

Why does the synthesis of N-phenylbenzamide from benzenesulfinate and phenylisocyanate via the palladium-mediated Extrusion–Insertion pathway not work? A mechanistic exploration
Yang Yang A , Allan J. Canty B and Richard A. J. O’Hair A *
A *
A School of Chemistry, Bio21 Institute of Molecular Science and Biotechnology, The University of Melbourne, Vic. 3010, Australia.
B School of Natural Sciences - Chemistry, University of Tasmania, Private Bag 75, Hobart, Tas. 7001, Australia.
Australian Journal of Chemistry 76(1) 49-57 https://doi.org/10.1071/CH22209
Submitted: 27 September 2022 Accepted: 3 November 2022 Published: 16 December 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing.
Abstract
The gas-phase extrusion–insertion (ExIn) reactions of the palladium complexes [(phen)nPd(O2SC6H5)]+ (phen = 1,10-phenanthroline, n = 1 or 2), were investigated in the gas phase by multistage mass spectrometry (MSn) experiments consisting of electrospray ionisation and a linear ion trap combined with density functional theory (DFT) calculations. Desulfination of palladium sulfinate cations under collision-induced dissociation (CID) generates the organopalladium intermediates [(phen)nPd(C6H5)]+. Of these two organometallic cations, only [(phen)Pd(C6H5)]+ reacts with phenyl isocyanate via insertion to yield [(phen)Pd(NPhC(O)C6H5)]+. The formation of a coordinated amidate anion is supported by DFT calculations. In exploring this reactivity in the solution phase, we found that heating a mixture of benzenesulfinic acid, phenylisocyanate and palladium trifluoroacetate under a range of different conditions (ligand free versus with ligand, different solvents, addition of acid or base) failed to lead to the formation N-phenyl-benzamide in all cases. Instead, biphenyl was formed and could be isolated in a yield of 46%. DFT calculations using a solvent continuum reveal that the barrier associated with the insertion reaction lies above the competing sequential reactions of desulfination of a second phenyl sulfinate followed by reductive elimination of biphenyl.
Keywords: biaryl coupling, desulfination, DFT calculations, extrusion, insertion, mass spectrometry, palladium mediated reactions, reaction mechanisms.
Introduction
There has been considerable interest in developing transition metal catalysed reactions for organic synthesis that avoid a transmetalation step requiring the use of stoichiometric and often toxic organometallic/organometalloid reagents.[1] Thus alternative reagents that allow for formation of the key organotransition metal intermediate have been sought. Two key classes of alternative reagents have emerged as front runners: carboxylic acids, which undergo metal-catalysed decarboxylation reactions,[2–9] and sulfinic acids or their salts, which undergo related desulfination reactions.[10–13] Based on our gas-phase studies over the past two decades, where we have examined a wide range of metal-catalysed decarboxylation reactions[14–22] and some metal-catalysed desulfination reactions,[23,24] recent efforts have focussed on combining gas-phase (Scheme 1a) and solution-phase mechanistic studies coupled with DFT calculations to develop a new class of reactions for the synthesis of amides, thioamides, amidines and alkenes. These studies involve palladium-mediated/catalysed extrusion of CO2 to form an organopalladium intermediate followed by insertion of an appropriate (hetero)cumulene (Scheme 1b).[25–28] Given that these synthetic methods were limited to the use of 2,6-dimethoxybenzoic acid as a substrate, we recently explored the use of silver carbonate for the synthesis of N-phenyl-benzamide starting from benzoic acid and phenyl isocyanate.[29] While the desired ExIn mechanism operates in the gas-phase (Scheme 1c, Eqns 1, 2), a different base-catalysed condensation mechanism not requiring silver operates in solution. Given that phenylsulfinic acid has been shown to readily undergo desulfination by palladium complexes in both the gas-phase[24] and in synthetic protocols,[30,31] here we demonstrate that while the desired palladium-mediated ExIn mechanism operates in the gas-phase (Scheme 1c, Eqns 1, 2), a palladium-mediated biaryl coupling side reaction dominates in solution.[32–35]
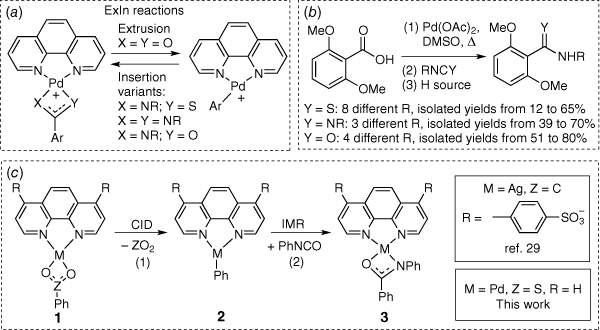
|
Results and discussion
Gas-phase formation of [(phen)nPd(C6H5)]+ via desulfination reactions and their reactions with phenyl isocyanate
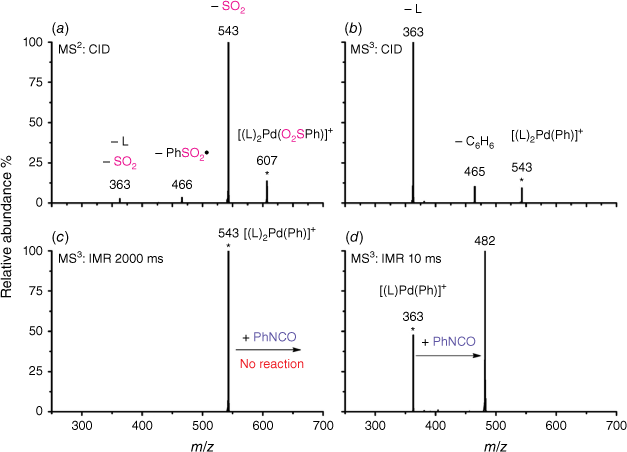
Electrospray ionisation (ESI) of a methanolic solution of 1,10-phenanthroline, palladium trifluoroacetate and benzoic acid gave rise to the cationic complexes, [(phen)nPd(O2SC6H5)]+. As noted previously, these complexes undergo desulfination under collision-induced dissociation (CID) conditions to form [(phen)nPd(C6H5)]+, as illustrated for [(phen)2Pd(O2SC6H5)]+ in Fig. 1a, Eqn 3. Another alternative route to prepare [(phen)Pd(C6H5)]+ in the gas phase is via ligand loss from [(phen)nPd(C6H5)]+ (Fig. 1b, Eqn 4). While [(phen)2Pd(C6H5)]+ was found to be unreactive towards phenylisocyanate in the gas phase (Fig. 1c), [(phen)Pd(C6H5)]+ undergoes an ion–molecule reaction (IMR) with phenylisocyanate to yield a product ion at m/z 482 (Fig. 1d, Eqn 5), a reaction previously observed for [(phen)Pd(C6H5)]+ formed via decarboxylation instead. The resultant [(phen)Pd(NPhC(O)C6H5)]+ (m/z 482) fragments undergo both deinsertion (Eqn 6) and loss of benzene (Eqn 7), as previously reported for the ExIn product formed via decarboxylation.[27]



|
Investigation of the palladium ExIn pathway for the solution-phase synthesis of amides
Encouraged by the gas-phase studies, suggesting a palladium-mediated stepwise extrusion of SO2 followed by insertion of phenyl isocyanate could be a viable approach to synthesise amides, the palladium-catalysed approach was explored using a one-pot method under ligand free conditions and in the presence of neutral ligands using different solvents (DMSO or NMP) and with and without additives (base or acid). As in our previous work, the crude reaction mixtures were analysed via GC-MS. No amide product was observed under a range of reaction conditions (all attempts are listed in Table 1). Instead, in all cases the dominant side product was the biaryl species arising from double desulfination followed by homocoupling. The formation of the desulfinated intermediate was detected by electrospray ionisation HRMS (entry 4 in Table 1, Fig. 2), which showed the arylpalladium complexes coordinated with one acetonitrile and one 6mbpy ligand and one with two 6mbpy ligands (at low abundance). However, the coordination with phenyl isocyanate was not detected, nor was the [M + H]+ ion of N-phenyl-benzamide. The GC-MS data revealed that there was still a significant amount of unreacted phenyl isocyanate while the ESI-HRMS revealed that some phenyl isocyanate has transformed into its urea analogue.

|

|
The formation of biaryl products is likely due to coordination of a second phenyl sulfinate to the arylpalladium followed by desulfination and reductive elimination of biphenyl. It is worth noting that related sulfinate coordinated arylpalladium intermediates have been formed via oxidative addition of aryl iodide into PCy3 ligated palladium(0) followed by coordination with a sulfinate anion.[35] The resultant binuclear palladium complex has sulfinates as bridging ligands and could be transformed to a mononuclear palladium complex via the addition of another equivalent of the ligand PCy3 (Scheme 2a). Both the binuclear and mononuclear palladium complexes were structurally characterised via X-ray crystallography (Scheme 2b, c). Unfortunately such species were not detected using ESI-MS as they have no net charge.

|
DFT exploration of the competition between insertion and the alternative side reaction involving desulfination of a second phenyl sulfinate followed by reductive elimination of biphenyl in the condensed phase
Having established that the gas-phase ExIn reaction occurs but that the biphenyl side product is formed in solution, we were interested in using DFT calculations to explore the mechanistic aspects (reaction pathways and energetics) associated with the biphenyl side product and to compare the energetics to that for the insertion of phenylisocyanate (Fig. 3). We first examined the relative stabilities of the three coordinate complex [(phen)Pd(C6H5)]+, 4, and the DMSO solvated complex, [(phen)Pd(C6H5)(S(O)Me2)]+, 5. The latter was found to be more stable and was thus used as the key complex to calculate the insertion versus biphenyl side reaction pathways. The key energy barriers for the insertion manifold are associated with the transition states for displacement of the DMSO ligand by the isocyanate ligand TS5–6 (17.8 kcal mol–1) and the insertion reaction TS6–7 (13.4 kcal mol–1). In contrast, replacement of the coordinated DMSO ligand with the anionic sulfinate proceeds with a low barrier of 2.4 kcal mol–1 (TS9–10) and is a highly exothermic reaction to form the highly stable S-coordinated palladium complex, 10. Even though the desulfination reaction through TS10–11 has a relatively high energy barrier of 29.4 kcal mol–1, it is lower than the energetics required for formation of 6 from 10 (33.9 kcal mol–1). Thus desulfination is the favoured pathway. After losing SO2, the homocoupling reaction of the double desulfinated palladium complex, 12, leads to the formation of a biaryl coordinated complex, 13, via a low energy barrier (8.9 kcal mol–1). The fact that both key transition states in the homocoupling pathway are lower in energy than the one from the insertion pathway supports the hypothesis regarding the failure for the amide synthesis in the condensed phase.

|
Why do we not observe the biaryl side product in the ExIn reaction using 2,6-dimethoxybenzoic acid as a substrate?
While we have observed the protodecarboxylation side reaction in our previous studies of ExIn reactions involving the 2,6-dimethoxybenzoic acid substrate,[25–28] we never observed the formation of the biaryl side product. Thus we were interested in establishing the energetics of the biaryl side reaction relative to the insertion of phenylisocyanate (Fig. 4). The key energy barriers for the insertion manifold are associated with the transition states for displacement of the solvent ligand by the isocyanate ligand TS5–6b (17.9 kcal mol–1) and the insertion reaction TS6–7b (10.9 kcal mol–1). In contrast, replacement of the coordinated solvent ligand with the anionic carboxylate requires more energy (TS9–10b, 20.6 kcal mol–1 relative to 5b), while the decarboxylation step through TS10–11 has an energy barrier of 28.6 kcal mol–1. After losing CO2, the homocoupling reaction of the palladium complex, 13b, leads to the formation of a biaryl coordinated complex, 14b, via an energy barrier (19.3 kcal mol–1). The fact that both key transition states in the homocoupling pathway are higher in energy than those from the insertion pathway is consistent with the lack of biaryl formation in the experiments.

|
Conclusions
Gas-phase studies provide valuable information on elementary steps relevant to organometallic chemistry used in organic synthesis. Here we have shown that desulfination reactions can be used to form the organopalladium intermediates [(phen)nPd(C6H5)]+ and that in the case of n = 1, phenyl isocyanate inserts to yield [(phen)Pd(NPhC(O)C6H5)]+. A key challenge we have found in translating these gas-phase ExIn reactions to solution phase protocols which produced the desired product in high yield is the formation of unwanted side products. Thus while gas-phase studies provide exquisite control in a ‘pristine environment’, they do not capture the rich milieux of the condensed phase where other reagents that are absent in the gas-phase can facilitate the formation of side products. In the case of decarboxylation of benzoates, a side reaction often encountered in the condensed phase is protodecarboxylation in which the arylorganometallic intermediate is protonated by an acid to form the arene. Here we have encountered a different side reaction in the desulfination ExIn approach: biaryl formation via double desulfination followed by reductive elimination. This series of reactions cannot be observed in our gas-phase studies since the organopalladium intermediate cation [(phen)Pd(C6H5)]+ cannot react with a second phenylsulfinate, C6H5SO2−. The DFT calculations on the solution phase competition between insertion of isocyanate and homocoupling via desulfination confirmed the experimental results. Lower energy barriers identified in the homocoupling reaction via double desulfination on the palladium centre account for the biaryl species being the dominant product in the condensed phase.
Experimental
Reagents
Reagents, purchased from various commercial sources, were used as received. Chromatographic silica media (Davisil, 40−63 μm), was used as the stationary phase in flash column chromatography.
Preparation of samples for mass spectrometry experiments
Ligated palladium cations, [(L)nPd(O2XR)]+, (n = 1 or 2, X = C or S) were subjected to CID to form aryl-palladium cations [(L)nPd(R)]+, which were then mass selected for subsequent ion–molecule reaction studies with phenyl isocyanate. We followed the protocols outlined in previous work.[20,21] For instance, methanolic solutions of palladium(ii) salt (10 mM), carboxylic acid or sodium benzenesulfinate (10 mM) and 1,10-phenanthroline (10 mM) were mixed based on a ratio of 1:1:2 and then diluted to 10 µM in palladium salt. A syringe pump (flowrate of 5 μL min−1) was used to inject the diluted solution into a modified linear ion-trap mass spectrometer (Thermo Finnigan LTQ) via the ESI source. The modified system allows ion–molecule reactions between mass selected ions and neutral molecules such as phenylisocyanate within the linear ion trap.[36,37] All spectroscopic data were acquired between 20 and 100 duplicate spectra with 3–5 microscans in each scan.
Source conditions used in MSn experiments
Source settings (where AU = arbitrary units)
The sheath gas setting was 10 AU, auxiliary gas was set to 5 AU, the sweep gas was 0 AU, the spray voltage was 4 kV, the capillary temperature was set to 250°C, the capillary voltage was 2 V and the tube lens voltage was set to 75 V.
CID conditions
The precursor ion was mass selected with a window of 1 m/z and subjected to collisional activation via collisions with the helium bath gas using a 10 ms activation time. The normalised collision energy (NCE) was set so as to achieve a precursor ion depletion to 10%.
IMR conditions
The precursor ion was mass selected with a window of 1 m/z and subjected to IMRs with phenylisocyanate. The NCE was set to 0% so as not to activate the precursor ion.
Synthetic procedures
General methods
1H and 13C NMR spectra were recorded using a Jeol 400 MHz NMR spectrometer at 298 K, and were referenced to the 1H shift in CDCl3 (7.24 ppm) and 13C shift in CDCl3 (77 ppm). All the NMR spectra are reported in parts per million (ppm) and coupling constants (J) are reported in Hertz (Hz). Multiplicities are recorded as: t = triplet, d = doublet, s = singlet.
High-resolution mass spectra (ESI-HRMS) were obtained on a Thermo Scientific Exactive Plus Orbitrap mass spectrometer (Thermo, Bremen, Germany) via positive ion ESI and were used to examine species present in reactions mixtures and to confirm the molecular formulas of purified isolated products.
GC-MS (Agilent 7890A/5975C GC-MS) analyses were carried out in an HP-5ms capillary column (Agilent Technologies, phenyl methyl siloxane, 30 m × 0.25 mm × 0.25 μm). To achieve a good separation, the time program was used by beginning with 5 min at 70°C, followed by a 15°C min–1 ramp to 300°C and then 10 min at this temperature.
General procedure for ExIn attempts
To a solution of palladium trifluoroacetate (0.1 mmol, 0.5 equiv.) and bidentate ligand (0.11 mmol, 0.55 equiv.) in DMSO or NMP (2 mL) was added sodium benzenesulfinate (0.2 mmol, 1 equiv.), phenyl isocyanate (0.4 mmol, 2 equiv.) and additive. The mixture was heated at 110°C for 2 h and quenched with 1 M HCl (1 mL) and water (50 mL), followed by liquid–liquid extraction with diethyl ether (3 × 50 mL). The combined organic fractions were washed with water (100 mL) and brine (100 mL) and dried over anhydrous MgSO4. The sample was then subjected to analysis via GC-MS as described above. In the case of entry 12 of Table 1, the solvent was removed, and the residue was purified by column chromatography to give the biaryl.
1,1′-Biphenyl (B)
1,1′-Biphenyl (B) was prepared using the general method (entry 12 of Table 1): yield 7 mg (46%) as white solid. Column chromatography (silica gel, diethyl ether/n-hexane: 1/10). 1H NMR (400 MHz, CDCl3): δ 7. 59 (d, J = 6.9 Hz, 4H), 7.44 (t, J = 7.3 Hz, 4H), 7.34 (d, J = 7.5 Hz, 2H). 13C NMR (400 MHz, CDCl3): δ141.34, 128.84, 127.34, 127.26. GC-MS (EI): m/z [M]+ calcd. for C12H10 154.1, found 154.1.
Molecular modelling
The Gaussian 16 suite of programs was used to fully optimise all reactants, intermediates, transition states and products at the M06 level of density functional theory (DFT).[38,39] The effective-core potential of Hay and Wadt with a double-ξ valence basis set (LANL2DZ) was used to describe Pd[40,41] and the 6-31G(d) basis set was chosen for the other atoms.[42] In addition, a polarisation function (ξf = 1.472) was solely added for Pd.[43,44] BS1 was used to designate this combination of basis sets. In order to account for the solvation effects (DMSO in Fig. 3 and DMA in Fig. 4) on the optimised structures the CPCM model was used.[45] Frequency calculations were carried out at the same level of theory as those for the structural optimisation. The Berny algorithm was used to locate each of the transition structures. Intrinsic reaction coordinate (IRC) calculations were used to confirm the connectivity between structures of transition states and minima.[46,47]
Single-point energy calculations were carried out to further refine the energies. Thus the energies of the structures obtained from the M06/BS1 calculations were recalculated with a larger basis set (BS2) at the B3LYP-D3BJ or CAM- B3LYP-D3BJ level of theory.[48–51] BS2 utilises def2-TZVP11 for all atoms along with the effective core potential including scalar relativistic effects for Pd.[52] The solvation effect of DMSO and DMA were also considered in the single-point calculations using the CPCM model. Relative enthalpy (ΔH) and Gibbs energies (ΔG) at the BS2 level of theory were calculated using the correction values calculated from M06/BS1. Based on the method reported by Okuno, extra corrections for entropy calculations were considered in the solvent system.[53] When DMSO or DMA participate in the equilibrium of a certain transformation step, an additional correction was considered based on the concentration of the DMSO or DMA using the method proposed by Keith and Carter (Eqn 6 of their paper was used).[54] Unless otherwise stated, all the enthalpy and Gibbs free energies were calculated and corrected from the B3LYP-D3BJ/BS2//M06/BS1 level of theory.
Supplementary material
Supplementary material is available online.
Data availability
The data that support this study are available in the article and accompanying online supplementary material.
Conflicts of interest
The authors declare no conflicts of interest.
Declaration of funding
The authors thank the ARC for financial support (DP180101187 funding to AJC and RAJO), and acknowledge a generous allocation of time from the National Computing Infrastructure.
Acknowledgements
The authors thank Prof. Uta Wille for access to the GC-MS. They thank the Bio21 Mass Spectrometry and Proteomics Facility for access to the Thermo Scientific Exactive Plus Orbitrap mass spectrometer. YY thanks The University of Melbourne for the award of a Ph.D. scholarship.
References
[1] O Baudoin, New Approaches for Decarboxylative Biaryl Coupling. Angew Chem Int Ed 2007, 46, 1373.| New Approaches for Decarboxylative Biaryl Coupling.Crossref | GoogleScholarGoogle Scholar |
[2] LJ Gooßen, K Gooßen, N Rodríguez, M Blanchot, C Linder, B Zimmermann, New catalytic transformations of carboxylic acids. Pure Appl Chem 2008, 80, 1725.
| New catalytic transformations of carboxylic acids.Crossref | GoogleScholarGoogle Scholar |
[3] LJ Gooßen, N Rodríguez, K Gooßen, Carboxylic acids as substrates in homogeneous catalysis. Angew Chem Int Ed 2008, 47, 3100.
| Carboxylic acids as substrates in homogeneous catalysis.Crossref | GoogleScholarGoogle Scholar |
[4] LJ Goossen, F Collet, K Goossen, Decarboxylative Coupling Reactions. Isr J Chem 2010, 50, 617.
| Decarboxylative Coupling Reactions.Crossref | GoogleScholarGoogle Scholar |
[5] JD Weaver, A Recio, AJ Grenning, JA Tunge, Transition Metal-Catalyzed Decarboxylative Allylation and Benzylation Reactions. Chem Rev 2011, 111, 1846.
| Transition Metal-Catalyzed Decarboxylative Allylation and Benzylation Reactions.Crossref | GoogleScholarGoogle Scholar |
[6] N Rodríguez, LJ Goossen, Decarboxylative coupling reactions: a modern strategy for C-C-bond formation. Chem Soc Rev 2011, 40, 5030.
| Decarboxylative coupling reactions: a modern strategy for C-C-bond formation.Crossref | GoogleScholarGoogle Scholar |
[7] J Cornella, I Larrosa, Decarboxylative Carbon-Carbon Bond-Forming Transformations of (Hetero)aromatic Carboxylic Acids. Synthesis 2012, 44, 653.
| Decarboxylative Carbon-Carbon Bond-Forming Transformations of (Hetero)aromatic Carboxylic Acids.Crossref | GoogleScholarGoogle Scholar |
[8] K Park, S Lee, Transition metal-catalyzed decarboxylative coupling reactions of alkynyl carboxylic acids. RSC Adv 2013, 3, 14165.
| Transition metal-catalyzed decarboxylative coupling reactions of alkynyl carboxylic acids.Crossref | GoogleScholarGoogle Scholar |
[9] XT Yin, WJ Li, BL Zhao, K Cheng, Research Progress on Silver-Catalyzed Decarboxylative Coupling Reaction. Chinese J Org Chem 2018, 38, 2879.
| Research Progress on Silver-Catalyzed Decarboxylative Coupling Reaction.Crossref | GoogleScholarGoogle Scholar |
[10] J Aziz, S Messaoudi, M Alami, A Hamze, Sulfinate derivatives: dual and versatile partners in organic synthesis. Org Biomol Chem 2014, 12, 9743.
| Sulfinate derivatives: dual and versatile partners in organic synthesis.Crossref | GoogleScholarGoogle Scholar |
[11] K Yuan, J-F Soulé, H Doucet, Functionalization of C–H Bonds via Metal-Catalyzed Desulfitative Coupling: An Alternative Tool for Access to Aryl- or Alkyl-Substituted (Hetero)arenes. ACS Catal 2015, 5, 978.
| Functionalization of C–H Bonds via Metal-Catalyzed Desulfitative Coupling: An Alternative Tool for Access to Aryl- or Alkyl-Substituted (Hetero)arenes.Crossref | GoogleScholarGoogle Scholar |
[12] DH Ortgies, A Hassanpour, F Chen, S Woo, P Forgione, Desulfination as an Emerging Strategy in Palladium-Catalyzed C–C Coupling Reactions. Eur J Org Chem 2016, 2016, 408.
| Desulfination as an Emerging Strategy in Palladium-Catalyzed C–C Coupling Reactions.Crossref | GoogleScholarGoogle Scholar |
[13] S Sun, J-T Yu, Y Jiang, J Cheng, Copper(I)-Catalyzed Desulfinative Carboxylation of Sodium Sulfinates using Carbon Dioxide. Adv Synth Catal 2015, 357, 2022.
| Copper(I)-Catalyzed Desulfinative Carboxylation of Sodium Sulfinates using Carbon Dioxide.Crossref | GoogleScholarGoogle Scholar |
[14] RAJ O’Hair, Dimethylargenate is a stable species in the gas phase. Chem Commun 2002, 38, 20.
| Dimethylargenate is a stable species in the gas phase.Crossref | GoogleScholarGoogle Scholar |
[15] PF James, RAJ O’Hair, Dimethyl cuprate undergoes C–C bond coupling with methyliodide in the gas phase but dimethyl argenate does not. Org Lett 2004, 6, 2761.
| Dimethyl cuprate undergoes C–C bond coupling with methyliodide in the gas phase but dimethyl argenate does not.Crossref | GoogleScholarGoogle Scholar |
[16] N Rijs, GN Khairallah, T Waters, RAJ O’Hair, Gas-phase synthesis of the homo and hetero organocuprate anions [MeCuMe]−, [EtCuEt]−, and [MeCuR]−. J Am Chem Soc 2008, 130, 1069.
| Gas-phase synthesis of the homo and hetero organocuprate anions [MeCuMe]−, [EtCuEt]−, and [MeCuR]−.Crossref | GoogleScholarGoogle Scholar |
[17] NJ Rijs, RAJ O’Hair, Gas-Phase Synthesis of Organoargenate Anions and Comparisons with Their Organocuprate Analogues. Organometallics 2009, 28, 2684.
| Gas-Phase Synthesis of Organoargenate Anions and Comparisons with Their Organocuprate Analogues.Crossref | GoogleScholarGoogle Scholar |
[18] NJ Rijs, GB Sanvido, GN Khairallah, RAJ O’Hair, Gas phase synthesis and reactivity of dimethylaurate. Dalton Trans 2010, 39, 8655.
| Gas phase synthesis and reactivity of dimethylaurate.Crossref | GoogleScholarGoogle Scholar |
[19] K Vikse, GN Khairallah, JS McIndoe, RAJ O’Hair, Fixed-charge phosphine ligands to explore gas-phase coinage metal-mediated decarboxylation reactions. Dalton Trans 2013, 42, 6440.
| Fixed-charge phosphine ligands to explore gas-phase coinage metal-mediated decarboxylation reactions.Crossref | GoogleScholarGoogle Scholar |
[20] MJ Woolley, GN Khairallah, G da Silva, PS Donnelly, BF Yates, RAJ O’Hair, Role of the Metal, Ligand, and Alkyl/Aryl Group in the Hydrolysis Reactions of Group 10 Organometallic Cations [(L)M(R)]+. Organometallics 2013, 32, 6931.
| Role of the Metal, Ligand, and Alkyl/Aryl Group in the Hydrolysis Reactions of Group 10 Organometallic Cations [(L)M(R)]+.Crossref | GoogleScholarGoogle Scholar |
[21] M Woolley, A Ariafard, GN Khairallah, KH Kwan, PS Donnelly, JM White, AJ Canty, BF Yates, RAJ O’Hair, Decarboxylative-Coupling of Allyl Acetate Catalyzed by Group 10 Organometallics, [(phen)M(CH3)]+. J Org Chem 2014, 79, 12056.
| Decarboxylative-Coupling of Allyl Acetate Catalyzed by Group 10 Organometallics, [(phen)M(CH3)]+.Crossref | GoogleScholarGoogle Scholar |
[22] RAJ O’Hair, NJ Rijs, Gas Phase Studies of the Pesci Decarboxylation Reaction: Synthesis, Structure, and Unimolecular and Bimolecular Reactivity of Organometallic Ions. Acc Chem Res 2015, 48, 329.
| Gas Phase Studies of the Pesci Decarboxylation Reaction: Synthesis, Structure, and Unimolecular and Bimolecular Reactivity of Organometallic Ions.Crossref | GoogleScholarGoogle Scholar |
[23] LO Sraj, GN Khairallah, G da Silva, RAJ O’Hair, Who Wins: Pesci, Peters, or Deacon? Intrinsic Reactivity Orders for Organocuprate Formation via Ligand Decomposition. Organometallics 2012, 31, 1801.
| Who Wins: Pesci, Peters, or Deacon? Intrinsic Reactivity Orders for Organocuprate Formation via Ligand Decomposition.Crossref | GoogleScholarGoogle Scholar |
[24] Z Wang, Y Yang, PS Donnelly, AJ Canty, RAJ O’Hair, Desulfination versus decarboxylation as a means of generating three- and five-coordinate organopalladium complexes [(phen)nPd(C6H5)]+ (n = 1 and 2) to study their fundamental bimolecular reactivity. J Organomet Chem 2019, 882, 42.
| Desulfination versus decarboxylation as a means of generating three- and five-coordinate organopalladium complexes [(phen)nPd(C6H5)]+ (n = 1 and 2) to study their fundamental bimolecular reactivity.Crossref | GoogleScholarGoogle Scholar |
[25] A Noor, JW Li, GN Khairallah, Z Li, H Ghari, AJ Canty, A Ariafard, PS Donnelly, RAJ O’Hair, A one-pot route to thioamides discovered by gas-phase studies: palladium-mediated CO2 extrusion followed by insertion of isothiocyanates. Chem Commun 2017, 53, 3854.
| A one-pot route to thioamides discovered by gas-phase studies: palladium-mediated CO2 extrusion followed by insertion of isothiocyanates.Crossref | GoogleScholarGoogle Scholar |
[26] Y Yang, A Noor, AJ Canty, A Ariafard, PS Donnelly, RAJ O’Hair, Synthesis of Amidines by Palladium-Mediated CO2 Extrusion Followed by Insertion of Carbodiimides: Translating Mechanistic Studies to Develop a One-Pot Method. Organometallics 2019, 38, 424.
| Synthesis of Amidines by Palladium-Mediated CO2 Extrusion Followed by Insertion of Carbodiimides: Translating Mechanistic Studies to Develop a One-Pot Method.Crossref | GoogleScholarGoogle Scholar |
[27] Y Yang, AJ Canty, AI McKay, PS Donnelly, RAJ O’Hair, Palladium-Mediated CO2 Extrusion Followed by Insertion of Isocyanates for the Synthesis of Benzamides: Translating Fundamental Mechanistic Studies To Develop a Catalytic Protocol. Organometallics 2020, 39, 453.
| Palladium-Mediated CO2 Extrusion Followed by Insertion of Isocyanates for the Synthesis of Benzamides: Translating Fundamental Mechanistic Studies To Develop a Catalytic Protocol.Crossref | GoogleScholarGoogle Scholar |
[28] Y Yang, B Spyrou, JM White, AJ Canty, PS Donnelly, RAJ O’Hair, Palladium-mediated CO2 Extrusion Followed by Insertion of Allenes: Translating Mechanistic Studies to Develop a One-Pot Method for the Synthesis of Alkenes. Organometallics 2022, 41, 1595.
| Palladium-mediated CO2 Extrusion Followed by Insertion of Allenes: Translating Mechanistic Studies to Develop a One-Pot Method for the Synthesis of Alkenes.Crossref | GoogleScholarGoogle Scholar |
[29] Y Yang, B Spyrou, PS Donnelly, AJ Canty, RAJ O’Hair, The role of silver carbonate as a catalyst in the synthesis of N-phenylbenzamide from benzoic acid and phenyl isocyanate: A mechanistic exploration. Aust J Chem 2022, 75, 495.
| The role of silver carbonate as a catalyst in the synthesis of N-phenylbenzamide from benzoic acid and phenyl isocyanate: A mechanistic exploration.Crossref | GoogleScholarGoogle Scholar |
[30] B Skillinghaug, C Sköld, J Rydfjord, F Svensson, M Behrends, J Sävmarker, PJR Sjöberg, M Larhed, Palladium(II)-Catalyzed Desulfitative Synthesis of Aryl Ketones from Sodium Arylsulfinates and Nitriles: Scope, Limitations, and Mechanistic Studies. J Org Chem 2014, 79, 12018.
| Palladium(II)-Catalyzed Desulfitative Synthesis of Aryl Ketones from Sodium Arylsulfinates and Nitriles: Scope, Limitations, and Mechanistic Studies.Crossref | GoogleScholarGoogle Scholar |
[31] M Behrends, J Sävmarker, PJR Sjöberg, M Larhed, Microwave-Assisted Palladium(II)-Catalyzed Synthesis of Aryl Ketones from Aryl Sulfinates and Direct ESI-MS Studies Thereof. ACS Catal 2011, 1, 1455.
| Microwave-Assisted Palladium(II)-Catalyzed Synthesis of Aryl Ketones from Aryl Sulfinates and Direct ESI-MS Studies Thereof.Crossref | GoogleScholarGoogle Scholar |
[32] K Garves, Coupling, Carbonylation, and Vinylation Reactions of Aromatic Sulfinic Acids via Organopalladium Intermediates. J Org Chem 1970, 35, 3273.
| Coupling, Carbonylation, and Vinylation Reactions of Aromatic Sulfinic Acids via Organopalladium Intermediates.Crossref | GoogleScholarGoogle Scholar |
[33] DH Ortgies, F Chen, P Forgione, Palladium and TEMPO as Co-Catalysts in a Desulfinative Homocoupling Reaction. Eur J Org Chem 2014, 2014, 3917.
| Palladium and TEMPO as Co-Catalysts in a Desulfinative Homocoupling Reaction.Crossref | GoogleScholarGoogle Scholar |
[34] B Rao, W Zhang, L Hu, M Luo, Catalytic desulfitative homocoupling of sodium arylsulfinates in water using PdCl2 as the recyclable catalyst and O2 as the terminal oxidant. Green Chem 2012, 14, 3436.
| Catalytic desulfitative homocoupling of sodium arylsulfinates in water using PdCl2 as the recyclable catalyst and O2 as the terminal oxidant.Crossref | GoogleScholarGoogle Scholar |
[35] A de Gombert, AI McKay, CJ Davis, KM Wheelhouse, MC Willis, Mechanistic Studies of the Palladium-Catalyzed Desulfinative Cross-Coupling of Aryl Bromides and (Hetero)Aryl Sulfinate Salts. J Am Chem Soc 2020, 142, 3564.
| Mechanistic Studies of the Palladium-Catalyzed Desulfinative Cross-Coupling of Aryl Bromides and (Hetero)Aryl Sulfinate Salts.Crossref | GoogleScholarGoogle Scholar |
[36] WA Donald, CJ McKenzie, RAJ O’Hair, C–H Bond Activation of Methanol and Ethanol by a High-Spin FeIVO Biomimetic Complex. Angew Chem Int Ed 2011, 50, 8379.
| C–H Bond Activation of Methanol and Ethanol by a High-Spin FeIVO Biomimetic Complex.Crossref | GoogleScholarGoogle Scholar |
[37] AKY Lam, C Li, G Khairallah, BB Kirk, SJ Blanksby, AJ Trevitt, U Wille, RAJ O’Hair, G da Silva, Gas-phase reactions of aryl radicals with 2-butyne: experimental and theoretical investigation employing the N-methyl-pyridinium-4-yl radical cation. Phys Chem Chem Phys 2012, 14, 2417.
| Gas-phase reactions of aryl radicals with 2-butyne: experimental and theoretical investigation employing the N-methyl-pyridinium-4-yl radical cation.Crossref | GoogleScholarGoogle Scholar |
[38] Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams; Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery Jr JA, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ. Gaussian 16 Rev. C.01. Wallingford, CT; 2016.
[39] Y Zhao, DG Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc 2008, 120, 215.
| The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals.Crossref | GoogleScholarGoogle Scholar |
[40] M Dolg, U Wedig, H Stoll, H Preuss, Energy-Adjusted Ab initio Pseudopotentials for the first Row Transition Elements. J Chem Phys 1987, 86, 866.
| Energy-Adjusted Ab initio Pseudopotentials for the first Row Transition Elements.Crossref | GoogleScholarGoogle Scholar |
[41] D Andrae, U Häußermann, M Dolg, H Stoll, H Preuß, Energy-Adjusted Ab initio Pseudopotentials for the second and third row transition elements. Theor Chim Acta 1990, 77, 123.
| Energy-Adjusted Ab initio Pseudopotentials for the second and third row transition elements.Crossref | GoogleScholarGoogle Scholar |
[42] PC Harihara, JA Pople, The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies. Theor Chim Acta 1973, 28, 213.
| The Influence of Polarization Functions on Molecular Orbital Hydrogenation Energies.Crossref | GoogleScholarGoogle Scholar |
[43] AW Ehlers, M Böhme, S Dapprich, A Gobbi, A Höllwarth, V Jonas, KF Köhler, R Stegmann, A Veldkamp, G Frenking, A set of f-polarization functions for pseudo-potential basis sets of the transition metals Sc, Cu, Y, Ag and La, Au. Chem Phys Lett 1993, 208, 111.
| A set of f-polarization functions for pseudo-potential basis sets of the transition metals Sc, Cu, Y, Ag and La, Au.Crossref | GoogleScholarGoogle Scholar |
[44] A Höllwarth, M Böhme, S Dapprich, AW Ehlers, A Gobbi, V Jonas, KF Köhler, R Stegmann, A Veldkamp, G Frenking, A set of d-polarization functions for pseudo-potential basis sets of the main group elements AlBi and f-type polarization functions for Zn, Cd, Hg. Chem Phys Lett 1993, 208, 237.
| A set of d-polarization functions for pseudo-potential basis sets of the main group elements AlBi and f-type polarization functions for Zn, Cd, Hg.Crossref | GoogleScholarGoogle Scholar |
[45] V Barone, M Cossi, Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model. J Phys Chem A 1998, 102, 1995.
| Quantum calculation of molecular energies and energy gradients in solution by a conductor solvent model.Crossref | GoogleScholarGoogle Scholar |
[46] K Fukui, Formulation of the Reaction Coordinate. J Phys Chem 1970, 74, 4161.
| Formulation of the Reaction Coordinate.Crossref | GoogleScholarGoogle Scholar |
[47] K Fukui, The Path of Chemical Reactions — the Irc Approach. Acc Chem Res 1981, 14, 363.
| The Path of Chemical Reactions — the Irc Approach.Crossref | GoogleScholarGoogle Scholar |
[48] AD Becke, Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior. Phys Rev A 1988, 38, 3098.
| Density-Functional Exchange-Energy Approximation with Correct Asymptotic-Behavior.Crossref | GoogleScholarGoogle Scholar |
[49] CT Lee, WT Yang, RG Parr, Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density. Phys Rev B 1988, 37, 785.
| Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron-Density.Crossref | GoogleScholarGoogle Scholar |
[50] AD Becke, Density-Functional Thermochemistry. III. The Role of Exact Exchange. J Chem Phys 1993, 98, 5648.
| Density-Functional Thermochemistry. III. The Role of Exact Exchange.Crossref | GoogleScholarGoogle Scholar |
[51] PJ Stephens, FJ Devlin, CF Chabalowski, MJ Frisch, Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J Phys Chem 1994, 98, 11623.
| Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields.Crossref | GoogleScholarGoogle Scholar |
[52] F Weigend, F Furche, R Ahlrichs, Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr. J Chem Phys 2003, 119, 12753.
| Gaussian basis sets of quadruple zeta valence quality for atoms H–Kr.Crossref | GoogleScholarGoogle Scholar |
[53] Y Okuno, Theoretical Investigation of the Mechanism of the Baeyer-Villiger Reaction in Nonpolar Solvents. Chem Eur J 1997, 3, 212.
| Theoretical Investigation of the Mechanism of the Baeyer-Villiger Reaction in Nonpolar Solvents.Crossref | GoogleScholarGoogle Scholar |
[54] JA Keith, EA Carter, Quantum Chemical Benchmarking, Validation, and Prediction of Acidity Constants for Substituted Pyridinium Ions and Pyridinyl Radicals. J Chem Theory Comput 2012, 8, 3187.
| Quantum Chemical Benchmarking, Validation, and Prediction of Acidity Constants for Substituted Pyridinium Ions and Pyridinyl Radicals.Crossref | GoogleScholarGoogle Scholar |