

Mapping of arsenic species and identification of a novel arsenosugar in giant clams Tridacna maxima and Tridacna derasa using advanced mass spectrometric techniques
Volker Nischwitz A B and Spiros A. Pergantis A CA University of Crete, Department of Chemistry, Environmental Chemical Processes Laboratory, 71003 Voutes-Heraklion, Crete, Greece.
B Current address: GSF National Research Center for Environment and Health, Institute of Ecological Chemistry, Ingolstädter Landstr. 1, 85764 Neuherberg, Germany.
C Corresponding author. Email: spergantis@chemistry.uoc.gr
Environmental Chemistry 4(3) 187-196 https://doi.org/10.1071/EN07009
Submitted: 6 February 2007 Accepted: 23 April 2007 Published: 22 June 2007
Environmental context. Arsenic is known to accumulate in various marine organisms. The high acute toxicity of inorganic arsenic species and the potential chronic toxicity of some organoarsenic species require detailed knowledge about the occurrence and metabolism of arsenic compounds in marine organisms. The application of advanced analytical techniques still allows, even after decades of arsenic speciation, the identification of novel species. In addition, comprehensive mapping of all arsenic species present in marine organisms may allow for a more detailed understanding of arsenic metabolism.
Abstract. Because of their symbiotic microalgae, giant clams (Tridacna species) exhibit a unique arsenic metabolism, which has been shown in previous studies to involve a large number of arsenic species. This study demonstrates the application of liquid chromatography (HPLC) online with electrospray tandem mass spectrometry (ES-MS/MS) as well as inductively coupled plasma mass spectrometry (ICP-MS) for arsenic speciation analysis in giant clam extracts. Selected reaction monitoring (SRM) was used for sensitive and selective detection of a large number of arsenic species in a single chromatographic run. Novel aspects are the analysis of 10 tissue fractions from one clam and the analysis of kidney extracts both from T. maxima and T. derasa with the same method thus offering the possibility for direct comparisons. Moreover, HPLC-ES-MS/MS in the precursor ion scan mode and product ion scan mode allowed the identification of a novel sulfonated dimethylarsenosugar and the partial characterisation of another unknown arsenic species. The results indicate that most arsenic species are accumulated in the kidneys. However, arsenobetaine was found at similar contents in all analysed tissue fractions of one T. maxima clam.
Additional keywords: arsenic, giant clams, mass spectrometry (MS), metabolites, selective detection, speciation (metals), Tridacna species.
Introduction
The biogeochemistry of arsenic in the marine environment is known to involve a variety of organoarsenic compounds apart from arsenate, which is the predominant arsenic species in seawater. Arsenic metabolism in marine algae leads mainly to the formation of dimethylarsinoylribosides (arsenosugars), while the most abundant arsenic species in marine animals is arsenobetaine.[1] However, giant clams of the species Tridacna constitute a link between both types of arsenic metabolism because they contain symbiotic microalgae in their mantle tissue.[2,3] Because of the accumulation of algal metabolic products in the clam kidneys,[4] earlier studies focused on the characterisation of arsenic species in the kidneys of Tridacna maxima and Tridacna derasa.[2,5–7] Significant concentrations of dimethylated arsenosugars with glycol, phosphate, sulfonate, sulfate, carboxy, carbamate, and adenine groups in the aglycone have been detected among other arsenic compounds, by use of repeated preparative liquid chromatography with offline nuclear magnetic resonance (NMR) spectrometry in the initial studies,[6] and multi-stage high-performance liquid chromatography (HPLC) with elemental and molecular mass spectrometric detection in recent work.[7] McSheehy et al. reported several arsenic species found for the first time in Tridacna kidney extracts: DMAsSugarCarboxyl-2 (referred to as Sugar-313 in reference [7]), dimethylarsinoyldihydroxyfuran, Sugar-442, 5-dimethylarsinoyl-2,3-dihydroxypentanoic acid, 4-dimethylarsinoyl-2,3-dihydroxybutanoic acid, and Sugar-469.[7] These novel species have not been studied in later investigations and their identification has not been confirmed by other researchers.
Detailed arsenic speciation results for other Tridacna tissues, e.g. mantle and muscle, have not yet been reported. However, Francesconi et al. compared quantitative arsenic speciation data from kidney extracts of T. maxima and T. derasa.[2] They state that dimethylarsinoyltrihydroxypentanoic acid, which they had identified as the major arsenic compound in T. derasa kidney (50% of water soluble arsenic) is apparently absent in T. maxima kidneys.[2] Considerable developments in analytical methods have been made in the nine years between the two studies, and the use of different analytical techniques makes this comparison difficult. However, the arsenic metabolism may vary significantly from one Tridacna species to another, not only in terms of the quantitative amounts of the arsenic species, but also in terms of the presence or absence of individual arsenic species. In addition, the concentration of non-arsenic-containing metabolic products in giant clam tissues was found to be dependent on the shell size, i.e. the age of the clams.[8] Most likely, the arsenic speciation pattern is also dependent on the age of the investigated giant clam specimen.
The mentioned examples indicate that more detailed investigations on the characterisation of arsenic species in giant clam tissues have the potential to provide valuable information about arsenic metabolism in marine organisms, especially with respect to the major metabolic pathways and intermediates in the formation and transformation of arsenosugars as well as arsenobetaine. In particular, the use of advanced analytical techniques like HPLC electrospray tandem mass spectrometry (ES-MS/MS) in the selected reaction monitoring mode (SRM) offers the possibility to confirm the results of earlier studies with independent analytical techniques. HPLC-ES-MS/MS in the precursor ion scan mode[9] and product ion scan mode has the potential to provide additional information, for example, about currently unknown arsenic species.
Therefore, HPLC with elemental (inductively coupled plasma, ICP) and molecular (electrospray, ES) mass spectrometric detection was applied in the current study for the characterisation of arsenic species in tissue extracts of T. maxima and T. derasa. First, the identification of arsenic species, which were previously reported in Tridacna kidney extracts but not yet included in our previously published HPLC-ES-SRM method,[10,11] was attempted in an extract from T. maxima kidney using HPLC-ES-MS/MS (the recently published method[12] with 50 arsenic species was developed after the experiments described here and includes the new SRM transitions obtained in this study). Second, the arsenic speciation pattern in ten tissue fractions from a T. maxima clam was characterised both with HPLC-ICP-MS and HPLC-ES-SRM. Third, HPLC-ES-MS/MS in precursor ion scan mode and product ion scan mode, as well as HPLC-ES-MS with accurate mass determination in the selected ion monitoring mode (SIM), were applied in an attempt to characterise two unknown arsenic species observed using HPLC-ICP-MS. Finally, kidney extracts from T. maxima and T. derasa clams were analysed with HPLC-ES-SRM for identification of their arsenic species content and for quantification of selected species using the method of standard addition.
Experimental
Chemicals and standards of arsenic species
Ammonium bicarbonate (puriss.), 2,3-dimercaptopropan-1-ol, and diethyl ether (puriss., p.a.) were obtained from Riedel-de Haen, Seelze, Germany. Ammonium hydroxide solution (puriss., p.a.), nitric acid (65%, puriss., p.a.), and iodomethane (puriss.) were purchased from Fluka, Buchs, Switzerland. Methanol (gradient grade for HPLC) and acetic acid (100%, GR for analysis) were supplied from Merck, Darmstadt, Germany. Glycine-betaine monohydrate (99%) was obtained from Acros Organics.
The source of the arsenic species (either purified standards or raw samples) has previously been reported[10–13] and is repeated in the supporting information (pages S1–S4) together with an explanation of the abbreviations and the structures of the investigated arsenic species.
Tridacna samples
Two giant clams Tridacna maxima and two Tridacna derasa, which were commercially cultured on a farm in the Coral Sea, were obtained live by air freight from a German supplier. First, all clams were frozen. After 3 h, one T. maxima (TM1, 13.5 cm length) was thawed and dissected. All tissue fractions (kidneys, mantle, muscle, heart, etc.) were rinsed with deionised water, dried with paper tissue, collected separately in polyethylene containers, freeze-dried, and stored at –20°C. The second T. maxima (TM2, 13.5 cm length) was stored frozen for 10 weeks, the first T. derasa (TD1, 7 cm length) for 7 weeks, and the second T. derasa (TD2, 7.5 cm length) for 10.5 weeks and then prepared in the same way as described above.
Extraction procedure
The dry tissue samples were ground and a portion was extracted with deionised water (solid–liquid ratio typically 1 : 40) by shaking at room temperature for 1.5 h. After centrifugation the supernatant was collected and the residue was re-extracted in the same way (solid–liquid ratio 1 : 20) for 0.5 h. The combined supernatants constitute the extract. The extract volume was determined gravimetrically.
Total arsenic contents
Total contents of arsenic were determined in tissue extracts with ICP-MS using external calibration with In as the internal standard. Extraction residues and aliquots of the tissue samples were analysed in the same way after microwave-assisted acid digestion with HNO3 (MWS-2, Berghof, Germany). All contents refer to the tissue dry weight. Extraction efficiencies were calculated as percentage ratio of the total extracted amount of arsenic to the total arsenic content in the tissue sample.
Instrumentation and general conditions for HPLC-ICP-MS
Anion exchange chromatography was performed on a PRP-X100 column (250 mm × 4.1 mm) with PRP-X100 pre-column using isocratic elution with 20 × 10–3 M NH4HCO3 and 3% methanol at pH 10 (adjusted with ammonia) (method A). A Marathon IV HPLC pump (Rigas Laboratories, Thessaloniki, Greece) delivered the mobile phase at a flow rate of 1 mL min–1. Dimethylthioarsenosugars were not considered in this method because these compounds do not elute under the conditions applied. Manual injection was performed with a 20 μL loop. Indium (10 μg L–1 in 5% HNO3) was added post-column as an internal standard before entering the nebuliser. As (m/z 75) and In (m/z 115) were monitored with an X-Series ICP-MS (Thermo Electron Elemental Analysis, Winsford, UK) equipped with concentric nebuliser and impact bead spray chamber. In addition, m/z 77 was monitored to check for ArCl+ interferences. Calibration was done with arsenate, DMA, DMAsSugar-Glycol and -Adenine as described earlier.[11]
Instrumentation and general conditions for HPLC-ES-MS/MS
A Surveyor HPLC system with quarternary gradient pump and an autosampler was used with a TSQ Quantum enhanced resolution triple quadrupole mass spectrometer (Thermo Finnigan, San Jose, CA, USA) equipped with an electrospray source. Measurements were performed in the positive ion mode using an ES voltage of 4.1 kV, a sheath gas pressure of 45 arbitrary units, an auxiliary gas pressure of 25 arbitrary units, a capillary temperature of 300°C, and no source collision induced dissociation (CID) voltage.
Two previously developed HPLC methods were applied:[10] anion exchange chromatography (method B) was performed on a PRP-X100 column (250 mm × 4.1 mm) using two PRP-X800 cation exchange pre-columns (Hamilton, Reno, NV, USA). Gradient elution was done with 20 × 10–3 M NH4HCO3, pH 10 (eluent A) and 20 × 10–3 M NH4HCO3, 40% methanol, pH 10 (eluent B): for 5 min at 25% B; in 1 min to 100% B; 24 min at 100% B; in 0.5 min to 25% B; for 4.5 min at 25% B (35 min total run time).
For combined cation–anion exchange chromatography (method C) the second precolumn of the anion exchange method was substituted by an IonPac CS10 cation exchange column (250 mm × 4.0 mm) (Dionex, Camberley, Surrey, UK). Gradient elution was employed using 10 × 10–3 M ammonium acetate at pH 3 (adjusted with acetic acid) (eluent A), 20 × 10–3 M NH4HCO3, 10% methanol, pH 10 (eluent B), and 10% methanol in deionised water (eluent C): 100% A for 3 min, in 0.3 min to 100% C, 1.2 min 100% C, in 0.5 min to 100% B, 9 min 100% B, in 1 min to 100% C, 1 min 100% C, in 0.5 min to 100% A, and for 13.5 min 100% A (30 min run time).
In both cases the HPLC flow rate was 1 mL min–1 and a post column split was used, which reduced the sample flow rate to the ES source to 240 μL min–1. Injection was done either with the autosampler (20 μL loop) or manually (50 μL). The DMThioAsSugars were only monitored with the anion exchange method (method B).
Accurate mass measurements were performed with the anion exchange HPLC (method B) in the selected ion monitoring mode (SIM) using signals of ammoniated poly(ethylene glycol) (PEG) as lock masses. The PEG solution was supplied post-column with a syringe pump via the sheath flow channel of the electrospray source at 2.5 μL min–1. The accurate mass of the protonated molecular ion was calculated as the average (± standard deviation) of at least 30 scans around the peak maximum.
Characterisation of unknown 2
Fractions of unknown 2 originating from Tridacna maxima were collected from anion exchange chromatography (PRP-X100, 100 mm × 4.1 mm; 10 × 10–3 M NH4HCO3, 3% methanol, pH 10; 1 mL min–1). The combined fractions were divided into two equal aliquots and lyophilised. One fraction was redissolved in deionised water, the other in methanol. The methanolic solution was treated with 2,3-dimercaptopropan-1-ol and iodomethane to convert possibly present dimethylarsinoyl species into their trimethyarsonio derivates.[14] Deionised water was added to the mixture followed by shaking for 1 h to decompose any excess iodomethane. Afterwards the mixture was partitioned between water and diethylether. The aqueous phase was used for analysis.
In addition to the bicarbonate anion exchange HPLC method, an acidic anion exchange method was developed using a PRP-X100 column (100 mm × 4.1 mm) in the isocratic mode with 10% methanol and 3% acetic acid (pH 2.6) at 1 mL min–1.
Quantitative results
Analyte contents with confidence intervals (statistical level P = 95%) were calculated for each measurement from the linear regression of the calibration. For replicates (n > 1) mean values were calculated. The uncertainty of the mean was derived by propagation of error from the uncertainties of the individual results if the confidence intervals of the replicates overlapped, otherwise the standard deviation was calculated as the uncertainty.
Results and discussion
Electrospray SRM detection of arsenic species
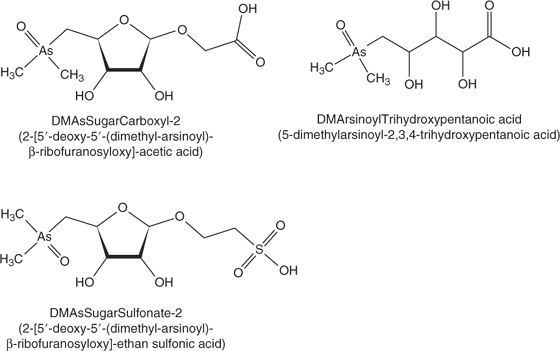
In our previous work, we developed an anion exchange HPLC-ES-SRM method for 30 arsenic species using a PRP-X100 column with cation exchange pre-columns and an ammonium bicarbonate mobile phase at pH 10 with methanol gradient.[10,11] The monitored SRM transitions including those for glycine betaine are given in the accessory publication (Table A). However, Francesconi et al.[2] and McSheehy et al.[7] have reported the identification of additional arsenic species in Tridacna extracts which we did not include in our previous methods because of lack of appropriate standards. Based on their published mass spectrometric data, we applied anion exchange HPLC-ES-MS/MS in the product ion scan mode and in the SRM mode in order to include these additional arsenic compounds in the method used for the analysis of kidney extracts from Tridacna maxima. With respect to these arsenic species, we were successful in identifying DMAsSugarCarboxyl-2 (named Sugar-313 in the study by McSheehy et al.[7]) and 5-dimethylarsinoyl-2,3,4-trihydroxypentanoic acid (DMArsinoylTrihydroxypentanoic acid, Fig. 1). Collision-induced dissociation curves for these compounds were constructed from the product ion spectra recorded at various collision energies in order to find the optimum conditions for their detection using SRM (see Fig. A in supporting information). The optimised SRM transitions for DMAsSugarCarboxyl-2 are 313 → 97 (20 eV) and 313 → 237 (15 eV), and for DMArsinoylTrihydroxypentanoic acid 271 → 253 (18 eV) and 271 → 195 (25 eV). The identification of DMAsSugarCarboxyl-2 in Tridacna kidney extracts confirms the results from McSheehy et al. obtained after multidimensional HPLC separation with ICP-MS and off-line ES-MS/MS detection.[7] The remaining arsenic species dimethylarsinoyldihydroxyfuran, Sugar-442, 5-dimethylarsinoyl-2,3-dihydroxypentanoic acid, 4-dimethylarsinoyl-2,3-dihydroxybutanoic acid, and Sugar-469, which were up to now only reported by McSheehy et al.[7] could not be detected with our one-dimensional HPLC-ES-MS/MS method. This may be attributable to low concentrations of those species in the extract and possibly to matrix suppression of the signal when using the crude extract for analysis.
|
Arsenic speciation in 10 tissue fractions from one Tridacna maxima clam
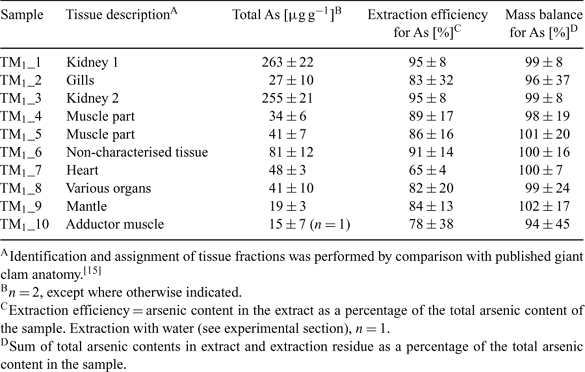
The dissection of one T. maxima clam resulted in 10 tissue fractions, which were assigned according to published giant clam anatomical details.[15] The description of the tissue fractions, total arsenic contents, extraction efficiencies, and mass balance for arsenic are summarised in Table 1. The high total arsenic contents in the two kidney samples compared to the other tissues clearly indicate the accumulation of arsenic species in the T. maxima kidney as reported by Benson and Summons.[4] Good extraction efficiencies for arsenic in the range of 78 to 95% were achieved with the water extraction with exception of the heart tissue (65%). Arsenic mass balances for the extractions ranged from 94 to 102%. The extracts of all tissue fractions were analysed with anion exchange HPLC-ICP-MS (method A) and HPLC-ES-SRM using anion exchange chromatography (method B) as well as combined cation–anion exchange chromatography (method C).
|
The applied isocratic anion exchange HPLC-ICP-MS method (method A) was not able to separate all arsenic species present in the tissue extracts. Peak assignment refers to the most abundant arsenic species at the respective retention times based on the analysis of standards and information from the analysis of the same sample with HPLC-ES-SRM. In spite of the co-elution of some arsenic species, this method provided a quantitative overview of the arsenic speciation pattern in the various tissue extracts and also revealed the presence of two unknown species (see Table B in accessory publication). The characterisation of unknown 1 and unknown 2 is discussed in detail in the next section. The recovery of arsenic from the anion exchange HPLC was between 84 and 94%.
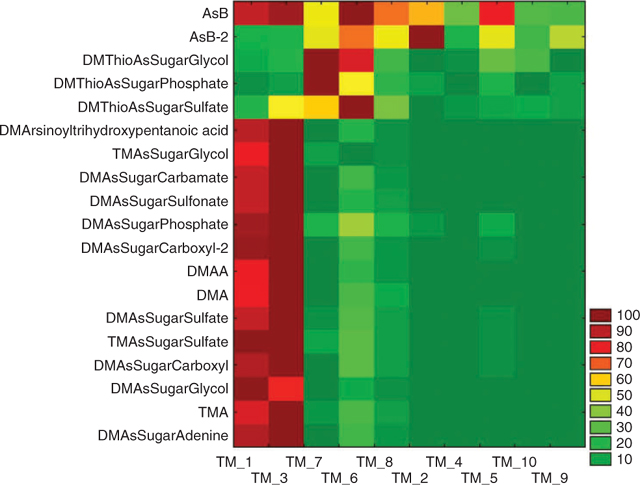
Anion exchange HPLC (method B) online with ES-SRM identified 19 arsenic species in the tissue extracts on the basis of two transitions monitored for each species and comparison of retention times with available standards. Quantification was not performed with this method because standards were not available for some species and the method of standard addition, which is required for accurate quantification because of matrix effects, is excessively time consuming for such a large number of samples and analytes. Therefore, comparison of the intensities of the identified analytes in the 10 tissue extracts was performed on the basis of peak areas (Fig. 2). Peak areas were corrected for extract volume and sample weight to calculate the contents in counts per g (dry weight). For each analyte, normalisation was performed on the sample with the highest content of this analyte. The graphical display in Fig. 2 reveals a clear pattern of the arsenic speciation data. The majority of arsenic species is present at high concentrations in the kidney samples and at much lower concentrations in all other tissue fractions. However, AsB, AsB-2, and glycine betaine were detected at similar levels in most tissue fractions. The highest intensities of DMThioAsSugar-Glycol, -Phosphate, and -Sulfate were found in the heart (TM1_7) and another non-characterised tissue fraction (TM1_6).
|
The combined cation–anion exchange HPLC (method C) allowed the identification of 17 arsenic species with ES-SRM detection. The DMThioAsSugars were not included in this method. However, TMAO was also detected compared with the anion exchange HPLC-ES-SRM method. The graphical display (not shown) in analogy to Fig. 2 exhibits the same pattern.
These results are meaningful in two ways. First, the good agreement of the results from three independent HPLC-MS methods demonstrates that HPLC-ES-SRM provides not only qualitative information but also semiquantitative information, which is representative for the analysed samples in spite of possibly occurring matrix effects. Second, the close correlation of the contents of most detected arsenic species in spite of their different chemical properties (neutral, anionic, cationic), which includes dimethylated and trimethylated arsenosugars, in a variety of tissue fractions suggests a common pathway for the biosynthesis of these compounds in the giant clam Tridacna maxima. However, the interpretation of these results is difficult because the location of the biosynthesis and transport processes may also have a significant influence on the concentration profiles in the various tissues. The different behaviour of the DMThioAsSugars demonstrates that the formation and presence of these compounds is dependent on particular conditions, for example, possibly the availability of hydrogen sulfide. Of interest is the distribution of AsB, and partly that of AsB-2, which correlate well with the nitrogen analogue glycine betaine. The presence of AsB in all tissue fractions at similar concentration levels supports the hypothesis that AsB is taken up as an osmolyte as a result of its structural similarity with glycine betaine, as suggested in earlier studies.[16,17] Low contents of AsB could be accumulated from food sources (marine algae[18]) or even from the seawater.[19] This would explain the high contents of AsB in many marine animal tissues and the lack of an obvious biosynthetic precursor of AsB in these tissues.
Characterisation of two unknown arsenic species in T. maxima kidney extract
Because the highest contents of most detected arsenic species were found in the kidney extracts, the following investigations focused on these samples. For direct correlation of results from elemental mass spectrometric detection (HPLC-ICP-MS) and molecular mass spectrometric detection (HPLC-ES-SRM), the same anion exchange HPLC method (method A) was coupled with both detection systems. Fig. 3 shows an overlay of the HPLC-ICP-MS chromatogram monitoring 75As+ with the summed HPLC-ES-SRM chromatogram for all detected arsenic species. The peaks of the well-separated species DMAsSugarSulfonate, -Sulfate, and -Adenine can easily be correlated. The early eluting species are partly co-eluting so it is not possible to unambiguously assign the peaks from HPLC-ICP-MS to individual arsenic species. Most interesting, however, are two peaks in the HPLC-ICP-MS chromatogram, which are not covered by matching HPLC-ES-SRM signals. These peaks are referred to as unknown 1 and unknown 2 (Fig. 3). For more detailed characterisation, fractions were collected at the elution times of both unknowns from the anion exchange separation. After freeze drying and re-dissolution in deionised water, the purity and stability of the fractions were verified with HPLC-ICP-MS (method A), as shown in Fig. 4 for unknown 1 (lower chromatogram). Subsequently, the fractions were analysed with HPLC-ES-MS/MS using the same anion exchange HPLC method. A precursor ion scan that monitored the product ion m/z 97, which is characteristic for arsenosugars, at a collision energy of 40 eV, was successful for unknown 1. A peak was obtained at the same retention time as the arsenic peak in HPLC-ICP-MS detection (Fig. 4). The precursor ion mass spectrum averaged for the peak shows signals at m/z 363 and 380 (Fig. 4 insert). Subsequently, product ion spectra were recorded for both precursor ions (Fig. B in the supporting information). In both cases, the characteristic product ions for dimethylarsinoyl sugars at m/z 97 and 237 are clearly detected. For the known arsenosugars, DMAsSugarSulfonate and DMAsSugarSulfate, there is an intense signal for [M + NH4]+ in the precursor ion spectra when using the ammonium bicarbonate mobile phase in which the pH was adjusted with ammonia.[9] The presence of m/z 363 in the single MS mode without any source CID voltage and the close retention times of the unknown 1 and DMAsSugarSulfonate suggest that unknown 1 is a dimethylarsinoyl sugar with a sulfonate-containing aglycone and a molecular ion of m/z 363 which forms m/z 380 with ammonium ions. In analogy to DMAsSugarCarboxyl-2 we propose the structure in Fig. 1 for unknown 1 and refer to it as DMAsSugarSulfonate-2. In both cases, the new compounds have one >CHOH unit less than the well-known DMAsSugarCarboxyl and -Sulfonate. This identification is further supported by accurate mass determination for the protonated molecular ion of DMAsSugarSulfonate-2 using anion exchange HPLC-ES-SIM with an enhanced resolution triple quadrupole mass spectrometer. The experimental molecular mass 363.0106 ± 0.0055 (mean ± standard deviation from three replicate runs obtained on two days) agrees within 3.1 ppm with the molecular mass calculated for the element composition of the proposed structure (most abundant isotopic composition). The collision-induced dissociation curve of DMAsSugarSulfonate-2 was recorded (see Fig. A in supporting information) to optimise the parameters for the SRM detection of this new compound: 363 → 97 (28 eV) and 363 → 237 (17 eV).

|

|
The same approach was used in an effort to identify unknown 2. However, precursor ion scans that monitored characteristic product ions for arsenosugars such as m/z 97 and 237 did not result in a peak that correlated with the arsenic peak for unknown 2 from HPLC-ICP-MS. Incubation of a fraction of unknown 2 with 1% nitric acid for 40 h, did not lead to a significant change of the HPLC-ICP-MS chromatogram of this fraction. This strongly indicates that unknown 2 is not an arsenosugar, because otherwise acid hydrolysis of the molecule would have been expected at low pH. After treatment of the fraction of unknown 2 with H2S, the peak was no longer detected at its original retention time and no other peak eluted using only 10% methanol in the mobile phase. This behaviour points towards a dimethylarsinoyl species that forms a strongly retained thio-arsinoyl derivative with H2S. The long retention time on the anion exchange HPLC with bicarbonate mobile phase at pH 10, which was not reduced when increasing the methanol content from 3 to 10%, indicates that unknown 2 has at least one functional group with a strong negative charge. This would also explain the low sensitivity for this compound in the positive ion mode ES-MS/MS. For enhanced sensitivity with the bicarbonate mobile phase, the methylated derivate of unknown 2 was prepared (see experimental section for details). The reaction mixture afforded only one arsenic peak in the anion exchange HPLC-ICP-MS chromatogram, and in full scan HPLC-ES-MS mode a peak was found in the mass chromatogram for m/z 366 at the same retention time. Product ion spectra of m/z 366 show characteristic signals for trimethylarsonio derivatives at m/z 193 and 163 for a collision energy of 40 eV (Fig. 5a), and at m/z 120 and 105 for a 60 eV collision energy (not shown). Analysis of the underivatised fraction of unknown 2 with anion exchange HPLC-ES-MS using an acetic acid-containing mobile phase resulted in a peak in the mass chromatogram for m/z 368 in the full scan mode. Collision-induced dissociation of m/z 368 led to the detection of product ions at m/z 271, 253, 225, 195, and 165 (Fig. 5b). Compared to Fig. 5a the spectrum is shifted by 2 m/z units towards higher masses, which matches with the presence of a dimethylarsinoyl species and its corresponding trimethylarsonio derivative. Remarkable is the formation of intense product ions at m/z 269 and 271, respectively, following the loss of a neutral fragment with 97 mass units. Because the product ion spectrum of unknown 2 at 40 eV is nearly identical with the product ion spectrum of dimethylarsinoyltrihydroxypentanoic acid (Fig. 5c), unknown 2 is most likely a derivative of this known arsenic species. The loss of a neutral fragment with mass 97 may point towards a sulfonate or sulfate-containing derivative. The obtained data from the positive ion mode ES-MS/MS were not sufficient to prove this. Negative ion mode was also attempted, but this was not compatible with the mobile phases used in this study. The development of an HPLC method optimised for negative ion mode ES-MS/MS may be successful for the final identification of unknown 2 in the future.

|
Comparison of arsenic speciation results for T. maxima kidneys and T. derasa kidneys
The comparison of the pattern of arsenic species detected in kidney extracts from T. maxima and T. derasa may provide additional information about the metabolism of arsenic compounds in giant clams. Francesconi and Edmonds also conclude that it is relevant to make such a comparison.[2] Our study is the first to analyse kidneys both from T. maxima and T. derasa for arsenic species. The T. maxima specimen (TM1 and TM2) had two kidneys each, while from the smaller T. derasa clams (TD1 and TD2) only one kidney each could be isolated. The published results from three previous studies are summarised in Table 2. Our results were obtained with anion exchange HPLC-ES-SRM (Table 3). The major focus was on the identification of arsenic species present in the extracts on the basis of matching retention times with standards and co-eluting peaks for both SRM transitions monitored for each compound. For selected species, quantification was performed using the method of standard addition. Significant differences were found for the T. derasa kidneys compared to T. maxima. TMA, DMAA, DMAsSugar-Adenine, -Glycol, -Sulfonate, -Carboxyl-2, and -Sulfonate-2 were detected in all T. maxima kidney extracts but not in T. derasa kidney extracts. In addition, DMAsSugar-Carbamate was detected in only one T. derasa sample at a far lower content compared to T. maxima. The latter was confirmed by the HPLC-ICP-MS chromatograms shown in the supporting information (Fig. C). However, McSheehy et al. found DMAA, DMAsSugar-Glycol, -Sulfonate, and -Carboxyl-2 in T. derasa.[7] This indicates that the differences observed in our study may be partly explained by the younger age of the T. derasa clams, i.e., their earlier stage of development, as reported to occur for non-arsenic-containing clam metabolites.[8]

|

|
Nearly all clearly identified arsenic species found by McSheehy et al. in T. derasa kidney[7] were identified by us in kidneys from two T. maxima clams (Tables 2 and 3). Therefore, the arsenic metabolism of adult T. maxima and T. derasa clams seems to be very similar in terms of the arsenic species present. In addition, we detected TMA, TMAsSugarSulfate, DMAsSugarAdenine, DMThioAsSugarGlycol, and DMAsSugarSulfonate-2 in all T. maxima kidney samples. TMAsSugarSulfate and DMAsSugarAdenine have been reported earlier in T. maxima kidney.[6] The not fully identified unknown 2 has a similar strong retention behaviour on anion exchange chromatography as the dimethylarsinoyl-taurine reported by Francesconi et al.[6]
While the currently available data are not sufficient for making firm and detailed conclusions about similarities and differences in the arsenic metabolism of both clam species, the emerging trends do allow a limited amount of informed speculation. Remarkable is the fact that the tissue extract with the lowest intensities of most arsenic species was the mantle tissue. This result seems to contradict the hypothesis that the symbiotic microalgae that are located in the mantle tissue have an important function in the arsenic metabolism within the giant clams. Based on the data from HPLC-ICP-MS, 97% of the total quantified arsenic species in the mantle tissue extract is arsenobetaine, while in the kidney extracts arsenobetaine accounts only for 5%. Thus, it seems unlikely that arsenic species formed in the mantle tissue could solely account for the accumulation of high concentrations of most arsenic species in the kidneys, because in this case arsenobetaine should have also been accumulated in a similar way. On the contrary, the results suggest that the generation of the arsenic species in the clam kidneys involves common metabolic products supplied from the microalgae in the mantle tissue. The arsenic source could be arsenate, which was detected in the clam kidney extracts but not in the mantle tissue extract. An uptake of arsenate from the seawater by the clams is likely.
A more detailed study including several Tridacna clam species varying in origin, shell size (age), etc. could reveal Tridacna species- and age-dependent changes in the metabolism and accumulation of arsenic species.
Conclusion
The application of HPLC online with ES-SRM allowed the sensitive, selective, and rapid detection of known arsenic species in the analysed giant clam extracts. In addition, the combined use of HPLC with ICP-MS and ES-MS/MS detection enabled the characterisation of unknown arsenic species.
The results for various tissues from a T. maxima sample revealed close correlation of the contents of most arsenic species and a different distribution pattern for arsenobetaine.
The large number of detected arsenic compounds and the fact that even after more than 20 years of arsenic speciation analysis in giant clams new arsenic species are still being found, clearly demonstrates the complexity of arsenic metabolism in these organisms. By analogy to other fields, the term arsenomics may be appropriate to refer to the analytical characterisation of the growing number of arsenic species known to occur as natural metabolites in order to unravel their potential role in biological systems. Now, the availability of advanced tandem mass spectrometric detection techniques offers the chance for a detailed study of the arsenic compounds present in various Tridacna species and an investigation of the dependence of the arsenic speciation pattern on the age of the analysed giant clams and possibly other environmental and biochemical parameters.
Accessory publication
The sources of the arsenic species standards, structures, abbreviations, and SRM transitions of the arsenic species, quantitative data from HPLC-ICP-MS, CID breakdown curves, product ion spectra of DMAsSugarSulfonate-2, and HPLC-ICP-MS chromatograms of T. maxima and T. derasa kidney extracts.
Acknowledgement
The authors acknowledge the funding of a Marie Curie Excellence Grant by the European Commission (Contract MEXT-CT-2003-002788) and thank Professor K. A. Francesconi for kindly donating some of the organoarsenic standards that were used in this study.
[1]
W. R. Cullen,
K. J. Reimer,
Chem. Rev. 1989, 89, 713.
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
| Crossref | GoogleScholarGoogle Scholar |
