

The cyclobutene diester approach to alkyl citrate natural products
Nikolai P. Rossouw A and Mark A. Rizzacasa A *
A *
A
Abstract
This review outlines the synthesis of alkyl citrate natural products using cyclobutene diester precursors. The approach is efficient and stereoselective and provides the correct oxidation state of the citrate core of these compounds. The synthesis of a number of alkyl citrates along with some higher oxidised members of this family is detailed.
Keywords: alkyl citrates, cyclobutenes, natural products, oxidation, rearrangement, total synthesis.
The alkyl citrate family of natural products
The alkyl citrate family of natural products are a group of secondary fungal metabolites that share a common citric acid core (Fig. 1). Alkyl citrate natural products contain two contiguous asymmetric centres at the C3 and C4 positions and an alkyl chain, ranging from simple aliphatic chains as found in (−)-CJ-13,9821 and L-731,1202 to more highly oxidised compounds such as viridiofungin A3 and trachyspic acid.4 Additionally, the C4 carboxylic acid can be incorporated into a lactone or amide as found in citrafungin A5 and viridiofungin A. Further oxidation of the citrate core at positions C2 and C4 gives compounds such as cinatrins C1 and C36,7 as well as the zaragozic acid family.8–12 Alkyl citrates exhibit a broad range of biological properties. This, coupled with their complex and densely functionalised structures has resulted in a number total syntheses of a range of alkyl citrates.13–15 The most well known are the zaragozic acids, which are potent squalene synthase (SSase) inhibitors. SSase is a key cholesterol biosynthesis enzyme and a target of interest for the treatment of cardiovascular disease resulting from hypercholesteremia. The key challenge in the synthesis of this class of natural product is the highly oxidised triacid moiety in a step and redox economical manner.16 This review details our efforts towards the synthesis of alkyl citrates using cyclobutene diesters as key intermediates.
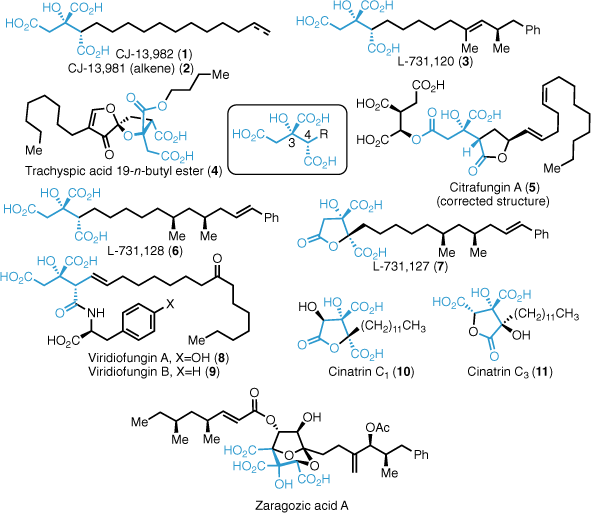
Challenges in alkyl citrate natural product total synthesis
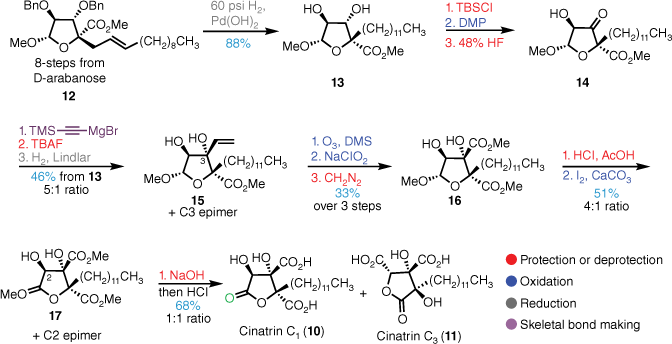
The main challenge encumbering the total syntheses of these natural products is the generation of the highly oxidised triacid core with appropriate protecting group strategy. Additionally, the stereoselective installation of the two contiguous asymmetric centres is difficult. A number of strategies have been employed to overcome this, but the oxidative manipulations required increase the number of steps resulting in low quantities of synthetic material for biological testing. An analysis of our earlier synthesis of cinatrins C1 and C3 exemplifies some of these issues (Scheme 1).17 Most steps in the partial sequence shown are protecting group manipulations, non-strategic oxidative or reductive manipulations and few skeletal or strategic bond disconnections. Obviously, it is not possible to do away with non-strategic steps entirely, but we sought a more direct approach to accessing the highly oxidised triacid citrate core using a common approach to access all members of this class of interesting natural products.
Previous total synthesis of cinatrins C1 and C3 (TBSCl, tert-butyldimethylsilyl chloride; DMP, Dess–Martin periodinane; TBAF, tetrabutylammonium fluoride; TMS, trimethylsilane; DMS, dimethylsulfide).
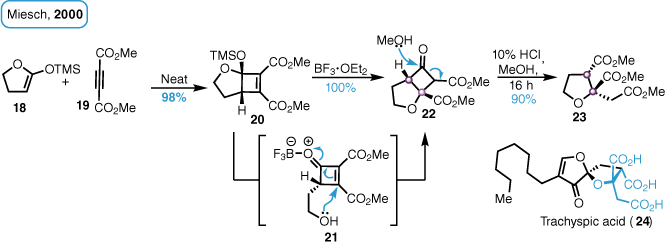
Miesch et al.18 and Miesch and Wendling19 reported that the cyclic ketene silyl acetal 18 undergoes an uncatalysed formal [2 + 2]-cycloaddition with dimethylacetylene dicarboxylate 19 (DMAD) to furnish the cyclobutene diester 20 in high yield. The resultant adduct then underwent a Lewis-acid-mediated rearrangement to the corresponding cyclobutanone 22 by a mechanism involving an intramolecular oxa–Michael reaction. Subsequent methanolysis of the fused cyclobutanone then afforded the trimethyl ester tetrahydrofuran 23 (Scheme 2), which maps onto the core of trachyspic acid. This sequence would be particularly valuable in providing direct access to the triacid core of the alkyl citrates such as trachyspic acid (24) from simple precursors and we wondered whether this cascade could deliver triester 23 directly from the corresponding cyclobutene diester 20.
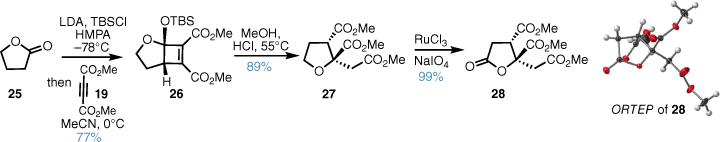
A model study20 found that the more stable tert-butyldimethylsilyl (TBS) ketene silyl acetal was superior and this underwent the formal [2 + 2]-cycloaddition in acetonitrile solvent to deliver the diester 26 in good yield. An acid-mediated acetal cleavage, oxa–Michael addition and methanolysis cascade by simple refluxing a solution of 26 in methanolic HCl delivered the triester 27 in one pot. THF oxidation then gave crystalline lactone 28 and X-ray analysis confirmed the relative configuration of the triester core (Scheme 3).
Several formal [2 + 2] cycloadditions were then performed with the chiral ketene silyl acetal derived from optically pure lactone 29 and a range of acetylene diesters (Scheme 4). This gave the adducts 30–32 in good yields with stereoselectivities ranging from 4.5:1 to 20:1. The diastereoselectivity is proposed to arise from top face approach of the Michael acceptor involving the higher energy transition state, thus the addition occurs from the bottom face, which rapidly undergoes ring closure from the zwitterionic intermediate. Cyclobutene diesters 30–32 were then utilised in the syntheses of all the alkyl citrates shown above (Fig. 1).
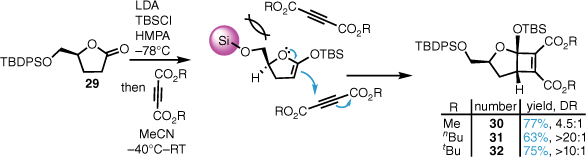
Total synthesis of CJ-13,982 and CJ-13,981
In 2001 researchers at Pfizer Japan isolated CJ-13,982 (1) and CJ-13,981(2) from an unidentified fungus (CL15036) (Fig. 2).1 They are the least complex examples of alkyl citrates, with a simple C2 alkyl chain and the citrate triacid moiety and are fairly potent inhibitors of squalene synthase (SQS).
To date, Calo et al.21 and our group20,22 are the only ones to have synthesised these compounds. Barrett reported the synthesis of the enantiomer of natural CJ-13,982 and CJ-13,981, which confirmed the absolute configuration of this compound. Both total syntheses required over 20 steps to reach the desired natural products and thus an improved route was investigated by a cyclobutene diester intermediate.
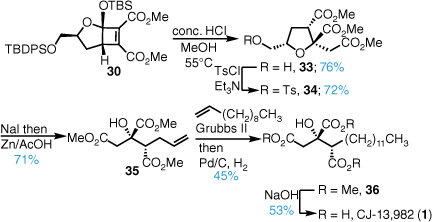
The synthesis began with exposure of cyclobutene diester 30 to methanolic HCl, which induced a silyl acetal cleavage, oxa–Michael, methanolysis cascade to furnish trimethyl ester 33 in excellent yield as a single stereoisomer (Scheme 5). Trimethyl ester 33 was converted into the corresponding iodide in good yield over two steps and treatment with zinc powder in acetic acid induced a Boord olefination and gave the citrate triester core 35 in excellent yield over a total of seven steps with no oxidative adjustments required. We have also established a flow chemistry protocol for the synthesis of this key intermediate.23 Grubbs II catalysed cross metathesis with 1-undecene followed by hydrogenation gave the trimethyl ester 36 and base-promoted hydrolysis furnished the natural product CJ-13,982 (1).
The synthesis of CJ-13,981 (2) commenced from primary alcohol 37, which was protected as the TBS silyl ether, and underwent Grubbs II catalysed cross metathesis with triester 35 followed by hydrogenation (Scheme 6). After deprotection, Dess–Martin periodinane (DMP) oxidation and Wittig olefination installed the terminal alkene in good yield. Base-promoted ester hydrolysis gave CJ-13,981 (2) in good yield following reverse-phase–high-performance liquid chromatography (RP-HPLC).
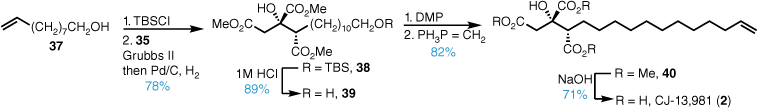
Total synthesis of citrafungin A
In 2004 a group at Merck (Rahway) isolated citrafungins A (5) and B (41) from cow dung-derived sterile mycelia MF6339 (Fig. 3).5 CJ-15,183 (42)24 is a proposed isoester of citrafungin B; however, the published spectroscopic data are identical, suggesting they are the same compound. Citrafungin A is an inhibitor of the membrane promotor GGTase I and thus exhibits antifungal properties. In the Merck group’s chemical degradation studies, a Mosher ester analysis led to an incorrect assignment of the natural absolute configuration. There are two total syntheses by Amer et al.25 and Calo et al.26 and a formal total synthesis27 of the original incorrect stereoisomer was reported by our group. We then corrected the absolute stereochemistry by the total synthesis of the correct isomer some 15 years later.28
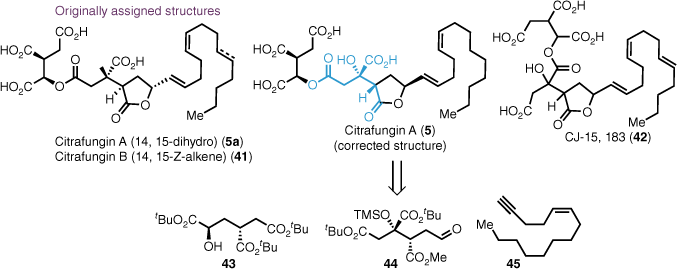
Our synthesis began with cyclobutene di-t-butyl ester 32, which, upon treatment with a HF·pyridine complex, furnished bicyclic lactone 46 as a crystalline solid that allowed for X-ray analysis (Scheme 7). The mechanism of this reaction involved desilylation, intramolecular oxa–Michael addition and concomitant cyclobutanone ring opening by the pendant primary alcohol. The intermediate bicyclic lactone 46 proved pivotal for the synthesis of a number of alkyl citrates by allowing for orthogonal protection of the C4 carboxylic acid. Methanolysis under basic conditions afforded THF intermediate 47 in high yield.
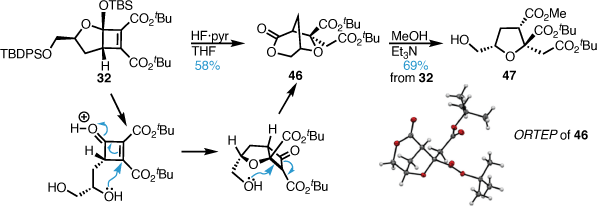
Functional group manipulation to the corresponding iodide, followed by Boord olefin synthesis furnished alkene 48 in high yield over three steps (Scheme 8). Protection of the C3 tertiary alcohol as the TMS silyl ether and oxidative cleavage of the terminal alkene yielded aldehyde 44 in good yield.
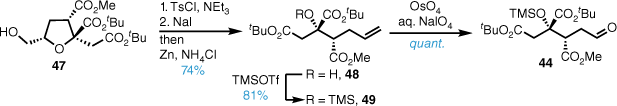
A (+)-MIB (2-methylisborneol)29-mediated diastereoselective vinylzinc (formed from alkyne 45) addition reaction with aldehyde 44 afforded allylic alcohol 50 in good yield and high diastereoselectivity that was converted into the corresponding lactone 51 in high yield (Scheme 9). Deprotection of 51 with formic acid gave citrafungin A diacid degradation product 52 that had identical spectral and chiroptical properties to the naturally derived material, and this confirmed the absolute configuration of the lactone fragment was enantiomeric to that originally proposed.
Diastereoselective vinyl zinc addition (PPTS, pyridinium p-toluenesulfonate; DR, diastereomeric ratio).
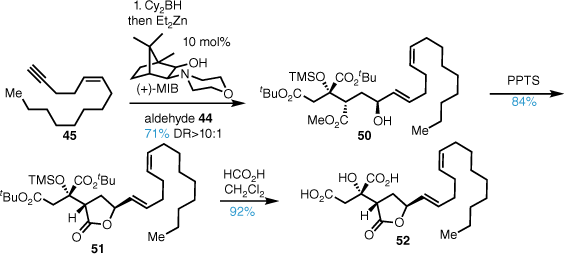
Selective C1 tert-butyl ester deprotection of 51 was mediated by brief exposure to trifluoroacetic acid (TFA), and the crude mono acid was coupled with the tri-tert-butyl isocitrate fragment 43 in good yield (Scheme 10). Exposure to formic acid cleanly afforded citrafungin A in high yield with identical spectroscopic properties and optical rotation to the reported material, confirming the stereochemical reassignment.
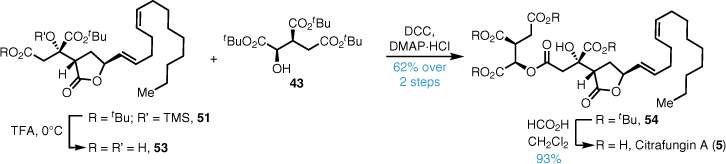
Total synthesis of viridiofungins A and B
The viridiofungins are a large group of alkyl citrate-type natural products. Viridiofungins A (8) and B (9) were isolated from Trichoderma viride in 1993 by a group at Merck (Rahway) while screening for novel SQS inhibitors (Fig. 4).3
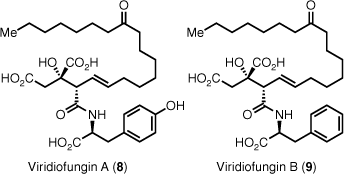
They were found to be broad spectrum anti-fungal compounds as well as inhibitors of SQS and serine palmitoyl transferase. Their structures were determined by 1- and 2-D 1H- and 13C-NMR spectroscopy and mass spectrometry fragmentation, which located the amino acid residue on C4. Several syntheses of the viridiofungins and their derivatives have been reported.30–38
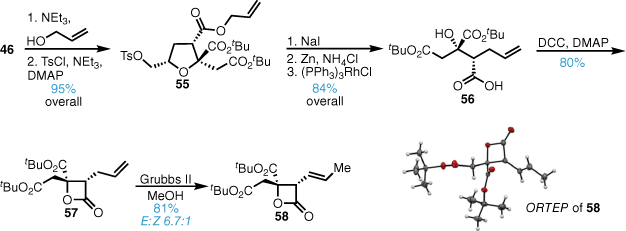
Our synthesis39 began with bicyclic lactone 46 (Scheme 11). Ring-opening with allyl alcohol in the presence of Et3N followed by tosylation gave tosylate 55. Iodide displacement followed by zinc-mediated reductive elimination gave the corresponding alkene and after allyl ester removal using Wilkinson’s catalyst the C4-carboxylic acid 56 was obtained. Intramolecular Steglich reaction gave β-lactone 57 in high yield and the terminal double bond was then isomerised with Grubbs II in refluxing methanol to give the disubstituted alkene 58, the structure of which was confirmed by X-ray crystallography.
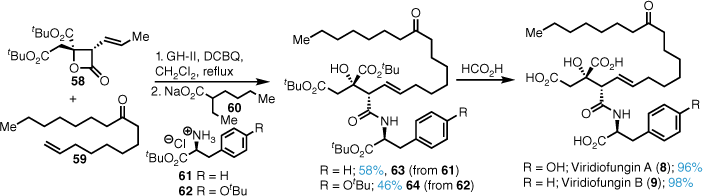
Cross metathesis of lactone 58 with alkene 59 using Grubbs–Hoveyda II in the presence of 2,6-dichloro-1,4-benzoquinone (DCBQ) (to retard undesired alkene isomerisation)40 proceeded in good yield and stereoselectivity (Scheme 12). Immediate treatment of the intermediate alkene with either the tert-butyl ester of tyrosine hydrochloride (61) or 62 in the presence of base 60 effected ring opening of the lactone to form the amides 63 and 64 high yield. Formic-acid-mediated global deprotection gave either viridiofungin A (8) or B (9) in high yield.
Total synthesis of trachyspic acid 19-n-butyl ester
Trachyspic acid (24) was isolated in 1995 from the fermentation broth of Talaromyces treachyspermus during a search for new heparinase inhibitors (Fig. 5).4 Later, in 2017 a group at RIKEN in Japan re-isolated trachyspic acid as well as trachyspic acid 19-butyl ester from the uncharacterised fungus RKGS-F2684 while screening for polo-like kinase 1 inhibitors (Plk1).41
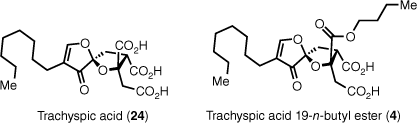
Both trachyspic acid (24) and trachyspic acid 19-n-butyl ester (4) are weak inhibitors of Plk1, whereas trachyspic acid is also active against heparanase, thus both molecules show potential as anti-cancer compounds. The total synthesis of trachyspic acid has been completed by Zammit et al.,42,43 Hirai et al.44 and Morokuma et al.,45 whereas a formal synthesis was completed by Calo et al.46 and the Johnson group has reported a synthesis of trachyspic acid dimethyl ester.47
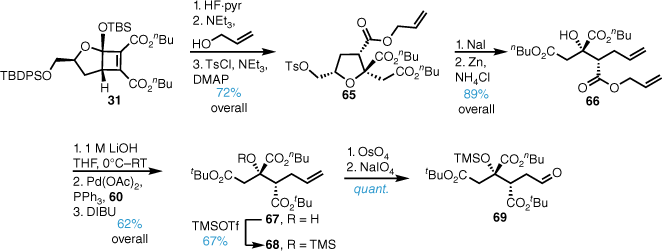
Our total synthesis48 of trachyspic acid 19-n-butyl ester began with di-n-butyl cyclobutene diester 31, which underwent the HF·pyridine-mediated rearrangement to give the corresponding bicyclic lactone (Scheme 13). Lactone ring-opening with allyl alcohol and tosylation gave intermediate 65. Functional group manipulation to the iodide, followed by zinc-mediated reductive elimination gave alkene 66 in high yield. Base-promoted cleavage of the C1 n-butyl ester, palladium-mediated allyl ester cleavage and reprotection as the di-tert-butyl ester gave the orthogonally protected di-tert-butyl-mono-n-butyl ester 67. Trimethylsilane (TMS) protection of the C3 hydroxy group and oxidative cleavage of the terminal double bond gave aldehyde 69 in high yield.
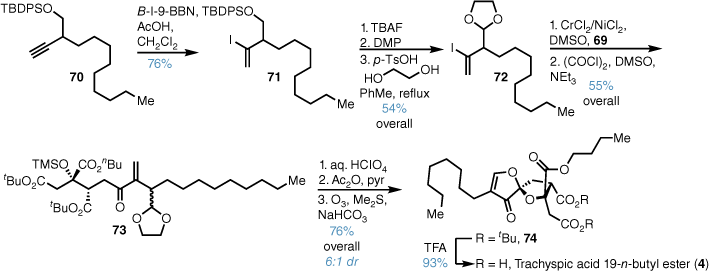
Synthesis of the sidechain began with Markovnikov iodoboration alkyne 70, and protodeborylation gave vinyl iodide 71 (Scheme 14). Fluoride-mediated desilylation, DMP oxidation and acetalisation with ethylene glycol then gave protected aldehyde 72. Kishi–Nozaki coupling with aldehyde 69 and Swern oxidation afforded enone 73 in good yield as a mixture of diastereoisomers. Acidic cleavage of the ketal and TMS ether caused spiroacetal formation and following acetate formation, ozonolysis and base-promoted elimination, 74 was obtained in high yield and in good diastereoselectivity for the desired spiroisomer. Exposure to TFA cleaved the tert-butyl esters to give the trachyspic acid 19-n-butyl ester in good yield.
Total synthesis of L-731,120, L-731,128 and L-731,127
In 1995, Harris et al. at Merck research laboratories reported three new alkyl citrates, L-731,120 (3), L-731,128 (6) and L-731,127 (7) (Fig. 6).2

Both 6 and 7 are from the fermentation broth of Sporormiella intermedia and 3 was isolated from the broth of Phoma sp. MF5453. L-731,120 and L-731,128 are also inhibitors of SQS but no half maximal inhibitory concentration (IC50) value was quoted for L-731,127. To date, the total synthesis of these compounds has only been reported by our group.20,49
The synthesis of the alkene sidechain for L-731,120 began with known alcohol 75,50 which was converted into the corresponding aldehyde by DMP oxidation. Grignard addition of isopropenylmagnesium bromide gave secondary alcohol 76 as a mixture of diastereomers and subsequent Johnson–Claisen rearrangement gave the methyl ester 77. Ester reduction, oxidation and Wittig homologation then gave alkene 78 in good yield over three steps (Scheme 15).
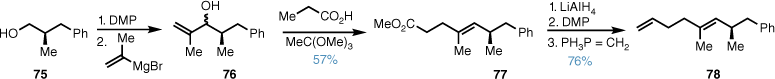
Cross metathesis between the trimethyl citrate 35 and alkene 78 gave a mixture of alkene isomers and regioselective alkene reduction using a modified Myers diimide reduction51 gave the trisubstituted alkene (Scheme 16). Methyl ester deprotection then afforded the desired natural product L-731,120 (3) and this confirmed the absolute configuration.
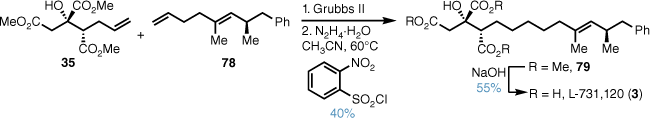
Since the stereochemistry of the sidechain for L-731,128 was unknown, we synthesised both syn- and anti- diastereomers. This began with a Myers diastereoselective alkylation52 of propionamide 80 with iodide 81, which afforded the syn-alkylated product 82 in excellent yield and diastereomeric ratio (DR) (Scheme 17). Reductive removal of the chiral auxiliary and conversion into the tosylate followed by cuprate displacement with allyl magnesium bromide gave alkene 83 in high yield over three steps. The anti-stereodiad 84 was also synthesised in good yield from ent-80 utilising a similar sequence.

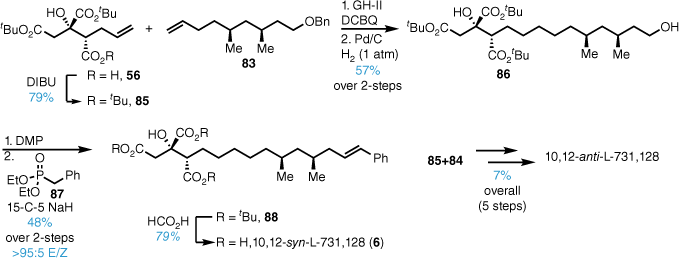
Cross metathesis of tri-tert-butyl ester 85 and alkene 83 followed by alkene reduction and benzyl ester hydrogenolysis gave the diol 86 (Scheme 18). Installation of the styrene moiety was achieved by primary alcohol oxidation followed by Horner–Wadsworth–Emmons (HWE) reaction using the anion derived from phosphonate 87. Formic-acid-mediated deprotection gave 10,12-syn-L-731,128 (6) that possessed identical spectroscopic data to that reported for the natural compound. The synthesis was repeated with anti-alkene 84 and the final 10,12-anti-L-731,128 showed significant deviations in the spectroscopic data, thus confirming the stereochemistry as 10,12-syn.
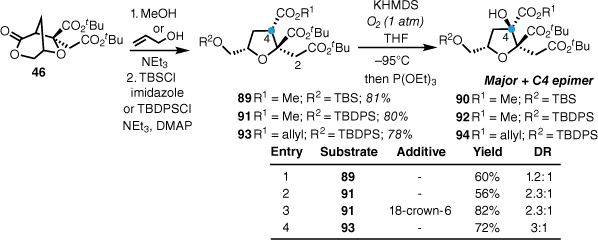
With the stereochemistry of L-731,128 determined, the synthesis of the C4 oxidised congener L-731,127 commenced with bicyclic lactone 46 (Scheme 19). Conversion into the corresponding methyl or allyl ester and protection with either a TBS or TBDPS silyl ether gave substrates 89, 90 and 91. Treatment of the silyl ethers with potassium hexamethyldisilazide (KHMDS) at −95°C under an oxygen atmosphere resulted in formation of the C4 tertiary alcohol in good yields with some diastereoselectivity. Improvements in yield were observed with the addition 18-crown-6 (Scheme 19). Remarkably, this oxidation was completely regioselective, with no C2 oxidation observed. This reaction enabled access to higher oxidised members of the alkyl citrates, such as L-731,127.
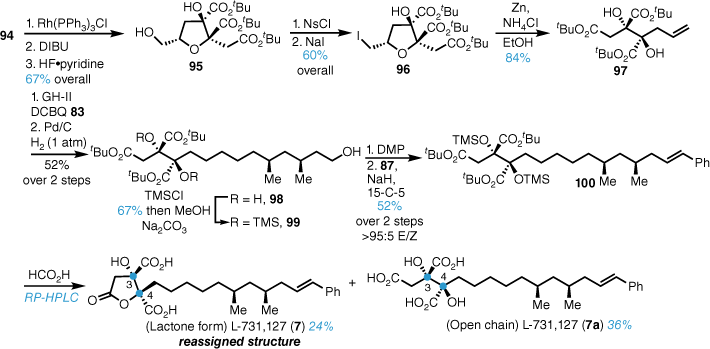
Conversion of 94 into the corresponding tri-tert-butyl ester and silyl deprotection gave diol 95 in good yield (Scheme 20). Functional group manipulation to the iodide 96 and reductive elimination gave tri-tert-butyl ester 97 in good yield over three steps. Cross metathesis of triester 95 and alkene 83 followed by concomitant alkene reduction and benzyl ether hydrogenolysis gave triol 98. Exhaustive TMS silylation and primary deprotection gave alcohol 99, which was homologated to the corresponding styrene in a manner similar to that described for L-731-128 above. Formic-acid-mediated deprotection and purification by RP-HPLC gave both the open chain triacid 7a, which was the assigned structure for L-731,127, as well lactone 7. The data for lactone 7 matched more closely to the partial data reported L-731,127 and thus the structure was reassigned as 7.
Total synthesis of cinatrins C1 and C3
The cinatrin family of alkyl citrates were first isolated in 1992 during screening for phospholipase A2 (PLA2) inhibitors (Fig. 7).6,7
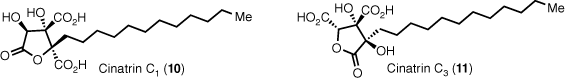
The cinatrins were isolated from the fermentation broth of Circinotrichum falcatisporum and were found to be weak inhibitors of PLA2. The first total synthesis of cinatrins C1 and C3 was reported by the Evans group53 and this confirmed their structures as enantiomeric to those originally proposed. The Rizzacasa group also completed the total synthesis of cinatrins C1, C317,49 and B,54 whereas the Hatakeyama group completed the synthesis of C155 and the Yukura group completed the synthesis of C3-epi-dimethyl cinatrin C1.56
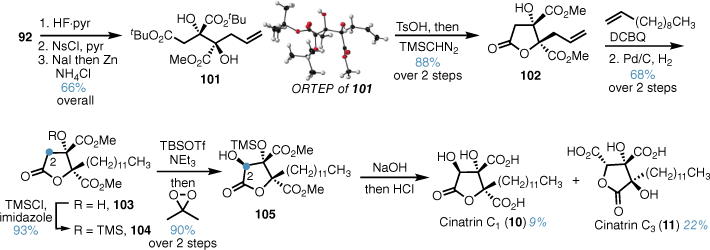
Our synthesis proceeded with deprotection of alcohol 92, conversion into the iodide and reductive elimination gave crystalline alkene 101, which confirmed the stereochemical outcome of the oxidation (Scheme 21). Acid catalysed lactonisation of alkene 101 and reprotection afforded lactone 102, which underwent cross metathesis with 1-undecene and reduction installed the chain to give lactone 103. Protection of the C3 tertiary alcohol and formation of the corresponding silyl ketene acetal under soft enolisation conditions enabled efficient and diastereoselective Rubottom type oxidation at the C2 position giving 105 in high yield. Subsequent base-mediated ester hydrolysis and acidification gave cinatrins C1 and C3 in moderate yield after RP-HPLC.
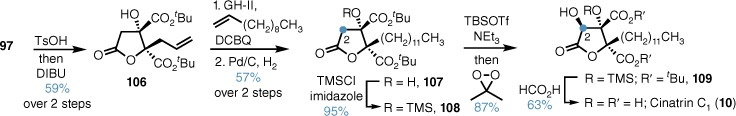
A more direct route to cinatrin C1 was undertaken, and this began with tri-tert-butyl ester 97 (Scheme 22). Lactone formation followed by cross metathesis and reduction using a similar sequence to that described above gave di-tert-butyl-lactone 106 in good yield. C2 oxidation proceeded in high yield and DR to give 109, and subsequent formic-acid-mediated deprotection gave cinatrin C1 in high yield.
Conclusion
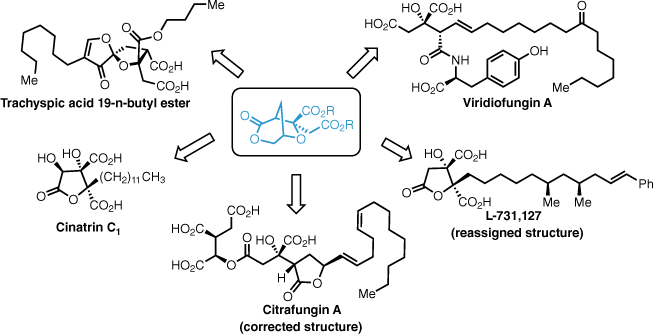
Cyclobutene diesters serve as useful precursors for the synthesis of the core of the alkyl citrate natural products. These are easily accessed from a simple silyl ketene acetal and various acetylene diesters by formal [2 + 2]-cycloadditions. More specifically, the derived bicyclic lactones are key intermediates for the synthesis of a large number of these natural products including the higher oxidised metabolites (Fig. 8). This robust methodology can provide the citrate core with no further oxidative manipulations required and, in essence, access to the entire family of these interesting natural products.
Data availability
Data sharing is not applicable as no new data were generated or analysed during this study.
Declaration of funding
The authors thank the Australian Research Council Discovery Grants Scheme for funding (DP DP200100722 to M. A. Rizzacasa).
References
1 Watanabe S, Hirai H, Kambara T, Kojima Y, Nlshida H, Sugiura A, Yamauchi Y, Yoshikawa N, Harwood HJ, Huang LH, Kojima N. CJ-13,981 and CJ-13,982, new squalene synthase inhibitors. J Antibiot 2001; 54: 1025-1030 54.
| Crossref | Google Scholar | PubMed |
2 Harris GH, Dufresne C, Joshua H, Koch LA, Zink DL, Salmon PM, Göklen KE, Kurtz MM, Rew DJ, Bergstrom JD, Wilson KE. Isolation, structure determination and squalene synthase activity of L-731,120 and L-731,128, alkyl citrate analogs of zaragozic acids A and B. Bioorg Med Chem Lett 1995; 5: 2403-2408.
| Crossref | Google Scholar |
3 Harris GH, Jones ETT, Meinz MS, Nallin-Omstead M, Helms GL, Bills GF, Zink D, Wilson KE. Isolation and structure elucidation of viridiofungins A, B and C. Tetrahedron Lett 1993; 34: 5235-5238.
| Crossref | Google Scholar |
4 Shiozawa H, Takahashi M, Takatsu T, Kinoshita T, Tanzawa K, Hosoya T, Furuya K, Takahashi S, Furihata K, Seto H. Trachyspic acid, a new metabolite produced by Talaromyces trachyspermus, that inhibits tumor cell heparanase: taxonomy of the producing strain, fermentation, isolation, structural elucidation, and biological activity. J Antibiot 1995; 48: 357-362.
| Crossref | Google Scholar | PubMed |
5 Singh SB, Zink DL, Doss GA, Polishook JD, Ruby C, Register E, Kelly TM, Bonfiglio C, Williamson JM, Kelly R. Citrafungins A and B, two new fungal metabolite inhibitors of GGTase I with antifungal activity. Org Lett 2004; 6: 337-340.
| Crossref | Google Scholar | PubMed |
6 Itazaki H, Nagashima K, Kawamura Y, Matsumoto K, Nakai H, Terui Y. Cinatrins, a novel family of phospholipase A2 inhibitors. I. Taxonomy and fermentation of the producing culture; isolation and structures of cinatrins. J Antibiot 1992; 45: 38-49.
| Crossref | Google Scholar | PubMed |
7 Tanaka K, Itazaki H, Yoshida T. Cinatrins, a novel family of phospholipase A2 inhibitors. II. Biological activities. J Antibiot 1992; 45: 50-55.
| Crossref | Google Scholar | PubMed |
8 Bergstrom JD, Dufresne C, Bills GF, Nallin-Omstead M, Byrne K. Discovery, biosynthesis, and mechanism of action of the zaragozic acids: potent inhibitors of squalene synthase. Annu Rev Microbiol 1995; 49: 607-639.
| Crossref | Google Scholar | PubMed |
9 Bergstrom JD, Kurtz MM, Rew DJ, Amend AM, Karkas JD, Bostedor RG, Bansal VS, Dufresne C, VanMiddlesworth FL, Hensens OD. Zaragozic acids: a family of fungal metabolites that are picomolar competitive inhibitors of squalene synthase. Proc Natl Acad Sci USA 1993; 90: 80-4.
| Crossref | Google Scholar | PubMed |
10 Dawson MJ, Farthing JE, Marshall PS, Middleton RF, O’Neill MJ, Shuttleworth A, Stylli C, Tait RM, Taylor PM, Wildman HG. The squalestatins, novel inhibitors of squalene synthase produced by a species of Phoma. I. Taxonomy, fermentation, isolation, physico-chemical properties and biological activity. J Antibiot 1992; 45: 639-647.
| Crossref | Google Scholar | PubMed |
11 Sidebottom PJ, Highcock RM, Lane SJ, Procopiou PA, Watson NS. The squalestatins, novel inhibitors of squalene synthase produced by a species of Phoma. II. Structure elucidation. J Antibiot 1992; 45: 648-658.
| Crossref | Google Scholar | PubMed |
12 Jones CA, Sidebottom PJ, Cannell RJ, Noble D, Rudd BA. The squalestatins, novel inhibitors of squalene synthase produced by a species of Phoma. III. Biosynthesis. J Antibiot 1992; 45: 1492-1498.
| Crossref | Google Scholar | PubMed |
13 Rizzacasa MA, Sturgess D. Total synthesis of alkyl citrate natural products. Org Biomol Chem 2014; 12: 1367-1382.
| Crossref | Google Scholar | PubMed |
14 Nadin A, Nicolaou K. Chemistry and biology of the zaragozic acids (squalestatins). Angew Chem Int Ed Engl 1996; 35: 1622-1656.
| Crossref | Google Scholar |
15 Armstrong A, Blench TJ. Recent synthetic studies on the zaragozic acids (squalestatins). Tetrahedron 2002; 58: 9321-9349.
| Crossref | Google Scholar |
16 Newhouse T, Baran PS, Hoffmann RW. The economies of synthesis. Chem Soc Rev 2009; 38: 3010-3021.
| Crossref | Google Scholar | PubMed |
17 Cuzzupe AN, Di Florio R, White JM, Rizzacasa MA. Enantiospecific synthesis of the phospholipase A2 inhibitors (−)-cinatrin C1 and (+)-cinatrin C3. Org Biomol Chem 2003; 1: 3572-3577.
| Crossref | Google Scholar | PubMed |
18 Miesch M, Wendling F, Franck-Neumann M. Uncatalyzed [2+2] cycloaddition of cyclic ketenetrimethylsilylacetals with electrophilic acetylenes. Tetrahedron Lett 1999; 40: 839-842.
| Crossref | Google Scholar |
19 Miesch M, Wendling F. Uncatalyzed, solvent-free [2+2] cycloaddition of cyclic ketene trimethylsilyl acetals with electrophilic acetylenes. Eur J Org Chem 2000; 2000: 3381-3391.
| Crossref | Google Scholar |
20 Atkin L, Chen Z, Robertson A, Sturgess D, White JM, Rizzacasa MA. Synthesis of alkyl citrates (−)-CJ-13,981, (−)-CJ-13,982, and (−)-L-731,120 via a cyclobutene diester. Org Lett 2018; 20: 4255-4258.
| Crossref | Google Scholar | PubMed |
21 Calo F, Bondke A, Richardson J, White AJP, Barrett AGM. Total synthesis and determination of the absolute stereochemistry of the squalene synthase inhibitors CJ-13,981 and CJ-13,982. Tetrahedron Lett 2009; 50: 3388-3390.
| Crossref | Google Scholar |
22 Sturgess D, Chen Z, White JM, Rizzacasa MA. Enantiospecific total synthesis of the squalene synthase inhibitors (–)-CJ-13,982 and its enantiomer from a common intermediate. J Antibiot 2018; 71: 234-239.
| Crossref | Google Scholar | PubMed |
23 Rossouw NP, Rizzacasa MA, Polyzos A. Flow-assisted synthesis of alkyl citrate natural products. J Org Chem 2021; 86: 14223-14231.
| Crossref | Google Scholar | PubMed |
24 Watanabe S, Hirai H, Ishiguro M, Kambara T, Kojima Y, Matsunaga T, Nishida H, Suzuki Y, Sugiura A, Harwood HJ, Huang LH, Kojima N. CJ-15,183, a new inhibitor of squalene synthase produced by a fungus, Aspergillus aculeatus. J Antibiot 2001; 54: 904-910.
| Crossref | Google Scholar | PubMed |
25 Amer MFA, Takahashi K, Ishihara J, Hatakeyama S. Total synthesis of citrafungin A. Heterocycles 2007; 72: 181-185.
| Google Scholar |
26 Calo F, Richardson J, Barrett AGM. Total synthesis of citrafungin A. J Org Chem 2008; 73: 9692-9697.
| Crossref | Google Scholar | PubMed |
27 Tsegay S, Hügel H, Rizzacasa MA. Formal total synthesis of (+)-citrafungin A. Aust J Chem 2009; 62: 676-682.
| Crossref | Google Scholar |
28 Chen Z, Robertson A, White JM, Rizzacasa MA. Total synthesis and stereochemical reassignment of citrafungin A. Org Lett 2019; 21: 9663-9666.
| Crossref | Google Scholar | PubMed |
29 Nugent WA. MIB: an advantageous alternative to DAIB for the addition of organozinc reagents to aldehydes. Chem Commun 1999; 1999(15): 1369-1370.
| Crossref | Google Scholar |
30 Kumagai N, Shibasaki M. Synthetic studies of viridiofungins, broad-spectrum antifungal agents and serine palmitoyl transferase inhibitors. J Antibiot 2018; 71: 53-59.
| Crossref | Google Scholar | PubMed |
31 Pollex A, Millet A, Müller J, Hiersemann M, Abraham L. Ester dienolate [2,3]-Wittig rearrangement in natural product synthesis: diastereoselective total synthesis of the triester of viridiofungin A, A2, and A4. J Org Chem 2005; 70: 5579-5591.
| Crossref | Google Scholar | PubMed |
32 Goldup SM, Pilkington CJ, White AJP, Burton A, Barrett AGM. A simple, short, and flexible synthesis of viridiofungin derivatives. J Org Chem 2006; 71: 6185-6191.
| Crossref | Google Scholar | PubMed |
33 Ghosh AK, Kass J. A stereoselective synthesis of (-)-viridiofungin A utilizing a TiCl4-promoted asymmetric multicomponent reaction. Org Lett 2012; 14: 510-512.
| Crossref | Google Scholar | PubMed |
34 Esumi T, Iwabuchi Y, Irie H, Hatakeyama S. Synthesis of viridiofungin A trimethyl ester and determination of the absolute structure of viridiofungin A. Tetrahedron Lett 1998; 39: 877-880.
| Crossref | Google Scholar |
35 Morokuma K, Takahashi K, Ishihara J, Hatakeyama S. Total synthesis of viridiofungin A. Chem Commun 2005; 2005(17): 2265-2267.
| Crossref | Google Scholar | PubMed |
36 Takechi S, Yasuda S, Kumagai N, Shibasaki M. A direct catalytic asymmetric aldol reaction of α-sulfanyl lactones: efficient synthesis of SPT inhibitors. Angew Chem Int Ed 2012; 51: 4218-4222 Takechi.
| Crossref | Google Scholar | PubMed |
37 Haneishi T, Kato Y, Fukuda H, Shimamura T, Tanokura T, Hiraide A, Koyama K, Fudesaka M, Maeda K, Nakata N, Nagase M, Yabuzaki T, Takao H, Kigawa M, Shimizu H, Shimizu M. Development of a kilogram-scale synthesis of a novel anti-HCV agent, CH4930808. Org Process Res Dev 2018; 22: 236-240.
| Crossref | Google Scholar |
38 Murakata M, Ikeda T. Stereoselective synthesis of the viridiofungin analogue NA808 from a chiral tetrahydrofuran-carboxylic acid. Org Biomol Chem 2017; 15: 6632-6639.
| Crossref | Google Scholar | PubMed |
39 Atkin L, Robertson A, White JM, Rizzacasa MA. Total synthesis of viridiofungins A and B. Org Lett 2021; 23: 3557-3560.
| Crossref | Google Scholar | PubMed |
40 Hong SH, Sanders DP, Lee CW, Grubbs RH. Prevention of undesirable isomerization during olefin metathesis. J Am Chem Soc 2005; 127: 17160-17161.
| Crossref | Google Scholar | PubMed |
41 Nogawa T, Ogita N, Futamura Y, Negishi S, Watanabe N, Osada H. Trachyspic acid 19-butyl ester, a new inhibitor of Plk1 polo box domain-dependent recognition from uncharacterized fungus RKGS-F2684. J Antibiot 2017; 70: 705-707.
| Crossref | Google Scholar | PubMed |
42 Zammit SC, White JM, Rizzacasa MA. Enantiospecific synthesis of (-)-trachyspic acid. Org Biomol Chem 2005; 3: 2073-2074.
| Crossref | Google Scholar | PubMed |
43 Zammit SC, Ferro V, Hammond E, Rizzacasa MA. Enantiospecific synthesis of the heparanase inhibitor (+)-trachyspic acid and stereoisomers from a common precursor. Org Biomol Chem 2007; 5: 2826-2834.
| Crossref | Google Scholar | PubMed |
44 Hirai K, Ooi H, Esumi T, Iwabuchi Y, Hatakeyama S. Total synthesis of (+/-)-trachyspic acid and determination of the relative configuration. Org Lett 2003; 5: 857-859.
| Crossref | Google Scholar | PubMed |
45 Morokuma K, Taira Y, Uehara Y, Shibahara S, Takahashi K, Ishihara J, Hatakeyama S. Asymmetric synthesis of (+)-trachyspic acid. Tetrahedron Lett 2008; 49: 6043-6045.
| Crossref | Google Scholar |
46 Calo F, Richardson J, White AJP, Barrett AGM. Enantioselective formal total synthesis of (−)-trachyspic acid. Tetrahedron Lett 2009; 50: 1566-1567.
| Crossref | Google Scholar |
47 Schmitt DC, Lam L, Johnson JS. Three-component coupling approach to trachyspic acid. Org Lett 2011; 13: 5136-5139.
| Crossref | Google Scholar | PubMed |
48 Rafaniello AA, Rizzacasa MA. Total synthesis of (+)-trachyspic acid 19-n-butyl ester. Org Lett 2020; 22: 1972-1975.
| Crossref | Google Scholar | PubMed |
49 Rossouw NP, Chen Z, White JM, Rizzacasa MA. Synthesis of more highly oxidized alkyl citrates via direct regio- and stereoselective oxidation. Org Lett 2023; 25: 8010-8015.
| Crossref | Google Scholar | PubMed |
50 Evans DA, Ennis MD, Mathre DJ. Asymmetric alkylation reactions of chiral imide enolates. A practical approach to the enantioselective synthesis of ɑ-substituted carboxylic acid derivatives. J Am Chem Soc 1982; 104: 1737-1739.
| Crossref | Google Scholar |
51 Marsh BJ, Carbery DR. One-pot o-nitrobenzenesulfonylhydrazide (NBSH) formation-diimide alkene reduction protocol. J Org Chem 2009; 74: 3186-3188.
| Crossref | Google Scholar | PubMed |
52 Myers AG, Yang BH, Chen H, McKinstry L, Kopecky DJ, Gleason JL. Pseudoephedrine as a practical chiral auxiliary for the synthesis of highly enantiomerically enriched carboxylic acids, alcohols, aldehydes, and ketones. J Am Chem Soc 1997; 119: 6496-6511.
| Crossref | Google Scholar |
53 Evans DA, Wesley Trotter B, Barrow JC. Aldol reactions of ketal-protected tartrate ester enolates. Asymmetric syntheses and absolute stereochemical assignments of phospholipase A2 inhibitors cinatrin C1 and C3. Tetrahedron 1997; 53: 8779-8794.
| Crossref | Google Scholar |
54 Cuzzupe AN, Di Florio R, Rizzacasa MA. Enantiospecific synthesis of the phospholipase A2 inhibitor (-)-cinatrin B. J Org Chem 2002; 67: 4392-4398.
| Crossref | Google Scholar | PubMed |
55 Urabe F, Nagashima S, Takahashi K, Ishihara J, Hatakeyama S. Total synthesis of (-)-cinatrin C1 based on an In(OTf)3-catalyzed Conia–ene reaction. J Org Chem 2013; 78: 3847-3857.
| Crossref | Google Scholar | PubMed |
56 Yakura T, Ozono A, Matsui K, Yamashita M, Fujiwara T. Application of a stereoselective rhodium(II)-catalyzed oxonium ylide formation–[2,3]-sigmatropic rearrangement of an α-diazo-β-keto ester to the synthesis of 2-epi-cinatrin C1 dimethyl ester. Synlett 2013; 24: 65-68.
| Crossref | Google Scholar |