

Evaluation of the 5-ethynyl-1,3,3-trimethyl-3H-indole ligand for molecular materials applications
David Jago A , David C. Milan B , Alexandre N. Sobolev C , Simon J. Higgins B , Andrea Vezzoli B , Richard J. Nichols B and George A. Koutsantonis A *
A , David C. Milan B , Alexandre N. Sobolev C , Simon J. Higgins B , Andrea Vezzoli B , Richard J. Nichols B and George A. Koutsantonis A *
A Department of Chemistry, School of Molecular Sciences, The University of Western Australia, Crawley, WA 6009, Australia.
B Department of Chemistry, University of Liverpool, Liverpool, UK.
C Centre for Microscopy, Characterisation and Analysis, The University of Western Australia, Crawley, WA 6009, Australia.
Australian Journal of Chemistry 76(4) 209-230 https://doi.org/10.1071/CH23069
Submitted: 12 April 2023 Accepted: 22 May 2023 Published: 6 July 2023
© 2023 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
The modification of conjugated organic compounds with organometallic moieties allows the modulation of the electronic and optoelectronic properties of such compounds and lends them to a variety of material applications. The organometallic complexes [M(Cp′)(L)n] (M = Ru or Fe; Cp′ = cyclopentadiene (Cp) or pentamethylcyclopentadiene (Cp*); (L)n = (PPh3)2 or 1,2-bi(diphenylphosphino)ethane (dppe)) and [M(L)n] (M = Ru; (L)n = (dppe)2 or (P(OEt)3)4; or M = Pt; (L)n = (PEt3)2, (PPh3)2 or tricyclohexylphosphine, (PCy3)2) modified with a 5-ethynyl-1,3,3-trimethyl-3H-indole ligand were prepared and characterised by NMR spectroscopy, IR and single-crystal X-ray diffraction. Cyclic voltammetry and IR spectroelectrochemistry of the ruthenium systems showed a single-electron oxidation localised over the M–C≡C–aryl moiety. The N-heteroatom of the indole ligand showed Lewis base properties and was able to extract a proton from a vinylidene intermediate as well as coordinate to CuI. Examples from the wire-like compounds were also studied by single-molecule break junction experiments but molecular junction formation was not observed. This is most likely attributable to the binding characteristics of the substituted terminal indole groups used here to the gold contacts.
Keywords: cyclic voltammetry, metal alkynyl, molecular materials, molecular electronics, organometallic chemistry, ruthenium, single-molecule electronics, single-molecule junction.
Introduction
The ability to incorporate molecules as functional building blocks in molecular systems has gained the attention of chemists for the best part of a century. Organic π-conjugated compounds possess properties that lend themselves to use in various electronic and optoelectronic materials.[1–3] Incorporating organometallic fragments allows the fine-tuning of electronic structure and function in organic molecules.[4] The capacity to modulate the properties of the ligand has seen metal complexes used in a variety of applications including molecular electronics, non-linear optical materials,[5] surface-based catalysis,[6] photovoltaics,[7] and luminescent and responsive materials.[8]
The design and optimisation of new materials based on organometallic complexes relies on the exploration of new metal complexes and the study of their chemical, physical and optoelectronic features. N-heterocyclic aromatic compounds are used ubiquitously as σ-donor ligands in inorganic chemistry, particularly pyridine-based ligands, which are used to construct large metal–organic frameworks and have gained interest in a variety of fields.[9] In addition, analogies between coordination and molecular-surface interactions provide a basis for motif testing for binding groups in large-area ensembles and molecular junctions.[10] Indole and indole derivatives are found both in nature and pharmacology.[11–13] In particular, the 3H-indole is an important structural motif and has been used as a scaffold to form other indoline alkaloids.[12,14] Further functionalisation of the 3H-indole moiety is attractive for expanding its synthetic facility.
Metal alkynyl complexes have become building blocks for the synthesis and study of ligand-bridged polymetallic systems.[15,16] They are ideal candidates for studying mixed-valence systems and other intramolecular electron-transfer processes. In particular, the complexes containing the d8 and d10 metal centres are well known organometallic systems with the ability to tune the oxidation site using different metals and ancillary ligands, which can moderate the opto-electronic properties of the ligand.[8,17,18]
We have previously investigated the electro-optical properties of pyridyl and bipyridyl moieties in σ-alkynyl, vinylidene and allenylidene organometallic complexes.[19–23] Herein, the synthesis of a series of organometallic complexes derived from the 5-ethynyl-3H-indole moiety is reported and their physical and chemical properties are described.
Results and discussion
Synthesis
The 3H-indole ligand 2 was obtained in high yield by the KOH-promoted desilylation of 1 (Scheme 1). The ethene 3 was also obtained from this reaction. The mechanism for the formation of 3 is not well understood.
A similar ethene compound from the reaction of an alkyllithium reagent and a 3H-indole has been reported and was proposed to form by a radical mechanism (Supplementary Scheme S1).[24] Electron Spin Resonance (ESR) spectroscopy confirmed the presence of a radical intermediate from the reaction of methylmagnesium iodide or organolithium reagents and a 3H-indole.[24,25] Alternative oxidation mechanisms are proposed in the absence of such alkyl reagents in the current study (Supplementary Scheme S2).
Following the oxidation of 2 to the aldehyde, in a manner similar to that in the literature,[26–28] an aldol condensation with another molecule of 2 and the elimination of water gives the ethene dimer 3. The propensity of molecules similar to 2 to undergo aldol condensation is known.[29] Alternatively, the oxidation of 2 to the N-oxide is also plausible. As similar self-condensation of indolium salts has previously been observed,[30,31] which involves the activation of the indole to the N-oxide for further reactions such as with another molecule of 3H-indole. Subsequent deprotonation and elimination of water yield the dimeric indole molecule. Schemes for these proposed mechanisms are shown in the supporting information.
The 1H NMR spectrum of 2 shows a resonance assigned to the terminal alkyne proton at 3.07 ppm. Sharp absorptions at 3176 (ν(CH)) and 2099 (ν(C≡C)) cm−1 in the IR spectrum further evince the terminal alkyne functional group. Compound 3 is fluorescent under UV light. The singlet at 3.07 and 128.51 ppm in the 1H and 13C NMR spectra respectively was assigned to the alkene bridge. A weak absorption at 1607 cm−1 in the IR provided further evidence for the alkene bridge (ν(C=C)). The atmospheric-pressure chemical ionisation (APCI) mass spectrum showed a peak at m/z 363.1851 consistent with the molecular ion. Single-crystal X-ray diffraction studies confirmed the connectivity of 2 and 3.
The half-sandwich monometallic σ-alkynyl complexes were synthesised using established or slightly modified literature methodologies. The complexes 4–7 were synthesised by the vinylidene route (Scheme 2).[32] The appropriate half-sandwich ruthenium or iron halides and 2 were refluxed in methanol with NH4PF6 present to produce a vinylidene intermediate. After the reaction mixture had cooled to room temperature, the vinylidene species was then deprotonated by carefully adding 1,8-diazabicycloundec-7-ene (DBU) and leaving the mixture undisturbed overnight. The slow diffusion of DBU over a long period afforded well-formed crystals of the targeted complexes. Copper-assisted transmetallation[33] of Ru(CO)2(Cp)Cl (Cp, cyclopentadiene) and 2 in THF and NEt3 afforded 8 in good yield (Scheme 2), whereas complex 9 was synthesised in a basic methanolic solution containing 2 and the gold chloride precursor, PPh3AuCl (Scheme 2).[34] Both 8 and 9 showed light sensitivity. The gold complex is of interest owing to its potential use as transmetallation reagent and catalyst.[35–37]
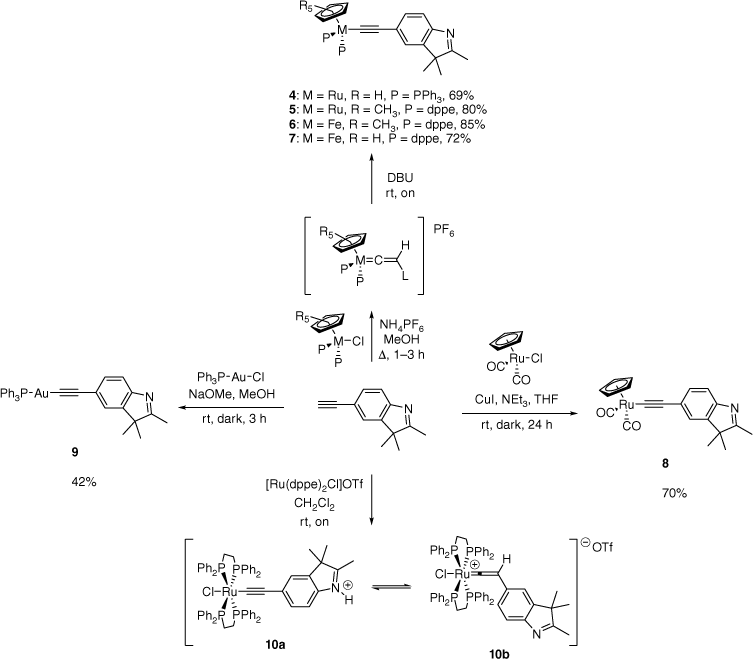
The ruthenium complexes 4 and 5 showed absorptions at ~2060 cm−1 in their IR spectra assigned to the alkynyl functional group. This assigned ν(C≡C) absorption appeared at a slightly lower wavenumber, ~2050 cm−1, for the iron complexes 6 and 7. Comparatively, the alkynyl stretch for 8 was assigned to the IR absorption at 2119 cm−1. Strong absorbances for the symmetric and asymmetric carbonyl stretches for 8 were assigned to the IR bands at 2035 and 1994 cm−1 respectively.
A single peak in the 31P NMR spectrum for 4–7 and 9 further evinced the synthesis of the single metal σ-alkynyl complexes. Resonances for the respective cyclopentadienyl ligands were assigned to the peaks at δ 4.51 (4), 1.68 (5), 1.56 (6), 4.32 (7), 5.48 (8). Resonances for the cyclopentadienyl and phosphine ligands are within the typical range of half-sandwich ruthenium and iron complexes.[20,35,38,39]
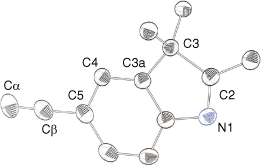
The 1H and 13C NMR spectra contained resonances consistent with the indole ligand for all complexes. Supplementary Fig. S1 shows the general atom numbering and labelling for the indole and phosphine ligands of the complexes. In general, resonances for Cα, Cβ and C5 are deshielded on metal–carbon bond formation of the free ligand. The extent of this deshielding is dependent on the electron density of the metal centre.[40] Peaks for the Cα carbon in 4, (δ 113.8) 5 (δ 127.1), 7 (δ 122.3 ppm) and 8 (δ 81.3 ppm) and the Cβ carbon in 4 (δ 116.1 ppm), 5 (δ 111.4 ppm), 6 (δ 121.1), 7 (δ 122.7) and 8 (δ 111.0) show close agreement with expected resonances.[20,41] The signals for Cα for complexes 4–7 appear as triplets owing to coupling to the phosphorus ligand on the metal centre. The peak for Cα in complex 9 appears as a doublet at δ 135.8 with a JCP coupling of 146 Hz, whereas the peak for Cβ appears at δ 104.1 with a JCP coupling of 26 Hz. The chemical shifts and coupling constants are similar to previously synthesised gold(I) alkynyl complexes.[34] The observation of these alkynyl carbons is in contrast to a previous report that suggests these analogous resonances were not observed as a consequence of quadrupolar broadening and quenching in those gold type complexes.[42]
The electrospray ionisation (ESI) mass spectra of complexes 4–9 showed ions consistent with an [M + H]+ species, except for 6, which showed a peak assigned to the [M]+ species. In addition, the mass spectra of complexes 4, 5 and 7 contained an ion consistent with an [M + 2H]2+ species, whereas the mass spectrum of complex 5 also showed an ion at m/z 663, consistent with the oxidative cleavage of the alkyne ligand to give the carbonyl cation complex [Ru(dppe)(Cp*)CO]+ (dppe, 1,2,-bis(diphenylphosphino)ethane).[43] Most complexes also showed fragmentation consistent with the loss of the alkyne ligand and incomplete desolvation. The spectrum of gold complex 9 exhibited an ion at m/z 721 for the [Au(PPh3)2]+ species also seen in previous studies.[42]
The synthesis of the alkynyl complex of 2 containing the [Ru(dppe)2Cl]+ moiety by a number of alternative, established literature procedures was unsuccessful.[44] However, the reaction of [Ru(dppe)2Cl]OTf and 2 afforded a mixture of complexes 10a and 10b, as evinced by spectroscopic data. We suggest that the basicity of the nitrogen on the indole ligand can deprotonate the intermediate vinylidene species; however, singlet peaks in the 31P NMR spectrum at ~48 and 37 ppm suggest two trans configurations are in equilibrium (Scheme 2).[45] This basic proligand effect has been seen in the syntheses of complexes from 5-ethynyl-2,2′-bipyridine using both the Ru(dppe)2Cl and RuCp(PPh3)2 moieties.[19,45] Similar effects have been noted in the formation of alkynyl complexes from Ru(dppe)2Cl2 and 4-ethynylaniline,[44] but not with the half-sandwich analogues using the Ru(dppe)(Cp*) moiety.[46] Contrarily, the syntheses of ruthenium pyridyl alkynyl complexes also report this deprotonation when the alkynyl is formed from RuCp(PPh3)2 halides,[47] but not Ru(dppm)2Cl2 (dppm, 1,2-bis(diphenylphosphino)methane).[48] The 19F NMR spectrum was consistent with the presence of the triflate anion. The ES mass spectrum showed a peak at m/z 1121 consistent with [M–Cl + MeCN]+ species. A presumed impurity, unassigned, was seen at 41 ppm in the 31P NMR spectrum and could not be removed by further purification steps. Attempts to deprotonate 10a/10b resulted in mixtures of decomposition and oxidation products evinced by the numerous peaks observed in the 31P NMR spectra obtained.
The cross-coupling of 2 with 1,4-di-iodobenzene in NEt3 gave 11 (Scheme 3). The 1H NMR spectrum shows resonances consistent with the geminal methyl groups and imine methyl group at δ 1.32 and 2.30 respectively. The multiplet at δ 7.46–7.50 in the 1H NMR spectrum was assigned to the 1,4-substituted benzene and indole. The 13C NMR shows peaks assigned to the alkynyl carbons at 91.9 and 89.1 ppm, shifted downfield from the parent alkyne. The alkynyl bond is also evinced by a band at 2203 cm−1 in the IR spectrum. Mass spectroscopy (APCI) shows a peak consistent with the protonated molecular ion at m/z 441.2338.
Preparation of the compounds 11–18. Conditions: (i) NEt3, 70°C, 19 h; (ii) HNiPr2, reflux, 3 h, (iii) 60°C, 11 days; (iv) 55°C, 21 days (on, overnight; rt, room temperature).
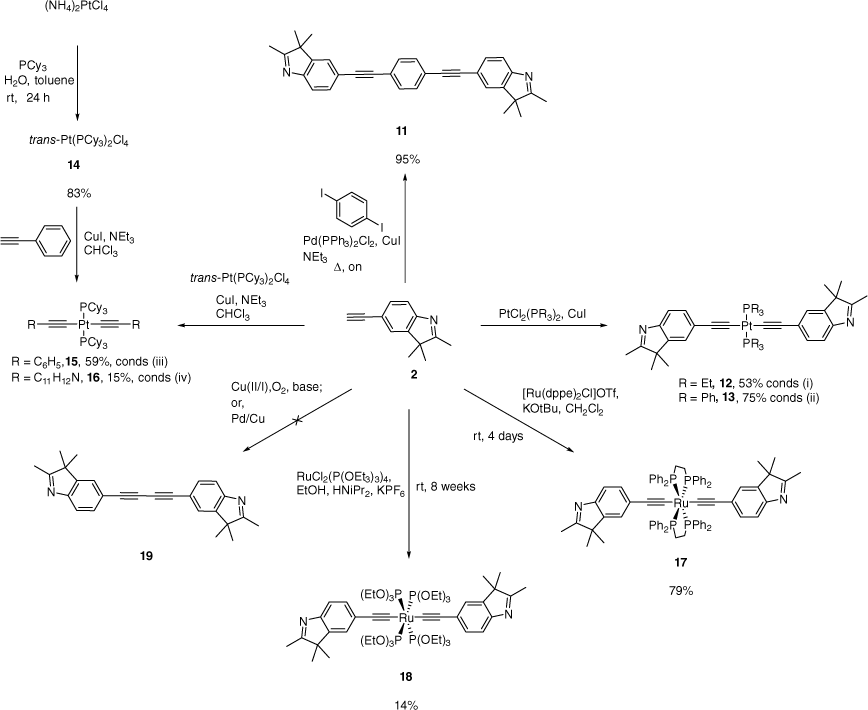
Access to the platinum metal complexes trans-[Pt(L)2(PR3)2]}, where L = CC-2,3,3-trimethyl-3H-indole, and R=Et (12), Ph (13) or Cy (16) (Scheme 3) was achieved by CuI-catalysed dehydrohalogenation of Pt(PR3)2Cl2 and 2 in the presence of an amine base.[49] The use of cis-Pt(PR3)2Cl2 conserves the cis-orientation of the complex[50,51]; however, heating under reflux allows access to the more thermodynamically stable trans isomer.
Spectral characteristics of 12 and 13 are comparable with other bis-alkynyl PtII complexes.[49] The 31P NMR spectrum of 12 and 13 respectively shows a single resonance (with 195Pt satellites) at δ 11.62 and 19.18, affirming a trans orientation of the phosphine ligands. Resonances in the 1H NMR spectra for the alkyl groups in the triethylphosphines in 12 and phenyl groups in the triphenylphosphines in 13 are assigned to signals at δ 2.17–2.20 (CH2) and 1.20–1.27 (CH3), and δ 7.40–7.84 (30H) respectively. The 1H NMR spectrum also shows singlets consistent with the geminal methyl groups and imine methyl group of the alkynyl indoles in both Pt complexes. The 13C NMR spectrum shows resonances assigned to the respective Cα and Cβ alkynyl carbons at δ 107.4 and 109.8 in 12. These alkynyl signals for 13 appear at δ 118.8 and 113.7. The IR spectrum of 12 shows a band at 2099 cm−1 also confirming an alkynyl ligand. An absorption at 2105 cm−1 in the IR spectrum of 13 is assigned to the ν(C≡C) . The HRMS contains peaks consistent with the protonated molecular ion at m/z 796.3510 and 1084.3472 for 12 (ES) and 13 (APCI) respectively.
In the synthesis of 12, extraction of the reaction residue with benzene and subsequent recrystallisation gave the coordination complex 12b (Supplementary Fig. S8). The complex is a polymeric compound consisting of 12 and CuI, with coordination at the nitrogen of the indole ligand. This compound’s connectivity was confirmed by single-crystal X-ray diffraction studies (Supplementary Fig. S9). Both 1H and 31P NMR spectra were unremarkable compared with the parent 12. Examples of coordination complexes of 3H-indoles are rare, with only a few iridium, platinum and gold complexes reported.[52–54]
Complex trans-[Pt(PCy3)2Cl2]} 14 has previously been synthesised by the reaction of [Pt(COD)Cl2] (COD, 1,5-cyclooctadiene) with PCy3 in THF.[55] Adapting the synthetic procedure for cis- and trans-PtCl2(PEt3)2,[56] the reaction of commercially available (NH4)2PtCl4 and PCy3 in toluene and water gives 14 in 83% yield (Scheme 3). Formation of the trans orientation is evinced by a single peak at δ 17.18 (JPPt 2398 Hz) in the 31P{1H} NMR spectrum. The increased steric bulk of PCy3 hinders formation of the cis orientation.
Owing to the scarcity of bis-alkynyl complexes of the type trans-[Pt(PCy3)2(CC–Ar)2], a brief optimisation synthesis of trans-[Pt(PCy3)2(CC–C6H5)2] 15 was undertaken. The reaction of 14 and phenylacetylene in the presence of CuI, triethylamine and co-solvent CHCl3 gave 15 in 59% yield (Scheme 3). The co-solvent was required owing to solubility issues of 14 in neat amine solutions. Other co-solvents tested were THF and CH2Cl2. The steric bulk of the PCy3 ancillary ligands renders this reaction sluggish even at elevated temperatures. This observation is mirrored in a study that found only 50% conversion (30% isolated yield) to the bis-alkynyl Pt complexes trans-[Pt(PCy3)2(CC–C6H4SAc)2] after 4 days at 60°C by similar methodology.[57] The 31P NMR spectrum of 15 shows a single peak at δ 24.06 (JPPt 2408 Hz). These conditions were employed to synthesise the Pt complex 16; however, this reaction took 21 days to complete and was low-yielding (Scheme 3).
A peak at δ 23.22 (JPPt 2424 Hz) in the 31P NMR spectrum for 16 evinced a single trans-orientated complex. The 1H NMR spectrum showed three resonances assigned to the aryl protons on the 3H-indole ligand at δ 7.34, 7.21 and 7.14. Broad unresolved resonances in the range δ 1.20–2.89 were assigned to signals from the cyclohexyl groups and the methyl groups on the 3H-indole. The ES mass spectrum shows a peak consistent with the protonated molecular ion at m/z 1120.6332.
The ruthenium complex 17 was synthesised by the reaction of [RuCl(dppe)2]OTf with excess 2, in the presence of KOtBu (Scheme 3). This reaction is analogous to established methodology using NaPF6 and NEt3[44,58–60]; however, the base and halide abstracting reagent are combined as a single compound, KOtBu. Another ruthenium complex, 18, was synthesised by reaction of trans-[RuCl2(P(OEt)3)4] with 2, KPF6 and HNiPr2 in ethanol for 8 weeks (Scheme 3). The low yield is comparable with that of previous bis(alkynyl) complexes synthesised by this method.[61] Although lithium alkynyl species can be used to obtain this type of alkynyl complexes in low to moderate yields,[62] a simple extraction and crystallisation workup makes this method favourable.
The IR spectra of complexes of 17 and 18 showed absorptions assigned to ν(C≡C) bands at 2048 and 2088 cm−1 respectively. Singlets in the 31P NMR spectrum of both complexes support trans arrangement of the ligands and are comparable with similar complexes.[61,63] The 1H NMR spectrum of 17 shows a slight upfield shift of the assigned signals for the geminal methyl groups at 1.12 ppm and the imine methyl group at 2.03 ppm compared with free ligand 2. This upfield shiftof the methyl groups was also seen in complex 18, but not to the same extent. Resonances assigned to the ancillary phosphine ligands in 17 were observed between δ 6.94–7.76 and 2.56 (m, 8H, PPh2CH2CH2PPh2) in the 1H NMR spectrum. Resonances for the ethyl fragments on the phosphite groups for 18 were assigned to the quartet at δ 4.33 (CH2) and the triplet at δ 1.21 (CH3) (J 7.2 Hz) in the 1H NMR spectrum. The mass spectrum (APCI for 17, ES for 18) of both complexes showed fragmentation consistent with the loss of one alkynyl ligand. In addition, 17 also showed a peak consistent with the protonated molecular ion at m/z 1263.3789 in the mass spectrum.
Numerous attempts to synthesise the buta-1,3-diyne bridged putative molecular wire 19 were unsuccessful (Scheme 3). These compounds are candidates for applications in molecular electronics, supramolecular chemistry, optoelectronics and conjugated polymers.[64–66] Hay,[67] Eglinton and Galbraith,[68] Glaser[69] and Pd-type[70] coupling reactions failed to produce the homo-coupled product, returning starting material or unidentified decomposed species.
Electronic spectroscopy
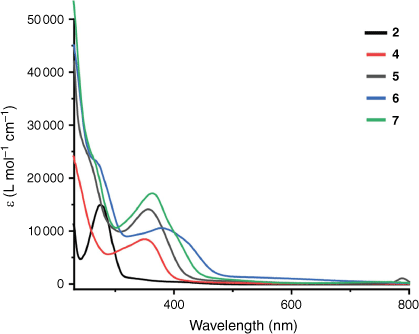
The UV-Vis spectra obtained for complexes 4–7 all displayed similar features (Fig. 1). All complexes show a metal-to-ligand charge transfer (MLCT) just below 400 nm[40] as well as a higher-energy transition below 300 nm. This higher-energy band is assigned to a π–π* (intraligand, IL) transition.
The UV-Vis spectra of 11 and complexes 12, 13, 17 and 18 are shown in Fig. 2. Compound 11 shows absorption maxima at 343 and 365 nm. The platinum complexes 12 and 13 show a high-energy transition at ~300 nm and a lower-energy transition between 350 and 370 nm. The lower-energy transition was assigned as an LMCT from the π(C≡CAr) to the mixed π(C≡CAr)* and metal p orbitals.[57,71,72] The ruthenium alkynyl complexes also exhibited a band of similar intensity between 350 and 370 nm, with a very intense band appearing below 300 nm for complex 17. The lower energy band for these and similar complexes was assigned as an MLCT and the high-energy transition as an IL charge transfer band.[73–76]
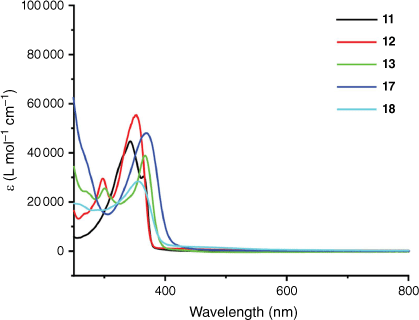
Molecular structures
The molecular structures of several compounds in this study were determined using single-crystal X-ray diffraction studies. Plots are shown in the figures below, and Tables 1 and 2 show selected bond lengths and angles.
| Compound | M–P1 | M–P2 | M–Cα | Cα–Cβ | Cβ–C5 | N1–C2 | C21–C22 | M–Cα–Cβ | Cα–Cβ–C5 | P–M–P | θ | θ′ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 1.198(4) | 1.439(4) | 1.289(3) | 178.0(3) | ||||||||
| 3 | 1.199(1) | 1.438(1) | 1.312(1) | 1.352(1) | 177.5(1) | 175.4(1) | ||||||
| 4 | 2.293(2) | 2.281(2) | 2.034(6) | 1.161(10) | 1.444(14) | 1.277(10) | 174.4(6) | 176.2(8) | 100.27(7) | 173.18 | ||
| 5 | 2.2578(7) | 2.2556(6) | 2.012(2) | 1.218(3) | 1.442(3) | 1.280(4) | 178.3(2) | 174.3(2) | 83.36(2) | 72.78 | ||
| 6 | 2.1795(6) | 2.1741(5) | 1.900(1) | 1.225(2) | 1.437(2) | 1.286(3) | 178.3(1) | 174.1(2) | 85.89(2) | 69.72 | ||
| 8 | 1.867A | 1.146(5)A | 2.033 | 1.182 | 1.461 | 1.278 | 175.8 | 179.3 | 91.9 | 0 | ||
| 9 | 2.2717(7) | 2.006(3) | 1.208(4) | 1.438(4) | 1.287(4) | 173.2(2) | 176.0(2) | 177.87(7)B |
ARu–C.
BP–Au–Cα. θ is the dihedral angle between the centroid of the Cp ring and the aryl plane for the alkynyl ligand for the half-sandwich complexes. θ′ is the torsion angle between the two indoline rings.
| Compound | Bond lengths (Å) and bond and torsion angles (°) | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M–P | M–Cα | Cα–Cβ | Cβ–C5′ | N1–C2′ | P–M–P (cis) | P–M–P (trans) | Cα–M–Cα | M–Cα Cβ | Cα Cβ–C5′ | C7A′–N1-C2′ | θ | θ′ | θ″ | |
| 11 | 1.428(3) | 1.209(3) | 1.437(3) | 1.301(3) | 178.3(3) | 176.2(3) | 106.3(2) | 35.0(3) | 180.0(2) | |||||
| 12 | 2.2934(3) | 1.9987(12) | 2.2185(17) | 1.4355(17) | 1.2917(19) | 180.0 | 180.0 | 178.81(12) | 176.49(14) | 106.51(11) | 53.1 | 180.0(1) | ||
| 13 | 2.3193(7) | 1.994(3 | 1.218(5) | 1.441(4) | 1.287(5) | 180.0(3) | 180.0 | 178.9(3) | 177.9(3) | 106.5(3) | 50.0 | 180.0(3) | ||
| 15 | 2.3138(5) | 2.003(2) | 1.214(3) | 1.443(3) | 178.708(17) | 177.51(8) | 172.92(19) | 174.7(2) | 61.7(2) | 39.4(3) | ||||
| 2.3232(5) | 178.02(19) | 172.6(2) | 58.8(2) | |||||||||||
| 16 | 2.3175(7) | 2.034(3) | 1.170(4) | 1.440(4) | 1.298(4) | 180.0 | 180.0 | 176.9(2) | 177.0(3) | 106.6(3) | 84.9 | 180.0(3) | ||
| 17 | 2.3539(12) | 2.092(5) | 1.186(7) | 1.434(7) | 1.282(8) | 82.37(4) | 180.00(6) | 180.0 | 176.0(4) | 175.8(5) | 106.3(5) | 77.2 | 180.0(5) | |
| 2.3592(12) | ||||||||||||||
| 18 | 2.3107(7) | 2.075(3) | 1.207(3) | 1.440(3) | 1.281(4) | 89.27(3) | 174.33(2) | 178.04(10) | 178.4(2) | 178.4(2) | 106.6(2) | 44.4(2) | 175.2(2) | |
| 2.3114(7) | 2.067(2) | 1.207(3) | 1.438(3) | 1.284(4) | 90.98(2) | 173.31(3) | 178.7(2) | 177.4(3) | 106.2(2) | 41.2(2) | ||||
| 2.3202(7) | 90.47(3) | |||||||||||||
| 2.3177(7) | 89.95(3) | |||||||||||||
θ is the torsion angle between the plane bisecting the dppe ligands and the plane of the arylethynyl ligand. θ′ is the lowest torsion angle between the C4′C5–Ru–P (any). θ″ is the angle between the planes of the indole rings.
The organic ligand 2 crystallised with two crystallographically distinct molecules in the unit cell (Fig. 3). The difference between the molecules lies in their clockwise or anticlockwise rotation along the C7–C7a–N1–C2 dihedral angle. However, given the near-planar arrangement of both molecules in the unit cell, this is unremarkable. This phenomenon was reported in a recent communication.[77] The ethene compound 3 shows elongation of the N1–C2 bond and shortening of the C2–C21 bond consistent with conjugation along the N1–C2–C21–C22–N2 backbone (Supplementary Fig. S6). Both indole rings are essentially coplanar and arranged in an antiperiplanar arrangement.
ORTEP representation of 2, and atom numbering scheme. Ellipsoids at the 50% probability level. Hydrogens removed for clarity.
Complex 4 crystallised with two CH2Cl2 solvate molecules (Fig. 4). One of the solvent molecules was modelled as disordered over one site. The additional solvate was hydrogen-bonded between C21–H21–Cl and CHCl2–H–N1 with respective distances of 3.68(1) and 3.54(2) Å between the donor and acceptor atoms.
ORTEP representations of (a) 4; (b) 5; (c) 6; (d) 8; and (e) 9, and atom numbering scheme. Ellipsoids at the 50% probability level. Hydrogens and solvates removed for clarity where appropriate.
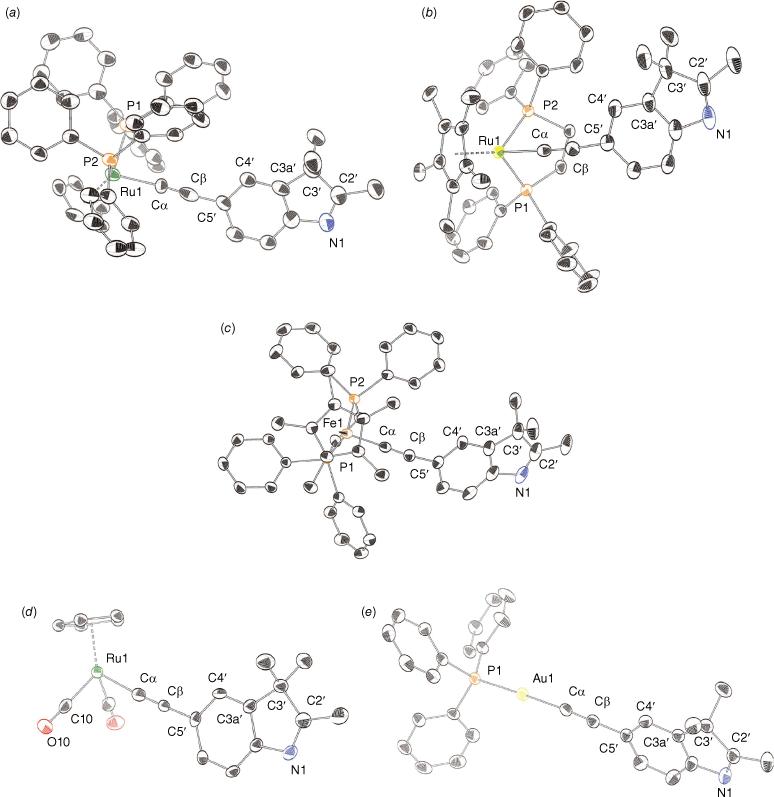
The bond lengths and angles around the metal core of the complexes are consistent with previously reported analogous alkynyl complexes.[20,39,41] Bond lengths along Ru–CC–C5 are not significantly different between complexes 4 and 5 (Fig. 4). A smaller Fe–C bond length in 6 (Fig. 4) can be ascribed to a smaller atomic radius.[17] The usual linear arrangement along the ruthenium–alkynyl backbone is observed, with a maximum deviation from 180° observed for 5 by −5.7°. The angles subtended by the phosphine atoms at the ruthenium atoms exemplify the pseudo-octahedral environment around the metal centres. The P–Ru–P angles are 100.27(7)° for the bis-triphenylphosphine and 83.36(2)° for the dppe. The shorter Ru–P bonds in 5 are consistent with the increased electron density at the metal centre, favouring back-bonding with the phosphine ligands. Bond lengths and angles for complex 8 (Fig. 4) are also unremarkable compared with similar complexes.
The Au–C and CC bond lengths and the bond angles about the P–A–CC chain for 9 (Fig. 4) are within the ranges for reported AuI alkynyl complexes.[34,42,78–81] A near-linear arrangement of alkynyl and phosphine ligands at the gold centre is observed. The slightly increased deviation from linearity at the Au–CC moiety has been associated with crystal packing forces.[79,81]
The relative orientation of the indole ring is influenced by the steric encumbrance of the ancillary ligands in the solid state. The torsion angle between the centroid of the cyclopentadienyl ligand, metal atom, and the C5 and C4 atoms is used to highlight the orientation of the indole ring (Supplementary Fig. S10). In complex 8, the indole ring lies perpendicular to the cyclopentadienyl ligand. In complex 4, introducing the bulky triphenylphosphine ligands causes the indole to rotate close to 180°. In complex 5, the indole ligand lies in the plane of the cyclopentadienyl ligand owing to the increased bulk of the permethylated Cp ligand and less sterically demanding dppe. Similar parallel conformations for functionalised ethynyl aryl rings in ruthenium σ-alkynyl complexes have been reported.[46] This is also reflected in the orientation of the indole ligand in complex 6.
Compound 11 (Fig. 5) mirrors the solid-state structure of the recently reported 1,4-bis(5-ethynyl-3,3-dimethyl-2,3-dihydrobenzo[b]thiophenyl)benzene.[60] The central phenyl ring possesses an inversion centre tilted by 35.0(3)° from the outer indole ring. Simpler phenylene-ethynylene structures seem to adopt a more coplanar orientation in the solid state.[60,82] Intermolecular hydrogen bonding between the N1 and imine methyl group dominates the crystal packing.
ORTEP representation of (a) 11; (b) 12; (c) 13; (d) 15; (e) 17; and (f) 18, and atom numbering scheme. Ellipsoids drawn at 50% probability. Hydrogens and solvate omitted for clarity where appropriate.
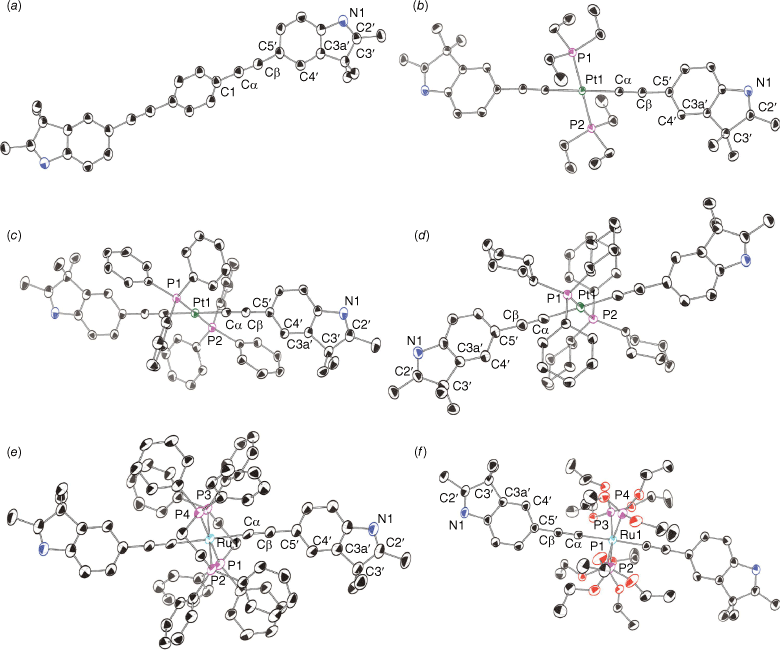
Solid-state structures showed the expected square planar geometry around the metal centre in the platinum complexes 12, 13 and 16 (Fig. 5) as well as 15 (Supplementary Fig. S7). All complexes except 15, which exhibits a slight distortion, sit on crystallographic inversion centres and have similar bond angles that deviate only slightly from idealised square planar geometry. Hydrogen bonding between N1 and methanol solvent dominates the crystal lattice of 16. The M–P bond length is shorter in 12 compared with the other Pt complexes in this study. In general, characteristic bonds lengths and angles along the metal–alkyne fragment were unremarkable and were within the range reported in the literature.[83–88]
Complex 17 possesses a centre of inversion about the ruthenium centre (Fig. 5). The plane of the arylethynyl rings of the indole lies between the dppe bidentate ligands. The angle between the plane of the arylethynyl ligand and the plane bisecting the dppe ligand, θ, as described recently,[63] is 76.8°. In this orientation, there is a better overlap of the ligand π-system with the metal centre. All other interatomic parameters are not significantly different from previously described trans-Ru bis(alkynyl) complexes.
Complex 18 shows the rod-like structure and pseudo-octahedral metal environment consistent with tetrakis(triethylphosphite) complexes (Fig. 5). Moreover, the bond distances and angles are in the range previously described for these complexes.[61] Interestingly, the angle between planes of the arylethynyl rings shows only a slight deviation from a coplanar orientation. Most complexes of this type adopt a twisted conformation between the aromatic groups.[60,61] The M–P bond lengths are shorter in 18 than in 17 as a result of Ru–P π back-bonding.
Bonds around the metal centres show slight variations. M–P bond lengths are longer in 17 than the Pt complexes, comparable with the bond lengths in 18. There are slight geometric differences about the respective metal centres; however, these are less remarkable moving away from the metal. Bond lengths and angles in the indole rings show no significant differences between the metal complexes. All indole rings of the arylethynyl complexes lie in the same plane, allowing delocalisation along the metal–alkyne backbone.
Electrochemistry
The electrochemical properties of the indole complexes were studied to compare the redox potentials as a function of the metal–ligand fragment (Table 3). The cyclic voltammograms of all complexes show single reversible oxidations at potentials between −640 and −60 mV (v. Fc/Fc+) and additional irreversible oxidations between 580 and 780 mV (v. Fc/Fc+) . The oxidation potentials are consistent with the indoline moiety being electron-donating.[39] The Cp* ligand makes the oxidation potential 200–300 mV more cathodic in both systems. The iron complexes undergo the first oxidation at potentials 300–400 mV more cathodic than their isostructural ruthenium complex. The second oxidation, presumably at the nitrogen of the indoline, is more dependent on the metal in the ruthenium complexes than in the iron complexes. This behaviour is predictable given the larger degree of delocalisation of electron density over the ruthenium acetylide.
| Compound | E1/2 (V) | ΔEp (V) | ipc/ipa | Epa (V) |
|---|---|---|---|---|
| 4 | −0.06 | 0.09 | 0.92 | 0.74 |
| 5 | −0.30 | 0.10 | 0.93 | 0.58 |
| 6 | −0.64 | 0.10 | 1.03 | 0.77 |
| 7 | −0.44 | 0.09 | 0.97 | 0.77 |
| 17 | −0.12 | 0.09 | 1.01 | 0.71 |
All experiments are in CH2Cl2 solutions containing 0.1 M Bu4NPF6 supporting electrolyte using a Pt working electrode and Pt/Ti counter and pseudo-reference electrode. The decamethylferrocene/decamethylferricenium (Fe(Cp*)2/[Fe(Cp*)2]+) couple was used as an internal reference, with all potentials reported relative to the ferrocene/ferricenium couple (FeCp2/[FeCp2]+ = 0 V, such that Fe(Cp*)2/[Fe(Cp*)2]+ = −0.55 V). Scan rate of 100 mV s−1 at room temperature. No correction for IR compensation was used. ipc/ipa is the ratio of cathodic and anodic current. Epa is anodic potential peak.
Single-molecule conductance studies
Normalised conductance histograms (on the left) and density plots (on the right) are shown in Fig. 6 and 7 for compounds 11 and 12 respectively. The conductance histograms show clear quantum of conductance G0 ((G0 = 2e2/h = 77.5 μS) (e, elementary charge; h, Planck constant)) peaks at log(G/G0) = 0, which correspond to the single channel Au–Au metal atom point contact. The 2-D density plots display log(conductance) v. electrode separation during the scanning tunnelling microscopy (STM) tip retraction from the metal point contact to 2.0 nm extension. As seen from these plots, there is little evidence of extended molecular junction formation and, hence, it is not possible to attribute a conductance peak to these compounds. Possible reasons for the absence of an apparent junction formation for these compounds could be a low junction formation probability or conductance values falling below the noise floor of ~10−5.5 G0. A low junction probability could result from steric hindrance from the vicinal methyl group to the proposed nitrogen dative binding site of the specific indole terminal groups employed here. Low binding strength of amine contacting groups has been attributed to the steric influence of hydrogens.[89] However, if a junction is forming, then the conductance itself could be lower than the noise floor (10−5.5 G0), which would mean that it would lie outside the measurement window of the conductance determination. The molecular backbone is chemically similar to previous studied compounds (and their conductance): trans-Pt(CC-CC6H4-CC-SiMe3)2(PEt3)2 (3.2 ± 1.3 × 10−5 G0),[56] trans-Pt(CC-cyclo-3-C4H3S)2(PEt3)2 (2.70 ± 0.66 × 10−4 G0) and 1,4-C6H4(CC-cyclo-3-C4H3S)2 (2.83 ± 0.65 × 10−4 G0),[90] all of which displayed molecular conductance values greater than 10−5 G0 (i.e. greater than the noise floor of our present measurements). Additionally, oligophenylethylenes (OPEs) with a similar backbone structure to compound 12 show conductance values of 2.6 ± 1.6 × 10−5 to 3.6 ± 1.6 × 10−5 G0.[91] Therefore, it seems rational to assume the primary cause for no apparent molecular junction formation is inherent to the new contact group. This lack of junction formation may be related to insufficient dative bonding between the N of the indole terminal groups and the gold electrodes or may arise from steric hindrance of this bonding as a result of the vicinal methyl group.
Normalised 1-D conductance histogram (left) and 2-D conductance-displacement density plot (right) for 11 using STM-BJ at 100 mV bias. More than 3000 individual breaking traces were compiled with no data selection.
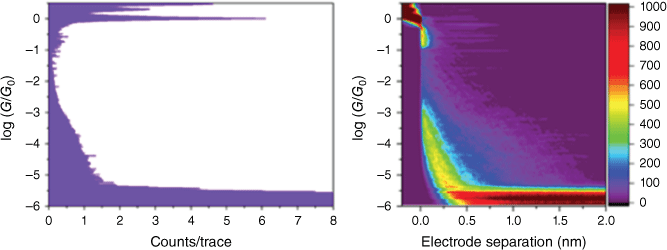
Normalised 1-D conductance histogram (left) and 2-D conductance-displacement density plot (right) for 12 using scanning tunnelling microscopy break junction (STM-BJ) at 100 mV bias. More than 3000 individual breaking traces were compiled with no data selection.
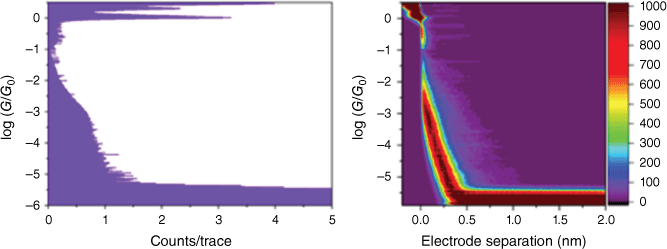
Steric hindrance can impede potential anchoring groups binding to gold contacts; for example, previous studies have noted that the high conductance hollow geometric formation seen in bipyridyl ethyne is inhibited by introducing vicinal C–F bonds.[92] It is instructive to consider how junctions might form in the STM-BJ experiment for compounds 11 and 12. Analogous pyridyl anchoring groups (e.g. in 4,4′ bipyridine molecular bridges with gold contacts[93]) evolve from a face-on alignment of the pyridyl ring to an end-on dative binding of the terminal N atom to a Au apex atom. As an anchor group slides across the pyramidal tip through a side configuration, it can jump to an apex position to form the final molecular junction.[94] If a similar mechanism is assumed here for our indole contact group, this last jump could be impeded by the vicinal methyl group, leading to a complete rupture. Another possible issue might be the chemical stability of the molecules under the experimental conditions needed to form Au–molecule–Au junctions. A pyrrolic-type indole-terminated diaryltetrayne has been shown not to exhibit junction formation owing to the chemical and thermal instability of the moiety under the employed experimental conditions.[95] However, given the ambient stability of the compounds synthesised in the present study, we believe that that degradation during single-molecule conductance experiments is less likely to cause absence of junction formation features. The last reason we postulate for the absence of junctions for compounds 11 and 12 is the slight increase in basicity of the indole contact group (pKa of the conjugated acid 6.33) compared with the pyridine contact group (pKa of the conjugated acid 5.25). Hard–soft acid–base (HSAB) theory has been shown to predict the formation of metal–molecule junctions with lone pairs on the anchoring group (e.g. pyridyl, amine) and gold electrodes.[96] Although pyridine is of an ‘intermediate’ character on the hard–soft continuum, it seems possible that the increase in basicity of the 3H-indole reduces its affinity for the gold electrodes if there is a less favourable HSAB match.
Spectroelectrochemistry
Spectroelectrochemical methods have become useful tools to determine the nature of redox processes and redox products for metal alkynyl complexes.[97] The complexes 4, 7 and 17 were chosen for spectroelectrochemical investigations to act as models across the series of compounds in this study owing to the ease of comparison with other similar species studied in the literature.
The infrared spectroelectrochemical spectra of complexes 4 and 7 during the first oxidation are illustrated in Fig. 8. The IR spectra of the neutral complex 4 shows a single absorption at 2065 cm−1 assigned to the ν(Ru–C≡C) band. On oxidation, the band decreases in intensity. A small IR absorption for the ν(Ru–C≡C) band was observed for the oxidised complex [4]+ at 1920 cm−1 (Fig. 8a).[39] Complex [4]+ was unstable on the timescale of spectroelectrochemical experiments, with decay of the band assigned to the ν(Ru–C≡C) at 1920 cm−1 over a short period of time. However, we can confidently assign this band to ν(Ru–C≡C) as reduction exchanges this band for the ~2060 cm−1 of complex 4 (Fig. 8b). We note also the possible formation of the [Ru(Cp)(PPh3)2(CO)]+ cation on the appearance of the band at ~1970 cm−1 (Fig. 8b). For the iron complex 7, two active ν(Fe–CC) modes were observed on oxidation. A shift of Δ80 cm−1 in the ν(Fe–CC) absorption band is in agreement with iron complexes of the type Fe(CCR)(dppe)Cp, and is consistent with a large degree of metal character in the first oxidation process (Fig. 8c).[39] The appearance of two broad active ν(Fe–C≡C) modes can be rationalised as a population of local states on the potential energy surface, where several accessible conformers differ by the orientation of the arylethynyl ligand with respect to the metal orbitals.[98]
IR spectra of complexes (a, b) [4]n+ (n = 0, 1); (c) [7]n+ (n = 0, 1); and (d) [17]n+ (n = 0, 1) recorded in a spectroelectrochemical cell (CH2Cl2/0.1M Bu4NPF6).
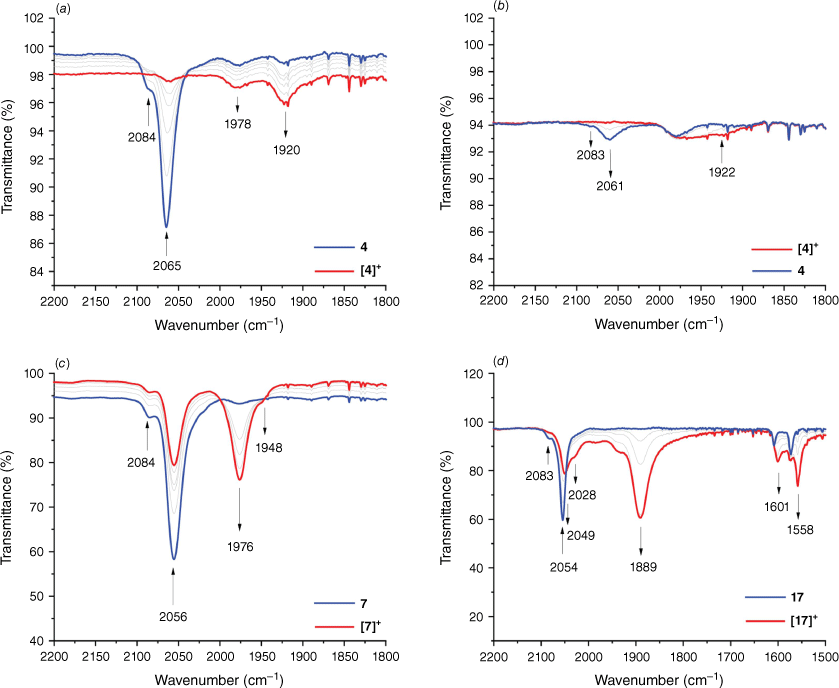
The IR spectra on oxidation of 17 to [17]+ is illustrated in Fig. 8d. Complex 17 is characterised by an absorption at 2054 cm−1 assigned to the ν(Ru–C≡C) stretch. On oxidation to the monocations, [17]+, the ν(Ru–C≡C) assigned absorption band shifts by 165–1889 cm−1. This large shift is indicative of the large contribution of the alkynyl ligand to the oxidation process. The broad and asymmetric nature of the ν(Ru–C≡C) envelope in [17]+ is consistent with a range of conformers that vary as a result of the relative orientation the arylethynyl ligands with respect to the metal centre.[63] A weak feature between 2049 and 2028 cm−1 is consistent with additional conformers with electronic structures localised over one arylethynyl ligand.[60] The additional absorptions at 1601 and 1558 cm−1 are likely assignable to aryl breathing modes, further consistent with the large contribution of the ethynyl ligand in the oxidation process.
Conclusion
The synthesis of new 3H-indole-functionalised σ-alkynyl complexes was achieved. These new ligands protonate the imine nitrogen atom and can form multicomplex architectures with copper. The complexes show reversible electrochemical behaviour that is localised over the metal–alkynyl component. The results suggest a broad scope of future work can be explored for these ligands in polymetallic complexes and frameworks and moderation of opto-electrical properties from such complexes.
Experimental
General
All reactions were carried out under high-purity argon or nitrogen using Schlenk techniques and oven-dried glassware. All solvents for reactions were dried by distillation under argon over an appropriate drying agent or by an Innovative Technologies solvent purification system (SPS) and deoxygenated (freeze/pump/thaw or sparging) before use. Hexanes and EtOAc for column chromatography were distilled before use. All other solvents were of AR grade and used without further purification. Unless otherwise stated, no special precautions were taken to exclude air or moisture during workup and purification. All other reagents were used as received from their supplier. Flash column chromatography was carried out using silica gel 60 (0.04–0.063 mm) or alumina (Brockmann activity IV, basic). Thin-layer chromatography was carried out using Merck silica gel 60 F254 pre-coated aluminium sheets.
Instruments
1H, 13C{1H}, 31P{1H} and 19F NMR spectra were recorded at 25°C on a Bruker 600, 500 or 400 MHz spectrometer. Chemical shifts (δ) are referenced to the internal undeuterated residual solvent signal (1H), deuterated solvent signal (13C) or external standard (31P, 85% H3PO4). First-order multiplets are as follows: s, singlet; d, doublet; t, triplet; q, quartet. Apparent multipets are labelled with the prefix ‘app’. Broad signals are labelled with the prefix ‘br’. Multiplets of higher order or that were undeterminable are labelled as m. Coupling constants (J) are reported in hertz (Hz). Signal assignments were made with help from HSQC (heteronuclear single quantum coherence), HMBC (heteronuclear multiple bond correlation) and 1H COSY (correlation spectroscopy) experiments, spectrum prediction software and literature precedent. Infrared spectroscopy was carried out with neat products using an ATR module-fitted FTIR spectrometer. ESI or APCI mass spectrometry was carried out on a Waters LCT Premier time-of-flight (TOF) spectrometer. Electron-impact ionisation was carried out using a Waters GCT Premier or Shimadzu GCMS QP2010. In most cases, HPLC grade acetonitrile was used as the solvent. If needed, a couple of drops of dichloromethane were used to help solubilise the compound for analysis. Melting points were collected on a Buchi M-565. Elemental analyses were performed by Stephen Boyer, London Metropolitan University, London, United Kingdom.
Synthesis
Compounds 1, [RuCl(dppe)2]OTf,[44] Ru(dppe)(Cp*)Cl,[99] CpRu(PPh3)2(Cp)Cl,[100] Fe(dppe)(Cp*)Cl,[99] Fe(dppe)(Cp)Cl,[99] PPh3AuCl[101] and Ru(CO)2(Cp)Cl[102] were synthesised according to literature procedures.
In a round-bottom flask, 2,3,3-trimethyl-5-trimethysilyl-alkynyl-3H-indole (3.01 g, 0.012 mol) (1) was dissolved in THF (20 mL). This solution was sparge-deoxygenated with argon for 20 min. Simultaneously, KOH (750 mg, 0.013 mol) was dissolved in MeOH (20 mL) and sparged with argon. The THF solution was transferred by cannula into the methanolic solution with stirring. The solution was stirred for 3 h. The solvent was removed to dryness under reduced pressure. The residue was dissolved in Et2O (50 mL) and washed with water (3 × 50 mL). Additionally, the aqueous solution was back-extracted with Et2O (50 mL). The combined organic phases were washed with brine (1 × 50 mL), dried over MgSO4, and evaporated to dryness. The yellow oily residue was subjected to chromatography (silica; 30% EtOAc/hexanes) to give yellow oil that solidified on standing (1.90 g, 86%). Crystals for X-ray diffraction (XRD) were grown by slow evaporation of an Et2O solution; mp: 50–51°C. 1H NMR (CDCl3, 500 MHz): δ 7.40–7.47 (m, 3H), 3.07 (s, 1H, CC–H), 2.28 (s, 3H, C2-CH3), 1.29 (s, 6H, gem-CH3). 13C{1H} NMR (CDCl3, 125 MHz): δ 189.7 (s, C2), 154.3 (s, C7a), 145.9 (s, C3a), 132.3 (s, C6), 125.3 (s, C4), 120.0 (s, C7), 118.7 (s, C5), 84.3 (s, Cβ), 77.0 (s, Cα), 53.9 (s, C3), 23.1 (s, C2-CH3), 15.6 (s, gem-CH3). IR (ATR, neat) νmax/cm−1: 3176 (C–H), 2958, 2972, 2926, 2099 (C≡C), 1571 (C=N), 1458. HRMS (APCI(+), MeCN) m/z: calcd for C13H14N+ 184.1121 [M + H]+, found 184.1118. Anal. calcd for C13H13N: C, 85.21; H, 7.15; N, 7.64. Found: C 85.15; H, 7.03; N 7.57%.
Isolated as a yellow powder. Crystals were grown by slow evaporation of an Et2O solution. 1H NMR (CDCl3, 600 MHz): δ 7.61–7.63 (m, 4H, H4 & H7), 7.52 (dd, J = 1.2, 7.8 Hz, 2H, H6), 7.46 (br, s, 2H, H8), 3.15 (s, 2H, C≡C–H), 1.48 (s, 12H, gem-CH3). 13C{1H} NMR (CDCl3, 126 MHz): δ 183.8 (s, C2), 154.2 (C7a), 147.1 (C3a), 132.7 (C6), 128.5 (C8), 125.3 (C4), 121.4 (C7), 120.3 (C5), 84.1 (Cβ), 78.0 (Cα), 53.2 (C3), 23.3 (gem-CH3). IR (ATR, neat) νmax/cm−1: 3302 (H–C≡), 3243 (H–C≡), 2104 (C≡C), 1607 (C=C), 1508 (C=N), 1454. HRMS (APCI(+), MeCN) m/z: calcd for C26H23N2+ 363.1856 [M + H]+, found 363.1851.
A solution of Ru(PPh3)2(Cp)Cl (199 mg, 0.274 mmol), 2 (100 mg, 0.546 mmol) and NH4PF6 (219 mg, 1.34 mmol) was refluxed in MeOH (20 mL) for 1 h, giving a dark red solution. The solution was cooled to ambient temperature and stirring ceased. A slight excess of DBU (0.15 mL) was slowly added dropwise and the solution was left undisturbed overnight and then cooled in an ice bath. The yellow crystals were collected by filtration and washed with cold MeOH and cold Et2O, dried in air, and then dried under vacuum (165 mg, 69%). Crystals for XRD were grown by diffusion on pentane in a concentrated CH2Cl2 solution. 1H NMR (C6D6, 400 MHz): δ 7.80 (d, J = 8.0 Hz, 1H, H7), 7.73–7.78 (m, 12H, Hmeta), 7.60 (dd, J = 1.6, 7.9 Hz, 1H, H6), 7.42 (d, J = 1.5 Hz, 1H, H4), 6.92–6.99 (m, 18H, Hpara and Hortho), 4.51 (s, 5H, Cp), 2.00 (s, 3H, NCCH3), 1.01 (s, 6H, gem-CH3). 13C{1H} NMR (C6D2, 100 MHz): δ 184.6 (C2), 151.5 (C7a), 146.0 (C3a), 139.3 (m, Cipso), 134.4 (t, J = 5 Hz, Cmeta), 130.4 (C6), 128.7 (Cpara), 128.1 (C5), 127.6 (t, J = 5 Hz, C), 124.3 (C4), 120.2 (C7), 116.1 (Cβ), 113.8 (t, J = 26 Hz, Cα) 85.7 (Cp), 53.2 (C3), 23.2 (gem-CH3), 15.1 (NCCH3). 31P{1H} NMR (CD2Cl2, 202 MHz): δ 50.6 (s, dppe). IR (ATR, neat) νmax/cm−1: 2060 (C≡C). HRMS (ES(+), MeCN) m/z: calcd for C54H49NP2Ru2+ [M + 2H]2+ 437.6187, found 437.6174; calcd for C43H38NP2Ru+ [M – L + MeCN]+ 732.1518, found 732.1575; calcd for C54H48NP2Ru+ [M + H]+ 874.2300, found 874.2360.
A solution of Ru(dppe)(Cp*)Cl (103 mg, 0.154 mmol), 2 (89 mg, 0.486 mmol) and NH4PF6 (150 mg, 0.920 mmol) was refluxed in MeOH (10 mL) for 3 h, giving a dark red solution. The solution was cooled to room temperature and stirring was ceased. An excess of DBU (0.15 mL) was slowly added dropwise and the solution was left undisturbed overnight and then cooled in an ice bath. The yellow needles were collected by filtration and washed with cold MeOH and cold pentane, dried in air, and then dried under high vacuum (101 mg, 80%). Crystals for XRD were grown by cooling an Et2O/pentane solution. 1H NMR (C6D6, 600 MHz): δ 7.92–7.95 (m, 4H, Hpara), 7.67 (d, J = 7.8 Hz, 1H, H7), 7.07–7.28 (m, 17H, Hortho, Hmeta and H6), 6.95 (d, J = 2 Hz, 1H, H4), 2.60–2.70 (m, 2H, dppe-CH2), 1.96 (s, 3H, NCCH3), 1.84–1.88 (m, 2H, dppe-CH2), 1.68 (s, 15H, Cp*), 0.96 (s, 6H, gem-CH3). 13C{1H} NMR (C6D6, 100 MHz): δ 184.3 (C2), 151.2 (C7a), 145.8 (C3a), 139.8 (m, Cipso), 137.8, 137.5, 134.3 (t, J = 5 Hz, Cortho), 133.7 (t, J = 5 Hz, Cortho), 130.3 (C6), 129.2 (PPh2) 129.0 (PPh2), 128.8 (PPh2), 128.7 (PPh2), 128.2 (PPh2), 128.0 (PPh2), 127.7 (PPh2) 127.6 (PPh2), 127.4 (PPh2), 127.1 (t, J = 25 Hz, Cα) 123.8 (C4), 120.0 (C7), 111.4 (Cβ), 92.8 (t, J = 2 Hz, Cp), 53.1 (C3), 29.9 (m, dppe-CH2), 23.2 (NCCH3), 15.1 (s, gem-CH3), 10.4 (s, Cp(CH3)5). 31P{1H} NMR (C6D6, 243 MHz): δ 81.6 (s, dppe). IR (ATR, neat) νmax/cm−1: 2058 (C≡C). HRMS (ES(+), MeCN) m/z: calcd for C49H53NP2Ru2+ [M + 2H]2+ 409.6343, found 409.6335; calcd for C37H39OP2Ru+ [M – L + CO]+ 663.1515, found 663.1531; calcd for C38H42NP2Ru+ [M – L + MeCN]+ 676.1831, found 676.1961; calcd for C49H52NP2Ru+ [M + H]+ 818.2613, found 818.2619.
A yellow solution of Fe(dppe)(Cp*)Cl (193 mg, 0.309 mmol), 2 (81 mg, 0.44 mmol) and NH4PF6 (100 mg, 0.613 mmol) was refluxed in MeOH (20 mL) for 2 h, giving a dark solution. The solution was cooled to room temperature and stirring was stopped. DBU (0.15 mL, excess) was slowly added dropwise and the solution was left undisturbed overnight and then cooled in an ice bath. The dark red needles were collected by filtration and washed with cold MeOH and cold pentane, dried in air, and then dried under vacuum (202 mg, 85%). Crystals for XRD were grown by diffusion of pentane into a concentrated CH2Cl2 solution. 1H NMR (C6D6, 600 MHz): δ 8.04–8.07 (m, 4H, Hpara), 7.70 (d, J = 7.9 Hz, 1H, H6), 7.01–7.34 (m, 18H, H7, H4, Hortho and Hmeta), 2.61–2.70 (m, 2H, dppe-CH2), 1.97 (s, 3H, NCCH3), 1.81–1.87 (m, 2H, dppe-CH2), 1.56 (s, 15H, Cp*), 0.97 (s, 6H, gem-CH3). 13C{1H} NMR (C6D6, 151 MHz): δ 184.4 (C2), 151.1 (C7a), 146.0 (C3a), 139.8 (PPh2), 138.3 (C5), 138.0 (PPh2), 134.8 (PPh2), 134.4 (PPh2), 130.1 (C6), 129.2 (PPh2), 129.0 (PPh2), 128.3 (PPh2), 128.1 (PPh2), 128.0 (PPh2), 127.5 (PPh2), 123.5 (C4), 121.1 (1H-13C{1H} HMBC, Cβ), 120.2 (C7), 87.8 (Cp(CH3)5), 53.2 (C3), 31.2 (m, dppe-CH2), 23.3 (s, NCCH3) 15.1 (gem-CH3), 10.48 (Cp(CH3)5), (Cα) not observed. 31P{1H} NMR (C6D6, 243 MHz): δ 100.87 (s, dppe). IR (ATR, neat) νmax/cm−1: 2045 (C≡C). HRMS (ES(+), MeCN) m/z: calcd for C49H52FeNP22+ [M + H]2+, found 386.142; calcd for C49H51FeNP+ [M]+ 771.2835, found 771.2824 [M]+.
A yellow solution of Fe(dppe)(Cp)Cl (196 mg, 0.353 mmol), 2 (86 mg, 0.47 mmol) and NH4PF6 (91 mg, 0.56 mmol) was refluxed in MeOH (15 mL) for 2 h, giving a dark brown solution. The solution was cooled to room temperature and stirring was ceased. A slight excess of DBU (0.15 mL) was slowly added dropwise and the solution was left undisturbed overnight and then cooled in an ice bath. The dark red needles were collected by filtration and washed with cold MeOH and cold pentane, dried in air, and then dried under vacuum (179 mg, 72%). Crystals for XRD were grown by diffusion of pentane into a concentrated CH2Cl2 solution. 1H NMR (C6D6, 600 MHz): δ 8.02 (br. s, 4H, Hpara), 7.55 (d, J = 7.9 Hz, 1H, H7), 7.20–7.27 (m, 10H, Hortho and Hmeta), 6.98–7.04 (m, 6H, Hortho and Hmeta), 6.92 (d, J = 7.9 Hz, 1H, H6), 6.70 (s, 1H, H4), 4.32 (s, 5H, Cp), 2.49–2.58 (m, 2H, dppe-CH2), 1.90–1.98 (m, 5H, NCCH3 and dppe-CH2), 0.92 (s, 6H, gem-CH3). 13C{1H} NMR (C6D6, 100 MHz): δ 184.4 (C2), 151.2 (C7a), 145.6 (C3a), 143.0 (m, PPh2), 138.9 (m, PPh2), 134.2 (t, PPh2), 132.2 (t, PPh2), 129.9 (C6), 129.3 (PPh2), 128.8 (PPh2), 128.7 (PPh2), 128.3 (PPh2), 128.2 (PPh2), 128.0 (PPh2), 124.1 (C4), 122.7 (Cβ), 122.3 (t, J = 42 Hz, Cα), 119.8 (C7), 79.5 (Cp), 53.0 (C3), 28.7 (m, dppe-CH2), 23.2 (s, NCCH3), 15.1 (s, gem-CH3), C5 obscured. 31P{1H} NMR (C6D6, 243 MHz): δ 106.94 (s, dppe). IR (ATR, neat) νmax/cm−1: 2055 (C≡C). HRMS (ES(+), MeCN) m/z: calcd for C44H42FeNP22+ [M + H]2+ 351.1063, found 351.1048; calc for C46H45FeN2P22+ [M + H + MeCN]2+ 371.6196, found 371.6192; calcd for C33H32FeNP2+ [M – L + MeCN]+ 560.1349, found 560.1346; calcd for C44H41FeNP2+ [M]+ 701.2053, found 701.2095 [M]+.
A Schlenk flask was charged with Ru(CO)2(Cp)Cl (102 mg, 0.393 mmol), 2 (79 mg, 0.431 mmol) and CuI (5 mg) and the solids were suspended in NEt3 (10 mL) and THF (10 mL). The light yellow solution was stirred for 24 h covered in foil. The solution was then evaporated under vacuum, and the residue was extracted with Et2O and filtered through cotton wool until the washings were colourless. The Et2O was removed by rotary evaporation and the orange residue was passed through a basic alumina column using CH2Cl2/CH3OH (99/1). The yellow band was collected and the solvent removed to give a yellow powder (111 mg, 70%). Crystals for XRD were obtained by slow cooling of an Et2O/pentane solution to yield yellow plates. 1H NMR (CDCl3, 600 MHz): δ 7.35 (d, J = 7.8Hz, 1H, H7), 7.28–7.29 (m, 2H, H4 and H6), 5.48 (s, 5H, Cp), 2.24 (s, 3H, NCCH3), 1.26 (s, 6H, gem-CH3). 13C{1H} NMR (CDCl3, 151 MHz): δ 196.9 (CO), 187.7 (C2), 151.6 (C7a), 145.4 (C3a), 131.3 (C6), 124.9 (C7), 124.2 (C5), 119.4 (C4), 111.0 (Cβ), 88.1 (Cp), 81.3 (Cα), 53.6 (C3), 23.3 (NCCH3), 15.51 (s, gem-CH3). IR (ATR, neat) νmax/cm−1: 2119 (C≡C), 2035 (C=O), and 1994 (C=O). HRMS (ES(+), MeCN) m/z: calcd for C20H18NO2Ru+ [M + H]+ 406.0376, found 406.0388. Anal. calcd for C20H24NO2Ru: C, 59.40; H, 4.24; N, 3.46. Found: C 59.11; H, 4.57; N 3.52%.
An argon-filled Schlenk flask was charged with (PPh3)AuCl (49 mg, 0.099 mmol), 2 (23 mg, 0.12 mmol) and NaOMe (8 mg, 0.1 mmol) and the mixture stirred in MeOH (20 mL) in the dark for 3 h. The reaction was concentrated and cooled in a freezer. The yellow powder formed was collected by filtration and washed with cold MeOH and Et2O (27 mg, 42%). Crystals for XRD were grown by slow evaporation of a THF/EtOH/heptane solution to yield yellow plates. 1H NMR (C6D6, 600 MHz): δ 7.86 (dd, J = 8.0, 1.3 Hz, 1H, H6), 7.72 (d, J = 1.3 Hz, 1H, H4), 7.61 (d, J = 8.0 Hz, 1H, H7), 7.25–7.29 (m, 3H, Hpara), 6.94–6.97 (m, 6H, Hmeta), 6.87–6.90 (m, 6H, Hortho), 1.88 (s, 3H, NCCH3), 0.82 (s, 6H, gem-CH3). 13C{1H} NMR (C6D6, 151 MHz): δ 186.6 (C2), 153.7 (C7a), 146.2 (C3a), 135.8 (d, J = 146 Hz, Cα), 134.5 (d, J = 14 Hz, Cortho), 132.6 (C6), 131.2 (d, J = 1.6 Hz, Cpara), 130.6 (d, J = 55 Hz, Cipso), 129.2 (d, J = 11 Hz, Cmeta), 125.8 (C4), 123.8 (C5), 120.4 (C7), 104.1 (d, J = 26 Hz, Cβ), 53.4 (C3), 22.8 (s, NCCH3), 15.0 (s, gem-CH3). 31P{1H} NMR (C6D6, 243 MHz): δ 42.2 (s, PPh3). IR (ATR, neat) νmax/cm−1: 1574, 1480, 1456, 1434. HRMS (ES(+), MeCN) m/z: calcd for C20H18AuNP+ [M – L + MeCN]+ 500.0838, found 500.0844;calcd for C31H28AuNP+ [M + H]+ 642.1620, found 642.1627; calcd for C36H30AuP2+ [Au(PPh3)2]+ 721.1483, found 721.1501; calcd for C49H42Au2NP2+ [M + (M – L)]+ 1100.2119, found 1100.2175.
A Schlenk tube charged with [Ru(dppe)2Cl]OTf (99 mg, 0.0915 mmol) and 2 (18 mg, 0.098 mmol) was placed under high vacuum for 30 min and refilled with argon. CH2Cl2 (5 mL) was then added and the solution was stirred overnight. Evaporation of the solvent yielded a yellow–red residue that was washed with diethyl ether. The residue was dissolved in a minimum amount of dry dichloromethane and dry diethyl ether was then added to form a biphasic system, and the vessel was placed in the freezer. A green–yellow solid was decanted from the mother liquors and washed with diethyl ether and pentane, and dried under high vacuum (91 mg). Further recrystallisation attempts could not eliminate side-products. 1H NMR (CD2Cl2, 500 MHz): δ 6.91–7.47 (m, 41H), 6.70 (s 1H), 6.25 (s, 1H), 2.69 (br app s, 11H, dppe and NCH3), 1.45 (s, 6H, gem-CH3). 13C{1H} NMR (CD2Cl2, 126 MHz): δ 136.6, 135.0, 134.5, 134.4, 131.8, 129.6, 129.2, 128.5, 127.6, 127.5, 30.8, 23.3, 15.70. 31P{1H} NMR (CD2Cl2, 202 MHz): δ 48.3 (s, 10a), 41.7 (s, unknown), 37.3 (s, 10b) (unreferenced). 19F{1H} NMR (CD2Cl2, 470 MHz): δ −79.0 (s, OTf) (unreferenced). HRMS (ES(+), MeCN) m/z: calcd for C67H64N2P4Ru2+ [M – L + MeCN + H]2+ 561.1526, found 561.1526; calcd for C69H67N3P4Ru2+ [M – L + 2MeCN + H]2+ 581.6659, found 581.6659; calcd for C67H63N2P4Ru+ [M – L + MeCN]+ 1121.2985, found 1121.3201.
Into an oven-dried and argon-filled reflux apparatus was placed anhydrous NEt3 (20 mL), which was sparged with argon for 1 h. To the degassed NEt3 were added 1,4-diiodobenzene (158 mg, 0.479 mmol), 2 (202 mg, 1.10 mmol), CuI (10 mg) and PdCl2(PPh3)2 (18 mg). The lemon-yellow solution was heated to reflux and stirred overnight. After cooling to ambient temperature, the solution was filtered and evaporated to dryness uncer vacuum. The orange residue was dissolved in EtOAc (40 mL) and washed with water (3 × 20 mL) and brine (20 mL). The organic layer was dried over MgSO4, filtered and evaporated to dryness. Flash silica chromatography using 1:1 EtOAc/hexanes yielded a yellow–orange powder (201 mg, 95%). Crystals were grown by diffusion of cyclohexane into a concentrated CHCl3 solution. 1H NMR (CDCl3, 500 MHz): δ 7.46–7.50 (m, 5H, Ar), 2.30 (s, 3H, CH3), 1.32 (s, 6H, gem-CH3). 13C{1H} NMR (CDCl3, 125 MHz): δ 189.6, 154.1, 146.0, 131.8, 131.6, 124.8, 123.2, 120.1, 119.7, 91.9, 89.1, 53.9, 23.4, 15.7. IR (ATR, neat) νmax/cm−1: 2964, 2203 (C≡C), 1736, 1568, 1510, 1461. HRMS (AP(+), MeCN) m/z: calcd for C32H29N2+ [M + H]+ 441.2331, found 441.2338.
An oven-dried and argon-filled reflux apparatus was charged with Pt(PEt3)2Cl2 (100 mg, 0.199 mmol), 2 (81 mg, 0.442 mmol) and CuI (4 mg, 0.02 mmol). The apparatus was evacuated for 30 min, then filled with argon and NEt3 (5 mL) was added. The lemon-yellow solution was heated to 70°C and stirred for 19 h. After cooling to ambient temperature, the solution was filtered and evaporated to dryness under vacuum. The yellow–orange residue was then subjected to flash chromatography (basic alumina) using 1:1 EtOAc/hexanes, yielding an orange powder (83 mg, 53%). Crystals were grown by slow evaporation of a concentrated diethyl ether/pentane solution. 1H NMR (CDCl3, 500 MHz): δ 7.15–7.36 (m, 3H, Ar), 2.23 (s, 3H, NCCH3), 2.17–2.20 (m, 6H, H1), 1.28 (s, 6H, gem-CH3), 1.20–1.27 (m, 9H, H2). 13C{1H} NMR (CDCl3, 126 MHz): δ 187.2, 151.2, 145.5, 130.5, 125.8, 123.8, 119.4, 109.8, 107.4 (t, J = 14 Hz), 53.5, 23.2, 16.4 (t, J = 17.6 Hz), 15.4, 8.4. 31P{1H} NMR (CDCl3, 243 MHz): δ 11.62 (s). IR (ATR, neat) νmax/cm−1: 2962, 2929, 2873, 2099 (C≡C), 1691, 1609, 1572, 1457, 1417. HRMS (ES(+), MeCN) m/z: calcd for C38H55N2P2Pt+ [M + H]+ 796.3488, found 796.3510.
Compound 12b was prepared in a similar manner to the above 12. After evaporation of the reaction solvent, the residue was extracted with benzene and filtered through a small alumina plug. A crystal for XRD studies was grown by diffusion of pentane into a concentrated chloroform solution. 1H NMR (CDCl3, 500 MHz): δ 7.39 (d, J = 8.0 Hz, 1H), 7.23 (dd, J = 1.5 Hz, 8.0 Hz, 1H), 7.16 (d, J = 1.5 Hz, 1H), 2.25 (s, 3H, NCCH3), 2.17–2.20 (m, 6H), 1.28 (s, 6H, gem-CH3), 1.20–1.27 (m, 9H). 31P{1H} NMR (CDCl3, 243 MHz): δ 11.13 (s, J = 1188 Hz) (unreferenced).
An oven-dried and argon-filled reflux apparatus was charged with cis-Pt(PPh3)2Cl2 (126 mg, 0.159 mmol), 2 (68 mg, 0.371 mmol) and CuI (6 mg, 0.03 mmol). The apparatus was evacuated for 30 min, then dry, degassed NHiPr2 (15 mL) was added. The yellow solution was heated to reflux and stirred for 3 h. After cooling to ambient temperature, the solution was filtered and evaporated to dryness under vacuum. The residue was triturated and washed with Et2O (5 × 2 mL) and EtOH (5 × 2 mL). Further filtration of a CH2Cl2 solution and subsequent evaporation gave a pale-yellow precipitate (130 mg, 75%). Crystals were grown by slow diffusion of pentane into a concentrated CDCl3 solution. 1H NMR (CDCl3, 500 MHz): δ 7.82–7.84 (m, 12H, PPh3), 7.45–7.48 (m, 6H, H4), 7.40–7.43 (m, 12H, PPh3), 7.04 (d, J = 7.8 Hz, 2H, H7′), 6.23 (dd, J = 1.8 Hz, 7.8 Hz, 2H, H6′), 6.06 (br. s, 2H, H4′), 2.14 (s, 6H, NCCH3), 1.10 (s, 12H, gem-CH3). 13C{1H} NMR (CDCl3, 126 MHz): δ 187.5, 151.2, 145.3, 135.4 (t, J = 7.5 Hz), 131.9 (t, J = 30 Hz), 130.7, 130.1, 128.3 (t, J = 4.5 Hz), 125.7, 124.5, 118.8 (C≡C), 113.7 (C≡C), 53.2, 23.2, 15.6. 31P{1H} NMR (CDCl3, 243 MHz): δ 19.18 (s). IR (ATR, neat) νmax/cm−1: 2960, 2105 (C≡C), 1573, 1481, 1458, 1434. HRMS (AP(+), MeCN) m/z: calcd for C62H55N2P2Pt+ [M + H]+ 1084.3488, found 1084.3472.
14 was synthesised by an adapted literature procedure.[56] (NH4)2PtCl4 (1.00 g, 2.68 mmol) was added to degassed water in a Schlenk flask. To this was added PCy3 (7.6 g 20% weight in toluene, 5.4 mmol) dropwise with vigorous stirring. The solution was stirred for 24 h at ambient temperature under argon. The white precipitate was collected by vacuum filtration and washed with water and ethanol, and hexanes, then dried in a vacuum desiccator (1.82 g, 83%). The NMR data are in agreement with the literature.[103] 1H NMR (CDCl3, 500 MHz): δ 2.60–2.65 (m, 3H), 1.64–1.98 (m, 20H), 1.22–1.30 (m, 10H). 31P{1H} NMR (CDCl3, 243 MHz): δ 17.18 (s, J = 2398 Hz).
Compound 14 (50 mg, 0.061 mmol) and CuI (2 mg) were suspended in CHCl3 (3 mL) and NEt3 (0.2 mL). Phenylacetylene (0.1 mL, 0.9 mmol) was added and the solution immediately became yellow. The reaction was heated at 55°C for 11 days. The solution was concentrated, and ethanol was added to precipitate the product. The yellow powder was then recrystallised from CHCl3 and ethanol (34 mg, 58%). 1H NMR (CD2Cl2, 500 MHz): δ 7.05–7.32 (m, 5H, Ph), 2.87–2.91 (m, 3H, Cy), 1.72–2.11 (m, 20H), 1.20–1.26 (m, 10H). 13C{1H} NMR (CD2Cl2, 126 MHz): δ 130.8, 128.2, 124.6, 33.9 (t, J = 14.3 Hz), 30.3, 28.2 (t, J = 6.5 Hz), 27.2. 31P{1H} NMR (CD2Cl2, 202 MHz): δ 24.06 (s, J = 2408 Hz). IR (ATR, neat) νmax/cm−1: 2927, 2848 2099 (C≡C), 1591, 1566, 1484, 1440. HRMS (ES(+), MeCN) m/z: calcd for C46H74NP2Pt+ [M – L + MeCN]+ 897.4944, found 897.4997; calcd for C52H77P2Pt+ [M + H]+ 958.5148, found 958.5185.
An oven-dried and argon-filled Schlenk tube was charged with Pt(PCy3)2Cl2 (100 mg, 0.121 mmol), 2 (81 mg, 0.442 mmol) and CuI (7 mg). The apparatus was evacuated for 30 min, then filled with argon, and dry CHCl3 (5 mL) and dry NEt3 (2 mL) were added. The solution was heated to 55°C and stirred for 3 weeks. After cooling to ambient temperature, the solution was evaporated to dryness under vacuum. The residue was then subjected to flash chromatography (basic alumina) using 1:1 EtOAc/hexanes, yielding orange microcrystals (21 mg, 15%). Crystals were grown by slow cooling of a concentrated methanol solution. 1H NMR (CDCl3, 500 MHz): δ 7.34 (d, J = 7.7 Hz, 2H), 7.21 (d, J = 7.7 Hz, 2H), 7.14 (s, 2H), 2.86–2.92 (m, 6H), 1.20–2.11 (m, 78H). 13C{1H} NMR (CDCl3, 126 MHz): δ 186.6, 150.5, 145.2, 129.7, 123.7, 119.2, 58.5, 53.3, 33.5 (t, J = 13 Hz), 29.9, 27.8 (t, J = 5.3), 26.8, 23.2, 18.4, 15.4. 31P{1H} NMR (CDCl3, 243 MHz): δ 23.22 (s, J = 2424 Hz). HRMS (ES(+). MeCN) m/z: calcd for C62H91N2P2Pt+ [M + H]+ 1120.6305, found 1120.6332.
[Ru(dppe)2Cl]OTf (107 mg, 0.099 mmol), 2 (49 mg, 0.267 mmol) and KOtBu (36 mg, 0.32 mmol) were placed in an oven-dried and argon-filled Schlenk tube and put under high vacuum for 30 min. The Schlenk tube was backfilled with argon, dry degassed CH2Cl2 (10 mL) was added, and the solution was stirred for 4 days at room temperature. The deep red solution was taken to dryness under vacuum. The crude purple residue was triturated and washed with dry Et2O (2 × 3 mL), CH3OH (2 × 3 mL) and pentane (3 mL). The residue was dissolved in CH2Cl2, filtered into a new Schlenk tube and evaporated to dryness, yielding a dark red powder (99 mg, 79%). Crystals were grown by diffusion of pentane into a CH2Cl2 solution. 1H NMR (C6D6, 600 MHz): δ 7.83 (d, J = 7.8 Hz, 2H, H7′), 7.51–7.76 (m, 16H, PPh2), 7.25 (dd, J = 1.8 Hz, 7.8 Hz, 2H, H6′), 6.94–7.02 (m, 24H, PPh2), 6.85 (d, J = 1.8 Hz, 2H, H4′), 2.56 (br app s, 8H, dppe), 2.03 (s, 6H, NCCH3), 1.12 (s, 12H, gem-CH3). 13C{1H} NMR (C6D6, 100 MHz): δ 184.6, 151.4, 145.8, 137.9 (m), 134.9, 130.3 (t, J = 15 Hz, Cα) 129.7, 129.0, 128.3, 128.0, 127.5, 123.8, 120.0, 117.9 (Cβ), 53.3, 31.8 (m, CH2CH2-dppe), 23.4 (gem-CH3), 15.2 (NCCH3). 31P{1H} NMR (C6D6, 243 MHz): δ 54.35 (s). IR (ATR, neat) νmax/cm−1: 2958, 2048 (C≡C), 1568, 1455, 1430. HRMS (AP(+), MeCN) m/z: calcd for C78H73N2P4Ru+ [M + H]+ 1263.3768, found 1263.3789.
Ru(P(OEt)3)4Cl2 (98 mg, 0.117 mmol), 2 (136 mg, 0.742 mmol) and KPF6 (127 mg, 0.690 mmol) were placed in an oven-dried and argon-filled Schlenk tube and put under high vacuum for 30 min. The Schlenk tube was backfilled with argon, dry degassed EtOH (5 mL) and dry degassed NHiPr2 (2 mL) were added, and the solution was stirred for 8 weeks at room temperature. The brown–orange solution was taken to dryness under vacuum. The crude brown oil was triturated with pentane and filtered through cotton wool until the extracts were colourless. This solution was reduced to dryness again by rotary evaporator. Recrystallisation of the extract with slow cooling of a pentane solution afforded light yellow crystals (19 mg, 14%). 1H NMR (CDCl3, 600 MHz): δ 7.27 (s, 2H, Ar), 6.93–6.95 (m, 4H, Ar), 4.33 (q, J = 7.2 Hz, 24H, P(OCH2CH3)3), 2.22 (s, 6H, NCCH3), 1.23 (s, 12H, gem-CH3) 1.21 (t, J = 7.2 Hz, 36H, P(OCH2CH3)3). 13C{1H} NMR (CDCl3, 126 MHz): δ 185.5, 149.2, 145.2, 128.8, 123.2, 119.1, 61.0, 53.2, 23.4, 22.5, 16.6, 15.4. 31P{1H} NMR (CDCl3, 243 MHz): δ 136.92 (s). IR (ATR, neat) νmax/cm−1: 2974, 2901, 2066 (C≡C), 1609, 1573, 1457. HRMS (ES(+), MeCN) m/z: calcd for C37H72NO12P4Ru+ [M–L]+ 948.3043, found 948.2996; calcd for C38H72NO13P4Ru+ [M – L + CO]+ 976.2993, found 976.2938.
Electrochemistry
Cyclic voltammetry was performed in 3 mL reaction vials (eDAQ) in a glovebox under an argon atmosphere using a Pt working disc electrode (1 mm diameter; eDAQ) and Pt-coated Ti rods as the counter and pseudo-reference electrodes (eDAQ). The surface of the working electrode was polished with an alumina paste (0.05 μm alumina powder) on a polishing cloth, rinsed with MilliQ water, ethanol and CH2Cl2 and dried under a stream of nitrogen before use. Ferrocene (Fc), decamethylferrocene (Fc*) or diacetylferrocene (Ac2Fc) were used as internal standard and all potentials are reported v. the Fc/[Fc]+ couple. Measurements were conducted in CH2Cl2 solution containing 0.1 M tetrabutylammonium hexafluorophosphate (Bu4NPF6) as the supporting electrolyte. The electrochemical potential within the cell was controlled using a PalmSens EmStat3+ potentiostat and PSTrace software.
Spectroelectrochemistry
All spectroelectrochemical (SEC) measurements were conducted using an OTTLE cell of Hartl design[104] using dry CH2Cl2 as the solvent and 0.1 M Bu4NPF6 as the supporting electrolyte. For each experiment, the SEC cell was filled with the analyte solution inside a glovebox and sealed before being removed for operation in the spectrometer. UV-Vis/NIR (NIR, near-infrared) spectra were recorded on an Agilent Cary 5000 spectrometer. IR spectra were recorded on an Agilent Cary 660 FT-IR/NIR spectrometer. The electrochemical potential within the cell was controlled using a PalmSens EmStat3+ potentiostat. For each measurement, the potential was incrementally increased, and a spectrum was recorded at each step.
Crystallography
X-ray diffraction measurements were carried out at 100 K on an Oxford Diffraction Gemini-R Ultradiffractometer fitted with a MoKα or CuKα radiation source at the University of Western Australia. Following analytical absorption corrections and solution by direct methods, the structures were refined against F2 with full-matrix least-squares using the SHELXL software suite. The .cif files have been deposited at the Cambridge Crystallographic Data Centre, numbers CCDC 2255621–2255635.
Single-molecule conductance
We performed STM-BJ[105] experiments to determine single-molecule conductance. In these experiments, the Au STM tip is driven into the Au STM substrate to form a metallic contact, and then it is withdrawn at a constant speed (13 nm s−1 in this study). As the tip is retracted, the metallic contact thins to a point contact having conductance of G0, which is then ruptured to yield two atomically sharp nanoelectrodes. The experiment is performed in a solution of the target molecule (1 mM in 1,2,5-trichlorobenzene in this study), which can self-assemble in the freshly formed nanogap to fabricate a molecular junction. The tip is further withdrawn so that the junction is stretched to its maximum length, and eventually ruptured. The tip is then driven again into the substrate and the process is repeated. Data (as G = I/V) (G, conductance; I, current; V, voltage) are continuously recorded and compiled in statistical 1-D conductance histograms and in 2-D conductance/electrode separation plots.
A modified commercial instrument (Keysight Technology 5500 SPM) was used in this study. The original electronics were used to apply the junction bias (V) and to drive the piezoelectric actuator. A custom current amplifier based on the design originally introduced by Mészáros et al.[106] was used as I/V converter, and the resulting four-channel data were acquired using a National Instruments DAQ (NI-9215). We used Au/Cr/glass slides (Arrandee) as substrate and 99.999+% Au wire (Goodfellow Cambridge) as tip.
Supplementary material
The Supplementary material is available free of charge: additional electrochemical, crystallography and spectroscopic data for all products. Supplementary material is available online.
Data availability
Single-molecule data collected at Liverpool are archived at https://datacat.liverpool.ac.uk/2181.
Conflicts of interest
G. A. Koutsantonis is a Co-Editor in Chief for the Australian Journal of Chemistry but did not at any stage have editor-level access to this manuscript while in peer review, as is the standard practice when handling manuscripts submitted by an editor to this journal. Australian Journal of Chemistry encourages its editors to publish in the journal and they are kept totally separate from the decision-making processes for their manuscripts. The authors have no further conflicts of interest to declare.
Declaration of funding
This research was supported under the Australian Research Council’s Discovery Projects funding scheme (Project numbers DP150104117 and DP200101659). The authors acknowledge the facilities and the scientific and technical assistance of the Australian Microscopy and Microanalysis Research Facility at the Centre for Microscopy, Characterisation, and Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments.
Acknowledgements
D. C. Milan gratefully acknowledges the School of Physical Sciences Postdoctoral Development Award of the University of Liverpool for financial support. D. Jago held an Australian RTP scholarship. A. Vezzoli thanks the Royal Society for support (University Research Fellowship URF\R1\191241).
References
1 Schwab PFH, Smith JR, Michl J. Synthesis and properties of molecular rods. 2. Zig-zag rods. Chem Rev 2005; 105(4): 1197-1280.
| Crossref | Google Scholar |
2 McQuade DT, Pullen AE, Swager TM. Conjugated polymer-based chemical sensors. Chem Rev 2000; 100(7): 2537-2574.
| Crossref | Google Scholar |
3 Tour JM. Conjugated macromolecules of precise length and constitution. Organic synthesis for the construction of nanoarchitectures. Chem Rev 1996; 96(1): 537-554.
| Crossref | Google Scholar |
4 Low PJ. Metal complexes in molecular electronics: progress and possibilities. Dalton Trans 2005; 17: 2821-2824.
| Crossref | Google Scholar |
5 Long NJ. Organometallic compounds for nonlinear optics — the search for en-light-enment. Angew Chem Int Ed 1995; 34(1): 21-38.
| Crossref | Google Scholar |
6 Sala X, Maji S, Bofill R, García-Antón J, Escriche L, Llobet A. Molecular water oxidation mechanisms followed by transition metals: state of the art. Acc Chem Res 2014; 47(2): 504-516.
| Crossref | Google Scholar |
7 Wong WY, Ho CL. Organometallic photovoltaics: a new and versatile approach for harvesting solar energy using conjugated polymetallaynes. Acc Chem Res 2010; 43(9): 1246-1256.
| Crossref | Google Scholar |
8 Yam VWW. Molecular design of transition metal alkynyl complexes as building blocks for luminescent metal-based materials: structural and photophysical aspects. Acc Chem Res 2002; 35(7): 555-563.
| Crossref | Google Scholar |
10 de Ruiter G, Lahav M, van der Boom ME. Pyridine coordination chemistry for molecular assemblies on surfaces. Acc Chem Res 2014; 47(12): 3407-3416.
| Crossref | Google Scholar |
11 He ZH, Li HR, Li ZP. Iodine-mediated synthesis of 3H-indoles via intramolecular cyclization of enamines. J Org Chem 2010; 75(13): 4636-4639.
| Crossref | Google Scholar |
12 Lim KH, Hiraku O, Komiyama K, Koyano T, Hayashi M, Kam TS. Biologically active indole alkaloids from Kopsia arborea. J Nat Prod 2007; 70(8): 1302-1307.
| Crossref | Google Scholar |
13 Kawasaki T, Higuchi K. Simple indole alkaloids and those with a non-rearranged monoterpenoid unit. Nat Prod Rep 2005; 22(6): 761-793.
| Crossref | Google Scholar |
14 Kam TS, Choo YM. New indole alkaloids from Alstonia macrophylla. J Nat Prod 2004; 67(4): 547-552.
| Crossref | Google Scholar |
15 Haque A, Al-Balushi RA, Al-Busaidi IJ, Khan MS, Raithby PR. Rise of conjugated poly-ynes and poly(metalla-ynes): from design through synthesis to structure–property relationships and applications. Chem Rev 2018; 118(18): 8474-8597.
| Crossref | Google Scholar |
16 Long NJ, Williams CK. Metal alkynyl σ complexes: synthesis and materials. Angew Chem Int Ed 2003; 42(23): 2586-2617.
| Crossref | Google Scholar |
17 Bruce MI, Ellis BG, Gaudio M, Lapinte C, Melino G, Paul F, Skelton BW, Smith ME, Toupet L, White AH. Preparation, structures and some reactions of novel diynyl complexes of iron and ruthenium. Dalton Trans 2004; 10: 1601-1609.
| Crossref | Google Scholar |
18 Miller-Clark LA, Ren T. Syntheses and material applications of Ru(II)(bisphosphine)2 alkynyls. J Organomet Chem 2021; 951: 122003.
| Crossref | Google Scholar |
19 Koutsantonis GA, Jenkins GI, Schauer PA, Szczepaniak B, Skelton BW, Tan C, White AH. Coordinating tectons: bipyridyl-terminated Group 8 alkynyl complexes. Organometallics 2009; 28(7): 2195-2205.
| Crossref | Google Scholar |
20 Koutsantonis GA, Low PJ, Mackenzie CFR, Skelton BW, Yufit DS. Coordinating tectons: bimetallic complexes from bipyridyl terminated Group 8 alkynyl complexes. Organometallics 2014; 33(18): 4911-4922.
| Crossref | Google Scholar |
21 Cifuentes MP, Humphrey MG, Koutsantonis GA, Lengkeek NA, Petrie S, Sanford V, Schauer PA, Skelton BW, Stranger R, White AH. Coordinating tectons: bipyridyl terminated allenylidene complexes. Organometallics 2008; 27(8): 1716-1726.
| Crossref | Google Scholar |
22 Schauer PA, Skelton BW, Koutsantonis GA. Coordinating tectons 4: coordination chemistry of the 4,5-diazafluoren-9-yl moiety as a metallo-ligand for allenylidene complexes. Organometallics 2015; 34(20): 4975-4988.
| Crossref | Google Scholar |
23 Bock S, Mackenzie CF, Skelton BW, Byrne LT, Koutsantonis GA, Low PJ. Clusters as ligands: synthesis, structure and coordination chemistry of ruthenium clusters derived from 4- and 5-ethynyl-2,2′-bipyridine. J Organomet Chem 2016; 812: 190-196.
| Crossref | Google Scholar |
24 Rodríguez JG, Urrutia A, Eugenio de Diego J, Paz Martínez-Alcazar M, Fonseca I. Synthesis of 2′-alkylspiro[2-X-cyclohexan-1,3′-3′ H-indole] (X = H; X = CH3) by an unexpected reaction between an organomagnesium halide and 2′-methylspiro[2-X-cyclohexan-1,3′-3′H-indole]. X-ray structure of a fluorescent dimeric compound. J Org Chem 1998; 63(13): 4332-4337.
| Crossref | Google Scholar |
25 Rodríguez JG, Urrutia A. Synthesis of sterically hindered 4a,9a-disubstituted 1,2,3,4,4a,9a-hexahydrocarbazoles from 4a-methyl-1,2,3,4-tetrahydro-4aH-carbazole with organolithium reagents. Tetrahedron 1998; 54(51): 15613-15618.
| Crossref | Google Scholar |
26 Fehlner JR, Borowski PJ, Pettinato PL, Freyer AJ. Condensation of 2,3,3-trimethyl-3H-indole with methylene iodide and oxidative coupling. J Org Chem 1984; 49(1): 170-172.
| Crossref | Google Scholar |
27 Kanaoka Y, Miyashita K, Yonemitsu O. 3H-Indoles. IV. Photo-and peroxide-induced oxygenation of 2-ethyl-3H-indoles. Chem Pharm Bull 1970; 18(3): 634-637.
| Crossref | Google Scholar |
28 Kanaoka Y, Miyashita K, Yonemitsu O. 3H-indoles—II: synthesis of 3-alkyl-3H-indoles by the alkylation of 2,3-disubstituted indoles with polyphosphate ester and some reactions of the 3H-indole system. Tetrahedron 1969; 25(14): 2757-2766.
| Crossref | Google Scholar |
29 Liu SY, Zhang WZ, Qu JP, Wang BM. Engaging 2-methyl indolenines in a tandem condensation/1,5-hydride transfer/cyclization process: construction of a novel indolenine–tetrahydroquinoline assembly. Org Chem Front 2018; 5(20): 3008-3012.
| Crossref | Google Scholar |
30 Wang Q-C, Qu DH, Ren J, Xu LH, Liu MY, Tian H. New benzo[e]indolinium cyanine dyes with two different fluorescence wavelengths. Dyes Pigm 2003; 59(2): 163-172.
| Crossref | Google Scholar |
31 Shachkus AA, Degutite RY. Reaction of 2,3,3-trimethyl-3H-indole salts with acrylamide. Synthesis of 1,2,3,4,10,10a-hexahydropyrimido[1,2-a]indol-2-one derivatives. Chem Heterocycl Compd 1986; 22(8): 852-855.
| Crossref | Google Scholar |
32 Bruce M, Koutsantonis G. Cyclopentadienyl-ruthenium and -osmium chemistry. XXXV. Some ethynyl, vinylidene and related complexes. Aust J Chem 1991; 44(2): 207-217.
| Crossref | Google Scholar |
33 Sonogashira K, Yatake T, Tohda Y, Takahashi S, Hagihara N. Novel preparation of σ-alkynyl complexes of transition metals by copper(I) iodide-catalysed dehydrohalogenation. J Chem Soc, Chem Commun 1977; 9: 291-292.
| Crossref | Google Scholar |
34 Vicente J, Gil-Rubio J, Barquero N, Jones PG, Bautista D. Synthesis of luminescent alkynyl gold metalaligands containing 2,2′-bipyridine-5-yl and 2,2′: 6′,2″-terpyridine-4-yl donor groups. Organometallics 2008; 27(4): 646-659.
| Crossref | Google Scholar |
35 Khairul WM, Fox MA, Zaitseva NN, Gaudio M, Yufit DS, Skelton BW, White AH, Howard JAK, Bruce MI, Low PJ. Transition metal alkynyl complexes by transmetallation from Au(C≡CAr)(PPh3) (Ar = C6H5 or C6H4Me-4). Dalton Trans 2009; 4: 610-620.
| Crossref | Google Scholar |
36 Rudolph M, Hashmi ASK. Gold catalysis in total synthesis – an update. Chem Soc Rev 2012; 41(6): 2448-2462.
| Crossref | Google Scholar |
37 Leyva-Pérez A, Doménech A, Al-Resayes SI, Corma A. Gold redox catalytic cycles for the oxidative coupling of alkynes. ACS Catal 2012; 2(1): 121-126.
| Crossref | Google Scholar |
38 Fox MA, Roberts RL, Khairul WM, Hartl F, Low PJ. Spectroscopic properties and electronic structures of 17-electron half-sandwich ruthenium acetylide complexes, [Ru(C≡CAr)(L-2)Cp′]+ (Ar = phenyl, p-tolyl, 1-naphthyl, 9-anthryl; L2 = (PPh3)2, Cp′ = Cp; L2 = dppe; Cp′ = Cp*). J Organomet Chem 2007; 692(15): 3277-3290.
| Crossref | Google Scholar |
39 Harrison DP, Kumar VJ, Noppers JN, Gluyas JBG, Sobolev AN, Moggach SA, Low PJ. Iron vs ruthenium: syntheses, structures and IR spectroelectrochemical characterisation of half-sandwich Group 8 acetylide complexes. New J Chem 2021; 45(33): 14932-14943.
| Crossref | Google Scholar |
40 Whittall IR, Humphrey MG, Hockless DCR, Skelton BW, White AH. Organometallic complexes for non-linear optics. 2. Syntheses, electrochemical studies, structural characterization, and computationally derived molecular quadratic hyperpolarizabilities of ruthenium σ-arylacetylides: X-ray crystal structures of Ru(C≡CPh)(PMe3)2(η-C5H5) and Ru(C≡CC6H4NO2-4)(L)2(η-C5H5) (L = PPh3, PMe3). Organometallics 1995; 14(8): 3970-3979.
| Crossref | Google Scholar |
41 Mackenzie CFR, Bock S, Lim CY, Skelton BW, Nervi C, Wild DA, Low PJ, Koutsantonis GA. Coordinating tectons. Experimental and computational infrared data as tools to identify conformational isomers and explore electronic structures of 4-ethynyl-2,2′-bipyridine complexes. Organometallics 2017; 36(10): 1946-1961.
| Crossref | Google Scholar |
42 Whittall IR, Humphrey MG, Houbrechts S, Persoons A, Hockless DCR. Organometallic complexes for non-linear optics. 8.1 Syntheses and molecular quadratic hyperpolarizabilities of systematically varied (triphenylphosphine)gold σ-arylacetylides: X-ray crystal structures of Au(C⋮CR)(PPh3) (R = 4-C6H4NO2, 4,4′-C6H4C6H4NO2). Organometallics 1996; 15(26): 5738-5745.
| Crossref | Google Scholar |
43 Bruce MI, Swincer AG, Wallis RC. Cyclopentadienyl–ruthenium and -osmium chemistry. Some reactions of substituted vinylidene complexes. J Organomet Chem 1979; 171(1): C5-C8.
| Crossref | Google Scholar |
44 Fox MA, Harris JE, Heider S, Pérez-Gregorio V, Zakrzewska ME, Farmer JD, Yufit DS, Howard JAK, Low PJ. A simple synthesis of trans-RuCl(C≡CR)(dppe)2 complexes and representative molecular structures. J Organomet Chem 2009; 694(15): 2350-2358.
| Crossref | Google Scholar |
45 Packheiser R, Ecorchard P, Walfort B, Lang H. Heterotrimetallic and heterotetrametallic transition metal complexes. J Organomet Chem 2008; 693(6): 933-946.
| Crossref | Google Scholar |
46 Paul F, Ellis BG, Bruce MI, Toupet L, Roisnel T, Costuas K, Halet JF, Lapinte C. Bonding and substituent effects in electron-rich mononuclear ruthenium σ-arylacetylides of the formula [(η2-dppe)(η5-C5Me5)Ru(C⋮C)-1,4-(C6H4)X][PF6]n (n = 0, 1; X = NO2, CN, F, H, OMe, NH2). Organometallics 2006; 25(3): 649-665.
| Crossref | Google Scholar |
47 Wu IY, Lin JT, Luo J, Sun SS, Li CS, Lin KJ, Tsai CT, Hsu CC, Lin JL. Syntheses and reactivity of ruthenium σ-pyridylacetylides. Organometallics 1997; 16(10): 2038-2048.
| Crossref | Google Scholar |
48 Ge Q, Hor TSA. Stepwise assembly of linearly-aligned Ru–M–Ru (M = Pd, Pt) heterotrimetallic complexes with σ-4-ethynylpyridine spacer. Dalton Trans 2008; 22: 2929-2936.
| Crossref | Google Scholar |
49 D’Amato R, Furlani A, Colapietro M, Portalone G, Casalboni M, Falconieri M, Russo MV. Synthesis, characterisation and optical properties of symmetrical and unsymmetrical Pt(II) and Pd(II) bis-acetylides. Crystal structure of trans-[Pt(PPh3)2(CC–C6H5)(CC–C6H4NO2)]. J Organomet Chem 2001; 627(1): 13-22.
| Crossref | Google Scholar |
50 Sonogashira K, Fujikura Y, Yatake T, Toyoshima N, Takahashi S, Hagihara N. Syntheses and properties of cis- and trans-dialkynyl complexes of platinum(II). J Organomet Chem 1978; 145(1): 101-108.
| Crossref | Google Scholar |
51 Xu H-B, Ni J, Chen K-J, Zhang L-Y, Chen Z-N. Preparation, characterization, and photophysical properties of cis- or trans-PtLn2 (Ln = Nd, Eu, Yb) arrays with 5-ethynyl-2,2′-bipyridine. Organometallics 2008; 27(21): 5665-5671.
| Crossref | Google Scholar |
52 Gimeno A, Rodríguez-Gimeno A, Cuenca AB, Ramírez de Arellano C, Medio-Simón M, Asensio G. Gold(I)-catalysed cascade reactions in the synthesis of 2,3-fused indole derivatives. Chem Commun 2015; 51(62): 12384-12387.
| Crossref | Google Scholar |
53 Takani M, Masuda H, Yamauchi O. Palladium(II) complex formation by indole-3-acetate. Mixed ligand complexes involving a unique spiro-ring formed by cyclopalladation. Inorg Chim Acta 1995; 235(1): 367-374.
| Crossref | Google Scholar |
54 Takani M, Takeda T, Yajima T, Yamauchi O. Indole rings in palladium(II) complexes. Dual mode of metal binding and aromatic ring stacking causing syn−anti isomerism. Inorg Chem 2006; 45(15): 5938-5946.
| Crossref | Google Scholar |
55 Barrett BJ, Iluc VM. An adaptable chelating diphosphine ligand for the stabilization of palladium and platinum carbenes. Organometallics 2017; 36(3): 730-741.
| Crossref | Google Scholar |
56 Al-Owaedi OA, Bock S, Milan DC, Oerthel MC, Inkpen MS, Yufit DS, Sobolev AN, Long NJ, Albrecht T, Higgins SJ, Bryce MR, Nichols RJ, Lambert CJ, Low PJ. Insulated molecular wires: Inhibiting orthogonal contacts in metal complex-based molecular junctions. Nanoscale 2017; 9(28): 9902-9912.
| Crossref | Google Scholar |
57 Schull TL, Kushmerick JG, Patterson CH, George C, Moore MH, Pollack SK, Shashidhar R. Ligand effects on charge transport in platinum(II) acetylides. J Am Chem Soc 2003; 125(11): 3202-3203.
| Crossref | Google Scholar |
58 Touchard D, Haquette P, Guesmi S, LePichon L, Daridor A, Toupet L, Dixneuf PH. Vinylidene-, alkynyl-, and trans-bis(alkynyl)ruthenium complexes. Crystal structure of trans-[Ru(NH3)(C⋮C−Ph)(Ph2PCH2CH2PPh2)2]PF6. Organometallics 1997; 16(16): 3640-3648.
| Crossref | Google Scholar |
59 Touchard D, Morice C, Cadierno V, Haquette P, Toupet L, Dixneuf PH. Novel allenylidene alkynyl and ammonia alkynyl metal complexes via selective synthesis of mono and bis alkynyl ruthenium(II) complexes; crystal structure of trans-[Ru(NH3)(C≡Cph)(Ph2PCH2CH2PPh2)2]PF6. J Chem Soc Chem Comm 1994; 1994(7): 859-860.
| Crossref | Google Scholar |
60 Naher M, Bock S, Langtry ZM, O’Malley KM, Sobolev AN, Skelton BW, Korb M, Low PJ. Synthesis, structure and physical properties of ‘wire-like’ metal complexes. Organometallics 2020; 39(24): 4667-4687.
| Crossref | Google Scholar |
61 Eaves SG, Skelton BW, Low PJ. Syntheses and molecular structures of trans-bis(alkynyl) tetrakis-triethylphosphite ruthenium complexes. J Organomet Chem 2017; 847: 242-250.
| Crossref | Google Scholar |
62 Albertin G, Autoniutti S, Bordignon E, Cazzaro F, Ianelli S, Pelizzi G. Preparation, structure, and reactivity of new bis(acetylide) and acetylide–vinylidene ruthenium(II) complexes stabilized by phosphite ligands. Organometallics 1995; 14(9): 4114-4125.
| Crossref | Google Scholar |
63 Marqués-González S, Parthey M, Yufit DS, Howard JAK, Kaupp M, Low PJ. Combined spectroscopic and quantum chemical study of [trans-Ru(C≡CC6H4R1-4)2(dppe)2]n+ and [trans-Ru(C≡CC6H4R1-4)(C≡CC6H4R2-4)(dppe)2]n+ (n = 0, 1) complexes: interpretations beyond the lowest energy conformer paradigm. Organometallics 2014; 33(18): 4947-4963.
| Crossref | Google Scholar |
64 Bryce MR. A review of functional linear carbon chains (oligoynes, polyynes, cumulenes) and their applications as molecular wires in molecular electronics and optoelectronics. J Mater Chem C 2021; 9: 10524-10546.
| Crossref | Google Scholar |
65 Casari CS, Tommasini M, Tykwinski RR, Milani A. Carbon-atom wires: 1-D systems with tunable properties. Nanoscale 2016; 8(8): 4414-4435.
| Crossref | Google Scholar |
66 Liu J, Lam JWY, Tang BZ. Acetylenic polymers: syntheses, structures, and functions. Chem Rev 2009; 109(11): 5799-5867.
| Crossref | Google Scholar |
67 Hay AS. Oxidative coupling of acetylenes. II. J Org Chem 1962; 27(9): 3320-3321.
| Crossref | Google Scholar |
68 Eglinton G, Galbraith AR. 182. Macrocyclic acetylenic compounds. Part I. Cyclotetradeca-1:3-diyne and related compounds. J Chem Soc (Resumed) 1959; 1959: 889-896.
| Crossref | Google Scholar |
69 Glaser C. Untersuchungen über einige Derivate der Zimmtsäure. [Studies on some derivatives of cinnamic acid.]. Justus Liebigs Ann Chem 1870; 154(2): 137-171 [In German].
| Crossref | Google Scholar |
70 Batsanov AS, Collings JC, Fairlamb IJS, Holland JP, Howard JAK, Lin Z, Marder TB, Parsons AC, Ward RM, Zhu J. Requirement for an oxidant in Pd/Cu co-catalyzed terminal alkyne homocoupling to give symmetrical 1,4-disubstituted 1,3-diynes. J Org Chem 2005; 70(2): 703-706.
| Crossref | Google Scholar |
71 Younus M, Long NJ, Raithby PR, Lewis J. Synthetic, spectroscopic and electrochemical characterisation of mixed-metal acetylide complexes. J Organomet Chem 1998; 570(1): 55-62.
| Crossref | Google Scholar |
72 Masai H, Sonogashira K, Hagihara N. Electronic spectra of square-planar bis(tertiary phosphine)dialkynyl complexes of nickel(II), palladium(II), and platinum(II). Bull Chem Soc Jpn 1971; 44(8): 2226-2230.
| Crossref | Google Scholar |
73 Choi MY, Chan MCW, Zhang SB, Cheung KK, Che CM, Wong KY. MLCT and LMCT transitions in acetylide complexes. Structural, spectroscopic, and redox properties of ruthenium(II) and -(III) bis(σ-arylacetylide) complexes supported by a tetradentate macrocyclic tertiary amine ligand. Organometallics 1999; 18(11): 2074-2080.
| Crossref | Google Scholar |
74 Wong CY, Che CM, Chan MCW, Han J, Leung KH, Phillips DL, Wong KY, Zhu NY. Probing ruthenium-acetylide bonding interactions: Synthesis, electrochemistry, and spectroscopic studies of acetylide–ruthenium complexes supported by tetradentate macrocyclic amine and diphosphine ligands. J Am Chem Soc 2005; 127(40): 13997-14007.
| Crossref | Google Scholar |
75 Powell CE, Cifuentes MP, McDonagh AM, Hurst SK, Lucas NT, Delfs CD, Stranger R, Humphrey MG, Houbrechts S, Asselberghs I, Persoons A, Hockless DCR. Organometallic complexes for nonlinear optics: Part 27. Syntheses and optical properties of some iron, ruthenium and osmium alkynyl complexes. Inorg Chim Acta 2003; 352: 9-18.
| Crossref | Google Scholar |
76 Powell CE, Cifuentes MP, Morrall JP, Stranger R, Humphrey MG, Samoc M, Luther-Davies B, Heath GA. Organometallic complexes for nonlinear optics. 30.1 Electrochromic linear and non-linear optical properties of alkynylbis(diphosphine)ruthenium complexes. J Am Chem Soc 2003; 125(2): 602-610.
| Crossref | Google Scholar |
77 Holliman PJ, Horton PN, Hursthouse MB. CCDC 1011855: experimental crystal structure determination; 2014. 10.5517/cc12yxhf
78 de Aquino A, Caparrós FJ, Aullón G, Ward JS, Rissanen K, Jung Y, Choi H, Lima JC, Rodríguez L. Effect of gold(I) on the room-temperature phosphorescence of ethynylphenanthrene. Chem Eur J 2021; 27(5): 1810-1820.
| Crossref | Google Scholar |
79 Hurst SK, Lucas NT, Humphrey MG, Isoshima T, Wostyn K, Asselberghs I, Clays K, Persoons A, Samoc M, Luther-Davies B. Organometallic complexes for non-linear optics. Part 29. Quadratic and cubic hyperpolarizabilities of stilbenylethynyl–gold and -ruthenium complexes. Inorg Chim Acta 2003; 350: 62-76.
| Crossref | Google Scholar |
80 Khairul WM, Albesa-Jové D, Yufit DS, Al-Haddad MR, Collings JC, Hartl F, Howard JAK, Marder TB, Low PJ. The syntheses, structures and redox properties of phosphine–gold(I) and triruthenium–carbonyl cluster derivatives of tolans. Inorg Chim Acta 2008; 361(6): 1646-1658.
| Crossref | Google Scholar |
81 Khairul WM, Porrès L, Albesa-Jové D, Senn MS, Jones M, Lydon DP, Howard JAK, Beeby A, Marder TB, Low PJ. Metal cluster terminated ‘molecular wires’. J Clust Sci 2006; 17(1): 65-85.
| Crossref | Google Scholar |
82 González MT, Zhao X, Manrique DZ, Miguel D, Leary E, Gulcur M, Batsanov AS, Rubio-Bollinger G, Lambert CJ, Bryce MR, Agraït N. Structural versus electrical functionalization of oligo(phenylene ethynylene) diamine molecular junctions. J Phys Chem C 2014; 118(37): 21655-21662.
| Crossref | Google Scholar |
83 Gagnon K, Mohammed Aly S, Brisach-Wittmeyer A, Bellows D, Bérubé J-F, Caron L, Abd-El-Aziz AS, Fortin D, Harvey PD. Conjugated oligomers and polymers of cis- and trans-platinum(II)-para- and ortho-bis(ethynylbenzene)quinone diimine. Organometallics 2008; 27(10): 2201-2214.
| Crossref | Google Scholar |
84 Mayor M, von Hänisch C, Weber HB, Reichert J, Beckmann D. A trans-platinum(II) complex as a single-molecule insulator. Angew Chem Int Ed 2002; 41(7): 1183-1186.
| Crossref | Google Scholar |
85 Parthey M, Vincent KB, Renz M, Schauer PA, Yufit DS, Howard JAK, Kaupp M, Low PJ. A combined computational and spectroelectrochemical study of platinum-bridged bis-triarylamine systems. Inorg Chem 2014; 53(3): 1544-1554.
| Crossref | Google Scholar |
86 Ravera M, D’Amato R, Guerri A. Probing delocalisation across highly ethynylated mono and dinuclear Pt(II) tethers containing nitro groups and organic models as redox active probes: X-ray crystal structure of trans-[Pt(C≡C–C6H4NO2)2(PPh3)2]. J Organomet Chem 2005; 690(9): 2376-2380.
| Crossref | Google Scholar |
87 Vicente J, Chicote M-T, Alvarez-Falcón MM, Jones PG. Platinum(II) and mixed platinum(II)/gold(I) σ-alkynyl complexes. The first anionic σ-alkynyl metal polymers. Organometallics 2005; 24(11): 2764-2772.
| Crossref | Google Scholar |
88 Zhou G, Wong W-Y, Poon S-Y, Ye C, Lin Z. Symmetric versus unsymmetric platinum(II) bis(aryleneethynylene)s with distinct electronic structures for optical power limiting/optical transparency trade-off optimization. Adv Funct Mater 2009; 19(4): 531-544.
| Crossref | Google Scholar |
89 Hong W, Manrique DZ, Moreno-García P, Gulcur M, Mishchenko A, Lambert CJ, Bryce MR, Wandlowski T. Single molecular conductance of tolanes: experimental and theoretical study on the junction evolution dependent on the anchoring group. J Am Chem Soc 2012; 134(4): 2292-2304.
| Crossref | Google Scholar |
90 Bock S, Al-Owaedi OA, Eaves SG, Milan DC, Lemmer M, Skelton BW, Osorio HM, Nichols RJ, Higgins SJ, Cea P, Long NJ, Albrecht T, Martín S, Lambert CJ, Low PJ. Single-molecule conductance studies of organometallic complexes bearing 3-thienyl contacting groups. Chem Eur J 2017; 23(9): 2133-2143.
| Crossref | Google Scholar |
91 González MT, Díaz A, Leary E, García R, Herranz MÁ, Rubio-Bollinger G, Martín N, Agraït N. Stability of single- and few-molecule junctions of conjugated diamines. J Am Chem Soc 2013; 135(14): 5420-5426.
| Crossref | Google Scholar |
92 Velizhanin KA, Zeidan TA, Alabugin IV, Smirnov S. Single molecule conductance of bipyridyl ethynes: the role of surface binding modes. J Phys Chem B 2010; 114(45): 14189-14193.
| Crossref | Google Scholar |
93 Quek SY, Kamenetska M, Steigerwald ML, Choi HJ, Louie SG, Hybertsen MS, Neaton JB, Venkataraman L. Mechanically controlled binary conductance switching of a single-molecule junction. Nat Nanotechnol 2009; 4(4): 230-234.
| Crossref | Google Scholar |
94 Moreno-García P, Gulcur M, Manrique DZ, Pope T, Hong W, Kaliginedi V, Huang C, Batsanov AS, Bryce MR, Lambert C, Wandlowski T. Single-molecule conductance of functionalized oligoynes: Length dependence and junction evolution. J Am Chem Soc 2013; 135(33): 12228-12240.
| Crossref | Google Scholar |
95 Gulcur M, Moreno-García P, Zhao X, Baghernejad M, Batsanov AS, Hong W, Bryce MR, Wandlowski T. The synthesis of functionalised diaryltetraynes and their transport properties in single-molecule junctions. Chem Eur J 2014; 20(16): 4653-4660.
| Crossref | Google Scholar |
96 Skipper HE, May CV, Rheingold AL, Doerrer LH, Kamenetska M. Hard–soft chemistry design principles for predictive assembly of single molecule–metal junctions. J Am Chem Soc 2021; 143(40): 16439-16447.
| Crossref | Google Scholar |
97 Low PJ, Bock S. Spectroelectrochemistry: a valuable tool for the study of organometallic-alkyne, -vinylidene, -cumulene, -alkynyl and related complexes. Electrochim Acta 2013; 110: 681-692.
| Crossref | Google Scholar |
98 Fox MA, Le Guennic B, Roberts RL, Brue DA, Yufit DS, Howard JAK, Manca G, Halet JF, Hartl F, Low PJ. Simultaneous bridge-localized and mixed-valence character in diruthenium radical cations featuring diethynylaromatic bridging ligands. J Am Chem Soc 2011; 133(45): 18433-18446.
| Crossref | Google Scholar |
99 Gluyas JBG, Brown NJ, Farmer JD, Low PJ. Optimised syntheses of the half-sandwich complexes FeCl(dppe)Cp*, FeCl(dppe)Cp, RuCl(dppe)Cp*, and RuCl(dppe)Cp. Aust J Chem 2017; 70(1): 113-119.
| Crossref | Google Scholar |
101 McAuliffe CA, Parish RV, Randall PD. Gold(I) complexes of unidentate and bidentate phosphorus-, arsenic-, antimony-, and sulphur-donor ligands. J Chem Soc, Dalton Trans 1979; 1730-1735.
| Crossref | Google Scholar |
102 Eisenstadt A, Tannenbaum R, Efraty A. Convenient synthetic routes to the cyclopentadienylruthenium dicarbonyl chloride and bromide. J Organomet Chem 1981; 221(3): 317-321.
| Crossref | Google Scholar |
103 Al-Najjar IM. 31P and 195Pt NMR characteristics of new binuclear complexes of [Pt2X4](PR3)2] cis/trans isomers and of mononuclear analogs. Inorg Chim Acta 1987; 128(1): 93-104.
| Crossref | Google Scholar |
104 Krejčik M, Daněk M, Hartl F. Simple construction of an infrared optically transparent thin-layer electrochemical cell: applications to the redox reactions of ferrocene, Mn2(CO)10 and Mn(CO)3(3,5-Di-t-Butyl-Catecholate)−. J Electroanal Chem Interfacial Electrochem 1991; 317(1–2): 179-187.
| Crossref | Google Scholar |
105 Xu BQ, Tao NJJ. Measurement of single-molecule resistance by repeated formation of molecular junctions. Science 2003; 301(5637): 1221-1223.
| Crossref | Google Scholar |
106 Mészáros G, Li C, Pobelov I, Wandlowski T. Current measurements in a wide dynamic range – applications in electrochemical nanotechnology. Nanotechnology 2007; 18(42): 424004.
| Crossref | Google Scholar |