

Antimony in the environment: knowns and unknowns
Montserrat Filella A B E , Peter A. Williams C and Nelson Belzile DA Department of Inorganic, Analytical and Applied Chemistry, University of Geneva, Quai Ernest-Ansermet 30, CH-1211 Geneva 4, Switzerland.
B SCHEMA, Rue Principale 92, L-6990 Rameldange, Luxembourg.
C School of Natural Sciences, University of Western Sydney, Locked Bag 1797, Penrith South DC, NSW 1797, Australia.
D Department of Chemistry and Biochemistry, Laurentian University, Sudbury, ON, P3E 2C6, Canada.
E Corresponding author. Email: montserrat.filella@unige.ch
Environmental Chemistry 6(2) 95-105 https://doi.org/10.1071/EN09007
Submitted: 5 January 2009 Accepted: 5 March 2009 Published: 27 April 2009
Environmental context. Antimony first attracted public attention in the mid-1990s amid claims that it was involved in Sudden Infant Death Syndrome. A substantial number of papers have now been published on the element and its behaviour in the natural environment. However, many key aspects of the environmental chemistry of antimony remain poorly understood. These include critical areas such as its ecotoxicology, its global cycling through different environmental compartments, and what chemical form it takes in different environments. More focussed research would help the situation. The present review highlights several areas of environmental antimony chemistry that urgently need to be addressed.
Abstract. The objective of the present article is to present a critical overview of issues related to the current state of knowledge on the behaviour of antimony in the environment. It makes no attempt to systematically review all published data. However, it does provide a list of the main published reviews on antimony and identifies subjects where systematic reviews are needed. Areas where our knowledge is strong – and the corresponding gaps – in subjects ranging from total concentrations and speciation in the various environmental compartments, to ecotoxicity, to cycling between compartments, are discussed, along with the underlying research. Determining total antimony no longer poses a problem for most environmental samples but speciation measurements remain challenging throughout the process, from sampling to analysis. This means that the analytical tools still need to be improved but experience shows that, to be useful in practice, this should be directly driven by the requirements of laboratory and field measurements. Many different issues can be identified where further research is required, both in the laboratory and in the field, the most urgently needed studies probably being: (i) long-term spatial and temporal studies in the different environmental compartments in order to collect the data needed to establish a global biogeochemical cycle; (ii) laboratory studies of antimony interactions with potential natural binders; (iii) reliable ecotoxicological studies.
Introduction
Antimony belongs to Group 15 of the periodic table of the elements, along with N, P, As and Bi. Antimony can exist in a variety of oxidation states (–III, 0, III, V) but in biological and environmental samples, it is mainly found in two (III and V). Antimony is ubiquitous throughout the environment as a result of natural processes and human activities. It has no known function in living organisms.
Some of the main topics of interest when evaluating the environmental chemistry of a trace element such as antimony are: (i) how much is present in the different environmental compartments; (ii) in which form it is actually present (i.e. speciation); (iii) its toxicity; and (iv) its cycling between the different compartments (i.e. fluxes). The current status of these issues regarding antimony will be discussed in the present article, which should be considered an attempt to highlight aspects that merit further research rather than a systematic review (see Sources of information section below for a definition). Therefore, the emphasis will be placed chiefly on unanswered questions and misleading ‘accepted facts’, while more established issues will be addressed by giving key references.
Sources of information
When trying to establish the state of the art in a particular field, it is useful to analyse the sources of information that contain relevant data. Various aspects of the three main sources of scientific information – original research articles, grey literature and reviews – concerning antimony are discussed below.
There is a widespread opinion that interest in antimony-related studies is growing and that the number of articles published on the element is rising. However, this is only partially true. Analysis of the literature shows that most articles belong to one of two categories, either: (i) studies in which this element is merely included with several other trace elements; this trend, which is not exclusive to antimony, is linked to the availability of multielement techniques such as neutron activation analysis (NAA), initially, and, more recently, inductively coupled plasma (ICP) techniques; or (ii) studies that describe new analytical methods or modified protocols. Usually a few ‘real’ samples are analysed, at the end of such papers, but, regrettably often, samples are spiked because the method is not sensitive enough. Even when this is not the case, the published data are of very limited interest because they are only one-off measurements and no attempt is made to provide any ancillary information about the system itself. At present, our antimony database contains 2900 articles covering a wide-range of analytical, geochemical, solution chemistry, ecotoxicological and toxicological aspects. The fact is that a quick inspection shows that articles devoted to the study of environmental antimony behaviour account at most for 300 articles.
The term ‘grey literature’ refers to any documentary material that is not commercially published, with typical examples being technical reports, working papers, business documents, and conference proceedings. These items can be difficult to locate and, although their reliability is always difficult to assess owing to the absence of peer-review control, they often contain useful information. For antimony, this is particularly the case for ecotoxicological issues (see Antimony ecotoxicity data section), a subject about which a great deal of data remains buried in non-refereed reports. To make such literature more easily accessible, a web-based repository has recently been created by one of the authors (www.schema.lu/Sb-grey.html, accessed 7 April 2009).
Reviews are usually published as articles, book chapters or reports. As mentioned, reports are categorised as grey literature, and are thus subject to the above-mentioned problems of quality control and availability. The importance of reviews has increased proportionally with the explosion in the amount of information available in the scientific literature. Reviews published on antimony the last 25 years are listed in Table A1 of the Accessory publication together with their contents, number of pages, number of references, etc. Unfortunately, many of the existing reviews are based on a limited number of references, with the criteria for choosing them going unstated. They are not ‘systematic reviews’ in the sense defined by Petticrew[1] (‘a review that strives to comprehensively identify, track down, and appraise all the literature on a topic’). It should be mentioned that reviews related to toxicological issues contain references that are older than those contained in reviews on other subjects. This might indicate a worrying lack of recent studies in this field. Systematic reviews are lacking on many environmental topics such as: antimony in the atmosphere, ecotoxicology, and soil–plant transfer, among others.
Factual information about production and uses of antimony is sometimes given in reviews and introductions of papers. However, very often data are already old when cited and, even where this is not the case, they will rapidly become outdated. Up-to-date and reliable information is available at http://minerals.usgs.gov/minerals/pubs/commodity/antimony/, accessed 7 April 2009. Citing and using such data from this source, rather than recopying outdated values, is recommended.
Another aspect that merits some comment is the manner in which published information is transmitted. For instance, there is a strong tendency to blindly reproduce some ‘well-known’ facts and references without looking them up or tracking down additional, more updated information. An example of bad, but common, referencing practices is given in the What is more toxic? section.
How much: total concentrations
A comprehensive review of antimony concentrations in surface waters, soils and sediments was published 7 years ago.[2] Typical total (but filtered) concentrations in unpolluted freshwaters are well below 1 μg L–1. In oceans, surface concentrations are of the order of 0.2 μg L–1. Concentrations of antimony in non-heavily polluted soils are of a few μg g–1. Antimony concentrations in unpolluted sediments are probably of the same order of magnitude, but most of the existing data concern heavily polluted systems and it is difficult to establish a ‘natural’ level from published data. Antimony concentrations in the atmosphere have not yet been the subject of a systematic review. Published aerosol concentrations are in the range <0.1 ng m–3 in the atmosphere over remote oceans to several ng m–3 over industrialised areas. Values of 0.2 ng m–3 and 0.45 pg m–3 were chosen by Austin and Millward[3] as representative of the continental and marine tropospheres, respectively, in their model of the global antimony cycle.
Nowadays, the problem of determining total antimony concentrations can be considered to be solved. The most common techniques for determining the total antimony concentrations include inductively coupled plasma–mass spectrometry (ICP-MS) and hydride generation (HG) coupled to an element-specific detector such as atomic absorption spectroscopy (AAS) or atomic fluorescence spectroscopy (AFS).[4,5] However, it must be pointed out that many freshwater systems have antimony concentrations close to the detection limit of these techniques. Shotyk et al.[6] showed that use of ICP-SMS (inductively coupled plasma–sector field mass spectrometry) and clean laboratory methods are required for determining antimony not only in polar snow and ice but also in pristine groundwaters. On-line preconcentration methods using specific sorbents have been proposed to overcome this problem, but so far none has emerged as a method of choice. Moreover, it must be remembered that, at such extremely low concentrations, not only is analytical sensitivity a challenge, but sample contamination is also a serious concern. It is also important to mention that the sample matrix may have a pronounced effect on the analytical signal, particularly when determining antimony in complex organic matrices.[7] Hydride generation techniques may also not give 100% recovery when non-hydride-forming species are present in the sample. This point is further discussed in the Soils section. Finally, the total and reliable extraction of antimony from solid matrices such as soils, sediments and plants cannot be considered to have been completely resolved.[4] The same is true of the digestion of aerosols retained in filters, as discussed by Smichowski.[5]
Antimony trioxide is extensively used by the polymer industry as a polycondensation catalyst in the production of polyethylene terephthalate (PET) and release of antimony from PET containers has been demonstrated.[6,8] Apart from its possible toxicological implications, these results suggest that sample contamination in the laboratory by antimony-bearing containers and sample-handling equipment could be more widespread than generally assumed. Migration of antimony has also been observed from PET containers into orange juice[9] and from food trays or PET materials for oven use to food.[10,11] Possible implications for sample conservation and treatment have not been studied.
Establishment of background concentration levels
Distinguishing between lithogenic (or natural) and anthropogenic contributions of the element of interest to a given compartment is a challenge that commonly arises in environmental studies. Establishing background concentrations is an unavoidable prerequisite for any study aiming to identify and evaluate sources of contamination. In the absence of human activity, elements are released to terrestrial, aquatic and atmospheric environments at rates that correspond to crustal rock composition, mineral presence and natural chemical and mechanical erosion times. However, human activity has significantly altered the rate of release of elements to the environment, with a subsequent cascading effect on the rate at which metals are exchanged between different reservoirs, which has rendered it extremely difficult to establish concentration levels for ideal pristine sites. This is particularly the case for antimony, where measurements in peat cores from ombrotrophic bogs in Switzerland, Scotland and the Faroe Islands have revealed atmospheric antimony contamination since Roman times.[12–16] Nevertheless, it should be still possible, and useful, to establish current background concentration values for ‘non-polluted’ sites linked to characteristics such as water hardness or lithogenic origin. This can be achieved through careful classification and evaluation of published data (currently lacking for antimony in soils and the atmosphere) and by performing new measurements in appropriately selected systems. For instance, in a recent study,[17] where large series of data on dissolved antimony concentrations from the hydrological network of Canton Geneva, Switzerland, were statistically analysed, significant differences were found as a function of the predominant lithology of the watershed, with lower concentrations in carbonated zones than in granitic ones.
Solubility issues
Trace element concentration in any environmental compartment is dependent on several factors, including the solubility of the solid phases it may form. Several simple Eh–pH and related diagrams for antimony in various oxidation states and involving oxide phases and aqueous or hydrolysed species have been reported in an effort to explain phase and solubility relationships in metallurgical and geochemical processes. With respect to geochemical applications, some of these diagrams have been elaborated to include the minerals stibnite, Sb2S3, and kermesite, Sb2S2O, for given activities of sulfate ion. Various examples of such diagrams can be found in the literature.[18–24] All of these diagrams suffer in part either from the fact that inaccurate thermochemical parameters were available at the time of their construction or that data were selected arbitrarily from one of several sources. In addition, metastable phases, notably Sb2O5(s), were sometimes modelled or species such as Sb(OH)3(s), which are not known to occur naturally, were included in the calculations. A particular drawback concerning inclusion of Sb2O5(s) is that calculations in conjunction with dissolved antimony species give rise to a misleading picture of the mobility of SbV under strongly oxidising conditions. It is apparent that this is in good part responsible for the often repeated view that antimony is highly mobile in the supergene environment, as mentioned for example by Vink.[22] In addition, other cation concentrations that may give rise to separate secondary mineral phases have never previously been taken into account.
A summary of ΔGf° (298.15 K) data is given in Table 1.[23,25–36] Reliable thermochemical data are available for stibnite,[25,26] but the accuracy and precision of data derived from Williams-Jones and Normand[23] and Babčan[27] for kermesite remain questionable. Fortunately, deficiencies associated with reliable thermochemical data for most of the common antimony oxides have been eliminated in the light of more recent experimental studies. Values reported by Zotov et al.[28] for valentinite and sénarmontite are consistent with earlier data listed by Wagman et al.[29] Values for the free energies of formation of valentinite and sénarmontite listed by Barin[37] are clearly erroneous, as noted by Vink.[22] The situation with respect to cervantite, α-Sb2O4 (SbIIISbVO4), has been somewhat more problematic. The generally accepted value for ΔGf° (298.15 K) is taken from Wagman et al.[29] but a range of values is found in the literature. Analysis of published values suggests that the Pankajavalli and Sreedharan value[30] is the more accurate. The complete lack of thermochemical data for the common mixed-valency species stibiconite, ideally SbIIISb2VO6OH, needs to be addressed urgently in order to provide a more complete picture of the behaviour of antimony under moderately oxidising conditions.
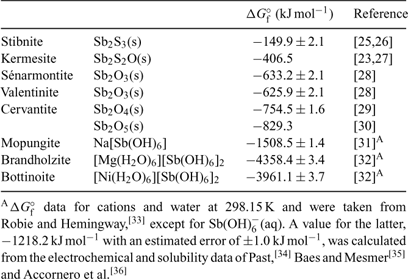
|
As mentioned above, Pourbaix diagrams for antimony based on the existence of Sb2O5(s) under the most oxidising conditions are misleading. The monoclinic phase can only be prepared under extreme conditions[38] and is metastable in aqueous solutions at ambient temperatures with respect to cubic ‘antimonic acid’, Sb2O5·4H2O.[31] No thermochemical data are available for the latter phase and this too represents a marked gap in our knowledge of the simple chemistry of antimony. More realistic proxies for the behaviour of SbV in aqueous systems are limited by the paucity of pertinent solubility data available in the literature.
Solubility data from 25 to 55°C for mopungite, Na[Sb(OH)6], have been reported,[31] from which a KSP at 298.15 K may be derived as being 8.89 × 10–6. Diemar et al.[32] reported solubility studies concerning brandholzite, [Mg(H2O)6][Sb(OH)6]2, and its NiII analogue, bottinoite. At 298.15 K, KSP values are 1.82 ± 0.15 × 10–8 and 1.29 ± 0.13 × 10–10, respectively. At this temperature, the solubilities of the minerals follow the series mopungite > brandholzite > bottinoite, but all three have appreciable solubility. Nevertheless, all of them are less soluble than the potassium salt, which is not known to occur naturally, and may be used in Pourbaix diagram calculations instead of Sb2O5(s). Under certain geochemical conditions, mopungite, brandholzite and bottinoite may serve to limit SbV concentrations in solution; these do indeed occur naturally.
Johnson et al.[39] reported unpublished values for the solubility products at (presumably) 25°C of Ca[Sb(OH)6]2(s) and Pb[Sb(OH)6]2(s) of 10–12.55 and 10–11.02, respectively, but it has been shown that these are amorphous materials that ultimately crystallise to yield roméite, ideally Ca2Sb2O7, and bindheimite, ideally Pb2Sb2O7.[32] Nevertheless, it is possible that the above data may represent the approximate upper limits of the solubility of SbV in the presence of Ca2+(aq) and Pb2+(aq) with short reaction times. With this in mind, it is noteworthy that Vitaliano and Mason[40] found that many of the natural specimens of stibiconite that they studied were X-ray-amorphous or poorly crystalline, and many contained appreciable amounts of calcium. Some solubility data are available for roméite and bindheimite.[32] These minerals do not dissolve congruently in acid solution.
How: speciation
In theory, speciation measurements aim at determining all chemical species formed by an element in a system. However, in practice this is seldom possible and speciation measurements only give operationally defined fractions as a function of the technique applied (i.e. size, redox state, lability, etc.). As discussed below, the fractions determined change depending on the environmental compartment considered.
Waters
In the case of antimony, speciation in waters has typically focussed on distinguishing between the two oxidation states of antimony, SbIII and SbV, and, to some extent, on determining alkylantimony species. In Filella et al.,[2] a comprehensive revision of data published to date gave 122 entries (one entry might contain data on more than one system) concerning freshwaters, seawater and estuaries. SbIII/SbV speciation data were present in 37% of these entries. Data showed that SbV was by far the predominant species present in oxic systems with a low amount of SbIII sometimes present but rarely representing more than 10% of total antimony. Data published since 2001 are shown in Table A2. Of the values published these last years for oxic systems (21 entries), 13 belong to the ‘application of an analytical method’ category (see comment in Sources of information section), six were published by the same research group but the articles contain few additional data and thus are of little interest, and only two papers[41,42] contain data obtained from studies specifically focussed on antimony behaviour in natural systems. This is disappointing. The situation is even worse for anoxic systems where only one study[43] has been published.
In recent years, many speciation methods for antimony have been, and are continuing to be, developed and published. However, when it comes to real environmental studies, few and well-known methods are used. This means that most of the methods being developed are never applied beyond the measurements that accompany the original publication. The reasons behind this blatant waste of research effort merit some thought.
As mentioned, antimony speciation studies focus nearly exclusively on determining the two oxidation states. Some methods can only determine them (e.g. voltammetry, HG methods), whereas in others (e.g. online coupling of high-performance liquid chromatography (HPLC) with an element-specific detector), the use of a mobile phase containing complexing ligands such as EDTA (ethylenediaminetetraacetic acid) and phthalic acid, which are needed to elute SbIII species from the column, prevent obtaining any information about the original antimony species present in samples because the complexing ability of the mobile-phase components change the original species during the chromatographic run. Some antimony-organic species such as SbV-citrate and SbV-lactate in yoghurt,[44] urine[45] and orange juice[9] as well as SbIII-(GS)3 and (CH3)SbIII-(GS)2 in sludge extracts[46] have been identified by electrospray-mass spectrometry (ES-MS), but most of these results have been obtained in antimony-enriched samples. Current techniques are not sensitive enough to allow for possible antimony-organic complexes to be detected in natural waters.
The use of other speciation methods, such as, for instance, those based on size (e.g. cross-flow-filtration, field flow fraction) has been rarely applied to the study of antimony. As discussed in Filella et al.,[47] current understanding is that antimony is mostly present as ‘dissolved’ in natural oxic waters.
Soils
It is common practice to determine concentrations extracted from soils subjected to extractions with reagents having different chemical properties. The extractants used range from water to relatively strong organic complexants such as citric and oxalic acids or EDTA. Because of the strong operational character of these methodological approaches, the extractant applied and the terminology used depend on the objective of the study. For instance, the fraction obtained by using mild extractants is often called ‘soluble’ or ‘reactive’ and, when the objective of the extractions is to mimic plant uptake, fractions are at times called ‘bioavailable’. Fractionation into operationally defined forms by sequential extraction with progressively stronger extractants, with the aim of investigating the components of the solids attached to the element of interest, is also widely applied. Extraction results for antimony have just recently been reviewed (M. Filella, unpubl. review). In the case of antimony, a very low percentage is extracted by using mild extractants and, when sequential extraction procedures are applied, some antimony is present in the ‘iron oxide fraction’ but most is present in the so-called residual fraction. Largely on the basis of such results, it has been established, and is generally accepted, that antimony is essentially immobile in soils. Very few studies contradict this statement (e.g. Gerritse et al.[48]). It must be mentioned, however, that most of the soils studied were heavily polluted systems, often located in the proximity of mines and smelters (M. Filella, unpubl. review), and that the form in which antimony is present in such soils might be very different to that in non-polluted ones. For instance, Aisnworth et al.[49] mentioned that most antimony was emitted as Sb2O3 from smelting operations and remained in that form after being deposited in the surface soils studied, and Hammel et al.[50] suggested, although did not prove, that antimony in contaminated soils is mostly immobile because much antimony remains as non-reactive oxides when it is deposited as Sb2O3. However, it is generally believed that antimony is mainly present as SbV in soils and Oorts et al.[51] found that more than 70% of antimony added as Sb2O3 to soils was present as SbV within 2 days. Knowing in which form antimony actually arrives in the soil – which may vary greatly among different soils – and how it is transformed in both polluted and non-polluted soils will surely help to understand the above-mentioned contradictions.
Sometimes, the objective is to obtain a maximum extraction yield without digesting the mineral matrix in order to keep the redox state of the element. In such cases, extractants that are supposed to preserve the redox state, such as citric acid, are used. Measuring techniques, already mentioned for waters, are then applied to the extracted solutions.[52,53] Interpreting these type of results remains extremely risky.
Biota
Extraction of antimony from biological matrices, including some of environmental interest, such as plants and fungi, has recently been discussed with remarkable insight by Hansen and Pergantis.[7] Among many other interesting issues, these authors mention the fact that, in contrast to extraction of naturally occurring antimony species, antimony-spiked samples are often successfully extracted, a possible reason being that spiked samples do not reach equilibrium with naturally occurring species within the sample matrix. The same observation applies to soils and possibly to waters. Therefore, the widespread practices of spiking samples when establishing analytical methods, testing sample conservation, etc., or of adding synthetic materials to natural matrices assuming that they behave like their natural counterparts (e.g. study by Oorts et al.[50] mentioned above) may need to be reconsidered.
Atmosphere
In the atmosphere, antimony speciation has been almost exclusively based on size considerations because of the well-known relationship between the size of airborne particles and intake into the lungs. Cascade impactors with different separation points (usually, 10 and 2 or 2.5 μm) have been used for particle fractionation, followed by the application of common methods for the determination of total antimony. As mentioned in the How much: total concentrations section, Smichowski[5] provides a good discussion of the methods available for sample digestion. Traditionally, antimony has been considered one of the ‘smallest’ particle size elements.[54] These elements are commonly associated with high-temperature anthropogenic processes, such as smelting of metals and combustion (coal, refuse incineration), which are still considered to be the main sources of antimony emission into the atmosphere.[55] It is the case that elements with low boiling points, such as antimony, are likely to be emitted as submicron particles or gases during combustion processes; many of the gases later condense onto submicron particles. Incidentally, so far, although data on aerosol concentrations are plentiful, the topic has not been the subject of any recent systematic review.
As antimony is very efficiently transported and redistributed through the atmosphere, the knowledge of the actual magnitude of the concentrations and deposition fluxes involved is of the utmost importance. However, of equal importance is the knowledge of the actual speciation of antimony in the atmosphere because it will affect its transport, but also, the form in which antimony is deposited onto a compartment will greatly influence its subsequent behaviour: the fate of antimony in waters and soils might be very different if the origin of the deposited aerosol is a combustion process rather than brake-wear particles.
After abundant research on the size distribution and nature of various elements, including antimony, on coal fly-ashes in the eighties, more recent studies on antimony have mainly been devoted to investigating the link between its use in brake linings (as a substitute for asbestos) and its presence in urban aerosols and soils close to roads.[56–58]
Extraction procedures similar to the ones described for soils have been applied to a limited extent to airborne particulate matter.[59,60] The operational character of such methods, and the limitations implied, remain the same as those discussed for other solid matrices.
Biomethylation
The biomethylation of antimony has been reviewed periodically either on its own or with other elements (Table A1). Most of the existing literature concerns the development of analytical methods and the study of laboratory cultures and landfill gases. Little data exist for natural waters and unpolluted soils and sediments.[61,62] Measured concentrations are low and antimony seems to be methylated less extensively than other elements, such as arsenic. However, recent results might change the current perception. For instance, the results of Duester et al.[63] showed a higher proportion of methylated antimony in soils compared with arsenic, which is contrary to what the well-known lower biomethylation of antimony v. arsenic in microbial incubations would lead one to expect. Conservative methylantimony profiles have been measured by Cutter and Cutter[42] in the Pacific Ocean, which suggests that antimony might behave less like arsenic than previously believed.
It should be pointed out that, with the exception of the few studies where methylated antimony species have been found in natural waters, most of the research on this subject has been carried out by a very small number of research groups that have produced the bulk of the existing data on the subject.
Conservation issues
Water samples
Irrespective of the analytical method used, SbIII/SbV ratios in natural waters may change substantially during storage and extraction procedures. No systematic study of antimony conservation procedures in speciation determinations exists, as is the case for arsenic.[64,65] All studies where the antimony oxidation state has been determined have been searched for observations or advice on storage. A certain number of studies plainly ignored the conservation issue (e.g. refs [66–69]). Observations have been summarised in Table A3. Those based on the previous spike of natural samples with unrealistically high concentrations of SbIII have not been considered. As expected, oxidation of SbIII is the most commonly observed change during storage. Its extent seems to be very variable but might be sufficient to invalidate many conclusions.
As shown in Table A3, most of the studies merely describe empirical observations without attempting to identify the causes. Although the same general causes of arsenic oxidation[64] can be imagined in the case of antimony, e.g. microbial activity, abiotic oxidation of SbIII by the addition of acid preservatives, photooxidation in response to exposure to light or UV radiation, and differential adsorption of antimony species on precipitated iron oxyhydroxides, it should be mentioned that antimony does not behave exactly as arsenic does with regard to oxidation. For instance, antimony is oxidised by hydrogen peroxide, a natural oxidant, in a narrower pH range compared with arsenic.[70] Standard strategies to preserve antimony redox speciation during storage include: (i) filtration (it suppresses speciation changes partly through the removal of iron oxyhydroxides and partly through the removal of some bacteria); (ii) refrigeration and/or freezing (they slow down rates of antimony species changes, presumably by slowing down microbial metabolism); (iii) addition of organic acids such as lactic, ascorbic, citric, tartaric acids, EDTA (they prevent SbIII oxidation by complexing it; EDTA also prevents iron oxyhydroxide precipitation). Acidification is not to be advised because it has been reported to lead to rapid oxidation of SbIII.[71] Addition of any exogenous substance (e.g. organic acids) may help to keep the oxidation state but will destroy existing natural complexes. Caution must be taken in the freezing and thawing processes to avoid formation of brines. Precipitation of arsenic oxide, irreversible on remelting, has been observed.[72] Acidification with HCl at pH < 2 is the method of choice for conserving methylated antimony species.[73]
Some authors have tested the effect of adding different organic acids on the stability of synthetic SbIII-containing solutions.[74] Although this type of experiment can give some indications of SbIII behaviour, the results obtained cannot be extrapolated directly to natural waters where the presence of a mixture of unknown oxidants cannot be excluded. Even in the case of synthetic solutions, Andreae et al.[75] mentioned that some diluted working SbIII solutions prepared from a stock solution of potassium antimonyl tartrate (1 mg L–1) could last for months whereas others were oxidised within hours. They attributed the effect to the quality of the water used to prepare the solutions.
Soils
The extraction of antimony compounds from soil samples without changing their chemical forms remains an analytical challenge. Moreover, the chemical composition of the extracting solution is not always compatible with the chromatographic separation conditions or with the detection system employed to detect the antimony species after their separation. The extracting agent could affect the chromatographic behaviour of the species by changing their retention times, in some cases making certain species undetectable by the HG-AFS detection system. For instance, tartaric, lactic, citric and oxalic acids are known to suppress the detection of SbV in HG methods.[60,76] Addition of a post-column photooxidation step may help to solve the problem.[77] Many of the insights of Hansen and Pergantis[7] on conservation of biological samples also apply to other solid matrices such as soils.
Common procedures used for soil sampling and pretreatment, such an open-air drying, grinding and sieving (Table A4), do not seem particularly well adapted to conserving antimony speciation.
Solid phase speciation
Few studies have examined the speciation of Sb in the solid phase by X-ray absorption spectroscopy (XAS) in real environmental samples. Only environmental systems containing antimony concentrations high enough for XAS analysis, such as soils around an antimony smelter,[78] affected by an antimony mine pit[79] and from shooting ranges[80] have been studied so far using these techniques. These studies found that antimony was present exclusively as SbV and that FeIII hydroxide was probably the host phase.
Thermodynamic calculations
It is generally accepted and repeated in many articles that thermodynamic predictions suggest that Sb should be present as SbV in oxic media and as SbIII in anoxic ones. Although existing thermodynamic data for SbV and SbIII equilibria are limited, they do allow us to consider this prediction reliable. Thermodynamic data for low molecular mass aqueous species of SbV and SbIII have been collected and evaluated by Filella and May.[81,82] Since then, a few valuable data have been published.[34] Filella and May studies made it possible to select thermodynamically consistent equilibrium constants for some key equilibria but also made it clear that reliable data are missing for many equilibria of interest in biological and environmental systems. In particular, constants for SbV are virtually non-existent. It should also be mentioned that most of the data for SbIII low molecular mass organic ligands are of rather limited applicability because titrations covered a very narrow pH range. Owing to solubility problems, most titrations were performed up to pH 5 at most. This means that the corresponding equilibrium data do not cover the pH domain of interest in environmental and biological systems. The situation concerning solid species has already been discussed in detail in the Solubility issues section.
Finally, it should be mentioned that there is some confusion concerning the notation used for the species formed. In brief, the common convention is to represent the hydrolytic species of antimony as hydroxide complexes (e.g. see Baes and Mesmer[35]) but in many early publications and some more recent papers, such as Vink,[22] these species are represented as oxyanions, analogous to those for arsenate/arsenite and phosphate. This should not pose any particular problem provided the chosen notation is used consistently. Unfortunately, this is not the case in the databases of some popular computer programs, leading some authors to think that, for instance, Sb(OH)3 and HSbO2 coexist in solution. See two notorious examples in Kawamoto and Morisawa[83] and Watkins et al.[84]; these latter authors even spent several paragraphs discussing the binding of both species onto goethite surfaces.
Even if some sparse data exist (M. Filella, unpubl. review), at present it is not possible to include the presence of complexants such as iron and manganese oxides, aluminosilicates and natural organic matter in thermodynamic calculations.
Presence of thermodynamically unstable species
In spite of thermodynamic predictions (see Section above), significant concentrations of thermodynamically unstable antimony species have been measured in oxic and in anoxic systems and to date this issue remains largely unexplained.[2] The presence of such species (SbIII in oxic systems and SbV in anoxic media) requires both a source and some kind of kinetic stabilisation. A possible external source of SbIII in oxic surface waters is atmospheric deposition (see Atmosphere section), whereas possible sources of SbV in anoxic media are the transport of SbV on sinking detritus from oxic waters or advection of oxic waters containing high SbV concentrations. As yet, however, experimental evidence is weak. Internal sources of SbIII include biotic and abiotic reduction of SbV. Biotic and abiotic SbIII oxidation and formation of SbV thiocomplexes can explain the presence of SbV in anoxic waters.
Biological activity has been invoked by different authors to explain the presence of SbIII in surface waters without any proof being provided. Experimental observations regarding biota-mediated antimony oxidation and reduction have been reviewed by Filella et al.[61] Soon after publication of their review, interesting results were reported by Lehr et al.[85] These authors documented SbIII oxidation by Agrobacterium tumefaciens and, for the first time, by a eukaryotic organism – an acidothermophilic alga belonging to the order Cyanidiales. Interestingly, Agrobacterium tumefaciens can also oxidise AsIII but the mechanisms are different for arsenic and antimony.
As they found no correlation between SbIII and biotic tracers, Cutter et al.[86] suggested that photochemical reactions might be a source of SbIII in surface waters of the subtropical and equatorial Atlantic Ocean as is the case for FeII. Ellwood and Maher[87] considered the hypothesis of SbV reduction by sunlight in ocean waters likely but did not provide any proof. However, photooxidants that are produced in sunlit natural waters might be also capable of inducing SbIII oxidation. Buschmann et al.[88] studied the effect of humic substances on SbIII oxidation in the laboratory and found that photooxidation was 9000 times faster than the dark reaction in the presence of 5 mg L–1 humic acids, a concentration that is relatively high for surface waters, particularly in marine systems. Li et al.[89] studied the photooxidation of SbIII through the combined effect of light and algae. Unfortunately, their study contains significant flaws in both experimental design and the interpretation of data.
Abiotic oxidation of SbIII by different natural oxidants has been studied: synthetic and natural Fe and Mn oxyhydroxides,[90,91] hydrogen peroxide[70,92–94] and iodate.[95] Oxidation of SbIII with hydrogen peroxide and iodate is pH-dependent: no oxidation is observed below pH 9 because formation of Sb(OH)4– is needed for oxidation to occur. Arsenic behaves in a similar way with hydrogen peroxide but, whereas Sb(OH)3 has a pK of 11.7, the pK of the corresponding arsenic species is 9.2. This pK difference confers kinetic resistance vis-à-vis oxidation over a larger pH range to SbIII compared with AsIII. Abiotic SbV reduction has been much less studied. It is well known that SbV is reduced by thiol-containing ligands[96,97] and, for instance, cysteine is widely used as a SbV pre-reductant in HG methods, but the extent of such reduction by thiol compounds in real environmental systems is unknown. Abiotic reduction of SbV by green rust[98,99] and magnetite and mackinawite[100] have been described recently. In spite of these new results, being able to predict antimony redox distribution in natural waters and soils probably lies far in the future.
Biogeochemical cycle
A first attempt to establish a global biogeochemical cycle for antimony was described by Austin and Millward[3] 20 years ago on the basis of data existing at that time. Since then, most efforts have gone into establishing atmospheric emissions,[55,101] with far less emphasis on integrating water, land and biota into a global cycle. However, integrating existing data into a biogeochemical cycle should be a priority task today. It would contribute to our understanding of how antimony moves through the different compartments, and in what amounts, and would help to identify the main gaps in our knowledge (e.g. content and mobility from rocks[17] and soils, antimony deposition in the atmosphere and oceans, biota contents, soil–plant transfer, etc.).
Ecotoxicity
What is more toxic?
In the introduction of many publications, great emphasis is placed on the danger of antimony, often giving the impression that the chance of being published increases in proportion with the risk involved. However, the real environmental risk posed by antimony is still largely in need of assessment and proof. Incidentally, careful reading of the literature shows that many authors copy other articles’ introductions, sometimes even word for word, and rarely verify the sources. The case of the comparison of SbIII v. SbV toxicity is paradigmatic. In the introduction of 98 papers, it is found that ‘SbIII is more dangerous than SbV’ and, of these, 32 even go further and state that ‘SbIII is 10 times more dangerous than SbV’. This statement can only be wrong because toxicity depends on many parameters such as the considered organism, the route of exposure, or the presence of other contaminants. Thus, in general, no chemical species is exactly x times more toxic than another. Moreover, when the sources given by the authors in support of their statement are analysed, the results are disheartening: the authors of 10 papers give no source at all, 12 give a variety of purely analytical studies that have no connection with toxicity issues, seven give various general reviews, two give two different toxicological papers and six cite Venugopal and Luckey,[102] who could be at the origin of the above-mentioned statement, although their data do not clearly support it.
Antimony ecotoxicity data
A comprehensive compilation, review and evaluation of ecotoxicological data of antimony has never been published in a peer-reviewed paper. However, a certain number of reports wherein data are compiled and compared, and sometimes used for environmental risk assessments, are available (e.g. Vangheluwe and Van Hyfte[103]). They have usually been prepared either by private consulting companies or by organisations such as the US Environmental Protection Agency, European Union or Organisation for Economic Co-operation and Development. The total number of original studies is low, some are relatively old and a significant amount of the existing data has been produced either by private companies commissioned by antimony producers or by the above-mentioned organisations. As a result, some studies are not easily available and the data are not always guaranteed to be reliable according to current scientific standards. The criteria that need to be applied to evaluate the reliability of ecotoxicological data are not simple and their application to the evaluation of published antimony data is beyond the scope of the current review. However, a quick assessment from the chemical point of view alone shows that many results are flawed. As an illustration, published studies on acute antimony ecotoxicity for aquatic organisms have been compiled and are shown in Table A5. All results (except one) are based on nominal concentrations and the authors do not take any account of speciation aspects (e.g. complexation by the media). None controls for antimony oxidation or reduction during the experiment. More importantly, the nominal concentrations used often clearly exceed antimony solubility. Consider the case of Sb2O3(s), where concentrations of 500 mg L–1 are claimed to have been used. Cubic Sb2O3(s) is the thermodynamically stable phase and it can be easily deduced from Zotov et al. data[28] that at 25°C, and at pH values from 4 to 10, the solubility of sénarmontite in water limits dissolved Sb concentrations to ~1.3 mg L–1. Yet there is a long-standing awareness of the solubility problem. For instance, more than 50 years ago, Bradley and Fredrick[104] concluded that the ‘toxicity is determined by the solubility of the compound in question’. However, to take one example, this did not impede the publication of a recent study[105] where, after the authors stated that SbIII sulfate is insoluble in water and showed that measured antimony concentrations were ~10% of the nominal ones, they nonetheless expressed the results in nominal terms.
Chronic toxicity tests have not been widely applied to antimony and some might not be exempted from the solubility problems mentioned above. Aspects such as genotoxicity[106–109] or metalloestrogenecity[110,111] will not be discussed here, but no firm conclusions can be drawn from the scant published studies. Initial data on the effect of antimony on the microbial growth and the activities of soil enzymes have recently become available.[112,113]
Conclusions
Each section of the present article discusses aspects of the environmental chemistry of antimony that require further research both in the laboratory and in the field. A large number of points have been raised and they will not all be repeated here. However, of these, the most urgent attention probably needs to be given to: (i) performing sound long-term spatial and temporal studies in the different environmental compartments in order to collect the data necessary to establish a global biogeochemical cycle for antimony; (ii) clarifying antimony interactions with potential natural binders under the relevant environmental conditions; (iii) completing reliable ecotoxicological studies. The extent of work that remains to be done is clearly not insignificant. This is not surprising. It should not be forgotten that, in recent years, much greater effort has been put into the study of arsenic, the companion element of antimony. Extensive research on arsenic has been driven by the need to solve very serious environmental and toxicological problems but, despite this effort, many unknowns remain. Although antimony poses less of an environmental threat than arsenic, and as such has been much less studied, there is no reason to think that the chemistry of antimony is not at least as complicated, and interesting, as arsenic’s. Well-focussed research, building on thorough knowledge of what is already known, will undoubtedly shorten the long path that lies ahead.
Abbreviations
AAS, atomic absorption spectroscopy; AFS, atomic fluorescence spectroscopy; EDTA, ethylenediaminetetraacetic acid; ES-MS, electrospray-mass spectrometry; GS, glutathione; HG, hydride generation; HG-AFS, hydride generation–atomic fluorescence spectroscopy; HPLC, high-performance liquid chromatography; ICP, inductively coupled plasma; ICP-MS, inductively coupled plasma–mass spectrometry; ICP-SMS, inductively coupled plasma–sector field mass spectrometry; NAA, neutron activation analysis; PET, polyethylene terephthalate; XAS, X-ray absorption spectroscopy.
Accessory publication
Five tables containing the following information: reviews on antimony, or that contain a significant part on this element, published in the last 20 years; speciation data on antimony in natural waters published after 2001; observations related to the preservation of antimony speciation during sampling and storage of natural waters; procedures described for soil sampling, pretreatment and conservation in studies where redox speciation measurements, determination of methylated species or solid speciation have been performed; chemical parameters in published antimony acute toxicity tests for aquatic organisms. This material is available free of charge via the Internet at http://publish.csiro.au/nid/188.htm.

|
[1]
M. Petticrew ,
Why certain systematic reviews reach uncertain conclusions.
BMJ 2003
, 326, 756.
| Crossref | GoogleScholarGoogle Scholar | PubMed |

[2]
M. Filella ,
N. Belzile ,
Y.-W. Chen ,
Antimony in the environment: a review focused on natural waters I. Occurrence.
Earth Sci. Rev. 2002
, 57, 125.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[3]
L. S. Austin ,
G. E. Millward ,
Simulated effects of tropospheric emissions on the global antimony cycle.
Atmos. Environ. 1988
, 22, 1395.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[4]
M. J. Nash ,
J. E. Maskall ,
S. J. Hill ,
Methodologies for determination of antimony in terrestrial environmental samples.
J. Environ. Monit. 2000
, 2, 97.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[5]
P. Smichowski ,
Antimony in the environment as a global pollutant: a review on analytical methodologies for its determination in atmospheric aerosols.
Talanta 2008
, 75, 2.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[6]
W. Shotyk ,
M. Krachler ,
B. Chen ,
J. Zheng ,
Natural abundance of Sb and Se in pristine groundwaters, Springwater Township, Ontario, Canada, and implications for tracing contamination from landfill leachates.
J. Environ. Monit. 2005
, 7, 1238.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[7]
H. R. Hansen ,
S. A. Pergantis ,
Analytical techniques and methods used for antimony speciation analysis in biological matrices.
J. Anal. At. Spectrom. 2008
, 23, 1328.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[8]
W. Shotyk ,
M. Krachler ,
B. Chen ,
Contamination of Canadian and European bottled waters with antimony from PET containers.
J. Environ. Monit. 2006
, 8, 288.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[9]
H. R. Hansen ,
S. A. Pergantis ,
Detection of antimony species in citrus juices and drinking water stored in PET containers.
J. Anal. At. Spectrom. 2006
, 21, 731.
| Crossref | GoogleScholarGoogle Scholar |
[10]
P. J. Fordham ,
J. W. Gramshaw ,
H. M. Crews ,
L. Castle ,
Element residues in food contact plastics and their migration into food simulants, measured by inductively-coupled plasma–mass spectrometry.
Food Addit. Contam. 1995
, 12, 651.
|
CAS |
PubMed |
[11]
M. Haldimann ,
A. Blanc ,
V. Dudler ,
Exposure to antimony from polyethylene terephthalate (PET) trays used in ready-to-eat meals.
Food Addit. Contam. 2007
, 24, 860.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[12]
W. Shotyk ,
Natural and anthropogenic enrichments of As, Cu, Pb, Sb, and Zn in ombrotrophic minerotrophic peat bog profiles, Jura Mountains, Switzerland.
Water Air Soil Pollut. 1996
, 90, 375.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[13]
W. Shotyk ,
A. K. Cheburkin ,
P. G. Appleby ,
A. Fankhauser ,
J. D. Kramers ,
Two thousand years of atmospheric arsenic, antimony, and lead deposition recorded in an ombrotrophic peat bog profile, Jura Mountains, Switzerland.
Earth Planet. Sci. Lett. 1996
, 145, E1.
| Crossref | GoogleScholarGoogle Scholar |
[14]
W. Shotyk ,
M. Krachler ,
B. Chen ,
Antimony in recent, ombrotrophic peat from Switzerland and Scotland: comparison with natural background values (5320 to 8020 14C yr BP) and implications for the global Sb cycle.
Global Biogeochem. Cycles 2004
, 18, GB1016.
| Crossref | GoogleScholarGoogle Scholar |
[15]
J. M. Cloy ,
J. G. Farmer ,
M. C. Graham ,
A. B. MacKenzie ,
G. T. Cook ,
A comparison of antimony and lead profiles over the past 2500 years in Flanders Moss ombotrophic peat bog, Scotland.
J. Environ. Monit. 2005
, 7, 1137.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[16]
W. Shotyk ,
B. Chen ,
M. Krachler ,
Lithogenic, oceanic and anthropogenic sources of atmospheric Sb to a maritime blanket bog, Myrarnar, Faroe Islands.
J. Environ. Monit. 2005
, 7, 1148.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[17]
P. Nirel ,
M. Filella ,
Dissolved antimony concentrations in contrasted watersheds: the importance of lithogenic origin.
J. Environ. Monit. 2008
, 10, 256.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[18]
[19]
H. D. Holland ,
Some applications of thermochemical data to problems of ore deposits. I. Stability relations among the oxides, sulfides, sulfates and carbonates of ore and gangue minerals.
Econ. Geol. 1959
, 54, 184.
|
CAS |
| Crossref |
[20]
D. G. Brookins ,
Geochemical behaviour of antimony, arsenic, cadmium and thallium: Eh–pH diagrams for 25°C, 1-bar pressure.
Chem. Geol. 1986
, 54, 271.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[21]
[22]
B. W. Vink ,
Stability relations of antimony and arsenic compounds in the light of revised and extended Eh–pH diagrams.
Chem. Geol. 1996
, 130, 21.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[23]
A. E. Williams-Jones ,
C. Normand ,
Controls on mineral parageneses in the system Fe–Sb–S–O.
Econ. Geol. 1997
, 92, 308.
|
CAS |
| Crossref |
[24]
[25]
L. T. Bryndzia ,
O. J. Kleppa ,
High temperature reaction calorimetry of solid and liquid phases in part of the quasi-binary system Cu2S–Sb2S3.
Am. Mineral. 1988
, 73, 707.
|
CAS |
[26]
R. R. Seal ,
R. A. Robie ,
B. S. Hemingway ,
P. B. Barton ,
Superambient heat capacities of synthetic stibnite, berthierite and chalcostibite: revised thermodynamic properties and implications for phase equilibria.
Econ. Geol. 1992
, 87, 1911.
|
CAS |
| Crossref |
[27]
J. Babčan ,
Enstehung und Stabilität von Antimonmineralen im System Sb3+–S2––H+–OH–.
Chem. Erde 1976
, 35, 281.
[28]
A. V. Zotov ,
N. D. Shikina ,
N. N. Akinfiev ,
Thermodynamic properties of the Sb(III) hydroxide complex Sb(OH)3(aq) at hydrothermal conditions.
Geochim. Cosmochim. Acta 2003
, 67, 1821.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[29]
D. D. Wagman ,
W. H. Evans ,
V. B. Parker ,
R. H. Schumm ,
I. Halow ,
S. M. Bailey ,
K. I. Churney ,
R. I. Nuttall ,
The NBS tables of chemical thermodynamic properties: selected values for inorganic and C1 and C2 organic substances in SI units.
J. Phys. Chem. Ref. Data 1982
, 11, 1.
[30]
R. Pankajavalli ,
O. M. Sreedharan ,
Thermodynamic stability of Sb2O4 by a solid oxide electrolyte EMF method.
J. Mater. Sci. 1987
, 22, 177.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[31]
M. J. Blandamer ,
J. Burgess ,
R. D. Peacock ,
Solubility of sodium hexahydroxoantimonate in water and in mixed aqueous solvents.
J. Chem. Soc., Dalton Trans. 1974
, 1084.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[32]
[33]
R. A. Robie ,
B. S. Hemmingway ,
Thermodynamic properties of minerals and related substances at 298.15 K and 1 bar (105 Pascals) pressure and at higher temperatures.
U.S. Geol. Surv. Bull. 1995
, 2131, 1.
[34]
[35]
[36]
M. Accornero ,
L. Marini ,
M. Lelli ,
The dissociation constant of antimonic acid at 10–40°C.
J. Sol. Chem. 2008
, 37, 785.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[37]
[38]
M. Jansen ,
Die Kristallstruktur von Antimon(V)-Oxid.
Acta Crystallogr. 1979
, B35, 539.
|
CAS |
[39]
C. A. Johnson ,
H. Moench ,
P. Wersin ,
P. Kugler ,
C. Wenger ,
Solubility of antimony and other elements in samples taken from shooting ranges.
J. Environ. Qual. 2005
, 34, 248.
|
CAS |
PubMed |
[40]
C. J. Vitaliano ,
B. Mason ,
Stibiconite and cervantite.
Am. Mineral. 1952
, 37, 982.
|
CAS |
[41]
O. Rouxel ,
J. Ludden ,
Y. Fouquet ,
Antimony isotope variations in natural systems and implications for their use as geochemical tracers.
Chem. Geol. 2003
, 200, 25.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[42]
G. A. Cutter ,
L. S. Cutter ,
Biogeochemistry of arsenic and antimony in the North Pacific Ocean.
Geochem. Geophys. Geosyst. 2006
, 7, Q05M08.
| Crossref | GoogleScholarGoogle Scholar |
[43]
Y.-W. Chen ,
T.-L. Deng ,
M. Filella ,
N. Belzile ,
Distribution and early diagenesis of antimony species in sediments and porewaters of freshwater lakes.
Environ. Sci. Technol. 2003
, 37, 1163.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[44]
H. R. Hansen ,
S. A. Pergantis ,
Identification of Sb(V) complexes in biological and food matrixes and their stibine formation efficiency during Hydride Generation with ICPMS detection.
Anal. Chem. 2007
, 79, 5304.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[45]
H. R. Hansen ,
S. A. Pergantis ,
Investigating the formation of an Sb(V)–citrate complex by HPLC-ICPMS and HPLC-ES-MS(/MS).
J. Anal. At. Spectrom. 2006
, 21, 1240.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[46]
S. Wehmeier ,
A. Raab ,
J. Feldmann ,
Investigations into the role of methylcobalamin and glutathione for the methylation of antimony using isotopically enriched antimony(V).
Appl. Organomet. Chem. 2004
, 18, 631.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[47]
M. Filella ,
N. Belzile ,
Y.-W. Chen ,
Antimony in the environment: a review focused on natural waters. II. Relevant solution chemistry.
Earth Sci. Rev. 2002
, 59, 265.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[48]
R. G. Gerritse ,
R. Vriesema ,
J. W. Dalenberg ,
H. P. de Roos ,
Effect of sewage sludge on trace element mobility in soils.
J. Environ. Qual. 1982
, 11, 359.
|
CAS |
[49]
N. Ainsworth ,
J. A. Cooke ,
M. S. Johnson ,
Biological significance of antimony in contaminated grassland.
Water Air Soil Pollut. 1991
, 57–58, 193.
| Crossref | GoogleScholarGoogle Scholar |
[50]
W. Hammel ,
R. Debus ,
L. Steubing ,
Mobility of antimony in soil and its availability to plants.
Chemosphere 2000
, 41, 1791.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[51]
K. Oorts ,
E. Smolders ,
F. Degryse ,
J. Buekers ,
G. Gascó ,
G. Cornelis ,
J. Mertens ,
Solubility and toxicity of antimony trioxide (Sb2O3) in soil.
Environ. Sci. Technol. 2008
, 42, 4378.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[52]
S. Amereih ,
T. Meisel ,
R. Scholger ,
W. Wegscheider ,
Antimony speciation in soil samples along two Austrian motorways by HPLC-ID-ICP-MS.
J. Environ. Monit. 2005
, 7, 1200.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[53]
G. Ceriotti ,
D. Amarasiriwardena ,
A study of antimony complexed to soil-derived humic acids and inorganic antimony species along a Massachusetts highway.
Microchem. J. 2009
, 91, 85.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[54]
J. B. Milford ,
C. I. Davidson ,
The sizes of particulate trace elements in the atmosphere – a review.
J. Air Pollut. Control Assoc. 1985
, 35, 1249.
|
CAS |
PubMed |
[55]
J. M. Pacyna ,
E. G. Pacyna ,
An assessment of global and regional emissions of trace metals to the atmosphere from anthropogenic sources worldwide.
Environ. Rev. 2001
, 9, 269.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[56]
J. Sternbeck ,
A. Sjödin ,
K. Andréasson ,
Metal emissions from road traffic and the influence of resuspension – results from two tunnel studies.
Atmos. Environ. 2002
, 36, 4735.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[57]
N. Furuta ,
A. Iijima ,
A. Kambe ,
K. Sakai ,
K. Sato ,
Concentrations, enrichment and predominant sources of Sb and other trace elements in size classified airborne particulate matter collected in Tokyo from 1995 to 2004.
J. Environ. Monit. 2005
, 7, 1155.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[58]
D. S. T. Hjortenkrans ,
B. G. Bergbäck ,
A. V. Häggerud ,
Metal emissions from brake linings and tires: case studies of Stockholm, Sweden 1995/1998 and 2005.
Environ. Sci. Technol. 2007
, 41, 5224.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[59]
J. Zheng ,
M. Ohata ,
N. Furuta ,
Studies on the speciation of inorganic antimony compounds in airborne particulate matter by HPLC-ICP-MS.
Analyst 2000
, 125, 1025.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[60]
J. Zheng ,
A. Iijima ,
N. Furuta ,
Complexation effect of antimony compounds with citric acid and its application to the speciation of antimony(III) and antimony(V) using HPLC-ICP-MS.
J. Anal. At. Spectrom. 2001
, 16, 812.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[61]
M. Filella ,
N. Belzile ,
M.-C. Lett ,
Antimony in the environment: a review focused on natural waters. III. Microbiota relevant interactions.
Earth Sci. Rev. 2007
, 80, 195.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[62]
[63]
L. Duester ,
R. A. Diaz-Bone ,
J. Kösters ,
A. V. Hirner ,
Methylated arsenic, antimony and tin species in soils.
J. Environ. Monit. 2005
, 7, 1186.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[64]
D. A. Polya ,
P. R. Lythgoe ,
F. Abou-Shakra ,
A. G. Gault ,
J. R. Brydie ,
J. G. Webster ,
K. L. Brown ,
M. K. Nimfopoulos ,
K. M. Michailidis ,
IC–ICP-MS and IC–ICP-HEX-MS determination of arsenic speciation in surface and groundwaters: preservation and analytical issues.
Mineral. Mag. 2003
, 67, 247.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[65]
R. B. McCleskey ,
D. K. Nordstrom ,
A. S. Maest ,
Preservation of water samples for arsenic(III/V) determinations: an evaluation of the literature and new analytical results.
Appl. Geochem. 2004
, 19, 995.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[66]
G. Gillain ,
C. Brihaye ,
A routine speciation method for a pollution survey of coastal sea water.
Oceanol. Acta 1985
, 8, 231.
|
CAS |
[67]
J. J. Middelburg ,
D. Hoede ,
H. A. Van Der Sloot ,
C. H. Van Der Weijden ,
J. Wijkstra ,
Arsenic, antimony and vanadium in the North Atlantic Ocean.
Geochim. Cosmochim. Acta 1988
, 52, 2871.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[68]
S. Garbos ,
E. Bulska ,
A. Hulanicki ,
N. I. Shcherbinina ,
E. M. Sedykh ,
Preconcentration of inorganic species of antimony by sorption on Polyorgs 31 followed by atomic spectrometry detection.
Anal. Chim. Acta 1997
, 342, 167.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[69]
B. Mohammad ,
A. M. Ure ,
J. Reglinski ,
D. Littlejohn ,
Speciation of antimony in natural waters: the determination of Sb(III) and Sb(V) by continuous flow hydride generation–atomic absorption spectrometry.
Chem. Spec. Bioavail. 1990
, 3, 117.
[70]
F. Quentel ,
M. Filella ,
C. Elleouet ,
C.-L. Madec ,
Kinetic studies on Sb(III) oxidation by hydrogen peroxide in natural waters.
Environ. Sci. Technol. 2004
, 38, 2843.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[71]
F. Quentel ,
M. Filella ,
Determination of inorganic antimony species in seawater by DPASV: stability of the trivalent state.
Anal. Chim. Acta 2002
, 452, 237.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[72]
J. Aggett ,
M. R. Kriegman ,
The extent of formation of arsenic(III) in sediment interstitial waters and its release to hypolimnetic waters in Lake Ohakuri.
Water Res. 1988
, 22, 407.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[73]
[74]
M. B. de la Calle-Guntiñas ,
Y. Madrid ,
C. Cámara ,
Stability study of total antimony, Sb(III) and Sb(V) at the trace level.
Fresenius J. Anal. Chem. 1992
, 344, 27.
| Crossref | GoogleScholarGoogle Scholar |
[75]
M. O. Andreae ,
J.-F. Asmode ,
P. Foster ,
L. Van’t Dack ,
Determination of antimony(III), antimony(V), and methylantimony species in natural waters by atomic absorption spectrometry with hydride generation.
Anal. Chem. 1981
, 53, 1766.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[76]
M. Yamamoto ,
K. Urata ,
Y. Yamamoto ,
Differential determination of antimony(III) and antimony(V) by hydride generation–atomic absorption spectrophotometry.
Anal. Lett. 1981
, 14, 21–26.
|
CAS |
[77]
M. Potin-Gautier ,
F. Pannier ,
Q. Quiroz ,
H. Pinochet ,
I. de Gregori ,
Antimony speciation analysis in sediment reference materials using high performance liquid chromatography coupled to hydride generation atomic fluorescence spectrometry.
Anal. Chim. Acta 2005
, 553, 214.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[78]
M. Takaoka ,
S. Fukutani ,
T. Yamamoto ,
M. Horiuchi ,
N. Satta ,
N. Takeda ,
K. Oshita ,
M. Yoneda ,
S. Morisawa ,
T. Tanaka ,
Determination of chemical form of antimony in contaminated soil around a smelter using X-ray absorption fine structure.
Anal. Sci. 2005
, 21, 769.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[79]
S. Mitsunobu ,
T. Harada ,
Y. Takahashi ,
Comparison of antimony behavior with that of arsenic under various soil redox conditions.
Environ. Sci. Technol. 2006
, 40, 7270.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[80]
A. C. Scheinost ,
A. Rossberg ,
D. Vantelon ,
I. Xifra ,
R. Kretzschmar ,
A.-K. Leuz ,
H. Funke ,
C. A. Johnson ,
Quantitative antimony speciation in shooting-range soils by EXAFS spectroscopy.
Geochim. Cosmochim. Acta 2006
, 70, 3299.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[81]
M. Filella ,
P. M. May ,
Computer simulation of the low-molecular-weight inorganic species distribution of antimony(III) and antimony(V) in natural waters.
Geochim. Cosmochim. Acta 2003
, 67, 4013.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[82]
M. Filella ,
P. M. May ,
Critical appraisal of available thermodynamic data for the complexation of antimony(III) and antimony(V) by low-molecular-mass organic ligands.
J. Environ. Monit. 2005
, 7, 1226.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[83]
Y. Kawamoto ,
S. Morisawa ,
The distribution and speciation of antimony in river water, sediment and biota in Yodo River, Japan.
Environ. Technol. 2003
, 24, 1349.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[84]
R. Watkins ,
D. Weiss ,
W. Dubbin ,
K. Peel ,
B. Coles ,
T. Arnold ,
Investigations into the kinetics and thermodynamics of Sb(III) adsorption on goethite (α-FeOOH).
J. Colloid Interface Sci. 2006
, 303, 639.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[85]
C. R. Lehr ,
D. R. Kashyap ,
T. R. McDermott ,
New insights into microbial oxidation of antimony and arsenic.
Appl. Environ. Microbiol. 2007
, 73, 2386.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[86]
G. A. Cutter ,
L. S. Cutter ,
A. M. Featherstone ,
S. E. Lohrenz ,
Antimony and arsenic in the western Atlantic Ocean.
Deep Sea Res. Part II Top. Stud. Oceanogr. 2001
, 48, 2895.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[87]
M. J. Ellwood ,
W. A. Maher ,
Arsenic and antimony species in surface transects and depth profiles across a frontal zone: the Chatham Rise, New Zealand.
Deep Sea Res. Part I Oceanogr. Res. Pap. 2002
, 49, 19711.
| Crossref | GoogleScholarGoogle Scholar |
[88]
J. Buschmann ,
S. Canonica ,
L. Sigg ,
Photoinduced oxidation of antimony(III) in the presence of humic acid.
Environ. Sci. Technol. 2005
, 39, 5335.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[89]
S.-X. Li ,
F.-Y. Zheng ,
H.-S. Hong ,
N.-S. Deng ,
X.-Y. Zhou ,
Photooxidation of Sb(III) in the seawater by marine phytoplankton–transition metals–light system.
Chemosphere 2006
, 65, 1432.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[90]
N. Belzile ,
Y.-W. Chen ,
Z. Wang ,
Oxidation of antimony(III) by amorphous iron and manganese oxyhydroxides.
Chem. Geol. 2001
, 174, 379.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[91]
A. K. Leuz ,
H. Monch ,
C. A. Johnson ,
Sorption of Sb(III) and Sb(V) to goethite: influence on Sb(III) oxidation and mobilization.
Environ. Sci. Technol. 2006
, 40, 7277.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[92]
C. Elleouet ,
F. Quentel ,
C. L. Madec ,
M. Filella ,
The effect of the presence of trace metals on the oxidation of Sb(III) by hydrogen peroxide in aqueous solutions.
J. Environ. Monit. 2005
, 7, 1220.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[93]
A. K. Leuz ,
S. J. Hug ,
B. Wehrli ,
C. A. Johnson ,
Iron-mediated oxidation of antimony(III) by oxygen and hydrogen peroxide compared to arsenic(III) oxidation.
Environ. Sci. Technol. 2006
, 40, 2565.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[94]
A. K. Leuz ,
C. A. Johnson ,
Oxidation of Sb(III) to Sb(V) by O2 and H2O2 in aqueous solutions.
Geochim. Cosmochim. Acta 2005
, 69, 1165.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[95]
F. Quentel ,
M. Filella ,
C. Elleouet ,
C.-L. Madec ,
Sb(III) oxidation by iodate in seawater: a cautionary tale.
Sci. Total Environ. 2006
, 355, 259.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[96]
S. Yan ,
F. Li ,
K. Ding ,
H. Sun ,
Reduction of pentavalent antimony by trypanothione and formation of a binary and ternary complex of antimony(III) and trypanothione.
J. Biol. Inorg. Chem. 2003
, 8, 689.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[97]
C. dos Santos Ferreira ,
P. S. Martins ,
C. Demicheli ,
C. Brochu ,
M. Ouellette ,
F. Frézard ,
Thiol-induced reduction of antimony(V) into antimony(III): a comparative study with trypanothione, cysteinyl-glycine, cysteine and glutathione.
Biometals 2003
, 16, 441.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
[98]
S. Mitsunobu ,
Y. Takahashi ,
Y. Sakai ,
Abiotic reduction of antimony(V) by green rust (Fe4(II)Fe2(III)(OH)12SO4·3H2O).
Chemosphere 2007
, 70, 942.
| Crossref | GoogleScholarGoogle Scholar | PubMed |
[99]
S. Mitsunobu ,
Y. Takahashi ,
Y. Sakai ,
K. Inumaru ,
Interaction of synthetic sulfate green rust with antimony(V).
Environ. Sci. Technol. 2009
, 43, 318.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[100]
R. Kirsch ,
A. C. Scheinost ,
A. Rossberg ,
D. Banerjee ,
L. Charlet ,
Reduction of antimony by nano-particulate magnetite and mackinawite.
Mineral. Mag. 2008
, 72, 185.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[101]
[102]
[103]
[104]
W. R. Bradley ,
W. G. Fredrick ,
The toxicity of antimony – animal studies.
Ind. Med. 1941
, 2, 15.
[105]
R. G. Kuperman ,
R. T. Checkal ,
M. Simini ,
C. T. Phillips ,
J. A. Speicher ,
D. J. Barcliff ,
Toxicity benchmarks for antimony, barium, and beryllium determined using reproduction endpoints for Folsimia candida, Eisenia fetida, and Enchytraeus crypticus.
Environ. Toxicol. Chem. 2006
, 25, 754.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[106]
N. Kanematsu ,
M. Hara ,
T. Kada ,
Rec assays and mutagenicity studies on metal compounds.
Mutat. Res. 1980
, 77, 109.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[107]
K. Kuroda ,
G. Endo ,
A. Okamoto ,
Y. S. Yoo ,
S. Horiguchi ,
Genotoxicity of beryllium, gallium and antimony in short-term assays.
Mutat. Res. 1991
, 264, 163.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[108]
N. Gurnani ,
A. Sharma ,
G. Talukder ,
Comparison of the clastogenic effects of antimony trioxide on mice in vivo following acute and chronic exposure.
Biometals 1992
, 5, 47.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[109]
S. Knasmüller ,
E. Gottmann ,
H. Steinkellner ,
A. Fomin ,
C. Pickl ,
A. Paschke ,
R. Göd ,
M. Kundi ,
Detection of genotoxic effects of heavy metal contaminated soils with plant bioassays.
Mutat. Res. 1998
, 420, 37.
| PubMed |
[110]
S.-Y. Choe ,
S.-J. Kim ,
H.-G. Kim ,
J. H. Lee ,
Y. Choi ,
H. Lee ,
Y. Kim ,
Evaluation of estrogenicity of major heavy metals.
Sci. Total Environ. 2003
, 312, 15.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[111]
P. D. Darbre ,
Metalloestrogens: an emerging class of inorganic xenoestrogens with potential to add to the oestrogenic burden of the human breast.
J. Appl. Toxicol. 2006
, 26, 191.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |
[112]
T. Murata ,
M. Kanao-Koshikawa ,
T. Takamatsu ,
Effects of Pb, Cu, Sb, In and Ag contamination on the proliferation of soil bacterial colonies, soil dehydrogenase activity, and phospholipid fatty acid profiles of soil microbial communities.
Water Air Soil Pollut. 2005
, 164, 103.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
[113]
Y.-J. An ,
M. Kim ,
Effect of antimony on the microbial growth and the activities of soil enzymes.
Chemosphere 2009
, 74, 654.
| Crossref | GoogleScholarGoogle Scholar |
CAS |
PubMed |