or they can react at the carbon–carbon double bond to form organonitrates. Because the GLY enol forms from monohydrated or unhydrated GLY, not dihydrated GLY, the Fig. 3a reaction is more likely to occur during droplet evaporation or in wet acidic aerosols.[58] The nitration of hydrated GLY (diols) with HNO3 (Fig. 3b) is also more likely to occur in very concentrated solutions, as might be present in evaporating droplets and in wet aerosols.
Likewise, although reactions with NH4, including formation of imidazole, occur in wet aerosols, these reactions are slow relative to OH• radical reactions. Our experimental conditions (1 mM GLY + 5 mM H2O2 + UV + 840 µM (NH4)2SO4) produce hydroxyl radical concentrations of ~10–12 M at pH 3 and the rate constant for organic nitrogen formation (k[GLY + NH4+]) given by Noziere et al.[34] is 3.6 × 10–7 M–1 s–1. This is much smaller than the rate constant for GLY + OH• (1.1 × 109 M–1 s–1).[21] This suggests that the presence of NH4+ at cloud-relevant concentrations will not affect GLY + OH• chemistry. In addition, we found no evidence of the imidazoles reported by Galloway et al.[6] by FT-ICR MS in the positive mode.
Organic nitrogen is detected in fine and course particles.[59] Previous studies have reported the formation of organonitrates in the gas phase or in the aerosol phase, where concentrations of ammonium and nitrate are quite high. For example, imine and imidazole formation have been observed in chamber studies and bulk aqueous aerosol mimics.[6,34,60] Ammonium and nitrate concentrations are several orders of magnitude lower in clouds and in these experiments, and we did not see evidence of such products in the experiments reported herein.
Oligomer formation mechanism for GLY + OH•
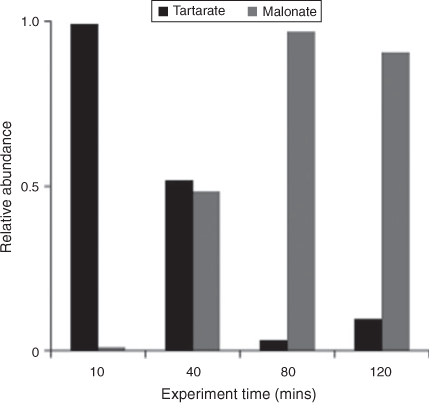
IC/ESI-MS analysis permits the identification of compounds that co-elute in the IC system, for example, tartarate and malonate. The IC peak at that retention time (21.5 min; tartarate + malonate) is quantified as tartarate throughout these experiments (Fig. 2). Peak E in the IC/ESI-MS of the mixed standard (Fig. 4a) is tartarate. The corresponding mass spectrum of peak E shows that the m/z –149 ion (tartarate) is the only major compound detected in the mixed standard. Peak E was not present in the blanks. Peak E in Fig. 4b–d is a product of GLY photooxidation and reveals the presence of both tartarate (m/z –149) and malonate (m/z –103). Thus, both are products formed during GLY photooxidation in the presence and absence of nitric acid and ammonium sulfate. According to the chemical mechanism of Lim et al.,[21] tartarate forms first and malonate forms more slowly during GLY photooxidation. Fig. 5 supports this claim by showing that the abundance of the m/z –149 ion in peak E (tartarate) is dominant early in the reaction. The abundance of the m/z –103 ion in peak E (malonate) reaches a maximum later in the experiment (between 40 and 120 min) when peak E (tartarate + malonate) is quite small (Fig. 2). Thus, we conclude that peak E is predominantly tartarate.
|
Verification that tartarate is the major first-generation radical–radical product of GLY + OH• provides critically important support for the oligomer formation mechanism proposed by Lim et al.[21] Although Tan et al.[33] identified a peak with the same IC retention time as tartarate–malonate, the IC/ESI-MS data presented here is the first confirmation that this peak is predominantly tartarate. The slower formation of malonate is also consistent with the organic radical–radical chemistry proposed by Lim et al.,[21] because malonate forms through radical–radical reactions followed by acid catalysed dehydration. Lim et al.[21] proposed that, at the high organic concentrations that are found in wet aerosols (1–10 M), organic radical–radical reactions dominate, producing higher carbon number products. Note that Lim et al.[21] used GLY as a surrogate species to represent total dissolved organics in wet aerosols, in order to better understand the differences in chemistry in clouds and in wet aerosols. It should be recognised that both organics and inorganics are present at high concentrations in wet aerosols. Although organic–inorganic interactions do not appear to play an important role in clouds, they can play an important role in droplet evaporation chemistry and in wet aerosols. The chemistry leading to SOA formation in evaporating droplets and in aerosols is clearly complex and remains poorly understood.
Atmospheric implications
The biogenic and anthropogenic source strengths of isoprene and aromatics and the water-solubility of GLY, a gas-phase product of these sources, make GLY a potentially important precursor to SOAaq formation. This research suggests that the currently understood chemistry leading to the formation of SOA through in-cloud OH• radical oxidation of GLY[21,52] is robust in the presence of ammonium sulfate and nitric acid. In contrast, others have demonstrated that organic nitrogen products can form at the much higher concentrations found in wet aerosols.[6,34] They might also form during cloud and fog droplet evaporation, where GLY may be present in its monohydrated form.[58] Detection of tartarate during the GLY + OH• experiments herein provides key verification of the organic radical–radical oligomer formation mechanism proposed by Lim et al.[21] This chemistry is detectable in experiments with 1 mM of organic, and is predicted to be dominant at the high (1–10 M) concentrations of organics found in wet aerosols. Thus, oxalate is the major product of dilute (in cloud) GLY oxidation and oligomers[21] and organic nitrogen[34] are major products of GLY chemistry in wet aerosols.
Supplementary material
Additional experimental measurements and modelling results are provided in the Supplementary material. This includes measured species (i.e. H2O2, oxalate, NO3–) and modelled species (i.e. H2O2, oxalate, NO3–, OH•, pH) for selected experiments and control experiments. The Supplementary material is available from the journal online (see http://www.publish.csiro.au/?act=view_file&file_id=EN13074_AC.pdf).
Acknowledgements
The authors gratefully acknowledge that the career advances of Richard Kamens and Harvey Jeffries in atmospheric organic photochemistry, organic gas-particle partitioning and secondary organic aerosol formation have helped to make the research described herein possible. Although there are some distinct differences between the gas and aqueous chemistry of organics (e.g. because of hydration and the behaviour of radicals), many insights into organic photooxidation in atmospheric waters have come from gas-phase organic photochemistry, including insights developed by Prof. Jeffries. Understanding factors that affect yields of SOA is a process that has taken many decades, to date. A major step forward came when it was recognised that semi-volatile products of gas-phase photochemistry partition into pre-existing particulate organic matter. Works conducted by Prof. Kamens and others on the gas-particle partitioning of polycyclic aromatic hydrocarbons played an important role in the evolving thought about organic partitioning and SOA formation. Prof. Kamens has been conducting smog chamber experiments throughout his career, examining the oxidation of individual compounds, urban air pollution mixtures spiked with individual compounds and complex mixtures like woodsmoke and mobile source emissions. Recently, a small number of smog chamber experiments have been conducted at high relative humidity (RH) to examine the effect of aqueous chemistry on SOA yields. Insights provided by Prof. Kamens’ group have helped to explain large differences in the yields obtained by different groups, namely that aqueous SOA formation depends on liquid water concentrations rather than on RH, and that liquid water concentrations depend on the number concentration and composition of particles as well as on RH. It is our pleasure to recognise the novel and creative contributions of Prof. Kamens and Prof. Jeffries through this contribution.
This research was supported by the US Environmental Protection Agency (EPA) Science to Achieve Results (STAR) program (#R833751), US Department of Commerce’s National Oceanic and Atmospheric Administration (NOAA) (#NA07OAR4310279), the New Jersey Agricultural Experiment Station (NJAES) and the US Department of Agriculture (USDA‐NIFA). The findings and conclusions presented here are expressed by the authors and do not necessarily reflect the views of the funding agencies. No endorsement should be inferred. The authors acknowledge Melissa Soule, Krista Longnecker, Elizabeth Kujawinski and the funding sources of the WHOI FT-MS Users’ Facility (National Science Foundation OCE-0619608 and the Gordon and Betty Moore Foundation). The authors also acknowledge the assistance of Sybil Seitzinger, Ron Lauck, Diana Ortiz and Natasha Hodas.
References
[1] A. G. Carlton, B. J. Turpin, H. J. Lim, K. E. Altieri, S. P. Seitzinger, Link between isoprene and SOA: pyruvic acid oxidation yields and low volatility organic acids in clouds. Geophys. Res. Lett. 2006, 33, L06822.| Link between isoprene and SOA: pyruvic acid oxidation yields and low volatility organic acids in clouds.Crossref | GoogleScholarGoogle Scholar |
[2] M. J. Perri, S. P. Seitzinger, B. J. Turpin, Secondary organic aerosol production from aqueous photooxidation of glycolaldehyde: laboratory experiments. Atmos. Environ. 2009, 43, 1487.
| Secondary organic aerosol production from aqueous photooxidation of glycolaldehyde: laboratory experiments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFemtLo%3D&md5=57222d3d34ec18f71120daada32ba2b8CAS |
[3] K. E. Altieri, A. G. Carlton, H. J. Lim, B. J. Turpin, S. P. Seitzinger, Evidence for oligomer formation in clouds: reactions of isoprene oxidation products. Environ. Sci. Technol. 2006, 40, 4956.
| Evidence for oligomer formation in clouds: reactions of isoprene oxidation products.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xmslaqsrg%3D&md5=223633d12584346acb444ec9baab548fCAS | 16955892PubMed |
[4] Y. Sun, Q. Zhang, C. Anastasio, J. Sun, Insights into secondary organic aerosol formed via aqueous-phase reactions of phenolic compounds based on high resolution mass spectrometry. Atmos. Chem. Phys. 2010, 10, 4809.
| Insights into secondary organic aerosol formed via aqueous-phase reactions of phenolic compounds based on high resolution mass spectrometry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXht12qsrrE&md5=d89767e6c4557ac81638b59e90402a43CAS |
[5] J. D. Surratt, A. W. H. Chan, N. C. Eddingsaas, M. N. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg, J. H. Seinfeld, Reactive intermediates revealed in secondary organic aerosol formation from isoprene. Proc. Natl. Acad. Sci. USA 2010, 107, 6640.
| Reactive intermediates revealed in secondary organic aerosol formation from isoprene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXltFSjsL0%3D&md5=219fe201c6efbe8bec7657581ad5c79fCAS | 20080572PubMed |
[6] M. M. Galloway, P. S. Chhabra, A. W. H. Chan, J. D. Surratt, R. C. Flagan, J. H. Seinfeld, F. N. Keutsch, Glyoxal uptake on ammonium sulphate seed aerosol: reaction products and reversibility of uptake under dark and irradiated conditions. Atmos. Chem. Phys. 2009, 9, 3331.
| Glyoxal uptake on ammonium sulphate seed aerosol: reaction products and reversibility of uptake under dark and irradiated conditions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXos1amsb4%3D&md5=6d92e66c31822e836c238347726bab66CAS |
[7] I. El Haddad, Y. Liu, L. Nieto-Gligorovski, V. Michaud, B. Temime-Roussel, E. Quivet, N. Marchand, K. Sellegri, A. Monod, In-cloud processes of methacrolein under simulated conditions – Part 2: formation of secondary organic aerosol. Atmos. Chem. Phys. 2009, 9, 5107.
| In-cloud processes of methacrolein under simulated conditions – Part 2: formation of secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFGhsr%2FL&md5=9733556f0599acd6e9bb7031d6709a0cCAS |
[8] A. K. Y. Lee, R. Zhao, S. S. Gao, J. P. D. Abbatt, Aqueous phase OH oxidation of glyoxal: application of a novel analytical approach employing aerosol mass spectrometry and complementary off-line techniques. J. Phys. Chem. A 2011, 115, 10 517.
| Aqueous phase OH oxidation of glyoxal: application of a novel analytical approach employing aerosol mass spectrometry and complementary off-line techniques.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtFelu7%2FO&md5=ffae0ee20ba40246ac13239466ba4b0aCAS |
[9] D. L. Ortiz-Montalvo, Y. B. Lim, M. J. Perri, S. P. Seitzinger, B. J. Turpin, Volatility and yield of glycolaldehyde SOA formed through aqueous photochemistry and droplet evaporation. Aerosol Sci. Technol. 2012, 46, 1002.
| Volatility and yield of glycolaldehyde SOA formed through aqueous photochemistry and droplet evaporation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XmtFagsL0%3D&md5=993d6da51f06760e3b1680a385253118CAS |
[10] Y. Zhou, H. Zhang, H. M. Parikh, E. H. Chen, W. Rattanavaraha, E. P. Rosen, W. Wang, R. M. Kamens, Secondary organic aerosol formation from xylenes and mixtures of toluene and xylenes in an atmospheric urban hydrocarbon mixture: water and particle seed effects (II). Atmos. Environ. 2011, 45, 3882.
| Secondary organic aerosol formation from xylenes and mixtures of toluene and xylenes in an atmospheric urban hydrocarbon mixture: water and particle seed effects (II).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmvFOgsrw%3D&md5=c9e1cc03e5d99df2c1fe20023a695aebCAS |
[11] R. M. Kamens, H. Zhang, E. H. Chen, Y. Zhou, H. M. Parikh, R. L. Wilson, K. E. Galloway, E. P. Rosen, Secondary organic aerosol formation from toluene in an atmospheric hydrocarbon mixture: water and particle seed effects. Atmos. Environ. 2011, 45, 2324.
| Secondary organic aerosol formation from toluene in an atmospheric hydrocarbon mixture: water and particle seed effects.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjvVWktbs%3D&md5=70dcfaa60ebb5bdf1b9cec81061bbb5dCAS |
[12] J. D. Blando, B. J. Turpin, Secondary organic aerosol formation in cloud and fog droplets: a literature evaluation of plausibility. Atmos. Environ. 2000, 34, 1623.
| Secondary organic aerosol formation in cloud and fog droplets: a literature evaluation of plausibility.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXitFOjt7c%3D&md5=0d4ea5e801d14c0a230234974956c55dCAS |
[13] B. Ervens, B. Turpin, R. Weber, Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field, and model studies. Atmos. Chem. Phys. 2011, 11, 11 069.
| Secondary organic aerosol formation in cloud droplets and aqueous particles (aqSOA): a review of laboratory, field, and model studies.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XisFOisLw%3D&md5=181a6701e42d0b51b62842e66c2a2585CAS |
[14] J. H. Seinfeld, J. F. Pankow, Organic atmospheric particulate material. Annu. Rev. Phys. Chem. 2003, 54, 121.
| Organic atmospheric particulate material.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXntFSgs7s%3D&md5=9937f341a2cdeed5352c5cf2bb1f5d1dCAS | 12524426PubMed |
[15] M. Jang, R. M. Kamens, K. B. Leach, M. R. Strommen, A thermodynamic approach using group contribution methods to model the partitioning of semivolatile organic compounds on atmospheric particulate matter. Environ. Sci. Technol. 1997, 31, 2805.
| A thermodynamic approach using group contribution methods to model the partitioning of semivolatile organic compounds on atmospheric particulate matter.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXlsFymtLw%3D&md5=98cef71f3f1db9184e8e18a03835a0e0CAS |
[16] T. M. Fu, D. J. Jacob, F. Wittrock, J. P. Burrows, M. Vrekoussis, M. V. Henze, Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols. J. Geophys. Res. 2008, 113, D15303.
| Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols.Crossref | GoogleScholarGoogle Scholar |
[17] S. Myriokefalitakis, K. Tsigaridis, N. Mihalopoulos, J. Sciare, A. Nenes, K. Kawamura, A. Segers, M. Kanakidou, In-cloud oxalate formation in the global troposphere: a 3-D modeling study. Atmos. Chem. Phys. 2011, 11, 5761.
| In-cloud oxalate formation in the global troposphere: a 3-D modeling study.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1WjtrvI&md5=8d434026d1ed25e883a05d9833fc95acCAS |
[18] G. Lin, J. E. Penner, S. Sillman, D. Taraborrelli, J. Lelieveld, Global modeling of SOA formation from dicarbonyls, epoxides, organic nitrates, and peroxides. Atmos. Chem. Phys. 2012, 12, 4743.
| Global modeling of SOA formation from dicarbonyls, epoxides, organic nitrates, and peroxides.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtFGkt7%2FN&md5=ed4744e4def6949c63f9f67b9e4dc1aaCAS |
[19] J. Liu, L. H. Horowitz, S. Fan, A. G. Carlton, H. Levy, Global in-cloud production of secondary organic aerosols: implementation of a detailed chemical mechanism in the GFDL atmospheric model AM3. J. Geophys. Res., D, Atmospheres 2012, 117, D15303.
| Global in-cloud production of secondary organic aerosols: implementation of a detailed chemical mechanism in the GFDL atmospheric model AM3.Crossref | GoogleScholarGoogle Scholar |
[20] A. G. Carlton, B. J. Turpin, K. E. Altieri, S. P. Seitzinger, R. Mathur, S. J. Roselle, R. J. Weber, CMAQ Model performance enhanced when in-cloud secondary organic aerosol is included: comparisons of organic carbon predictions with measurements. Environ. Sci. Technol. 2008, 42, 8798.
| CMAQ Model performance enhanced when in-cloud secondary organic aerosol is included: comparisons of organic carbon predictions with measurements.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlCiu7rM&md5=2da2af7a2b6b0cf9f5b15be289466bbeCAS | 19192800PubMed |
[21] Y. B. Lim, Y. Tan, M. J. Perri, S. P. Seitzinger, B. J. Turpin, Aqueous chemistry and its role in secondary organic aerosol (SOA) formation. Atmos. Chem. Phys. 2010, 10, 10 521.
| Aqueous chemistry and its role in secondary organic aerosol (SOA) formation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjs1Kjtb0%3D&md5=a1cf26b034855d2be78ffab4028190d1CAS |
[22] M. D. Petters, S. M. Kreidenweis, A single parameter representation of hygroscopic growth and cloud condensation nucleus activity. Atmos. Chem. Phys. 2007, 7, 1961.
| A single parameter representation of hygroscopic growth and cloud condensation nucleus activity.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXls1eitbY%3D&md5=726b170f5abbcb8906c5fc52c94579a4CAS |
[23] R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe, Evaluated kinetic and photochemical data for atmospheric chemistry: volume II – gas phase reactions of organic species. Atmos. Chem. Phys. 2006, 6, 3625.
| Evaluated kinetic and photochemical data for atmospheric chemistry: volume II – gas phase reactions of organic species.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtV2hs7bF&md5=024d153f9381c8cbdcdf5cd3c0a22fe1CAS |
[24] A. Guenther, T. Karl, P. Harley, C. Wiedinmyer, P. I. Palmer, C. Geron, Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature). Atmos. Chem. Phys. 2006, 6, 3181.
| Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtV2hs7vF&md5=a8ee5544f67ae8808632485303e1cb26CAS |
[25] L. Y. Yeung, M. J. Pennino, A. M. Miller, M. J. Elrod, Kinetic and mechanistic studies of the atmospheric oxidation of alkynes. J. Phys. Chem. 2005, 109, 1879.
| Kinetic and mechanistic studies of the atmospheric oxidation of alkynes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1ejs74%3D&md5=2cbe1da8adc233556ab3a0ee9af7a3c1CAS |
[26] F. Wittrock, A. Richter, H. Oetjen, J. P. Burrows, M. Kanakidou, S. Myriokefalitakis, R. Volkamer, S. Beirle, U. Platt, T. Wagner, Simultaneous global observations of glyoxal and formaldehyde from space. Geophys. Res. Lett. 2006, 33, L16804.
| Simultaneous global observations of glyoxal and formaldehyde from space.Crossref | GoogleScholarGoogle Scholar |
[27] M. Rinaldi, S. Decesari, C. Carbone, E. Finessi, S. Fuzzi, D. Ceburnis, C. D. O’Dowd, J. Sciare, J. P. Burrows, M. Vrekoussis, B. Ervens, K. Tsigaridis, M. C. Facchini, Evidence of a natural marine source of oxalic acid and a possible link to glyoxal. J. Geophys. Res. 2011, 116, D16204.
| Evidence of a natural marine source of oxalic acid and a possible link to glyoxal.Crossref | GoogleScholarGoogle Scholar |
[28] X. Zhou, K. Mopper, Apparent partition coefficients of 15 carbonyl compounds between air and seawater and between air and freshwater; implications for air–sea exchange. Environ. Sci. Technol. 1990, 24, 1864.
| Apparent partition coefficients of 15 carbonyl compounds between air and seawater and between air and freshwater; implications for air–sea exchange.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXlvFOgurs%3D&md5=675ecf25e8b356ddf3186e00f54546e1CAS |
[29] K. Matsumoto, S. Kawai, M. Igawa, Dominant factors controlling concentrations of aldehydes in rain, fog, dew water, and in the gas phase. Atmos. Environ. 2005, 39, 7321.
| Dominant factors controlling concentrations of aldehydes in rain, fog, dew water, and in the gas phase.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtFGht7rM&md5=fb5d22870fbccaf513ea7996b34d8b83CAS |
[30] M. Igawa, J. W. Munger, M. R. Hoffmann, Analysis of aldehydes in cloud- and fogwater samples by HPLC with a postcolumn reaction detector. Environ. Sci. Technol. 1989, 23, 556.
| Analysis of aldehydes in cloud- and fogwater samples by HPLC with a postcolumn reaction detector.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXhsl2itro%3D&md5=d65ad2ce5785a7d04283589d8afdf44bCAS |
[31] S. A. Epstein, S. A. Nizkorodov, A comparison of the chemical sinks of atmospheric organics in the gas and aqueous phase. Atmos. Chem. Phys. 2012, 12, 8205.
| A comparison of the chemical sinks of atmospheric organics in the gas and aqueous phase.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXjtlOls70%3D&md5=6d0e3a1c2d78fd3c101591cbb363a130CAS |
[32] A. G. Carlton, B. J. Turpin, K. E. Altieri, S. Seitzinger, A. Reff, H. J. Lim, B. Ervens, Atmospheric oxalic acid and SOA production from glyoxal: results of aqueous photooxidation experiments. Atmos. Environ. 2007, 41, 7588.
| Atmospheric oxalic acid and SOA production from glyoxal: results of aqueous photooxidation experiments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1yis7vJ&md5=3f93353ef136482c3558eac9b47e7e72CAS |
[33] Y. Tan, M. J. Perri, S. P. Seitzinger, B. J. Turpin, Effects of precursor concentration and acidic sulfate in aqueous glyoxal-OH radical oxidation and implications for secondary organic aerosol. Environ. Sci. Technol. 2009, 43, 8105.
| Effects of precursor concentration and acidic sulfate in aqueous glyoxal-OH radical oxidation and implications for secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1arur7J&md5=ce1cb1ea6cdb61deb7dd1fd88ad9b8bbCAS | 19924930PubMed |
[34] B. Nozière, P. Dziedzic, A. Cordova, Products and kinetics of the liquid-phase reaction of glyoxal catalyzed by ammonium ions (NH4+). J. Phys. Chem. A 2009, 113, 231.
| Products and kinetics of the liquid-phase reaction of glyoxal catalyzed by ammonium ions (NH4+).Crossref | GoogleScholarGoogle Scholar | 19118483PubMed |
[35] M. J. Perri, Y. B. Lim, S. P. Seitzinger, B. J. Turpin, Organosulfates from glycolaldehyde in aqueous aerosols and clouds: laboratory studies. Atmos. Environ. 2010, 44, 2658.
| Organosulfates from glycolaldehyde in aqueous aerosols and clouds: laboratory studies.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXnt12ltLs%3D&md5=3e7dbf2c685dfef0a50f566b735b2791CAS |
[36] B. Nozière, S. Ekstrom, T. Alsberg, S. Holmstrom, Radical-initiated formation of organosulfates and surfactants in atmospheric aerosols. Geophys. Res. Lett. 2010, 37, L05806.
| Radical-initiated formation of organosulfates and surfactants in atmospheric aerosols.Crossref | GoogleScholarGoogle Scholar |
[37] E. L. Shapiro, J. Szprengiel, N. Sareen, C. N. Jen, M. R. Giordano, V. F. McNeill, Light-absorbing secondary organic aerosol material formed by glyoxal in aqueous aerosol mimics. Atmos. Chem. Phys. 2009, 9, 2289.
| Light-absorbing secondary organic aerosol material formed by glyoxal in aqueous aerosol mimics.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmvVGjtL0%3D&md5=17916b4adcac944c9fd814804c96967fCAS |
[38] J. M. Waldman, J. W. Munger, D. J. Jacob, R. C. Flagan, J. J. Morgan, M. R. Hoffman, Chemical composition of acid fog. Science 1982, 128, 677.
| Chemical composition of acid fog.Crossref | GoogleScholarGoogle Scholar |
[39] Q. Zhang, J. L. Jimenez, M. R. Canagaratna, J. D. Allan, H. Coe, I. Ulbrich, M. R. Alfarra, A. Takami, A. M. Middlebrook, Y. L. Sun, K. Dzepina, E. Dunlea, K. Docherty, P. F. DeCarlo, D. Salcedo, T. Onasch, J. T. Jayne, T. Miyoshi, A. Shimono, S. Hatakeyama, N. Takegawa, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, P. Williams, K. Bower, R. Bahreini, L. Cottrell, R. J. Griffin, J. Rautiainen, J. Y. Sun, Y. M. Zhang, D. R. Worsnop, Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically influenced northern hemisphere midlatitudes. Geophys. Res. Lett. 2007, 34, L13801.
| Ubiquity and dominance of oxygenated species in organic aerosols in anthropogenically influenced northern hemisphere midlatitudes.Crossref | GoogleScholarGoogle Scholar |
[40] Q. Zhang, C. Anastasio, Chemistry of fog waters in California’s Central Valley. Part 3: concentrations and speciation of organic and inorganic nitrogen. Atmos. Environ. 2001, 35, 5629.
| Chemistry of fog waters in California’s Central Valley. Part 3: concentrations and speciation of organic and inorganic nitrogen.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXosVGjuro%3D&md5=fd48b311444f9100f5d9435c9f57881cCAS |
[41] K. E. Altieri, B. J. Turpin, S. P. Seitzinger, Oligomers, organosulfates, and nitrooxy organosulfates in rainwater identified by ultra-high resolution electrospray ionization FT-ICR mass spectrometry. Atmos. Chem. Phys. 2009, 9, 2533.
| Oligomers, organosulfates, and nitrooxy organosulfates in rainwater identified by ultra-high resolution electrospray ionization FT-ICR mass spectrometry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmvVGjtbc%3D&md5=f5aff397fb1b9917a42f02c807b0250dCAS |
[42] L. R. Mazzoleni, B. M. Ehrmann, X. Shen, A. G. Marshall, J. L. Collett, Water-soluble atmospheric organic matter in fog: exact masses and chemical formula identification by ultrahigh-resolution Fourier transform ion cyclotron resonance mass spectrometry. Environ. Sci. Technol. 2010, 44, 3690.
| Water-soluble atmospheric organic matter in fog: exact masses and chemical formula identification by ultrahigh-resolution Fourier transform ion cyclotron resonance mass spectrometry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXkvVaiu7o%3D&md5=034fdac097066014223c5fb12c4939eaCAS | 20397689PubMed |
[43] D. L. Jacob, Chemistry of OH in remote clouds and its role in the production of formic acid and peroxymonosulfate. J. Geophys. Res. 1986, 91, 9807.
| Chemistry of OH in remote clouds and its role in the production of formic acid and peroxymonosulfate.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL28XlsFKntLc%3D&md5=3f0ce66626bb768f3d3c5225e8bc47a2CAS |
[44] N. V. Klassen, D. Marchington, H. C. E. McGowan, H2O2 determination by the I3– method and by KMnO4 titration. Anal. Chem. 1994, 66, 2921.
| H2O2 determination by the I3– method and by KMnO4 titration.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXlsVOqsLw%3D&md5=99493d030395660e7493cd9c52b4c19fCAS |
[45] Y. Tan, A. G. Carlton, S. P. Seitzinger, B. J. Turpin, SOA from methylglyoxal in clouds and wet aerosols: measurement and prediction of key products. Atmos. Environ. 2010, 44, 5218.
| SOA from methylglyoxal in clouds and wet aerosols: measurement and prediction of key products.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlGntL7I&md5=4843c23d15d03b6e855164b831007bacCAS |
[46] S. P. Seitzinger, R. M. Styles, R. Lauck, M. A. Mazurek, Atmospheric pressure mass spectrometry: a new analytical chemical characterization method for dissolved organic matter in rainwater. Environ. Sci. Technol. 2003, 37, 131.
| Atmospheric pressure mass spectrometry: a new analytical chemical characterization method for dissolved organic matter in rainwater.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XovV2ltLo%3D&md5=ce8b825ad45c400a8f894a62ed8faabaCAS | 12542301PubMed |
[47] M. C. Kido Soule, K. Longnecker, S. J. Giovannoni, E. B. Kujawinski, Impact of instrument and experiment parameters on reproducibility of ultrahigh resolution ESI FT-ICR mass spectra of natural organic matter. Org. Geochem. 2010, 41, 725.
| Impact of instrument and experiment parameters on reproducibility of ultrahigh resolution ESI FT-ICR mass spectra of natural organic matter.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXosF2ltr0%3D&md5=99f7fb46bd5fed6b4a13ca132193f7c9CAS |
[48] A. D. Southam, T. G. Payne, H. J. Cooper, T. N. Arvanitis, M. R. Viant, Dynamic range and mass accuracy of wide-scan direct infusion nanoelectrospray Fourier Transform Ion Cyclotron Resonance mass spectrometry-based metabolomics increased by the spectral stitching method. Anal. Chem. 2007, 79, 4595.
| Dynamic range and mass accuracy of wide-scan direct infusion nanoelectrospray Fourier Transform Ion Cyclotron Resonance mass spectrometry-based metabolomics increased by the spectral stitching method.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXlsVSgurw%3D&md5=04162ae1c2b5e8b4bdb3089ac27718c0CAS | 17511421PubMed |
[49] D. Mantini, F. Petrucci, D. Pieragostino, P. Del Boccio, M. Di Nicola, C. Di Ilio, G. Federici, P. Sacchetta, S. Comani, A. Urbani, LIMPIC: a computational method for the separation of protein MALDI-TOF-MS signals from noise. BMC Bioinformatics 2007, 8, 101.
| LIMPIC: a computational method for the separation of protein MALDI-TOF-MS signals from noise.Crossref | GoogleScholarGoogle Scholar | 17386085PubMed |
[50] M. P. Bhatia, S. B. Das, K. Longnecker, M. A. Charette, E. B. Kujawinski, Molecular characterization of dissolved organic matter associated with the Greenland ice sheet. Geochim. Cosmochim. Acta 2010, 74, 3768.
| Molecular characterization of dissolved organic matter associated with the Greenland ice sheet.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmtlGjsL4%3D&md5=5defe6d25fadec4bd852448dc409ded6CAS |
[51] P. Warneck, The relative importance of various pathways for the oxidation of sulfur dioxide and nitrogen dioxide in sunlit continental fair weather clouds. Phys. Chem. Chem. Phys. 1999, 1, 5471.
| The relative importance of various pathways for the oxidation of sulfur dioxide and nitrogen dioxide in sunlit continental fair weather clouds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXos1aksw%3D%3D&md5=3b5862ba6dbd67d20296fc5f358a9dc1CAS |
[52] H. J. Lim, A. G. Carlton, B. J. Turpin, Isoprene forms secondary organic aerosol through cloud processing: model simulations. Environ. Sci. Technol. 2005, 39, 4441.
| Isoprene forms secondary organic aerosol through cloud processing: model simulations.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXktVWrsrg%3D&md5=555691f199f9159e51b9ddbac358d181CAS | 16047779PubMed |
[53] J. Mack, J. Bolton, Photochemistry of nitrite and nitrate in aqueous solution: a review. J. Photochem. Photobiol. Chem. 1999, 128, 1.
| Photochemistry of nitrite and nitrate in aqueous solution: a review.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXnvVyqsbY%3D&md5=20af1c71e648ace24f5b3a2aa6135d77CAS |
[54] P. Neta, R. E. Huie, Rate constants for reactions of NO3 radicals in aqueous solutions. J. Phys. Chem. 1986, 90, 4644.
| Rate constants for reactions of NO3 radicals in aqueous solutions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL28Xlt1WlsrY%3D&md5=2ba444882fba75b557fb94d26dd5e34cCAS |
[55] Y. B. Lim, P. J. Ziemann, Products and mechanism of secondary organic aerosol formation from reactions with n-alkanes with OH radicals in the presence of NOX. Environ. Sci. Technol. 2005, 39, 9229.
| Products and mechanism of secondary organic aerosol formation from reactions with n-alkanes with OH radicals in the presence of NOX.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtFKltr3O&md5=5f111704cfc078fa5fd88d3aafb6a09fCAS | 16382947PubMed |
[56] H. Gong, A. Matsunaga, P. J. Ziemann, Products and mechanism of secondary organic aerosol formation from reactions of linear alkenes with NO3 radicals. J. Phys. Chem. A 2005, 109, 4312.
| Products and mechanism of secondary organic aerosol formation from reactions of linear alkenes with NO3 radicals.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjsFCqsrY%3D&md5=077ff6c9ca41059b5459f7290d7aa067CAS | 16833761PubMed |
[57] B. M. Connelly, D. O. De Haan, M. A. Tolbert, Heterogeneous glyoxal oxidation: a potential source of secondary organic aerosol. J. Phys. Chem. A 2012, 116, 6180.
| Heterogeneous glyoxal oxidation: a potential source of secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xls12jsbs%3D&md5=7aabce6f32a27454613b464eb473f55bCAS | 22510110PubMed |
[58] D. O. De Haan, A. L. Corrigan, K. W. Smith, D. R. Stroik, J. J. Turley, F. E. Lee, M. A. Tolbert, J. L. Jimenez, K. E. Cordova, G. R. Ferrell, Secondary organic aerosol-forming reactions of glyoxal with amino acids. Environ. Sci. Technol. 2009, 43, 2818.
| Secondary organic aerosol-forming reactions of glyoxal with amino acids.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjtFyru7Y%3D&md5=b559159cd361dca190767101c9e49ac2CAS | 19475956PubMed |
[59] Q. Zhang, C. Anastasio, Free and combined amino compounds in atmospheric fine particles (PM2.5) and fog waters from northern California. Atmos. Environ. 2003, 37, 2247.
| Free and combined amino compounds in atmospheric fine particles (PM2.5) and fog waters from northern California.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXivVCrs7k%3D&md5=fbc09e3a0646ba8ddbdd8dec947d1497CAS |
[60] N. Sareen, A. N. Schwier, E. L. Shapiro, D. Mitroo, V. F. McNeill, Secondary organic material formed by methylglyoxal in aqueous aerosol mimics. Atmos. Chem. Phys. 2010, 10, 997.
| Secondary organic material formed by methylglyoxal in aqueous aerosol mimics.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjsl2rtLo%3D&md5=f7015cc52ff11993b98dcaa3e2e60de7CAS |