

Sources, nature and influence on climate of marine airborne particles
E. Keith BiggEnvironmental Chemistry 4(3) 155-161 https://doi.org/10.1071/EN07001
Submitted: 10 January 2007 Accepted: 3 May 2007 Published: 22 June 2007
Environmental context. Climate models are of considerable interest to scientists and the general public given the increasing awareness of global climate change. A large uncertainty in climate models is the influence of airborne particles on the amount of sunlight that clouds reflect back to space. Since oceans comprise 70% of the Earth’s surface, it is important that we gain an understanding of the factors that control the sources and nature of marine airborne particles. This work describes previously unexplored features of the marine aerosol at a clean site exposed to the Southern Ocean and its environmental importance, which will be of benefit to future climate models.
Abstract. Airborne particles (aerosol) collected at Cape Grim, Tasmania, in February 2006 in baseline conditions were examined by transmission electron microscopy. Particles recognised as marine exopolymer gels, and aggregates of insoluble organic particles that have diameters of ~40 nm, formed 9% of the particles larger than 200 nm. Once water-soluble compounds were removed by dialysis, the proportion rose to 30%. The gels and exopolymers were mainly of marine algal and bacterial origin. Their highly surface-active properties make them potentially environmentally important in the aerosol because of their ability to act as cloud condensation nuclei. The chemical constitution of particles in the 80–200-nm diameter size range is controversial, and widely varying estimates of the proportion of sea salt they contain have been published. Possible reasons for this are discussed. The present work supports the lowest estimate.
Additional keywords: aerosol (bio-), cloud condensation nuclei (CCN), exopolymers, sea salt.
Introduction
An important uncertainty in climate change models results from the poorly determined interaction between particles on which cloud drops form (cloud condensation nuclei, CCN) and their influence on radiation budget that results from their effect on cloud reflectivity. Feedback effects on climate would also result if CCN production was temperature dependent. Since more than 70% of the world’s surface is covered by oceans, the sources, nature and cloud nucleating properties of the marine aerosol need to be fully known as a first step in reducing the uncertainty.
There have been many analyses of the main chemical species found in marine aerosols, with sea salt and non-sea-salt sulfate usually dominating in terms of mass. Recently, much more attention has been paid to the submicrometer size range where most of the particle number resides, and to the organic content. In clean conditions on the west coast of Ireland,[1] during spring and autumn when phytoplankton blooms were prevalent, it was found that water-insoluble organic carbon was the dominant species by mass, with 0.66 ± 0.11 μg m–3, while sea salt only provided 0.39 ± 0.08 μg m–3, non-sea-salt sulfate 0.26 ± 0.04 μg m–3, and water-soluble organic carbon 0.25 ± 0.04 μg m–3. This was a far greater proportion of organic carbon than found in earlier studies and was considered to have been derived from the sea through bubble bursting at the surface. However, the physical and chemical properties of individual particles rather than a group average have to be known before their environmental implications can be assessed. A remote-sensing study of cloud properties above a phytoplankton bloom over the Southern Ocean[2] suggests that aerosol derived from this source has a profound effect on cloud properties, doubling the number of particles on which cloud drops form (CCN) and results in a substantial increase in short wave radiation reflected by the clouds.
Transmission electron microscopy (TEM) examination of submicrometer-sized aerosols in various remote oceanic situations[3] showed three main types of water-insoluble organic particles. These were: (a) amorphous lumps partially transparent to electrons with typical diameters of ~100 to 500 nm, (b) aggregates of up to several hundred insoluble particles with modal diameters of ~40 nm, and (c) particles plainly biological in nature, such as bacteria and fragments of diatoms. The first class was concluded to be gels formed from exopolymer (EP) molecules derived mainly from algae and bacteria and injected into the atmosphere by bursting bubbles. The process of gel formation[4] and the depolymerisation of EP by ultraviolet light and acidification[4,5] have been described previously. It has not been generally recognised how numerous these particles are, with one estimate[6] putting the global mass of marine gels as 50 times that of all marine organisms. The aggregates were shown to occur both in the surface microlayer of the oceans and in the atmosphere,[7,8] and were clearly the ‘microcolloids’ that had been previously described.[9] The very high concentrations of small particulates in the ocean, such as viruses and fragments of larger organisms, leads to coagulation into these aggregates, and EP molecules bind them together. In the absence of a better term, those that become airborne will be called ‘airborne marine aggregates’ or AMAs.
Like the EP gels and biological particles, the marine aggregates are apparently injected into the atmosphere by bubble bursting. The EP molecules that join the particles depolymerise in the atmosphere under the influence of ultraviolet light or acidification. The components then separate[8] and can act as centres for the deposition of sulfates or other supersaturated gases. The first aim of this paper is to see whether gels and AMAs were present in the submicrometer-sized aerosol in clean marine conditions during summer at the Cape Grim Baseline Monitoring Station on the north-west tip of Tasmania, and whether their concentrations and properties made them environmentally important.
A second objective was to try to resolve a discrepancy in the proportion of sea salt claimed to be in particles smaller than 200 nm in diameter. In the course of 6 years of almost continuous TEM study of the Cape Grim aerosol[10] it was noted that no particles smaller than this showed any signs of containing sea salt. Morphologically they resembled ammonium sulfate and most reacted with barium chloride, which indicates a composition of mainly sulfate. A similar result was found for aerosols collected during a lengthy cruise through the mid-Atlantic.[11] Scanning electron microscopy (SEM) with energy dispersive X-ray analysis (EDX) were used and showed that all particles < 200 nm in diameter contained only sulfates or organic compounds. However, during the ACE-1 campaign at Cape Grim, it was concluded from an automated SEM study of 19 640 impacted particles using EDX that more than half the particles as small as 130 nm consisted wholly of unreacted sea salt.[12] Laser mass spectrometry of single particles also showed the presence of sodium and chlorine in most particles. Laboratory studies[13,14] of sea salt production by bubbling artificial sea water also showed that sea salt particles as small as 10 nm were produced. High volume sampling at Cape Grim has also shown a substantial sea salt contribution in particles nominally smaller than 250 nm.[15] Because sea salt has a much higher hygroscopic growth factor than ammonium sulfate or most organic compounds it is a more effective CCN, and the discrepancy needs to be resolved if aerosol models used to calculate the indirect influence of aerosols on the radiation budget are to be accurate.
The site, atmospheric conditions and methods
The Cape Grim Baseline Atmospheric Monitoring Station is located at 40.7°S, 144.7° near the edge of a cliff overlooking the sea. Sampling was carried out on the roof top at a height of 97 m above sea level between 13 and 25 February 2006. The station has a full range of instruments to determine whether the air is affected by land influences, and samples were separated according to whether the air was or was not affected. The latter cases are termed ‘baseline conditions’. During this period there were 6 days with baseline conditions, with winds from the south-west or west, at a rate of typically ~11 m s–1.
Aerosol samples were collected directly onto the poly(vinyl formal) (‘formvar’) surfaces of 3-mm TEM grids. Three different impactors were used, with nozzle diameters of 1 mm, 100 nm, and 70 nm, the latter two using electron microscope apertures as nozzles and a vacuum pump for sampling. Estimated 50% collection efficiency cut-points were ~200, 70, and 50 nm diameter respectively. Samples were collected at 3-hourly intervals between 0900 and 1800 hours each day. The 1-mm impactor required 10 min for an adequate particle density on the surface, the 100-nm impactor required ~40 min and the 70-nm impactor about an hour. One in every three of the aerosol collections was examined without treatment and another was exposed to the vapour of an organic solvent. The point of using organic vapours is to check for the presence of organic compounds soluble in those vapours. If a particle contained a component soluble in an organic vapour to which it was exposed, a solution formed a continuous ring around the particle to leave a residue (sometimes crystalline) when the vapour was removed. No untreated particles were surrounded by such rings. Vapours that did not affect inorganic compounds were used, such as decane, xylene, or kerosene. Diethyl ether dissolves both organic and water-soluble inorganic compounds. The differing solubilities can cause components to separate as the solvent evaporates.
The third specimen of the group of three was floated on distilled water to remove soluble material and to expose hidden insoluble material. The formvar support for the specimens is permeable to ions, so in addition to using plain distilled water to expose the insoluble material, barium chloride could be added to detect sulfate through the formation of insoluble barium sulfate. Other solutions were also used but the results are not reported here. Following flotation on an ionic solution, the specimens were floated on distilled water to remove any soluble residues. Blank specimens were also floated to check for artefacts. A few stray irregular particles were found, to the extent of about one per twenty 40-μm grid squares. Since particle concentrations near the centres of impaction spots were typically about three orders of magnitude greater, this should not have led to any confusion.
Specimens left untreated were stored in a sealed jar that contained silica gel until examined. Those treated with vapours or flotation were stored in a similar fashion after treatment. Examination by TEM occurred 4 to 8-weeks later.
Artificial shadows using metals such as platinum are usually applied in order to deduce three-dimensional morphology of the particles, but this obscures any objects (‘inclusions’) within the particles that have a different electron density. In the Cape Grim collections most of the particles were left unshadowed since internal structure is an important unknown. Three-dimensional structures of the aerosol at this site were well known from an earlier 6-year study.[10] Altogether 770 particles were photographed using the charge-coupled camera on a Philips CM-12 TEM at Sydney University’s Electron Microscopy Unit. In this paper all TEM photos are negatives of bright-field emission photographs using an objective lens aperture. Electron-dense portions of particles appear brighter than electron-transparent ones in order to appear like an optical picture.
Removal of soluble material by flotation (dialysis) was used on both untreated and vapour-exposed specimens.
The most common particles observed in baseline conditions
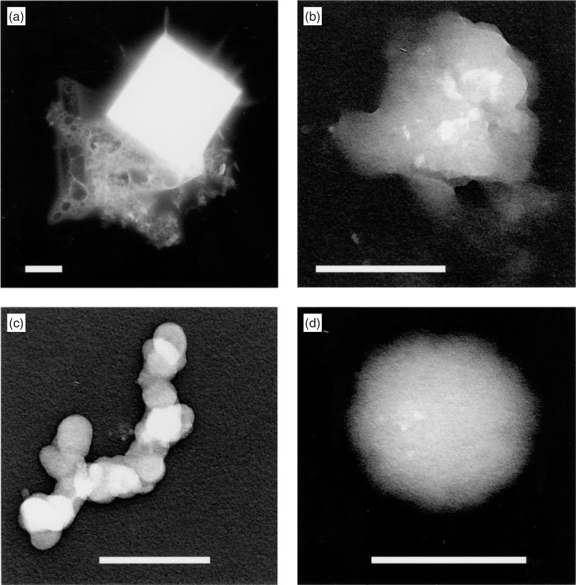
In untreated specimens collected with the 1-mm impactor, the most obvious particles were sea salt with typical diameters from 250 nm to several μm. Some of these had formed well-shaped crystals (Fig. 1a), associated with organic material. Others had less well-developed crystal shapes and were surrounded by smooth amorphous material and most had to be classed as unknown. Exopolymer secretion (EPS) gels (Fig. 1b) with embedded electron-dense inclusions and AMAs (Fig. 1c) were much less numerous in untreated specimens than at Lizard Island[16] in the Barrier Reef. Over the whole period they accounted for 9% of particles larger than 200 nm instead of ~50%. Fig. 1b was one of the largest encountered gels, most of those recognised in untreated samples being in the size range 250–500 nm. The components of Fig. 1c had an appearance and size distribution closely resembling those in AMAs found at other sites. Particles that were clearly biological (bacteria and fragments of organisms) comprised <5% of this size range and had no visible associated sea salt. This somewhat surprising fact, in view of their marine origin, has previously been observed at Cape Grim[17] and elsewhere.[18] Dialysis of specimens showed that proportions of EP gels and AMAs in the aerosol were very much underestimated in untreated specimens. Once the soluble material was removed their proportions rose to ~30%, but is still less than that observed at Lizard Island.
|
About 80% of particles smaller than 200 nm had a uniform circular outline and from the brightness contrast appeared to have a domed shape. Particles of pure ammonium sulfate also have a circular outline but are more nearly hemispherical on a formvar substrate and tend to evaporate in an electron beam. The remaining particles in this size range were of a varied and indeterminate nature. Numerically, particles between ~80 nm (the lower limit for good collection efficiency with the small nozzle impactors) and 200 nm accounted for ~65% of all particles >80 nm observed during this period. Size distributions obtained at the same time with a differential mobility particle sampler showed that those >80 nm were only a small proportion of the total particles in baseline conditions, and a modal size near 40 nm usually provided the majority.
Previous studies over the summer Arctic central ocean and at a tropical site[8,16] have shown that the majority of particles <200 nm that had been dialysed, or were sufficiently transparent to see internal structures, had what appeared to be AMA components within them. In this study approximately half the particles in this size range had sufficient associated water-insoluble material to prevent detection of inclusions of this nature. In the remainder, ~45% had definite inclusions of this nature and a further 20% had possible ones.
The first objective of this paper was to check that EP gels and AMAs formed a significant part of the Cape Grim aerosol and it seems that this has been achieved. Without the special test of dialysis they could easily be written off as a very minor component, but, if their properties have a direct relevance to cloud drop nucleating ability or to secondary aerosol formation, they are in sufficient concentrations to be important. It should be remarked, however, that the proportions of the different aerosol types given above refer to a limited period and only 770 particles (a substantial proportion of them remaining unclassified), which may be insufficient to give an accurate representation.
Possible environmental effects of gels and aggregates of small particles
EP gels are highly surface active and consist of 99% water[17] trapped within a polymer network. This perhaps explains why they have the ability to take up ether or water and swell to supermicrometer size. Some may also retain this liquid in the vacuum of the electron microscope. While none of the untreated gels was larger than 1 μm, all those exposed to ether vapour or relative humidities close to water saturation were found to be as large, or larger than 1 μm, or gave indications that they had reached that size. Fig. 2a shows a typical gel that had been subjected to ether vapour, one of twenty photographed, two of which had been photographed before being exposed to the vapour. Fig. 2b shows one exposed to water vapour that had shrunk in the vacuum of the electron microscope to leave a small halo of evaporated material, while Fig. 2c shows one that has evaporated much more completely to leave a halo of larger particles. It is known that gels can collapse under the influence of ultraviolet light or acidification[4,5] and a sequence of TEM morphologies has been suggested to represent progressive stages of this collapse.[16] The differing degrees to which evaporation of swollen gels occurs in the TEM could, therefore, be attributed to the fact that the gels had different lengths of atmospheric exposure before collection.

|
The relevance of these observations to cloud formation is that gels swollen to a large size will be especially active CCN, which leads to broad drop-size distributions in clouds and aids in the production of precipitation by the coalescence process. They will also provide very suitable sites for the oxidation of sulfur dioxide. At a site similar to Cape Grim it has been found[18] that aqueous phase oxidation mechanisms account for most of the substantial non-sea-salt sulfate component of coarse mode aerosols. Research will be needed to see whether the presence of EP gels influences this oxidation.
As mentioned earlier, it has been proposed that AMAs break down into individual ~40 nm particles or small groups of them. These can then act as centres for the deposition of supersaturated gases such as the oxidation products of sulfur-containing gases. At Cape Grim 45–65% of particles in which such centres could have been seen contained them. They, therefore, represent a potentially important source of secondary aerosol, either organic or inorganic.
Effects of organic compounds in the aerosol
Organic compounds present in the ocean other than EP and their gels and aggregates and biological particles will also become part of the primary aerosol by the bubble bursting process and will be difficult to distinguish from secondary organics that arise from photochemical processes and deposits onto existing aerosols. Another secondary source is the compounds produced by depolymerisation of EP and EP gels. The latter are of interest because their production could affect the ability of sea salt, EP gels, and AMAs to act as CCN. Completely amorphous transparent gels that are interpreted to have been recently produced[16] were unaffected by decane vapour. Fig. 3a shows how the material on a particle interpreted to be an aged gel has apparently collapsed into a flat particle that contains holes after dialysis and exposure to decane. Since EP gels are 99% water, little material is left after a collapse. The appearance of this particle suggests that water-soluble material was removed by dialysis and much of the remaining structure was concentrated into patches by the decane. When AMA particles were exposed to ether vapour, the components either grew much larger (Fig. 3b) or were completely bare. This suggests that those having short atmospheric residence times were covered with EP while those that were more aged (Fig. 3c) had lost it. If the interpretations are correct, then ageing will produce profound changes in the ability of both EP gels and AMAs to act as CCN. Such changes might be expected during the depolymerisation of the EP polysaccharides. Relatively insoluble long-chain compounds will first be released, then chain lengths will decrease and water-solubility will increase.

|
The time scale of changes will depend on the availability of ultraviolet light and on deposition of acidic compounds. There is one indication that it is not noticeable in an hour under conditions of strong sunlight. At Lizard Island,[16] the supposedly fresh amorphous gels formed a much greater proportion of the total than at Cape Grim. If the heavy surf on the outer Barrier Reef was their source, the hour of air travel was evidently insufficient for appreciable ageing.
All sea-salt particles appear to be accompanied by at least a small amount of organic material. In some this spreads out in a thin film to wet the surface, which suggests that it is surface-active EP. Decane or similar solvents do not affect this film. Other salt particles have a more concentrated organic material adjacent to the salt that is affected, and the vapours cause it to spread, which leaves a ring of material when the solvent evaporates. Thus sea-salt particles, like EP gels, may undergo a similar progressive decrease in their ability to act as CCN.
Do particles smaller than 200 nm in diameter contain sea salt?
As already stated, the Cape Grim particles <200 nm contained no obvious sea salt, which is consistent with the more definite X-ray analyses of Atlantic aerosol.[11] Flotation on a weak barium chloride solution produced a ring of insoluble barium sulfate (Figs 4a, b) with half the particles showing the presence of sulfate and otherwise only soluble material or organic inclusions. This does not rule out the presence of some sodium chloride, which would have dissolved into the solution. In the other half of the photographed particles floated on barium chloride solution the barium sulfate ring was confined to rings within the particle outlines. Figs 4c, d show examples with diameters larger than 200 nm, but they are apparently flat, so that their diameters in the atmosphere would have been below 200 nm. A suggested interpretation of these images is that aqueous oxidation of sulfur dioxide had occurred within the gels, and the acidification eventually caused their collapse to a small size. The barium chloride solution gained access to the sulfate through holes left by the collapse.

|
The observation that 50% of 130-nm particles were composed entirely of unreacted sea salt[12] may be a gross overestimate. The particles were collected by impaction and when the relative humidity is high, the temperature drop upon expansion in an impactor can cause salt particles to dissolve and then recrystallise after collection stops. Some published examples[11] show a ring of separate small crystals at a distance from the parent particle. An automated scanning system will register these as individual particles and because they crystallized from solution would find them to be mostly simple sodium chloride. However, the laser mass spectrometry also used in those experiments detected chlorine and sodium ions in a large proportion of particles. The single particle analysis has no obvious defects that could cause artefacts. X-Ray analysis of EP gels in the surface microlayer of the ocean[7] showed that chlorine and to a lesser extent sodium were prominent. Residues of those elements in particles such as those of Figs 4c, d, if they are collapsed gels, might explain the results.
High-volume samples that use Andersen cascade impactors usually show the presence of sea salt in the back-up filter at marine sites. At Cape Grim in summer it was found[15] that sea-salt concentrations were about two thirds that of non-sea-salt sulfate. This result is strongly influenced by instrumental characteristics. Filter paper was used as an impaction substrate and it smooths the nominal 0.25-μm cut-off of the last stage. Sufficient particles larger than 0.25 μm could reach the filter to completely dominate the mass. With the continuous sampling used in baseline conditions, deliquescence of salt particles can also lead to leakage of moisture to the back-up filter at times of high relative humidity and these are common at Cape Grim. The true proportion of sea salt in small particles is, therefore, not resolved by these measurements, but they do cast doubt on the claim[12] that 60% of particles of 0.25-μm diameter were unreacted sea salt and a further 20% were sea salt and sulfate, or sodium rich. Even ignoring other ions and organics and the collection of particles larger than 0.25 μm, the high volume samples showed only 40% of sea salt.
The thermal stability of particles at 300°C has often been used to identify sea-salt particles[20] and to show that in a marine environment they are present down to much smaller sizes than 200 nm. Some high-molecular-weight compounds such as polysaccharides and proteins can survive this temperature. If AMA components consist mainly of viruses, which seems possible since their concentrations in sea water are typically of the order of 107 mL–1[21] and they are likely to be enriched in the surface microlayer, this may be a mistaken identification.
There is, therefore, considerable doubt about the actual composition of particles <200 nm. Because they are numerous and likely to be potential CCN because of their sulfate content, if not because of their salt content, this doubt needs to be resolved.
Discussion and conclusions
TEM examination of aerosols is time consuming and, therefore, very limited numbers of particles can be studied in the time available. The interpretation of images relies heavily on the ability to separate and identify components of a mixed aerosol, and some of the interpretations given may need revision as new information is obtained. An example is the identification of a ‘soot’ particle by TEM at Cape Grim[22] which, judging from the predominantly angular components, was almost certainly an AMA. In spite of these drawbacks TEM images give details of the nature of particles that cannot be gained from automated systems. Airborne EP gels and AMAs are examples of particles that could be environmentally important but have until recently not received any attention.
Marine organisms or their fragments do not usually have any associated sea salt[18,19] as might be expected if they are injected into the atmosphere by bursting bubbles. A possible explanation comes from the observation[23] that bacteria and viruses were mainly transferred to the atmosphere in transparent gel-like particles when sea water from the surface microlayer of the ocean was bubbled. Collapse of gels can be sudden and irreversible[4] and bacteria and viruses may then be ejected with, and separated from, the trapped water.
The possibility that components of AMAs act as sites for the generation of secondary aerosol is indicated by the presence of similar small insoluble inclusions in about half of the particles < 200 nm at Cape Grim, which contain only sulfate and other soluble compounds. It seems possible that they may also play a part in maintaining Aitken particle concentrations over the oceans.
The observation[2] that cloud properties were substantially altered over a phytoplankton bloom in the Southern Ocean was suggested to be caused by the oxidation of isoprene produced by the phytoplankton. Phytoplankton blooms lead to a rapid multiplication of bacteria and viruses and will be prime sites for the generation of EP, EP gels, and marine aggregates. Oxygen supersaturations in the sea water that result from photosyntheses will lead to bubble production episodes, so that concentrations of airborne gels, sea salt, and associated EP and aggregates will increase. It may not be necessary to invoke a nucleation process to explain the alteration of cloud properties.
The first objective of this work, to determine whether the organic primary aerosols were present in sufficient quantities under Cape Grim baseline conditions to be potentially environmentally important, appears to have been met. However, much more comprehensive long-term research is needed to assess their impact. The title of a paper[24] on marine particulates: ‘Marine colloids. A neglected dimension’ would apply equally to their counterparts in the air and suggests a useful direction for future research.
The second aim of this paper, to find whether particles <200 nm contain a significant proportion of sea salt, has not been met. The discussion does appear to have raised doubts about the proportions of sea salt in those particles claimed by other experimenters but the present observations could not rule out that some was present. Again, much more research is needed on the properties of these particles because they usually comprise more than half of the potential CCN for marine clouds.
Acknowledgements
The author thanks the Director of the Cape Grim Baseline Station, Dr Jill Cainey, for the opportunity to take these measurements and for financial support with the electron microscopy. Sydney University’s Electron Microscope Unit and Dr Ian Kaplin are also thanked for the electron microscopy.
[1]
F. Cavalli ,
M. C. Facchini ,
S. Decesari ,
M. Mircea ,
L. Emblico ,
S. Fuzzi ,
D. Ceburnis ,
Y. J. Yoon ,
et al. Advances in characterization of size-resolved organic matter in marine aerosol over the North Atlantic.
J. Geophys. Res. 2004
, 109, D24215.
| Crossref | GoogleScholarGoogle Scholar |

[2]
N. Meskhidze ,
A. Nenes ,
Phytoplankton and cloudiness in the Southern Ocean.
Science 2006
, 314, 1419.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
[3]
C. Leck ,
E. K. Bigg ,
Evolution of the marine aerosol – A new perspective.
Geophys. Res. Lett. 2005
, 32, L19803.
| Crossref | GoogleScholarGoogle Scholar |
[4]
W.-C. Chin ,
M. V. Orellana ,
P. Verdugo ,
Spontaneous assembly of marine dissolved organic matter into polymer gels.
Nature 1998
, 391, 568.
| Crossref | GoogleScholarGoogle Scholar |
[5]
M. V. Orellana ,
P. Verdugo ,
Ultraviolet light blocks the organic carbon exchange between the dissolved phase and the gel phase in the ocean.
Limnol. Oceanogr. 2003
, 48, 1618.
[6]
P. Verdugo ,
A. L. Alldredge ,
F. Azam ,
D. I. Kirchman ,
U. Passow ,
P. H. Santschi ,
The oceanic gel phase: a bridge in the DOM-POM continuum.
Mar. Chem. 2004
, 92, 67.
| Crossref | GoogleScholarGoogle Scholar |
[7]
E. K. Bigg ,
C. Leck ,
L. Tranvik ,
Occurrence of small colloids in sea water.
Mar. Chem. 2004
, 91, 131.
| Crossref | GoogleScholarGoogle Scholar |
[8]
C. Leck ,
E. K. Bigg ,
Biogenic particles in the surface microlayer and overlying atmosphere in the central Arctic Ocean during summer.
Tellus 2005
, 57B, 305.
[9]
M. L. Wells ,
E. D. Goldberg ,
Occurrence of small colloids in sea water.
Nature 1991
, 353, 342.
| Crossref | GoogleScholarGoogle Scholar |
[10]
E. K. Bigg ,
Comparison of the aerosol at four baseline atmospheric monitoring stations.
J. Appl. Meteorol. 1980
, 19, 521.
| Crossref | GoogleScholarGoogle Scholar |
[11]
F. P. Parungo ,
C. T. Nagamoto ,
J. M. Harris ,
Temporal and spatial variations of marine aerosols over the Atlantic Ocean.
Atmos. Res. 1986
, 20, 23.
| Crossref | GoogleScholarGoogle Scholar |
[12]
D. M. Murphy ,
J. R. Anderson ,
P. K. Quinn ,
L. M. McInnes ,
F. J. Brechtel ,
S. M. Kreidenweis ,
A. M. Middlebrook ,
M. Posfai ,
et al. Influences of sea-salt on aerosol radiative properties in the Southern Ocean marine boundary layer.
Nature 1998
, 392, 62.
| Crossref | GoogleScholarGoogle Scholar |
[13]
E. M. Mårtensson ,
E. D. Nilsson ,
G. de Leeuw ,
L. H. Cohen ,
H.-C. Hansson ,
Laboratory simulations of the primary marine aerosol production.
J. Geophys. Res. 2003
, 108, 4297.
| Crossref | GoogleScholarGoogle Scholar |
[14]
K. Sellegri ,
C. D. O'Dowd ,
Y. J. Yoon ,
S. G. Jennings ,
G. de Leeuw ,
Surfactants and sub-micron sea-spray generation.
J. Geophys. Res. 2006
, 111, D22215.
| Crossref | GoogleScholarGoogle Scholar |
[15]
M. O. Andreae ,
W. Elbert ,
Y. Cai ,
T. W. Andreae ,
Non-sea-salt sulfate, methanesulfonate, and nitrate concentrations and size distributions at Cape Grim, Tasmania.
J. Geophys. Res. 1999
, 104 (D17), 21695.
| Crossref | GoogleScholarGoogle Scholar |
[16]
C. Leck ,
E. K. Bigg ,
Comparison of sources and nature of the tropical aerosol with the summer high Arctic aerosol.
Tellus B 2007
,
in press.
[17]
A. W. Decho ,
Microbial exopolymer secretions in ocean environments: their role(s) in food webs and marine processes.
Oceanogr. Mar. Biol. 1990
, 28, 73.
[18]
H. Sievering ,
J. Cainey ,
M. Harvey ,
J. McGregor ,
S. Nichol ,
P. Quinn ,
Aerosol non-sea-salt sulfate in the remote boundary layer under clear-sky and normal cloudiness conditions: Ocean-derived biogenic alkalinity enhances sea-salt sulfate production by ozone oxidation.
J. Geophys. Res. 2004
, 109, D19317.
| Crossref | GoogleScholarGoogle Scholar |
[19]
M. Pόsfai ,
J. Li ,
J. R. Anderson ,
P. R. Buseck ,
Aerosol bacteria over the Southern Ocean during ACE-1.
Atmos. Res. 2003
, 66, 231.
| Crossref | GoogleScholarGoogle Scholar |
[20]
A. D. Clarke ,
Submicrometer sea salt in the remote marine environment.
J. Aerosol Sci. 1999
, 30, S3.
| Crossref | GoogleScholarGoogle Scholar |
[21]
J. A. Fuhrman ,
Marine viruses and their biogeochemical and ecological effects.
Nature 1999
, 399, 541.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
[22]
M. Pósfai ,
J. R. Anderson ,
P. R. Busek ,
H. Sievering ,
Soot and aerosol particles in the remote marine troposphere.
J. Geophys. Res. 1999
, 104, 21685.
| Crossref | GoogleScholarGoogle Scholar |
[23]
J. Y. Aller ,
M. R. Kuznetsova ,
C. J. Jahns ,
P. F. Kemp ,
The sea surface microlayer as a source of viral and bacterial enrichment in marine aerosols.
J. Aerosol Sci. 2005
, 36, 801.
| Crossref | GoogleScholarGoogle Scholar |
[24]
M. L. Wells ,
Marine colloids. A neglected dimension.
Nature 1998
, 391, 530.
| Crossref | GoogleScholarGoogle Scholar |