

Competitive ligand exchange reveals time dependant changes in the reactivity of Hg–dissolved organic matter complexes
Carrie L. Miller A B , Liyuan Liang A and Baohua Gu AA Environmental Sciences Division, Oak Ridge National Laboratory, PO Box 2008, Oak Ridge, TN 37831, USA.
B Corresponding author. Email: millercl@ornl.gov
Environmental Chemistry 9(6) 495-501 https://doi.org/10.1071/EN12096
Submitted: 13 July 2012 Accepted: 26 October 2012 Published: 11 December 2012
Journal Compilation © CSIRO Publishing 2012 Open Access CC BY-NC-ND
Environmental context. Mercury, a globally important pollutant, undergoes transformations in the environment to form methylmercury that is toxic to humans. Naturally occurring dissolved organic matter is a controller in these transformations, and we demonstrate that its strength of interaction with mercury is time dependent. These changes in complexation with dissolved organic matter are likely to affect mercury’s reactivity in aquatic systems, thereby influencing how mercury is methylated and bioaccumulated.
Abstract. Mercury interactions with dissolved organic matter (DOM) are important in aquatic environments but the kinetics of Hg binding to and repartitioning within the DOM remain poorly understood. We examined changes in Hg–DOM complexes using glutathione (GSH) titrations, coupled with stannous-reducible Hg measurements during Hg equilibration with DOM. In laboratory prepared DOM solutions and in water from a Hg-contaminated creek, a fraction of the Hg present as Hg–DOM complexes did not react to GSH addition. This unreactive Hg fraction increased with time from 13 % at 1 h to 74 % after 48 h of equilibration with a Suwannee River DOM. In East Fork Poplar Creek water in Oak Ridge, Tennessee, ~58 % of the DOM-complexed Hg was unreactive with GSH 1 h after the sample was collected. This time-dependent increase in unreactive Hg suggests that Hg forms stronger complexes with DOM over time. Alternatively the DOM-complexed Hg may become more sterically protected from the ligand exchange reactions, as the binding environment changes within the DOM over time. These results have important implications to understanding Hg transformations in the natural environment, particularly in contaminated aquatic systems due to non-equilibrium interactions between Hg and DOM.
Additional keywords: complexation, kinetics, organic ligands, reactive mercury.
Introduction
The importance of dissolved organic matter (DOM) in the complexation of mercury in aquatic systems is well established.[1–4] DOM is the dominant ligand for Hg binding in fresh water ecosystems[1,4] and therefore Hg–DOM complexation is central to understanding many processes such as solid-phase partitioning,[4,5] oxidation–reduction reactions[6,7] and bioaccumulation.[8] Recent work has demonstrated that the formation of Hg–DOM complexes is kinetically controlled in both laboratory studies and natural aquatic systems.[3,9,10] Changes in the complexation of Hg and DOM may account for differences in behaviour between freshly added Hg and mercury that has been equilibrated with water. For example, mercuric (HgII) isotopes freshly added to lake mesocosms were reported to undergo reduction more readily than Hg already present in the system.[11] In another experiment in which an enriched isotope of Hg was added to surface lake water, the newly added and the existing Hg were partitioned into different size fractions of the DOM.[12] However, the details of Hg–DOM complexation, such as Hg repartition within the DOM, remain poorly understood. The time-dependent changes in reactivity of Hg complexed with DOM needs to be understood in order to predict how an ecosystem will respond to changes in Hg sources, either as point, industrial sources or diffuse sources such as wet and dry atmospheric deposition. Because complexation of Hg with DOM underpins many Hg transformation reactions, understanding changes in the reactivity of DOM-bound Hg will likely provide insights to the geochemical cycling and fate of Hg.
Previous studies have focussed on determining equilibrium complexation constants between Hg and DOM in aqueous solutions[1,2,13,14] and in natural waters.[10,15,16] Reported Hg–DOM formation constants range from 1021 to 1040 at environmentally relevant concentrations for Hg and DOM.[17] DOM is a mixture of complex macromolecules with varying molecular sizes, hydrophobicities and functional and structural properties. Of particular relevance in the DOM are the reduced sulfur or thiol functional groups, which are known to form strong bonds with Hg.[14,18–21] Although the thiol binding sites are not abundant relative to other functional groups on the DOM, they are in large excess of Hg in natural aquatic systems. Studies that use Hg at concentrations greater than the number of strong binding sites do not accurately reflect the binding mode of Hg with DOM under natural conditions. The excess Hg is likely to associate with the weaker binding sites such as carboxyl or amine moieties in the DOM under these conditions, resulting in weaker binding constants.[20,22] Thus the saturation of Hg strong binding sites in the DOM partially accounts for the variability of measured binding constants, but even in studies with very low Hg concentrations the measured Hg–DOM binding constants still vary by orders of magnitude.[17,22] Additional factors, such as the variability in the compositional and structural properties of DOM from different sources also affect Hg binding.
Recent studies have shown that complexation between Hg and DOM is not instantaneous,[3,9,10] and this kinetic effect may contribute to the variability in measured Hg–DOM binding constants. Slow reactions between Hg and DOM or rearrangements of the complexed Hg within DOM over time are particularly important to systems receiving fresh sources of Hg. For example, in the Hg-contaminated East Fork Poplar Creek (EFPC) in Oak Ridge, TN, USA, a fresh source of inorganic HgII is constantly discharged at the headwater, which is complexed with the DOM slowly as the water moves downstream.[9] It is unclear if both the strength of the Hg–DOM complexes and the Hg reactivity also change over time.
Therefore the primary objective of this study was to examine changes in the reactivity of Hg, once complexed with various sources of DOM, with competitive ligand exchange (CLE) titrations and assayed by measuring stannous-reducible Hg (HgR). The binding strength of Hg with DOM in EFPC water was also examined and compared with the results from experiments with a DOM isolate from the same creek. By examining changes in Hg complexation and reactivity as it equilibrates with DOM, the present study contributes to the understanding of geochemical cycling and fate of Hg in the natural aquatic environment.
Materials and methods
DOM ligand solutions
Competitive ligand exchange titrations were used to examine time-dependent changes in Hg complexation with several DOM isolates including the unfractionated Suwannee River natural organic matter (SR-NOM) and fractionated humic acid (SR-HA) from the International Humic Substance Society (IHSS) and East Fork Poplar Creek DOM (EFPC-DOM). The DOM collected from EFPC is a hydrophobic fraction that represents a major portion of the DOM retained by a XAD-8 resin under acidic conditions. The isolation procedure used for this DOM has previously been described.[13,23] The SR-HA is also a DOM fraction that was retained on XAD-8 but was further fractionated by precipitation at pH 2.[24] The SR-NOM was isolated by reverse osmosis.[25] Stock solutions (1000 mg C L–1) were prepared with DOM isolates dissolved in 10 mM phosphate buffer (pH 7). The dissolved organic carbon (DOC) concentration was determined with a Shimadzu TOC-5000A total organic carbon analyser after samples were acidified to pH ~2 with hydrochloric acid. All experimental solutions were prepared to obtain a DOC concentration of 5 mg C L–1 in 0.1-M sodium perchlorate and 5 mM MOPS (3-(N-morpholino)propanesulfonic acid) buffer adjusted to pH 7. MOPS buffer has been used in several laboratory experiments investigating the interaction of Hg with DOM because MOPS does not interfere with the Hg–DOM complexation.[9,15,26] The Hg concentration in the MOPS was below 0.01 nM. These solutions were equilibrated in the refrigerator under dark conditions overnight before the titrations were conducted. Background Hg concentrations in the DOM experimental solutions were low, typically less than 0.01 nM, except in the EFPC-DOM. Due to high Hg contamination in EFPC water, the DOM collected and purified from this site contained 0.55 nM Hg in the 5-mg C L–1 experimental solutions.
Creek water collection and characterisation
Time-dependent changes in Hg complexation were examined in filtered water from EFPC with and without the addition of Hg. The headwaters of EFPC are located in the Y-12 National Security Complex in Oak Ridge, TN, which has a history of Hg contamination. On average ~5–7 g Hg day–1 is discharged from a storm drain at the headwaters of EFPC,[27] and greater than 90 % of this Hg is stannous (SnII) reducible (HgR).[9] HgR is an operationally defined fraction of Hg that is converted into Hg0 by a stannous chloride solution (see details in Mercury analysis section) and this technique has been used to differentiate Hg that is associated with strong binding sites within DOM versus inorganic ligands.[9,16,28] We previously demonstrated that the association of Hg with EFPC creek DOM is kinetically controlled, with HgR decreasing from 90 to 27 % from the Hg source to a site 2.5-km downstream.[9] For the current study a sample was collected from this 2.5-km downstream location. The creek water was filtered (0.2-μm Supor filter) within 1 h of collection, and then titrated at 1, 4 and 24 h after the water collection. For the titrations in which additional Hg (0.25 nM) was added, the filtered water sample was held for 24 h before Hg spiking to allow the ambient Hg to reach a steady-state with the DOM in the water.
Competitive ligand titration
Competitive ligand exchange (CLE) titrations with reducible Hg measurements were conducted to examine changes in the interaction of Hg with DOM over time. Glutathione (GSH) was used as a DOM-competitive ligand because the binding strength of Hg–GSH is comparable to those of Hg–DOM (Table 1). Competitive complexation between GSH and DOM for Hg provides information on the strength of the interaction of Hg with the DOM. Because Hg–GSH complexes are readily reducible by SnII (at 96 ± 9 %), whereas strong Hg–DOM complexes are not, we used the HgR to quantify Hg–GSH (i.e. the GSH titratible Hg–DOM) thus examining reactivity changes in Hg–DOM complexes.

|
We prepared Hg–DOM solutions using different DOM sources and allowed the solutions to equilibrate from 1 to 48 h. GSH was then added to the Hg–DOM solutions as a competitive ligand to react with Hg (Reaction 1). The fraction of Hg that reacts with GSH and forms Hg(GSH)2 is referred to as ‘titratable Hg’ and the Hg that does not react with the GSH is ‘non-titratable’.
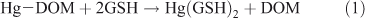
Hg was added to the DOM solution from a neutral pH, Hg stock solution to reach a concentration of 0.5 nM Hg (unless otherwise noted), and allowed to equilibrate for the desired amount of time (1–48 h). The Hg-spiked DOM solutions were then transferred to 20- or 40-mL glass vials before the GSH addition. A 10 mM GSH stock solution was prepared daily in the same MOPS buffer as the DOM solutions and dilutions of this stock were conducted before addition of the GSH to obtain concentrations ranging from 0.5 to 100 μM. Several preliminary experiments (details in the Supplementary material) were conducted to determine the required reaction time and stability of the Hg–DOM and Hg–GSH complexes in solutions, and based on these experiments a reaction time of GSH with the experimental solutions between 30 and 60 min was chosen. After the reaction of the GSH with the Hg–DOM solutions, HgR concentrations were immediately determined. The remaining sample was collected and preserved with bromine monochloride (BrCl) for total Hg (HgT) analysis.
Speciation calculations
Equilibrium speciation calculations were conducted with PHREEQC[29] to examine the competitive interaction of Hg with DOM and GSH. At the low ratio of Hg to DOM used in this study, the complexation of Hg with DOM is expected to result primarily from the interaction of Hg with reduced thiol functional groups within the DOM.[14,18–21] For speciation calculation the interaction of Hg with one and two thiol groups (RS–) were considered (Reactions 2, 3). The concentration of binding sites for Hg can be estimated assuming a DOM carbon content of 50 % and a total sulfur content of 0.86 %.[15,17] The sulfur is estimated to be 50 % reduced and the strong sites represent 2 % of the reduced sulfur.[17] This results in an estimated binding site concentration of 27 nM for the experimental solutions containing 5 mg C L–1 DOM. When only strong binding sites are considered, the binding constants between Hg and one or two thiol functional groups in DOM range from 1021 to 1040 (Table 1). The competitive interaction between DOM and GSH for Hg was examined as a function of the complexation constant. The large range of reported constants is a result of differences in DOM source and experimental conditions, including the equilibration time between the Hg and DOM, used to determine the constant. Slight differences in reported protonation constants for GSH (Table 1) did not affect the outcome of the speciation calculation at the pH of the experimental solutions so only the calculations from one set of GSH constants are shown.[15,30]
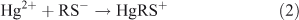
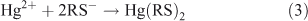
Mercury analysis
Methods for HgT and HgR analysis have been described in detail previously.[9] Briefly, Hg analysis was conducted by cold vapour atomic fluorescence spectroscopy (CVAFS) detection of Hg0.[28,31] At each titration point a sample was collected and preserved with HCl (0.5 %). Within 5 min of preservation an aliquot of the preserved sample was added to a gas washing bottle containing 100 mL of nanopure water for HgR analysis. SnCl2 (500 µL of a 20 % w/v solution in 10 % HCl) was added as a reducing agent. The reduced gaseous Hg0 was purged from solution with ultra high purity argon and collected on gold-coated sand traps. The Hg on the traps was thermally desorbed into the CVAFS detector. For HgT analysis, BrCl was added to samples for a minimum of 24 h. Hydroxylamine hydrochloride was then added to the sample just before the analysis with an automated CVAFS system (Tekran 2600). Sample duplicates, spikes and an acid-digested reference material (ERM-CC580) were routinely analysed for quality controls. For HgT analysis, relative standard deviations of duplicate samples were less than 5 % and average spike recoveries were 101 ± 6 %. Relative standard deviations of HgR samples were also less than 5 %. The loss of Hg to bottle walls during the experiment was estimated by examining changes in the HgT concentration over time. For all experiments this loss of Hg was less than 10 % during the course of the experiment.
Results and discussion
GSH – reducible Hg titration
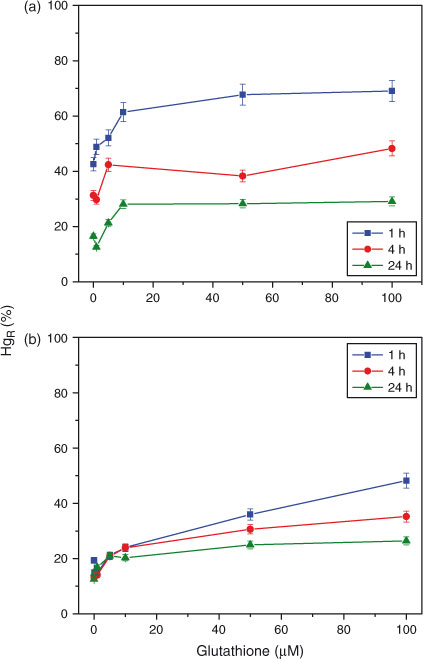
CLE HgR titrations coupled with HgR measurements were first used to examine changes in the interactions between Hg and SR-NOM or EFPC-DOM over time (Fig. 1). Titrations were conducted after Hg was equilibrated with SR-NOM (5 mg C L–1) for 1, 4, 24 and 48 h. When used independently, HgR measurements provide information on the association of Hg with DOM but not on changes in the complexation strength.[9,16] Without added GSH (GSH = 0 µM), HgR decreases over 48 h from 52 to 6.2 % (relative to the measured HgT concentration) as a result of Hg complexation with DOM (Fig. 1a). More information can be ascertained about the Hg–DOM complex when GSH titrations are coupled with the HgR measurements. For each time series, the amount of Hg reacting with the GSH increased with the increase in GSH concentration from 0.1 to 10 µM. No clear increase in HgR was observed at higher GSH concentrations, and the maximum GSH-titratable Hg was thus determined from the average of the 50 and 100 µM GSH titration data. After 1 h of Hg reaction with the SR-NOM, a maximum of ~87 % of the Hg was titratable, but as this complex aged for 4, 24 and 48 h the titratable Hg decreased (Fig. 1a). The maximum amount of titratable Hg dropped by 51 % within the first 24 h and this was followed by an additional change of only 8 % in the subsequent 24 h indicating that the majority of the changes in complexation occurred in the first 24 h.

|
The interaction of Hg with the EFPC-DOM was similar to the SR-NOM (Fig. 1b). Before adding the Hg spike the EFPC-DOM solution contained 0.55 nM HgT, of which 14.4 % (data not shown) was present as HgR. After spiking in 0.5 nM Hg but with no GSH addition, HgR decreased from 52 to 27 % in 24 h (Fig. 1b). As the GSH concentration increased from 0 to 10 µM, HgR substantially increased in each of the time series, but the HgR values levelled off at GSH concentrations above 10 µM and reached a maximum level at a GSH concentration of ~50 µM. Over time the maximum GSH titratable Hg decreased to 47 % for the 24-h reaction time series. This is similar to the results with the SR-NOM solutions in which a large fraction of the Hg did not react with the GSH, suggesting that the Hg is forming either very strong or unreactive complexes with EFPC-DOM as the reaction time increases.
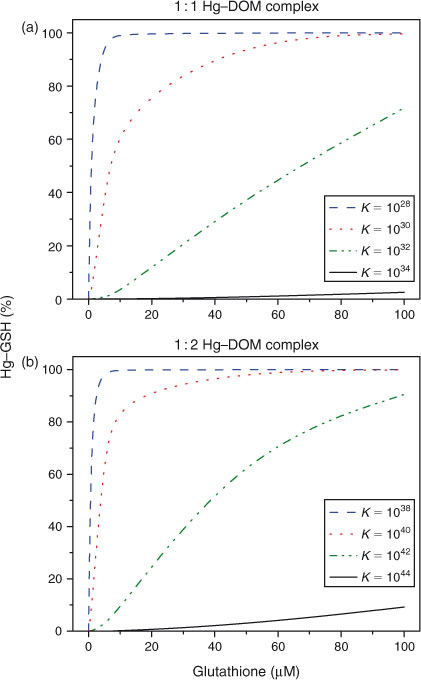
The decrease in GSH-titratable Hg with time indicates that the binding mode, or the binding strength, of Hg–DOM changed over time. The study was designed to examine time-dependent changes in the interaction of Hg with DOM. As a result equilibrium conditions were not established and equilibrium complexation constants could not be determined based on the data in this study. Speciation calculations with previously determined binding constants for the Hg–DOM and Hg–GSH complexes can provide insight into the GSH titration results. Equilibrium constants for GSH protonation and for complexation of Hg with GSH have been previously reported[15,30,32] and are used here (Table 1). Complexation constants for Hg and DOM binding range by many orders of magnitude[17] and have been determined for the formation of Hg with one and two thiol functional groups in the DOM (Reactions 2, 3). Due to the large range of the reported values, theoretical GSH titration curves were determined with a range of constants for the 1 : 1 and 1 : 2 Hg–thiol complexes (Fig. 2). The speciation calculations were performed with 0.5 nM Hg, 27 µM binding sites in 5 mg C L–1 DOM and the range of GSH concentrations used in the experiments. Under the experimental conditions, binding constants for the 1 : 1 and 1 : 2 Hg–DOM complexes would need to be greater than 1030 and 1040 respectively for the DOM to outcompete the GSH for binding.
|
A comparison of the experimental and modelling results indicates that the interaction of GSH with the Hg–DOM complex cannot be adequately predicted by equilibrium calculations. The increase in HgR with increasing GSH concentrations is theoretically predicted; GSH should be able to extract all the Hg from DOM, which is not demonstrated by experiments. This discrepancy suggests that either Hg is associated with multiple binding sites within the DOM or the DOM binding sites for Hg are inaccessible to GSH. The presence of multiple binding sites with different abundances and binding strengths in DOM is not unexpected[22] but the inability of GSH to compete with these binding sites on the DOM does not agree with previously published Hg–DOM binding constants. Presumably Hg is bound to a site that is in the hydrophobic moiety of the DOM, thus inaccessible to GSH. Alternatively, agglomeration of the DOM macromolecule could be occurring resulting in steric protection of Hg from reacting with GSH. It has been demonstrated that Hg forms complexes with organic matter in peat in which the Hg binds to thiolated aromatics.[33] These structures represent some examples of species that potentially form hydrophobic Hg–DOM complexes, and could potentially explain the lack of reactivity of the Hg–DOM complex with GSH.
A substantial decrease in GSH-titratable Hg is not observed with all DOM isolates. In solutions containing the SR-HA and in the absence of GSH, the fraction of HgR was 56 % after 1 h of Hg reaction with the DOM, and this fraction only decreased to 51 % after 24 h (Fig. 3a). The amount of GSH-titratable Hg was greater than 85 % and this did not decrease substantially over time (Fig. 3b). The large percentage of reducible Hg complex with the SR-HA isolate, regardless the equilibrium time, sets a strong contrast with the results of the unfractionated SR-NOM from the same water source. This does not suggest that the Hg is not complexed to SR-HA,[9] but indicates that the dominant complexes of Hg are likely different from those with SR-NOM. The SR-HA isolate was collected in 1982[24] whereas the unfractionated SR-NOM was collected in 1999 with different isolation methods[25] so variations in the DOM characteristics are expected. The composition of the DOM in the Suwannee River has been extensively studied, and the SR-HA fraction comprised >50 % of the total SR-NOM.[23] The SR-HA fraction is a hydrophobic fraction of SR-NOM isolated with XAD-8 resins. One possible reason for the differences observed between the unfractionated SR-NOM and the SR-HA isolates is that substantial oxidation of thiol functional groups may have occurred in SR-HA, because this DOM fraction was eluted with a strong base and has been stored for more than 30 years. On the contrary, SR-NOM was isolated by reverse osmosis and has never been exposed to strong base. The freshly isolated EFPC-DOM (also by XAD-8) exhibited similar increases in non-reducible Hg and non-titratable Hg over time as the SR-NOM. Nonetheless, the differences observed with the DOM isolates highlight the need to compare these results with experiments in which the unaltered natural water was used.
|
Complexation of Hg in EFPC
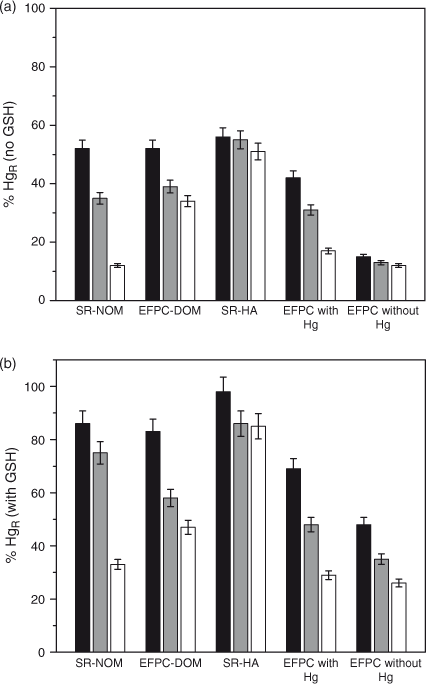
The GSH-titratable Hg in filtered EFPC water (Fig. 4) was similar to Hg spiked into laboratory solutions of SR-NOM and EFPC-DOM. Experiments were designed to simulate the addition of reactive Hg to natural water with DOM. Filtered water from EFPC, containing 1.6 mg C L–1, was held overnight to allow the ambient Hg to form non-reducible complexes with the DOM before new Hg was spiked into the water. The total Hg in the water before spiking was 0.35 nM, of which ~12 % was present as HgR. In the EFPC water spiked with Hg (0.25 nM) the maximum GSH-titratable Hg decreased over time (Fig. 4a) similar to what was observed in the laboratory solutions containing SR-NOM and EFPC-DOM. These results indicate that the interaction of Hg with DOM in the laboratory mimics the reaction of Hg in filtered creek water.
|
The GSH-titratable Hg was also examined in the unamended creek water from EFPC, and the HgR fraction decreased over time but to a lesser extent (Fig. 4b). The HgR concentration (without GSH addition) was 0.075 nM or 19 % of the filter-passing Hg 1 h after collection. The HgR fraction decreased to 12 % when the water was held for 24 h. With GSH addition up to 100 µM, the maximum GSH titratable Hg was only 42 % (Fig. 4b) analysed 1 h after sample collection, and the titratable fraction decreased to 25 % after 24 h. The 1-h data from the spiked and unspiked samples shows that the HgR fraction in EFPC water (Fig. 4b) is much lower than those in EFPC samples spiked with fresh Hg (Fig. 4a), suggesting that stronger or non-titratable Hg–DOM complexes have formed in EFPC. The EFPC water sample was collected at a location 2.5 km from the head water where Hg was discharged. Based on the average flow rates under base flow conditions, the water transit time for this 2.5-km section of the creek is ~1.5 h. Thus within this reaction time, the stronger Hg–DOM complex has formed in the creek, decreasing the reactivity of Hg. Data in Fig. 4b also suggest that the complexation of Hg with DOM is not at equilibrium and Hg speciation continues to evolve as water flows farther downstream.
Environmental implications
The complexation of Hg with DOM changes over time resulting in the formation of less reactive Hg–DOM complexes as they age. The presence of the GSH-non-titratable Hg complexes in EFPC creek indicates that the controlled laboratory studies with DOM isolates and Hg spikes can simulate processes occurring in the natural water. Changes in the reactivity of Hg towards GSH when the Hg is equilibrated with DOM suggest that the interaction of Hg and DOM is dynamic, and the rearrangement of the Hg within the DOM macromolecules occurs over time. Other studies have also noted the inability of a fraction of the Hg to react with GSH in natural water samples and in laboratory prepared solutions with DOM isolates.[3,15] In natural water this was attributed to the potential presence of inorganic Hg–sulfide complexes[15] and recent studies have demonstrated the stabilisation of nanoparticulate Hg–sulfides by DOM.[34] The presence of metal sulfides is possible in the experimental solutions and could potentially account for the less than predicted interaction of Hg with GSH. The decrease in reactivity of Hg towards GSH over time could also be explained as a result of the formation of Hg–DOM complexes that are in regions of the DOM that are not accessible to the GSH.
The presence of GSH-non-titratable Hg–DOM complexes in EFPC water can influence the transformation and fate of Hg in this and other aquatic ecosystems. Changes in the reactivity of Hg as it equilibrates with DOM provide an explanation for observed differences between the reactivity of freshly added Hg and the ambient Hg that has equilibrated with the DOM in natural aquatic systems. For example, the increase in dissolved Hg0 produced in the open ocean water after a rain event[35,36] or in mesocosm studies after the addition of a Hg spike[11,37–39] could be a result of the newly added Hg being more reactive than the Hg that has long been equilibrated with the surface water DOM. It is not surprising that such interactions are highly complex and dynamic because DOM comprises a complex mixture of organic compounds that vary in size, molecular structure and compositions.[40] Therefore to understand the role of DOM in aquatic Hg cycling, not only the sources of DOM need to be considered but the reaction kinetics, complexation strength and reactivity also need to be examined.
Supporting material
Influence of reaction time between the Hg–DOM solutions and GSH on the stannous chloride reducible Hg concentration results is provided (see http://www.publish.csiro.au/?act=view_file&file_id=EN12096_AC.pdf).
Acknowledgements
This research is part of the Mercury Science Focus Area Program at Oak Ridge National Laboratory (ORNL) supported by the Office of the Biological and Environmental Research, Office of Science, US Department of Energy (DOE). ORNL is managed by UT-Battelle LLC for US DOE under contract DE-AC05-00OR22725.
References
[1] M. Ravichandran, Interactions between mercury and dissolved organic matter – a review Chemosphere 2004, 55, 319.| Interactions between mercury and dissolved organic matter – a reviewCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhsFWgtbg%3D&md5=c55961ddb9e8bd30f7d595386ecb0599CAS |
[2] J. M. Benoit, R. P. Mason, C. C. Gilmour, G. R. Aiken, Constants for mercury binding by dissolved organic matter isolates from the Florida Everglades Geochim. Cosmochim. Acta 2001, 65, 4445.
| Constants for mercury binding by dissolved organic matter isolates from the Florida EvergladesCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXptFens7w%3D&md5=349bde5c0128992f921073e3e027458fCAS |
[3] J. D. Gasper, G. R. Aiken, J. N. Ryan, A critical review of three methods used for the measurement of mercury (Hg2+)-dissolved organic matter stability constants Appl. Geochem. 2007, 22, 1583.
| A critical review of three methods used for the measurement of mercury (Hg2+)-dissolved organic matter stability constantsCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXpt1ylsrY%3D&md5=2abbd9315de6dd8ff01541b39f041a09CAS |
[4] U. Skyllberg, Competition among thiols and inorganic sulfides and polysulfides for Hg and MeHg in wetland soils and sediments under suboxic conditions: illumination of controversies and implications for MeHg net production J. Geophys. Res. – Biogeosciences 2008, 113, G00C03.
| Competition among thiols and inorganic sulfides and polysulfides for Hg and MeHg in wetland soils and sediments under suboxic conditions: illumination of controversies and implications for MeHg net productionCrossref | GoogleScholarGoogle Scholar |
[5] C. R. Hammerschmidt, W. F. Fitzgerald, P. H. Balcom, P. T. Visscher, Organic matter and sulfide inhibit methylmercury production in sediments of New York/New Jersey Harbor Mar. Chem. 2008, 109, 165.
| Organic matter and sulfide inhibit methylmercury production in sediments of New York/New Jersey HarborCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXivFKkt7w%3D&md5=677337ca189d0753c8fa82c13d8c136bCAS |
[6] B. Allard, I. Arsenie, Abiotic reduction of mercury by humic substances in aquatic system – an important process for the mercury cycle Water Air Soil Pollut. 1991, 56, 457.
| Abiotic reduction of mercury by humic substances in aquatic system – an important process for the mercury cycleCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XhsFygsbk%3D&md5=4370090e37cc7eb7720bc82710e92babCAS |
[7] B. H. Gu, Y. R. Bian, C. L. Miller, W. M. Dong, X. Jiang, L. Y. Liang, Mercury reduction and complexation by natural organic matter in anoxic environments Proc. Natl. Acad. Sci. USA 2011, 108, 1479.
| Mercury reduction and complexation by natural organic matter in anoxic environmentsCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhs1Smt70%3D&md5=23df88dc7d12ecf1b16680a37144c93dCAS |
[8] P. C. Pickhardt, N. S. Fisher, Accumulation of inorganic and methylmercury by freshwater phytoplankton in two contrasting water bodies Environ. Sci. Technol. 2007, 41, 125.
| Accumulation of inorganic and methylmercury by freshwater phytoplankton in two contrasting water bodiesCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtlCit7nN&md5=54604fc61262a0aa6ee35f847c3cad16CAS |
[9] C. L. Miller, G. Southworth, S. Brooks, L. Y. Liang, B. H. Gu, Kinetic controls on the complexation between mercury and dissolved organic matter in a contaminated environment Environ. Sci. Technol. 2009, 43, 8548.
| Kinetic controls on the complexation between mercury and dissolved organic matter in a contaminated environmentCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1artb%2FP&md5=5bc8bf5637e02a157c8b1bf0615f162cCAS |
[10] F. J. Black, K. W. Bruland, A. R. Flegal, Competing ligand exchange-solid phase extraction method for the determination of the complexation of dissolved inorganic mercury(II) in natural waters Anal. Chim. Acta 2007, 598, 318.
| Competing ligand exchange-solid phase extraction method for the determination of the complexation of dissolved inorganic mercury(II) in natural watersCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXps1Grs70%3D&md5=3b9e26922ec71cd3cd8d6e8ad3491c5eCAS |
[11] A. J. Poulain, D. M. Orihel, M. Amyot, M. J. Paterson, H. Hintelmann, G. R. Southworth, Relationship to aquatic between the loading rate of inorganic mercury ecosystems and dissolved gaseous mercury production and evasion Chemosphere 2006, 65, 2199.
| Relationship to aquatic between the loading rate of inorganic mercury ecosystems and dissolved gaseous mercury production and evasionCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xht1CqsbrM&md5=0d9b9ca4ca9e820c0e82b9a3d146449cCAS |
[12] C. L. Babiarz, J. P. Hurley, S. R. Hoffmann, A. W. Andren, M. M. Shafer, D. E. Armstrong, Partitioning of total mercury and methylmercury to the colloidal phase in freshwaters Environ. Sci. Technol. 2001, 35, 4773.
| Partitioning of total mercury and methylmercury to the colloidal phase in freshwatersCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXotFOnsLY%3D&md5=cd1ff80441fa372935e36b5ae27335ffCAS |
[13] W. M. Dong, Y. R. Bian, L. Y. Liang, B. H. Gu, Binding constants of mercury and dissolved organic matter determined by a modified ion exchange technique Environ. Sci. Technol. 2011, 45, 3576.
| Binding constants of mercury and dissolved organic matter determined by a modified ion exchange techniqueCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjsVWmt7c%3D&md5=7f9113b468f340be4ed31da6f6246996CAS |
[14] M. Haitzer, G. R. Aiken, J. N. Ryan, Binding of mercury(II) to dissolved organic matter: the role of the mercury-to-DOM concentration ratio Environ. Sci. Technol. 2002, 36, 3564.
| Binding of mercury(II) to dissolved organic matter: the role of the mercury-to-DOM concentration ratioCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XltlWkurY%3D&md5=3940ab4225546541f4fc3fbdffd8105cCAS |
[15] H. Hsu, D. L. Sedlak, Strong HgII complexation in municipal wastewater effluent and surface waters Environ. Sci. Technol. 2003, 37, 2743.
| Strong HgII complexation in municipal wastewater effluent and surface watersCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjs1GgtL0%3D&md5=43dc30e4c4b5f1cfeeb2f11119764e2cCAS |
[16] C. H. Lamborg, C. M. Tseng, W. F. Fitzgerald, P. H. Balcom, C. R. Hammerschmidt, Determination of the mercury complexation characteristics of dissolved organic matter in natural waters with ‘reducible Hg’ titrations Environ. Sci. Technol. 2003, 37, 3316.
| Determination of the mercury complexation characteristics of dissolved organic matter in natural waters with ‘reducible Hg’ titrationsCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXksF2ktb4%3D&md5=876cd3eb848911d11588164890db1d3bCAS |
[17] W. M. Dong, L. Y. Liang, S. Brooks, G. Southworth, B. H. Gu, Roles of dissolved organic matter in the speciation of mercury and methylmercury in a contaminated ecosystem in Oak Ridge, Tennessee Environ. Chem. 2010, 7, 94.
| Roles of dissolved organic matter in the speciation of mercury and methylmercury in a contaminated ecosystem in Oak Ridge, TennesseeCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjt12jtLc%3D&md5=07fd655ec06c96f879d1057b3b3293a3CAS |
[18] K. Xia, U. L. Skyllberg, W. F. Bleam, P. R. Bloom, E. A. Nater, P. A. Helmke, X-ray absorption spectroscopic evidence for the complexation of HgII by reduced sulfur in soil humic substances Environ. Sci. Technol. 1999, 33, 257.
| X-ray absorption spectroscopic evidence for the complexation of HgII by reduced sulfur in soil humic substancesCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXns1ylsLk%3D&md5=f5f439020c05d0248bd3338640a2c72fCAS |
[19] D. Hesterberg, J. W. Chou, K. J. Hutchison, D. E. Sayers, Bonding of HgII to reduced organic sulfur in humic acid as affected by S/Hg ratio Environ. Sci. Technol. 2001, 35, 2741.
| Bonding of HgII to reduced organic sulfur in humic acid as affected by S/Hg ratioCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXjvFSlu7o%3D&md5=0bc3731a0573f67d8c9f6d43cc9cd249CAS |
[20] R. T. Drexel, M. Haitzer, J. N. Ryan, G. R. Aiken, K. L. Nagy, Mercury(II) sorption to two Florida Everglades peats: evidence for strong and weak binding and competition by dissolved organic matter released from the peat Environ. Sci. Technol. 2002, 36, 4058.
| Mercury(II) sorption to two Florida Everglades peats: evidence for strong and weak binding and competition by dissolved organic matter released from the peatCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XmsFSks78%3D&md5=ada8e551507dd9829ed28b10d39d0f69CAS |
[21] U. Skyllberg, P. R. Bloom, J. Qian, C. M. Lin, W. F. Bleam, Complexation of mercury(II) in soil organic matter: EXAFS evidence for linear two-coordination with reduced sulfur groups Environ. Sci. Technol. 2006, 40, 4174.
| Complexation of mercury(II) in soil organic matter: EXAFS evidence for linear two-coordination with reduced sulfur groupsCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XkvVGgt70%3D&md5=53a66d029391f34b7ab0f7add7e30d99CAS |
[22] S. H. Han, G. A. Gill, Determination of mercury complexation in coastal and estuarine waters using competitive ligand exchange method Environ. Sci. Technol. 2005, 39, 6607.
| Determination of mercury complexation in coastal and estuarine waters using competitive ligand exchange methodCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXmsVyit7o%3D&md5=5c9af7b2145dbe7057db813eb1563021CAS |
[23] G. R. Aiken, D. M. McKnight, K. A. Thorn, E. M. Thurman, Isolationof hydrophilic organic-acids frm water using nonionic macroporous resins Org. Geochem. 1992, 18, 567.
| Isolationof hydrophilic organic-acids frm water using nonionic macroporous resinsCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XlvFyjtb4%3D&md5=cf6ffa59bf13fab6f294fa47f3bd2bb3CAS |
[24] R. L. Malcolm, G. R. Aiken, E. C. Bowles, J. D. Malcolm, Isolation of Fulvic and Humic Acids from the Suwannee River, in Humic Substances in the Suwannee River, Georgia: Interactions, Properties, and Proposed Structures (Eds R. C. Averett, J. A. Leenheer, D. M. McKnight, K. A. Thorn) 1995 (United States Government Printing, US Geological Survey: Denver , CO).
[25] S. M. Serkiz, E. M. Perdue, Isolation of dissolved organic matter form the suwannee river using reverse osmosis Water Res. 1990, 24, 911.
| Isolation of dissolved organic matter form the suwannee river using reverse osmosisCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXlt1Smt7k%3D&md5=c44eba28608b3b1ec6df968227b23513CAS |
[26] H. Hsu-Kim, D. L. Sedlak, Similarities between inorganic sulfide and the strong HgII – complexing ligands in municipal wastewater effluent Environ. Sci. Technol. 2005, 39, 4035.
| Similarities between inorganic sulfide and the strong HgII – complexing ligands in municipal wastewater effluentCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXjsFCmt7w%3D&md5=dd20d1d2dcdb0958c053b27b523f1378CAS |
[27] S. C. Brooks, G. R. Southworth, History of mercury use and environmental contamination at the Oak Ridge Y-12 Plant Environ. Pollut. 2011, 159, 219.
| History of mercury use and environmental contamination at the Oak Ridge Y-12 PlantCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVahtbnF&md5=d44a3ed97d80e58354b03561cd00112dCAS |
[28] N. S. Bloom, E. A. Crecelius, Determination of mercury in seawater at sub-nanogram per liter levels Mar. Chem. 1983, 14, 49.
| Determination of mercury in seawater at sub-nanogram per liter levelsCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXht1eisQ%3D%3D&md5=e4175d28a4b31f558ad244922348c5edCAS |
[29] D. L. Parkhurst, C. A. J. Appelo, User’s Guide to PHREEQC (Version 2) – A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations 2004 (US Geological Survey: Denver, CO).
[30] P. D. Oram, X. J. Fang, Q. Fernando, P. Letkeman, D. Letkeman, The formation constants of mercury(II)-glutathione complexes Chem. Res. Toxicol. 1996, 9, 709.
| The formation constants of mercury(II)-glutathione complexesCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XivVOqtbY%3D&md5=c3840410a67eb5ddcb4274bcd22e3a24CAS |
[31] N. Bloom, W. F. Fitzgerald, Determination of volatile mercury species at the picogram level by low-temperature gas-chromatography with cold-vapor atomic fluorescence detection Anal. Chim. Acta 1988, 208, 151.
| Determination of volatile mercury species at the picogram level by low-temperature gas-chromatography with cold-vapor atomic fluorescence detectionCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXks1SmtrY%3D&md5=386b2ac3b97d928f0d0765f4262656f6CAS |
[32] A. E. Martell, R. M. Smith, R. J. Motekaitis, NIST Critically Selected Stability Constants of Metal Complexes Data Base, NIST Std. Ref. Database # 46 1998 (US Department of Commerce: Gaithersburg, MD).
[33] K. L. Nagy, A. Manceau, J. D. Gasper, J. N. Ryan, G. R. Aiken, Metallothionein-like multinuclear clusters of mercury(ii) and sulfur in peat Environ. Sci. Technol. 2011, 45, 7298.
| Metallothionein-like multinuclear clusters of mercury(ii) and sulfur in peatCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXps1Oit7k%3D&md5=f1279f153b9a4cf2ba084ccc16597dccCAS |
[34] C. A. Gerbig, C. S. Kim, J. P. Stegemeier, J. N. Ryan, G. R. Aiken, Formation of nanocolloidal metacinnabar in mercury-DOM-sulfide systems Environ. Sci. Technol. 2011, 45, 9180.
| Formation of nanocolloidal metacinnabar in mercury-DOM-sulfide systemsCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1ClsbzI&md5=ef072acbfd920f33e2735357cbb160c3CAS |
[35] M. Amyot, D. R. S. Lean, L. Poissant, M. R. Doyon, Distribution and transformation of elemental mercury in the St Lawrence River and Lake Ontario Can. J. Fish. Aquat. Sci. 2000, 57, 155.
| Distribution and transformation of elemental mercury in the St Lawrence River and Lake OntarioCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXjtVyqt74%3D&md5=e82522b4d070bbca2ac3437e05f0b904CAS |
[36] G. M. Vandal, W. F. Fitzgerald, K. R. Rolfhus, C. H. Lamborg, Modeling the elemental mercury cycle in Pallette Lake, Wisconsin, USA Water Air Soil Pollut. 1995, 80, 529.
| Modeling the elemental mercury cycle in Pallette Lake, Wisconsin, USACrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXnsVChtLo%3D&md5=8746491a4d07d96e5364db8cc2877128CAS |
[37] D. M. Orihel, M. J. Paterson, P. J. Blanchfield, R. A. Bodaly, H. Hintelmann, Experimental evidence of a linear relationship between inorganic mercury loading and methylmercury accumulation by aquatic biota Environ. Sci. Technol. 2007, 41, 4952.
| Experimental evidence of a linear relationship between inorganic mercury loading and methylmercury accumulation by aquatic biotaCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmsFGgtb0%3D&md5=97a26da86e5c21091f5dbcfb82748db4CAS |
[38] R. C. Harris, J. W. M. Rudd, M. Amyot, C. L. Babiarz, K. G. Beaty, P. J. Blanchfield, R. A. Bodaly, B. A. Branfireun, C. C. Gilmour, J. A. Graydon, A. Heyes, H. Hintelmann, J. P. Hurley, C. A. Kelly, D. P. Krabbenhoft, S. E. Lindberg, R. P. Mason, M. J. Paterson, C. L. Podemski, A. Robinson, K. A. Sandilands, G. R. Southworth, V. L. S. Louis, M. T. Tate, Whole-ecosystem study shows rapid fish-mercury response to changes in mercury deposition Proc. Natl. Acad. Sci. USA 2007, 104, 16586.
| Whole-ecosystem study shows rapid fish-mercury response to changes in mercury depositionCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1Wgs7rM&md5=c0b56fcb38ed76b920270428cf49951fCAS |
[39] G. Southworth, S. Lindberg, H. Hintelmann, M. Amyot, A. Poulain, M. Bogle, M. Peterson, J. Rudd, R. Harris, K. Sandilands, D. Krabbenhoft, M. Olsen, Evasion of added isotopic mercury from a northern temperate lake Environ. Toxicol. Chem. 2007, 26, 53.
| Evasion of added isotopic mercury from a northern temperate lakeCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXpsVGgtA%3D%3D&md5=72652a8bcbeaa0c60f062a8579bcf0acCAS |
[40] G. Aiken, H. Hsu-Kim, J. N. Ryan, Influence of dissolved organic matter on the environmental fate of metals, nanoparticles, and colloids Environ. Sci. Technol. 2011, 45, 3196.
| Influence of dissolved organic matter on the environmental fate of metals, nanoparticles, and colloidsCrossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXjtFKqt7k%3D&md5=976212de8b9db5d621f75e74acba9821CAS |