

Mutagenicity of N-acyloxy-N-alkoxyamides – QSAR determination of factors controlling activity
Stephen A. Glover A *
A *
A Department of Chemistry, School of Physical Sciences, University of New England, NSW 2350, Australia.
Australian Journal of Chemistry 76(1) 1-24 https://doi.org/10.1071/CH22205
Submitted: 23 September 2022 Accepted: 27 October 2022 Published: 19 January 2023
© 2023 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
This account describes the origins of our extensive investigations into the mutagenicity of N-acyloxy-N-alkoxyamides. Since their discovery as biologically active anomeric amides that mutate DNA in the Ames reverse mutation assay without the need for metabolic activation, we have used activities in the Ames test to understand the impact of structural variation on cellular access to, binding to and reactivity with DNA. We have developed an understanding of the roles played by hydrophobicity, electrophilic reactivity, steric effects and, importantly, intercalation on mutagenicity levels and therefore interactions with DNA. The evolution and application of meaningful quantitative structure–activity relationships is described, and examples of their utility in explaining molecule–DNA interactions are given. Their ability to explain previous mutagenicity data and, importantly, to predict meaningful mutagenic behaviour is also demonstrated.
Keywords: acyloxy alkoxyamides, ames mutagenicity, anomeric amides, bilinear QSAR, deamination, direct‐acting mutagenicity, DNA binding, groove binding, HERON reactions, intercalation, linear QSAR, mutagenic amides, nitrogen deletion, PAH, pyramidal amides, QSAR, quantitative structure‐activity relationship, skeletal editing, TA98, TA100.
Introduction
N-Acyloxy-N-alkoxyamides (NAAs), 2, are members of the unusual class of anomeric amides 1, which we defined as amides bearing two electronegative heteroatoms bonded to the amide nitrogen (Chart 1).[1] These have unusual properties. Firstly, through Bent’s Rule,[2,3] the electron demand of the electronegative substituents in 1 imparts more p character to the nitrogen hybrid orbitals bonded to them, with a concomitant increase in s character in the nitrogen lone pair orbital. Hence, these amides possess distinctly pyramidal nitrogens.[1,4–13] This, combined with the lower-energy lone pair, results in diminished amide resonance. Secondly, anomeric interactions between the nitrogen substituents results in unusual chemistry at the amide nitrogen.
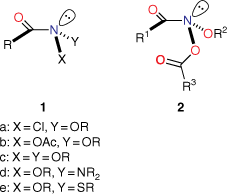
|
In the case of N-acyloxy-N-alkoxyamides 2, the nitrogen is bonded to two oxygen atoms and, as a result, they are strongly pyramidal at nitrogen, evidence for which stems from X-ray structures[7] as well as computational data.[8,9,13] Several NAAs we have made are the most pyramidal amides known.[7] The combined electronegativity results in a very significant reduction in resonance, estimated computationally to be ~50% that of the iconic standard amide N,N-dimethylacetamide, and manifests itself in longer amide bonds.[7,8,13] These structural properties are borne out spectroscopically, most notably by infrared spectroscopy where the amide carbonyl stretch frequencies are considerably higher than found in primary, secondary and tertiary amides. Typically, these are in the range of 1718–1742 cm−1 on average, some 50–54 cm−1 higher than their precursor hydroxamic esters.[8]
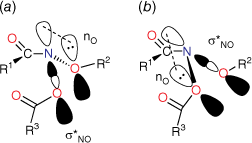
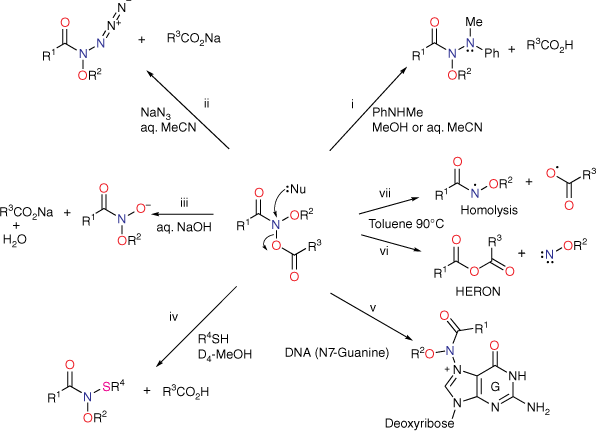
Although two anomeric interactions are possible in NAAs (Fig. 1), because the acyloxyl group would be more electron-demanding, the more favourable anomeric interaction at the amide nitrogen involves donation of the alkoxyl oxygen lone pair into the σ* orbital between nitrogen and the acyloxyl oxygen (nOR–σ*NOAcyl) for which there is structural and theoretical evidence.[7,8,13] The destablisation of the bond to the acyloxyl group renders NAAs susceptible to both unimolecular and bimolecular reactions at nitrogen. Under conditions of acid catalysis, they react by the unusual AAl1 mechanism, producing nitrenium ion intermediates.[14,15] However, they undergo SN2 reactions at nitrogen with a range of nucleophiles including amines (path i, Scheme 1),[16–20] azides (path ii, Scheme 1),[21] hydroxides (path iii, Scheme 1)[22] and thiols (path iv, Scheme 1),[23] each process producing new anomeric amide intermediates that react further. All these processes occur at ambient temperatures. At higher temperatures, NAAs undergo the novel HERON reaction, producing anhydrides and alkoxynitrenes in competition with homolysis (path vi and vii, Scheme 1).[24]
|
|
We originally investigated the chemical properties of NAAs as N-alkoxyl analogues of N-acetoxy-N-arylacetamides 4, one penultimate carcinogenic metabolite of aromatic amines 3 such as carcinogenic 2-acetylaminofluorene or 4-acetylaminobiphenyl (Scheme 2).[25–29] These produce by solvolysis N-acetyl-N-arylnitrenium ions 7, the ultimate electrophilic carcinogenic metabolites, which react with DNA.[30–33]

|
In foundation chemistry, we synthesised N-acetoxy-N-alkoxymides 5 for the first time from hydroxamic esters 6 by chlorination at nitrogen followed by reaction with silver acetate and found them to be mutagenic in the Ames test.[14,34,35] N-acyl-N-alkoxynitrenium ions 8 and N-acetyl-N-arylnitrenium ions 7 are similarly resonance-stabilised,[36,37] so by analogy with 4, we envisaged that 5 could undergo solvolysis to 8, which could react with nucleophilic centres in DNA (Scheme 2, dashed route). However, in vitro DNA damage studies demonstrated that NAAs bind and react intact with plasmid DNA primarily at G-N7 in the major groove and, to a lesser degree, at A-N3 in the minor groove (Scheme 2, solid arrow; path v, Scheme 1), rather than through the intermediacy of nitrenium ions 8.[38,39] Furthermore, both the acyloxyl and an alkoxyl substituent at nitrogen are determinants of reactivity with DNA.[38] Not surprisingly therefore, NAAs are direct-acting mutagens reverting the point mutation strain Salmonella typhimurium TA100 to wild type in the Ames reverse mutation assay without the need for metabolic activation.[34,40] Reactions with N-methylaniline have been widely studied kinetically to determine factors affecting SN2 reactivity, as this was deemed pivotal in their behaviour within the major groove of DNA.[16,18–20]
In the full-plate incorporation assay used throughout our Ames investigations, point mutations result in countable colonies,[41] and at the outset, we found that in nearly all cases the NAAs gave a linear dose response, the gradient of which afforded LogTA100, the log10 of induced revertants at 1 μmol per plate.[40] A comparison of activities at 1 μmol per plate enabled us to investigate factors that both promoted and inhibited their mutagenic activity. In all, we have determined comparative data for some 100 NAAs.
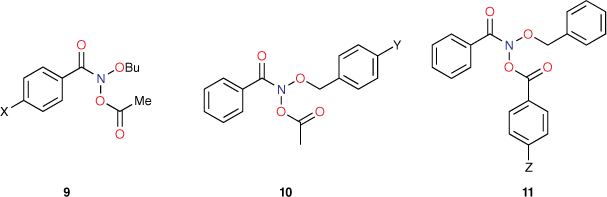
The Ames methodology involves some variability in testing medium and therefore background reversion rates. However, to counteract this, we tested every new compound in parallel with a single compound, N-butoxy-N-acetoxybenzamide (9, X = H). In this manner, all new data could be scaled to the activity of this standard. As a consequence, changes in LogTA100 could be sheeted home to alterations in structure rather than assay variability. This proved to be pivotal in determining structural influence on a range of cellular and intracellular processes. As the target giving rise to reverse mutation in bacterial TA100 must be cytosolic DNA, we have been able to use standardised Ames mutagenicity to deduce factors affecting the transport to, binding with and reactions with DNA from correlations between chemical structure and activity.
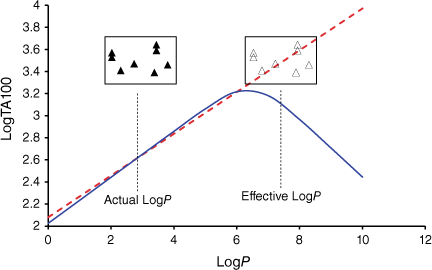
Fig. 2 illustrates the various intervening processes involved in the ultimate damage to DNA. Passage through the cell wall and associated equilibrium constant, KL, is governed by lipophilicity and is a function of logP, log10 of the octanol/water partition coefficient for the molecule. Reaction with cytosolic DNA must depend on binding in the major (G-N7) and minor (A-N3) grooves and be governed by binding constant KM, which must also depend, in part, on logP. However, KM must also be related to other factors such as steric bulk or the dimensions of the mutagen, or structural aspects that enhance binding to DNA such as intercalation. The final determinant of DNA damage is the rate constant for bimolecular reaction with the nucleophilic centres in DNA, kM, which can be dictated by factors affecting the electrophilicity of the amide nitrogen and conformation for SN2 attack at the amide nitrogen. In the course of our studies, we have encountered structural elements impacting on all of these processes.

|
A review of the mutagenicity of NAAs covering early correlations appeared in 2008 as part of a wider study of the structure, reactivity and biological activity of NAAs and led to the first working quantitative structure–activity relationship (QSAR).[8] This account serves to build on this and to demonstrate applications of higher-level QSARs to gain insights into transport, DNA binding and reactivity of NAAs with DNA.
Development of a linear QSAR
As with most drug interactions, the hydrophobicity of NAAs is a critical determinant of activity.[43–48] In this host–guest relationship, hydrophobicity is important for binding in the hydrophobic grooves of DNA. Hence, logP is an important molecular property. Throughout our studies on NAAs, we have computed this according to the Ghose–Crippen model,[49] values for which are readily determined in most computational chemistry packages.
|
The importance of steric effects will be discussed at a later point, but initial mutagenicity data from series of mutagens bearing para-substituted phenyl groups on all three side chains (9–11, Chart 2) indicated that, whereas logP for para-tert-butylphenyl-substituted systems (X, Y and Z = But) was much larger than for para-methylphenyl-substituted systems (X, Y and Z = Me), activities as determined by LogTA100 were lower in each case. Similarly, para-phenyl groups (X, Y and Z = Ph) with much larger logP afforded only modest increases in LogTA100.[50] In development of QSARs, Taft steric parameters for para-phenyl substituents were deemed to be representative of these steric effects and, where relevant, were incorporated as Es1, Es2 and Es3 for para substituents on benzamide, benzyloxy and benzoyloxy phenyl rings respectively.[43,51]
Reactivity at the amide nitrogen was seen to be a significant determinant of activity. As substituents on a benzamide or benzyloxyl sidechains are fairly remote from the reactive nitrogen, they were not expected to impact measurably on activity. In the case of a series of para-substituted N-acetoxy-N-butoxybenzamides 9, this was borne out by relative rates of SN2 reactivity with N-methylaniline, which correlated very weakly with Hammett σ constants (reaction constant ρ = 0.13).[18,19] The electronic effect of para-substituents on a benzoyloxyl group in N-alkoxy-N-benzoyloxyamides was very different. Bimolecular rate constants for the reaction of N-methylaniline with a series of N-benzyloxy-N-(para-substituted-benzoyloxy)benzamides 11 correlated strongly with Hammett σ constants (ρ = 1.69)[18,19] in support of the modelled charge-separated SN2 transition state, which invokes partial negative and positive charge on the leaving group and nucleophilic nitrogen respectively.[8,16,17,19] In addition, we demonstrated that rates constants for SN2 reaction of a range of N-alkanoyloxyl and benzoyloxyl amides with N-methylaniline, k303, correlate negatively with pKa of the departing carboxylic acid group (Eqn 1).[16] However, mutagenicity of a series of N-benzyloxy-N-(para-substituted-benzoyloxy)benzamides correlated negatively with Hammett σ constants (ρ = –0.57)[22] or positively with the pKa of the carboxylic acid (Eqn 2); the more reactive an NAA, the greater the probability that it will react with adventitious intracellular nucleophiles prior to binding to DNA, thereby lowering the effective concentration.
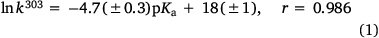
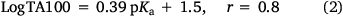
Several earlier QSARs based on limited data[8,50] culminated in a QSAR based on logP, pKa of the leaving group and Taft Es1, Es2 and Es3, which performed well for 50 analogues (Eqn 3, Fig. 3, open squares).[52] correlation coefficient, r, standard error, s, F-statistic and the leave-one-out cross validation index, Q2, all point to reasonable predictive ability.
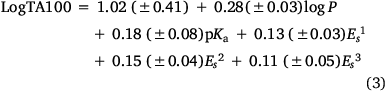
n = 50, r = 0.89, s = 0.16, F = 34, LOOCV Q2 = 0.70.
|
Hydrophobicity and development of a bilinear QSAR
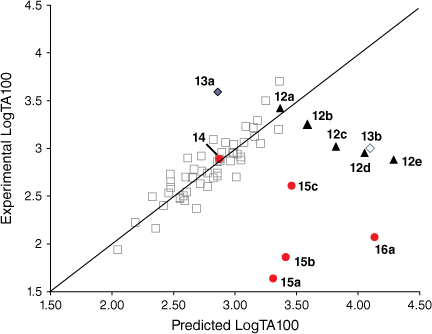
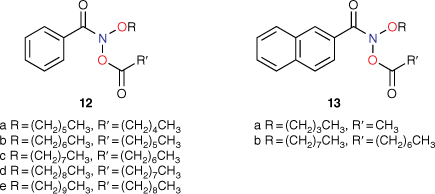
At the time of publication of such relationships, it was deemed unusual for direct-acting mutagens to show a logP dependence because binding to activating enzymes in metabolism was not important.[43,45,53] Where mutagens or other molecules require metabolic activation, the role of structural change and logP becomes less clear owing to its effect on binding to enzymes, which may be the determining factor. The modest dependence on logP is clear cut in the case of these direct-acting mutagens and relates to binding to DNA. These molecules are small and the bacterial strains developed by Ames are altered by the rfa mutation, rendering their cell walls more permeable to bulky molecules.[41,54] Drug-like compounds are more than likely to have a logP < 5 and, similarly, direct-acting mutagens acting on Salmonella typhimurium would be expected to conform to this rule.[55] However, in a series of NAAs bearing long aliphatic chains 12 (Chart 3), a significant deviation from the predicted LogTA100 was observed.[52] Moreover, the deviation was logP-dependent (Fig. 3, solid triangles, Table 1). Whereas the activity of 12a was well predicted by the QSAR in Eqn 3, activities of 12b–e deviated progressively from their predicted values with increasing logP. Though unrelated to the benzamides, the deviation from the predicted activity for the naphthamide 13b bearing long aliphatic chains on the alkoxyl and acyloxyl group was also significant (Fig. 3, open diamond).

|
|
The decrease in mutagenicity relative to their predicted activity can be ascribed to lipid entrapment through membrane localisation and a reduced KL (Fig. 2) resulting in lower concentrations of mutagens available to interact with cytosolic DNA.[43] With respect to the mutagenicity of N-acyloxy-N-alkoxybenzamides, there is clearly an optimum logP value in the range of logPs covered by mutagens 12a–e.
Several treatments are possible to generate QSARs catering for drugs covering a wide range of logP. To establish a QSAR that encompasses the activities of increasingly hydrophobic mutagens 12a–e, we chose Kubinyi’s bilinear model.[52,56–58] In Eqn 4, β is a non-linear term calculated by an iterative procedure. In cases where logP is sufficiently small, βP becomes negligible and therefore the log(βP + 1) term decreases to zero, converting the model to the linear form (Eqn 3). We determined that a logP0 of 6.4 is the optimum logP for activity and, by Kubinya’s method, can be derived from logP and log(βP + 1) coefficients, a and b, from the regression analysis using Eqn 5.[52,56–58] Experimental and predicted LogTA100 from the bilinear model in Eqn 4 are given in Table 1 and are in good agreement when compared with the result from the linear model in Eqn 3. It will be reported in the section on ‘Naphthalene as an intercalator’ that a naphthalene substituent increases binding to DNA through intercalation. As illustrated in Fig. 3, the activity of 13a (closed diamond), given here for comparison,[40] is significantly higher than predicted, whereas that of the lipophilic 13b is strongly suppressed (Fig. 3, open diamond). However, the activity of 13b is well reproduced by Eqn 4, indicating that the activity enhancement due to the naphthamide structure is largely negated owing to lipid entrapment. KL in Fig. 2 is dominant and limiting in this case.
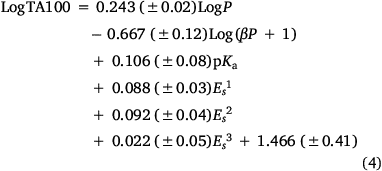
Logβ = −6.639, n = 55, r2 = 0.75, adj. r2 = 0.71, s = 0.18, F = 19.7; LOOCV Q2 = 0.60.
Steric effects on activity
Distal steric effects in triarylated mutagens bearing tert-butyl groups[59]
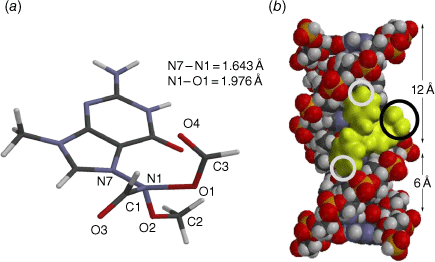
Steric effects would be expected to impact on activity levels in several ways; they could influence the capacity of NAAs to bind in the hydrophobic grooves of DNA (reducing KM) or, in the bound state, they could inhibit reaction with nucleotides (reducing kM). Previous investigations found that mono tert-butylated mutagens (9, X = But, 10, Y = But, 11, Z = But) were well predicted by an earlier QSAR.[59] Activity of the di-tert-butylated diaryl mutagen 14 (Chart 4) was also well predicted by the linear and bilinear QSARs in Eqns 3, 4 (Fig. 3, Table 2). For a series of di-tert-butylated triaryl mutagens 15a–c and 16a (Chart 4), we encountered significantly diminished activity with the linear QSAR in Eqn 3 (Fig. 3, filled circles, Table 2) and although dose responses were evident, with LogTA100 > 2, only 15c can be regarded as mutagenic. No dose response could be obtained with tri-tert-butylated substrates 15d and 16b, which must be regarded as non-mutagenic towards TA100. We originally ascribed this reduced activity to the dimensions of these triarylated systems, which from X-ray and computed structures are from end to end between 14 and 17 Å, at least of the order of the width of the major groove of DNA (12 Å).[8,59] However, these substrates have logP values of 8.24, larger than the logP0 for NAAs (logP0 = 6.4). The bilinear QSAR in Eqn 4 corrects predicted values in relation to their high hydrophobicity and it is found that 15c is well predicted (Table 2); the deviation from expected activity from Eqn 3 is entirely due to the increase in hydrophobicity in this case. On the other hand, activities of mutagens 15a–b and 16a still deviate strongly from predicted values after taking their increased hydrophobicity into account, which reinforces the groove exclusion argument for these substrates. As a tert-butylbenzyloxy group is common to all triarylated mutagens with greatly reduced activity, it would appear to be a limiting feature, whereas tert-butyl groups on the benzoyloxyl group may be less critical. From models, it is also possible that the transition state complex for SN2 reaction at G-N7 can only be achieved with one para tert-butyl group.[8] Fig. 4a depicts the AM1-optimised transition state for the reaction of model N-formyloxy-N-methoxyformamide with G-N7. Modelling this into the major groove of a segment of DNA with 15e (Fig. 4b) indicates severe restrictions for tert-butyl groups on the benzoyloxy and benzamide rings, which would need to make adverse contact with the wall of the major groove.

|
|

|
Proximal steric effects on the amide side chain[59]
Branching adjacent to the amide carbonyl in NAAs has a profound effect on the mutagenic response. Whereas acetamide 17a is well predicted, propanamide 17b, 2-methylpropanamide 17c, 1-adamantanecarboxamide 17e and neohexamide 17f gave weak responses deviating significantly from the predicted activities from the linear QSAR in Eqn 3. Deviations from predicted values increased through the series CH3 < Et < Pri < But. The tert-butyl side chain in 17d resulted in no response at all. With adamantyl and neopentyl side chains (17e and 17f), experimental activities deviated strongly from predicted activities (Fig. 5, filled diamonds; Table 3). The results are broadly in line with the rates of SN2 reactivity with N-methylaniline at 303 K in [D4]methanol where amides with branched amide side chains in 17c, 17d and 17e were unreactive (Table 3). Branching α to the carbonyl is also known to impede SN2 displacement of chlorine in α′-haloketones.[60] Mutagenicity, which must involve reaction of DNA at G-N7, was observed, albeit weak in all but the 2,2-dimethylpropanamide substrate (17d). We have attributed this difference to a greater ease of SN2 reaction in the hydrophobic groove of DNA; in solution, SN2 transition states for reaction with amines are strongly charge-separated, resulting in large negative entropies of activation due to solvent reorganisation.[8,16–18] However, in the hydrophobic grooves of DNA, a similar transition state would not be adversely affected by negative entropy of activation, making SN2 reactivity in the grooves of DNA easier than in solution. It is clear that in this series, mutagenic activity is limited by kM in Fig. 2

|
Proximal steric effects on the alkoxyl side chain[61]
Branching adjacent to the alkoxyl oxygen and the presence of bulky benzyloxy groups in series 18 has almost no effect on mutagenic activity and the activity of all mutagens was well predicted by the linear QSAR in Eqn 3 (Fig. 5 – filled squares, Table 3). Their activities are controlled by their logP and pKa values, which impact on KM and kM in Fig. 2. These are all small molecules, so groove binding would not be expected to be limiting and bulkiness at the alkoxyl group does not appear to impede SN2 reactivity at G-N7. The rates of SN2 reactions with N-methylaniline in [D4]methanol have also been measured for this series and rate constants at 303 K are somewhat smaller than their straight-chain n-butoxy analogue 19a (Table 3).[16] 18c, with a tert-butoxy group, was unreactive but the steric interference is not as severe as is found with branching on the amide side chain, which we attribute to greater flexibility on the alkoxyl side chain. Clearly though, a reduction in the ease of SN2 reactivity with branching does not manifest itself in the mutagenic response. Once again, the factors controlling reactivity in the hydrophobic grooves of DNA appear to be somewhat different from those governing SN2 reactions with amines in solution.
Steric effects on the acyloxyl side chain[61]
A series of mutagens bearing bulky substituents adjacent to the acyloxyl carbonyl in 19 demonstrated a systematic suppression of the mutagenic activity relative to the predicted activities from Eqn 3 (Fig. 5, filled triangles; Table 3). All have small logP values and ought not to be subject to lipid entrapment or restricted access to the major groove. Although for a range of such compounds rates of reactivity with N-methylaniline correlated negatively with pKa (Eqn 1),[16] mutagenic activities were suppressed by an average of 0.59 LogTA100. While mutagen 19a with an acetoxyl leaving group (in training set) and to a degree 19b with a butanoyloxyl group are predicted well, the effect of branching on LogTA100 as in 19c–g is significant, with the interference of isopropyl (19c) and tert-butyl groups (19g) being the greatest. This reflects steric hindrance to reaction with DNA, possibly through inability to achieve a conformation suitable for reaction at G-N7 (lowering kM in Fig. 2). It is possible that, unlike groups on the alkoxyl side chain, the acyloxyl group must make unavoidable contact with the wall of the major groove. A comparison of data for the benzoyloxy substrate 19j with the dimethylated analogues reinforces this. Although 19j was slightly overpredicted, the 2,6-dimethylphenyl side chain in 19i strongly inhibits activity (Table 3). The effect is reduced when the methyl groups are on the 3- and 5-positions in 19h. Clearly, in 19i the twisting that would be required to minimise steric interference with the carbonyl must have an influence (Fig. 6).
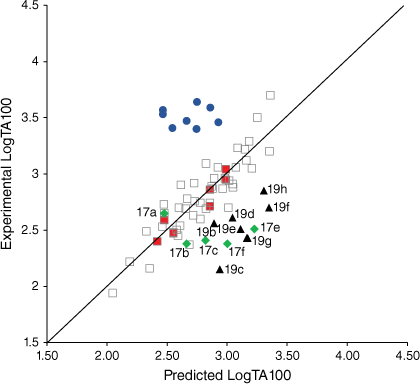
|
Intercalation of fused polycyclic aromatics
Naphthalene as an intercalator
N-Acetoxy-N-butoxy-2-naphthamide mutagen 13a was part of an earlier study in which we demonstrated an increased degree of DNA damage relative to other NAAs with similar logP.[38] Furthermore, 13a exhibited mutagenic activity nearly an order of magnitude higher than predicted by the QSAR in Eqn 3 (Fig. 3, solid diamond).[40] This, together with a series of naphthalene-containing NAAs 20a–g (Chart 5), all showed a remarkably consistent increased activity relative to predicted LogTA100 levels based on the QSAR in Eqn 3, with most differences between experimental and predicted activities close to 1 LogTA100 (Fig. 5, filled circles; Table 4).[42] Futhermore, the enhanced activity was evident irrespective of the regiochemistry, the locus of substitution on the naphthalene nucleus, or the length of aliphatic tethers where present. We attributed this enhancement to an increase in residence time on DNA through the capacity of the planar naphthalene to intercalate with the DNA bases.
|

|
|
Naphthalene has not generally been regarded as an intercalator, a property reserved for larger, fused polyaromatic systems with greater surface areas.[62–64] The ring system should possess a minimum of three rings to give an optimum surface area of more than 28 Å.[65] Various theories have been proffered regarding the capacity of planar aromatic systems to intercalate and the presence of positively charged side chains or acyl substitution was deemed important for naphthalene intercalation.[66,67] The intercalation of naphthalene mono- and di-imides, established threading intercalators, is well known.[68–70] Although in only two of the set studied, 13a and 20a, was naphthalene acyl-substituted, the remainder being bonded to methylene, it is hard to ascribe this increased activity to any process other than intercalation. These molecules have very modest computed logPs and no special features above those of similarly constituted benzamide systems in the training set. However, a modest, reversible ability to intercalate into DNA would increase residence time in the grooves of DNA, thereby increasing KM in Fig. 2.
TA98 is a frameshift strain derived from Salmonella typhimurium that is used to detect intercalation. In effect a base deletion results in a reading error and a failure to proliferate in the plate media used in Ames assays. The space requirement for an intercalated polycyclic aromatic hydrocarbon(PAH) (3.4 Å) is similar to that required by a single base pair,[63,71–76] and intercalation of a PAH between the bases of DNA reinstates the wild-type reading frame in S. typhimurium through an a + 1 frameshift.[41,76,77] Furthermore, such restoration is particularly effective (10–100-fold increase) when the intercalative group is associated with an electrophilic centre, reaction through which can localise the intercalating group.[76–79] Importantly, where the intercalator is dissociated in the reaction with DNA, the duality requirement is lost, resulting in a weak TA98 response. With their electrophilic amide nitrogens, NAAs appeared to be ideal candidates to effect such frameshifts. We subjected naphthalene-bearing mutagens 13a, 20a, 20d and 20g to TA98 tests and found linear dose responses and significant LogTA98 at 1 μmol per plate for 13a (490 revertants; average of six tests), 20d (287 revertants) and 20g (114 revertants), which was not evident in the control systems N-acetoxy-N-butoxybenzamide 19a and N-benzoyloxy-N-butoxyacetamide 17a, the phenyl analogues of 13a and 20a or naphthalene-bearing systems without electrophilic nitrogen such as 21a–b, both of which produced no dose response.[42] 20a, where the intercalator dissociates from the mutagen in reaction with DNA, resulted, predictably, in a weak response (15 revertants) in TA98.
We consider this clear evidence for intercalation of naphthalene moieties.
To account for this activity enhancement, we regenerated a linear (Eqn 6) and a corresponding bilinear (Eqn 7) QSAR to enable prediction of this intercalative ability by the introduction of an indicator variable, I, which is given the value 1 if a naphthalene is present, otherwise 0.
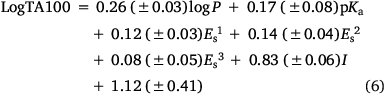
n = 58, R2 = 0.85, adj. R2 = 0.83, s = 0.16, F = 48.2; LOOCV Q2 = 0.85.
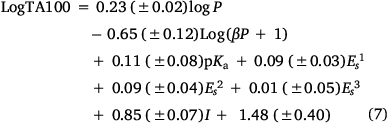
Logβ = −6.705, n = 63, R2 = 0.81, adj. R2 = 0.79, s = 0.18, F = 29.6; LOOCV Q2 = 0.76.
From Eqn 5 and the coefficients in Eqn 7, a revised LogP0 value of 6.44 can be calculated, slightly higher than 6.37 calculated from Eqn 3.
A comparison of QSAR results for 13a, 20a–g from Eqn 3 with those from revised linear and bilinear QSARs in Eqns 6, 7 using I = 1 is presented in Table 4. The differences between experimental and predicted mutagenicities are significantly diminished when compared with predictions from the linear QSAR in Eqn 3. Correlations between the predicted and experimental LogTA100, based on the new QSAR in Eqn 6, are illustrated in Fig. 7 (training set, filled squares).
As the correlations with logP and I in Eqn 6 are highly significant (P- values of 10−13 and 10−17 respectively), the respective coefficients of 0.26 and 0.83 indicate that the attachment of one naphthalene group increases activity equivalent to between 3 and 4 logP. Although the logP of 13a and 20a–g (logP 2.0–3.8) falls below the logP0 for NAAs (6.44 from Eqn 7), they exhibit mutagenic activity that is equivalent to mutagens with an ‘effective’ logP ranging between logP = 5 and 7. This can be readily seen from a plot of linear (Eqn 8) and bilinear logP dependence (Eqn 9) over the range logP = 0–10 (Fig. 8), where the boxes reflect actual and effective logP for this series of naphthalene-bearing NAAs.[42]
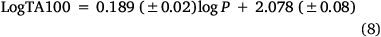
n = 55, R2 = 0.677, s = 0.19, F = 98.5.
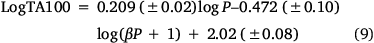
Logβ = −6.466, n = 55, R2 = 0.677, s = 0.19, F = 35.55954, logP0 = 6.37.
|
This finding that a naphthalene enhances the activity of NAAs to the tune of between 3 and 4 logP units by its hitherto unknown intercalative ability is an important new discovery. Presumably, the DNA targeting function of naphthalene is not unique to NAAs. The Lipinski Limit is a hydrophobicity above which lipid entrapment diminishes the activity of a drug.[55] A drug with logP close to the Lipinski Limit for that type of drug, but bearing a simple naphthalene substituent, can pass through the lipid membrane, yet exhibit binding to DNA equivalent to a molecule with a substantially higher hydrophobicity.
The role of naphthalene appears not to be unique. We have recently found that Eqn 6 (logP < 6.4) or Eqn 7 (logP > 6.4) can be used to differentiate between molecular substructure capable of intercalation into DNA and substructure that does not intercalate, based on the fit of measured LogTA100 with I = 1 or 0. If the fit to the QSAR is best with I = 0, the structure is a groove binder with mode of action similar to most of the mutagens we have studied. If the fit is best with I = 1, such molecules target DNA through a degree of intercalation akin to that of naphthalene-bearing mutagens.
Three substructures demonstrate this principle: NAAs bearing the biphenyl moiety, the fluorene ring system and the fluorenone group, which are related. In fluorenes, the biphenyl rings are bridged by a methylene, making fluorene groups largely planar but not cross-conjugated as is the case of fluorenones, where the rings are bridged by a carbonyl (Chart 6).
|
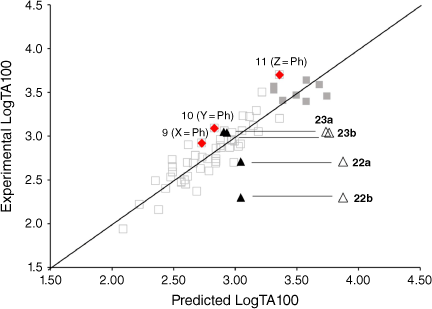
Measurement of TA100 activity for NAAs with potential intercalators incorporated into side chains was instructive. Firstly, biphenyl systems on all three side chains (Fig. 7, filled diamonds) have previously been shown to conform to linear QSARs and are part of the training set, which now incorporates eight naphthalene-bearing NAAs (Fig. 7, filled squares). The biphenyl group does not target DNA beyond its hydrophobic influence.
|
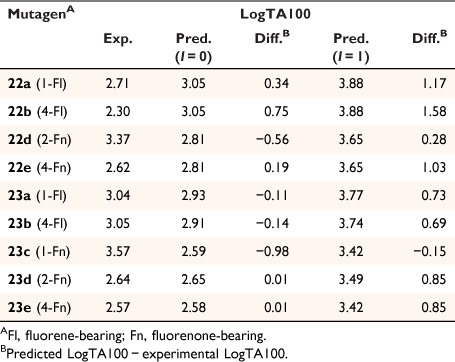
The activity of the four fluorene-bearing mutagens (Chart 7) is greatly over-predicted with I = 1 (Fig. 7, open triangles, Table 5).[80] Activities of 23a, 23b are well predicted and 22a is adequately predicted with I = 0. Activity of 22b would appear to be even lower than predicted with I = 0 (Fig. 7, filled triangles; Table 5). It would appear that all four NAAs bearing fluorene are not targeted to DNA through intercalation, as was the case with naphthalene. These structures possess an aliphatic bridge and no cross-conjugation, which may result in poor π–π stacking and a lack of polarisability. We ascribed the much diminished activity of 22b to the fact that the pendant group is directed into the bay region of the fluorene, resulting in more difficult access to nucleophilic DNA components. The same is true for 23b and both side chains would be twisted out of plane of the ring system. However, there is less flexibility in the pendant group in 22b.
|

|
22b, 23a and 23b gave negative results in the TA98 screen. Surprisingly, 22a gave a reasonably positive result (246 induced revertants at 1 μmol per plate) though it showed no enhancement in TA100 and in fact was underpredicted using the indicator variable I = 0. We have ascribed this to oxidation of the fluorene to the fluorenone 22c in the plate incubation or, alternatively, selective metabolism to 22c by bacterial S. typhimurium TA98.[80]
4-Aminobiphenyl and 2-aminofluorene are well known carcinogens where the metabolic route involves cytochrome P450 oxidation of the nitrogen, ultimately leading to electrophilic nitrenium ion formation, as illustrated in Scheme 2.[25–31] TA98 studies, and studies with other frameshift-sensitive strains, have implicated metabolites of these compounds as intercalators.[76,78] Our results point to the fact that such intercalation is a function of the binding of the derived metabolites, the aryl nitrenium ions. The biphenyl or fluorene aromatic ring systems do not appear to promote intercalation.
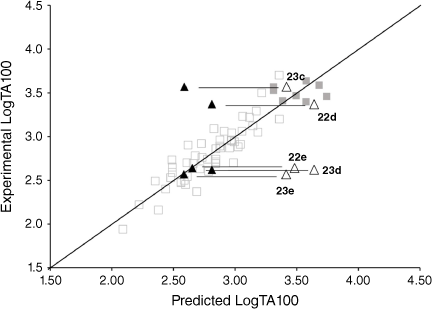
Fluorenone derivative 22c could not be synthesised by existing protocols,[80,81] but the remaining fluorenone-bearing compounds (Chart 8) yielded a clearcut result. Activities of two of the fluorenone substrates, 22d and 23c, are clearly well predicted when I = 1 and those of the other three, 22e and 23d–e, are greatly overpredicted (Fig. 9, Table 5). However, activities of these three are very well predicted with I = 0.[80] Clearly, two of the NAAs bearing fluorenone, like naphthalene derivatives, benefit from intercalation, whereas the remainder do not.
|
Substitution at the 2- (in 22d) and the 1-position (23c) appears to facilitate intercalation. In both these, the tricylic conjugated system can most probably penetrate adequately between base pairs. Of the three systems that did not report enhanced activity, 22e and 23e have the pendant active centre directed towards the distal ring in the bay region. In such systems, the intercalation would have to be edge-on rather than end-on and, presumably, would lead to a poor intra-base penetration and π–π stacking ability.
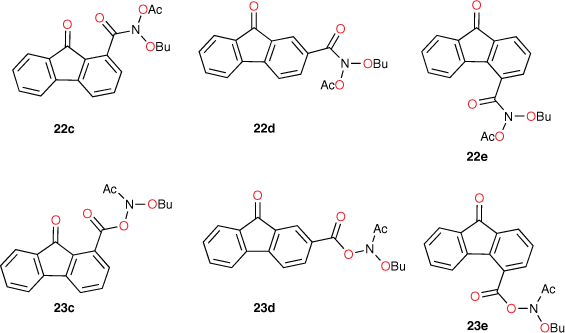
|
23d appears not to intercalate even though it could do so in an end-on fashion. We have argued that the difference between 23d and intercalating 22d may lie in different electronic effects owing to the fact that in 23d, the fluorenone is bonded through the 2-position to the ester carbonyl whereas in 22d, it is bonded to the amide carbonyl although the argument is by no means a compelling one.[80]
Fig. 10 shows AM1-optimised ground state geometries for 22c–e and 23c–e. It is clear, when comparing 22d with 22e, and also 23c with 23e that the accessible surface area of the tricyclic systems in 22e and 23e, where the pendant group is in the bay region, would be smaller than in 22d and 23c, which can bind in an end-on fashion.

|
TA98 studies confirmed that in keeping with its TA100 result, 22d is a strong intercalator (TA98 = 1458 revertants at 1 μmol per plate).[80] In accord with the dissociation arguments above, 23c (a strong intercalator from TA100 studies) gave a weak response through disconnection on reaction, as did 22e and 23d–e, which were not predicted to intercalate by TA100. Though we were unable to synthesise 22c, it is possible that it was generated from fluorene 22a by oxidation during the TA98 procedure.
1-Substituted naphthalenes and intercalation
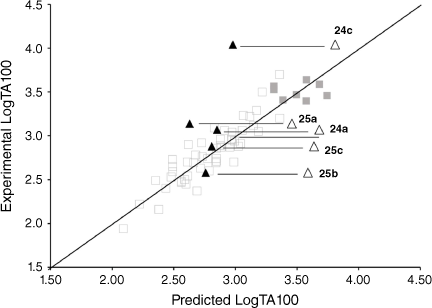
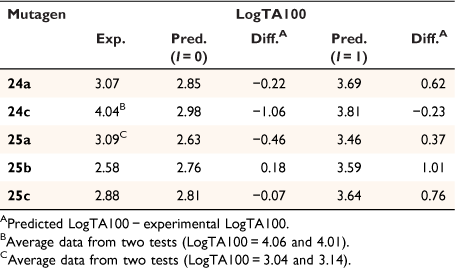
Application of our QSAR would appear to differentiate not only between substructure promoting intercalation or otherwise, but also between modes of intercalation. A study of the mutagenic activity of five other mutagens bearing a 1-naphthalene group (Chart 9) reinforced this concept. Although 24b could not be synthesised owing to steric impact of the 2-methyl group, LogTA100 data for five naphthalene-bearing mutagens 24a, 24c and 25a–c were acquired.[81]

|
The activities of 24a and 25b and 25c were clearly overpredicted from Eqn 6 with I = 1 (Fig. 11, open triangles; Table 6), but were not in excess of the predicted value with I = 0 (Fig. 11, closed triangles; Table 6). Such naphthalenes would need to intercalate in an edge-on rather than an end-on orientation as the pendant groups would be twisted out of the plane of the ring system, thereby blocking penetration of the naphthalene ring in a substantive fashion.[81]
|
|
25a gave a reproducible ambivalent result while 24c surprisingly and reproducibly correlated with I = 1. Of the five mutagens bearing 1-substituted naphthalenes, only 24c was active in TA98 (3803 revertants at 1 μmol per plate), confirming the TA100 study.[81] Although this was a seemingly incongruous result, there is some evidence from PAH intercalation that a methyl substituent can enhance intercalation, possibly by increasing polarisability of the ring system and enhancing electrostatic interactions and π–π stacking capability.[82] It is deemed that the electronic influence of aliphatic substituents that enhance intercalation are more important than the steric effects that inhibit intercalation.
There is fairly recent evidence that derivatives of the ‘left half’ of Azinomycin B modified to remove alkylating activity, 26, and that have an acyl side chain naphthalene motif similar to that of 25 bind intercalatively to DNA.[83] However, the unsubstituted naphthalene system in the same structure 27 was shown to be ineffective in DNA binding.[83] The impacts of substituents on π-electron density distribution are likely to make a significant contribution to the intercalative potential of the naphthalene group. It is not a clearcut situation though; computational studies by Alcaro et al. have been unable to clearly define the role of the naphthalene moiety in Azinomycin,[84,85] and Coleman et al.[86,87] and Casely-Hayford[88] suggest that there is no intercalation of the naphthalene moiety in 26 or 28. There is no doubt, though, that 24c intercalates and so too does 20g with a 1-naphthyl group on the alkoxyl side chain (Table 4).
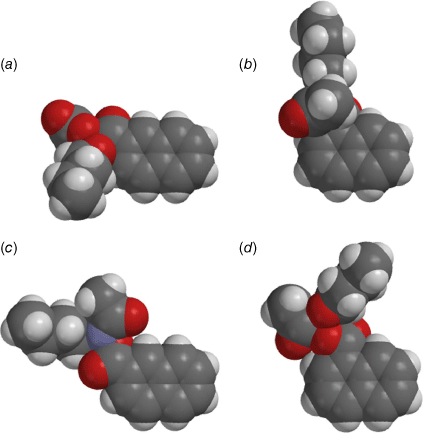
Once again, the difference between end-on and edge-on intercalative ability can clearly be seen in a comparison between the AM1-optimised structures of 13a and 20a, both 2-naphthalene derivatives, and their 1-naphthalene analogues 24a and 25a (Fig. 12).
|
Pyrene as an intercalator
Pyrene and its derivatives have long been known to bind to double-stranded DNA by intercalation and the property has been studied in detail through spectroscopic studies or binding studies.[89–95] Recently, kinetic studies have shown that the binding of a pyrene unit to DNA involves a two-step mechanism; the first step is the formation of a precursor complex, while the second step is the formation of the intercalated complex through penetration of the pyrene moiety between the base pairs of DNA.[90] Recently, the intercalation of a pyrene unit has been exploited as a photophysical probe,[93] in single-molecule atomic force microscopy (AFM) for double-stranded DNA mismatches,[96] and as a surface modification for the immobilisation of double-stranded DNA (ds-DNA) on a solid support.[91]
The capacity for pyrene to intercalate with ds-DNA with high binding constants spurred us to extend our QSAR work to NAAs bearing a pyrene substituent. On the premise that pyrene would localise the NAAs and the pendant electrophilic centre would need to access proximal guanines, 1-pyrene was attached directly and through a variable-length tether with a view to determining the impact of pyrene, or tethered pyrene, on mutagenic activity (Chart 10).
|
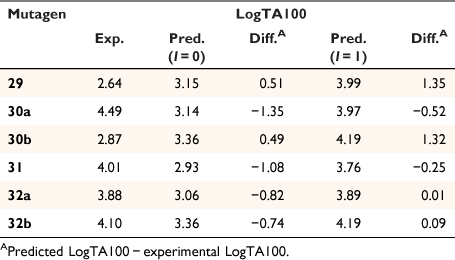
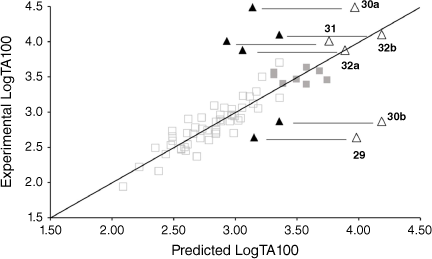
Table 7 gives experimental and predicted LogTA100 for pyrene-bearing mutagens, all of which have logP below 6, and the data are illustrated in Fig. 13. Four of the derivatives, 30a, 31, 32a and 32b, are better predicted with I = 1 (open triangles), although 30a, bearing a pyrene tethered through one methylene group on the amide side chain, is still underpredicted with I = 1. This was the most mutagenic of all the NAAs we have tested. Surprisingly, LogTA100 for 30b and 29 were better predicted with I = 0 although activities of both are somewhat overpredicted even with I = 0. 30a and 30b both bear the pyrene on the amide side chain but the propyl tether in 30b appears to render activity typical of a groove binder rather than an intercalator. All three substrates with pyrene on the acyloxyl group (31, 32a and 32b) are predicted to benefit from intercalation. 30a (9448 revertants at 1 μmol per plate), 31 (239 revertants at 1 μmol per plate) and 32a (385 revertants at 1 μmol per plate) gave a positive result in TA98, confirming their intercalative ability whereas 30b was negative in the TA98 test in accord with it reacting as a groove binder.
|
|
With pyrene-substituted mutagens, several scenarios are possible.
The pyrene could intercalate strongly, in which case the tether must be of suitable length to enable reaction at an N7 of an adjacent or distal guanine. Should this occur, the mutagen would register as benefiting from enhanced activity in TA100 and as a frameshift mutagen in TA98 through localisation of the pyrene. It would appear that 30a, 31 and 32a fall into this category. Two of these (30a and 31) bear a three-bond tether while 32a has a four-bond tether.
A mutagen could intercalate through pyrene but be unable to react at an adjacent or distal guanine. Intercalation is an equilibrium process, and such compounds could still damage DNA in a groove-bound situation and would register in TA100 with I = 0. However, they could also register as an intercalator in the TA98 test. 29 with a short, two-bond tether falls into this category. From Eqn 6, it correlates well with I = 0 but is moderately active in TA98 (281 revertants at 1 μmol per plate).
A mutagen could intercalate and facilitate a reaction of the tethered nitrogen at a proximal or distal G-N7 but if the tether becomes disconnected in the process, which, as described earlier, is the case where the pyrene is on the acyloxyl side chain, the pyrene may not localise sufficiently without the tether, and register weakly in TA98. 32b may fall into this category. The six-bond tether may inflict damage, probably at a distal G-N7, in the intercalated state and register as an intercalator in TA100 but disconnection through reaction at G-N7 results in poor pyrene retention; 32b is negative in TA98 (26 revertants at 1 μmol per plate)
A mutagen could register negatively for intercalation in both TA100 and TA98 if it reacts readily in the groove-bound state prior to intercalation but in the bonded state intercalation is difficult to achieve. 30b could be categorised as such. Its activity in TA100 does not benefit from intercalative enhancement and it is a very poor intercalator from TA98 (88 revertants at 1 μmol per plate).
Molecular modelling suggests that with a three-bond tether, an intercalated pyrene would be able to react at an adjacent guanine without severe disruption to the helix. Hence, 30a and 31 are of the most active forms.
Overall, though, throughout our investigations of intercalative ability, we have found that the result from the frameshift mutation strain TA98 was in agreement with the predictions from point mutation strain TA100 in 79% of the 24 compounds studied. In the majority of cases, those compounds showing enhanced TA100 mutagenicity were also mutagenic in TA98. In general, when I = 0, the mutagen was non-mutagenic in TA98, whereas with I = 1, the mutagen gave a positive result. Collectively, this adds credibility to the value of the indicator variable I being a reporter of intercalative ability of a PAH in the TA100 reverse mutation assay.[81]
Predictive power of QSAR
Activation by 4-nitrobenzyl substituents
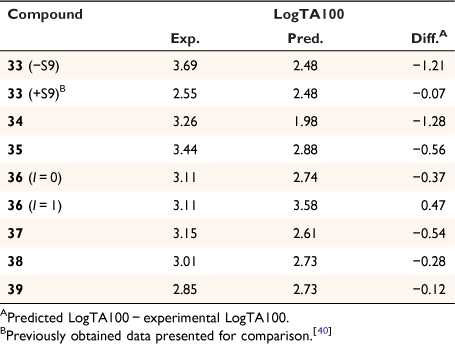
Early studies of substituent electronic effects for a series of N-acyloxy-N-benzyloxybenzamides 10 produced a result that at the time was little understood.[40] Activity for N-acetoxy-N-(4-nitrobenzyloxy)benzamide 33 in TA100 was substantially greater than expected but much lower in the presence of S9 liver homogenate, which is used to reproduce enzymic metabolic activation in the Ames test.[41] We subsequently investigated four other 4-nitrobenzyl-bearing NAAs, 34–37, as well as the 2-nitro- and 3-nitro- analogues of 33, 38 and 39 (Chart 11) (unpublished data, SA Glover, K Kavanagh, RR Schumacher).[81]
|
The results in Table 8 can be summarised as follows:
Although the activity of 33 in the absence of S9 was extremely high, the activity in the presence of S9 appeared to be well predicted by Eqn 6.
To varying degrees, the activities of all 4-nitrobenzyl-substituted NAAs 34–37 in the absence of metabolic activation are greater than predicted by Eqn 6, pointing to a general model for their mutagenicity.
The activity of the 3-methylated derivative 37 is underpredicted by Eqn 6, but not to the same extent as 33.
The 2- and 3-nitro analogues 38 and 39 were suitably predicted by Eqn 6.
|
We contend that the heightened activity of the 4-nitrobenzyl substrates 33–37 is most likely due to reduction of the nitro group to the hydroxylamine by nitro reductase present in bacterial TA100 (Scheme 3). Acetylase or sulfotransferase would lead to the ester or sulfate ester derivatives, which could react with DNA or solvolyse to an electrophilic phenylnitrenium ion, though these ions are predicted to have very short lifetimes in aqueous media.[30–33] Debnath et al. invoked these pathways in correlating the activities of a wide range of nitro aromatics in TA100 without metabolic activation.[46,97] Clearly, logP can relate to binding to nitroreductase or sulfotransferase as well as to DNA. In the case of 33 in the presence of S9, the cytochrome P450 oxidative enzymes most likely preserve the integrity of the nitro group. Hence, the LogTA100 in the presence of S9 is almost identical to the predicted value (Table 8). We demonstrated early on that S9 plays no role in the activity of NAAs and, in general, dose–response plots were extended linearly where toxicity was demonstrated at higher doses without S9.[40]

|
The 2- and 3-nitrobenzyl configurations 38 and 39 are well behaved, with LogTA100 close to the predicted value based on Eqn 6. It is apparent that in these cases, nitro reductase would appear to be inactive, which may be due to poor host–guest interactions relative to para-substituted benzyl groups in 33–36.
The 3-methyl-4-nitro analogue 37 appears not to be as mutagenic as 33 even though computations show that the nitro group in such configurations deviates minimally from planarity with the ring. This parallels the mutagenic activities of 4-nitrobiphenyl and its 3-methyl analogue, both of which require reduction and activation according to Scheme 3.[98] Boche showed that the reduced mutagenicity of the methylated derivative was more than likely due to interference with binding to nitroreductase in TA100 rather than disruption of coplanarity with the aromatic ring.[98,99]
Results for the naphthamide 36 indicate that, while the naphthyl group is present, it does not enhance activity through intercalation; LogTA100 is overpredicted with I = 1. With I = 0, the activity is significantly underpredicted as is the case for the other systems bearing a 4-nitrobenzyl group.
Activation of the 4-nitro substituent would appear not to be a uniform process across 33–37, which among other things could reflect different binding capacities with reductive enzymes in TA100. However, the molecules have capacity to react through two modes, leading to moderately enhanced activities relative to predicted levels.
The role of bis-naphthalene substitution
Prior to development of functional QSARs governing the mutagenic activity of NAAs, Clay synthesised two NAAs bearing two naphthalene substituents, for which we obtained mutagenicity data (unpublished data, AM Bonin, SF Clay, SA Glover).[81] Although a single naphthalene has been shown to elevate activity through intercalation, the role of multiple naphthalene units in the same molecule was unknown. 40 and 41 (Chart 12) were synthesised with the expectation that the presence of two naphthalene units would further elevate activity relative to mutagens bearing one naphthalene. The logP values of 41 (logP 6.17) approached the logP0 for NAAs (logP0 6.4) and application of both the linear and bilinear QSARs was appropriate.
|
From Table 9, it is apparent that neither mutagen exhibited activity in keeping with intercalative enhancement; differences between predicted and experimental LogTA100 values were small when activities were computed with I = 0 by both Eqns 6, 7. Both 40 and 41 behave as if they are groove binders and the activity is largely governed by their hydrophobicity. Although these are fairly large molecules, normal reaction with G-N7 in the major groove of DNA would appear to operate. Intercalation of either naphthalene must be prohibited by these configurations; intercalation of one planar group must inhibit reaction with a neighbouring guanine, presumably for steric reasons. Hence, reaction must occur in a groove-bound complex without intercalation.

|
It should be mentioned that dual intercalation is prohibited by the neighbouring exclusion principle, which states that, at most, intercalators can only bind every second possible base pair site and binding of additional intercalators adjacent to the first is prohibited.[63,71] Owing to the changes in the DNA backbone conformation associated with the helix unwinding that is involved in the base pair separation essential for intercalation, there are significant limitations on the proximity of intercalation sites to one another. Bis-naphthyl NAAs have both intercalator units within relatively close proximity to one another; thus, even if one naphthalene unit were capable of intercalative binding, the other naphthalene would be prohibited by the interaction of the first.
Potential mutagenicity of a new N-acyloxy-N-alkoxyamide reagent for skeletal editing
A recent paper by Levin and coworkers in Nature described a method of editing nitrogen from secondary benzylic amines and related systems.[100] The method is widely applicable to editing nitrogen from secondary amines and suitably constituted primary amines.[101] The reaction employs a novel NAA, N-benzyloxy-N-pivaloyloxy-4-(trifluoromethyl)benzamide 42 (Scheme 4). Based on our precedent,[102–104] 42 reacts with suitable secondary amines, 43, to produce an anomeric amide intermediate 44, which undergoes HERON rearrangement to give benzyl 4-(trifluoromethyl)benzoate 45 and a 1,1-diazene or amino nitrene 46. The reaction sequence was based on our discovery of both reactivity of NAAs with N-methylaniline,[8,16–19,50,59,105] and our discovery of the HERON reaction.[13,102–105] The 1,1-diazene 46 extrudes nitrogen, producing two alkyl radicals, one of which is conjugated, and solvent cage recombination yields alkane 47. Through this sequence, dibenzylamines generate bibenzyls in good yields, thereby editing nitrogen from the secondary amine.

|
The merit of Levin’s reaction and the value of the reagent has been recognised by both Unsworth[106] and Bräse.[107] Importantly, however, Bräse cautioned that we had much earlier proved NAAs to be chemical mutagens, a fact seemingly overlooked by Levin and coworkers. The author apprised Nature of potential mutagenicity of 42 and a correction to the paper appeared addressing this fact and stating that 42 belongs to the class of NAAs ‘some of which have been found to mutate genetic material’. However, the aforementioned sections indicate that almost all NAAs are classified as mutagenic; of the 100 variants we have made, only five structures were found not to be mutagenic in the Ames test.
Recently, we expanded our QSAR based on all published mutagens to date. Incorporation of three fluorene-bearing (22a and 23a–b) and five fluorenone-bearing mutagens (22d–e and 23c–e) yields Eqn 10:
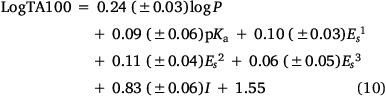
n = 67, R2 = 0.84, adj. R2 = 0.82 s = 0.17, F = 50.7; LOOCV Q2 = 0.83.
The predictive power of this QSAR, which applies to NAAs with logP < 6.4,[52] is demonstrated by the small standard error, high F-value and the LOOCV Q2 index of 0.83. Eqn 10 is similar to the preceding Eqn 6.[42,61]
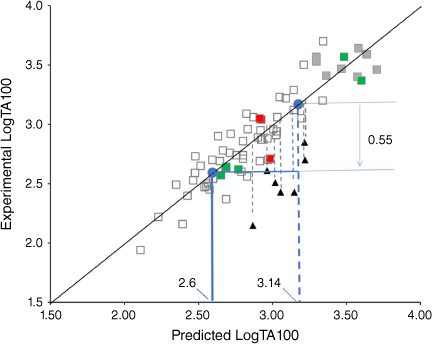
Our QSAR allows us to predict mutagenic activity of congeners with some accuracy, and employing the QSAR in Eqn 10 indicates that 42 has a very high probability of being a direct-acting mutagen. Initial application of the QSAR to 42 predicts a very high level of mutagenic activity (LogTA100 = 3.14, Fig. 14). From our studies of steric effects on the acyloxyl side chain in substrates 19a–g, we reported that branching at the α-position and other bulky groups reduces activity. Based on Eqn 10, the average suppression was 0.55 LogTA100 (Fig. 14, filled triangles).[61]
|
Correcting the initial activity of 42 yields an activity of LogTA100 = 2.6 (Fig. 14) and 42 would generate between 270 and 580 induced revertants at 1 μmol per plate. The QSAR points to 42 being at least as mutagenic as our standard, N-acetoxy-N-butoxybenzamide (9, X = H), which we have reproduced many times in the Ames studies.
Levin used other NAAs, 48, 49a–c, 50 and 51 (Chart 13), in the development of reagent 42. 48 is a proven mutagen; in an early study, we reported its activity at 1 μmol per plate to be LogTA100 = 2.63.[40] After steric correction for the pivaloyloxyl group, 49a–c and 50 are predicted to have very similar activity to that of 42 and 48 (Table 10) with activity of 51 slightly lower.

|

|
Of the wide range of some 100 NAAs for which we have mutagenicity data, which accounts for almost all known NAAs, only three, 15d, 16b and 17d, failed to generate a dose response in TA100 and can be considered to be non-mutagenic, while 15a and 15b generated very shallow dose responses and are weakly mutagenic according to the protocols (LogTA100 < 2).[41,59] As outlined in ‘Steric effects on activity’, non-mutagenicity of 15d and 16b has been attributed to their inability to enter the major groove of DNA on account of their overall dimensions.[52,59] The negative result for 17d in the Ames test was in accord with its resistance to SN2 reaction with N-methylaniline.[59] It is possible that 15d or 16b may be suitable candidates for nitrogen deletion from amines in spectral editing.
The QSAR we have developed would strongly suggest that 42 will be a direct-acting mutagen and must be handled very carefully by practitioners in the field. It is important that 42, or any analogues thereof, should not be declared to be safe reagents for wide use in skeletal editing, and for synthesis in large quantities for routine use, until it is proved not to produce a dose response in the Ames reverse mutation test. It is incumbent on the author to inform users of Levin’s skeletal editing method, those employing other NAAs for variations thereof or for other means, of the mutagenic properties of almost all known variants. Eqns 6, 7, 10 provide useful means of predicting a priori the likelihood and extent of mutagenic activity.
Conclusion
N-Acyloxy-N-alkoxyamides, as a class of anomeric amides, are mutagenic in the Ames reverse mutation assay with point mutation strain TA100, and select members bearing intercalating groups are active in frameshift strain TA98, in both cases without the need for metabolic activation. They have been shown to react with plasmid DNA, primarily at N7 of guanine in the major groove of DNA. As such, their mutagenic activity can be used to determine the factors controlling ease of access to bacterial DNA, binding modes with DNA and the ease of reaction with the guanosine nucleoside in the bound state. The Ames test requires minimal quantities of these compounds, which are easily synthesised in small amounts with a high degree of structural diversity. Almost all are heavy oils, which can be safely procured, purified, characterised and tested without exposure.
By measuring and comparing the mutagenicity at 1 μmol per plate with TA100 in a standardised manner, we have been able to establish how their activity is related to their structural features. For comparisons with our data, any new compound should be tested with the standard (9, X = H), which serves both as a cross-check of the test system as well as a means of relating activities. Quantitative structure–activity relationships have been established enabling identification of trends and processes of importance to small molecule–DNA interactions. We have developed QSARS catering for hydrophobic properties of the molecules, chemical reactivity at the electrophilic nitrogen, steric effects on all three side chains and intercalative ability of planar polycyclic aromatics. Based on the mutagenic activity of many structural and regio-isomers, the QSARs are accurate to the point of identifying impacts of small structural changes on ease of groove binding or of intercalation. They can also be used to focus on bacterial metabolic processes and mechanisms of DNA damage and to identify substructure potentially deleterious to DNA binding and reaction with DNA, as well as to predict, in advance, the mutagenic properties of new congeners.
All this is made possible on account of their anomeric substitution at nitrogen with both a donor alkoxyl and an acceptor acyloxyl group. The electronegativity of oxygen atoms at nitrogen results in greatly reduced amide resonance and nitrogens that are quite atypical of those in more common primary, N-alkyl secondary and N,N-dialkyl tertiary amides. Their pyramidality, combined with the anomeric interaction between the nitrogen substituents, both properties of which we have developed a thorough understanding, are entirely responsible for their reaction, directly, with nucleophilic sites in DNA.
Supplementary material
Supplementary material is available online.
Data availability
The data used in this account are supplied as Electronic Supplementary Material.
Conflicts of interest
The author declares no conflict of interest.
Declaration of funding
This work did not receive any specific funding.
Note in proof
The skeletal editing agent 42 has recently been shown to be a direct-acting mutagen in the Ames II assay.[108] Qualitative results from the Ames II method, which detects point mutations in a mix of S. typhimurium strains, are generally in good agreement with those from the full plate Ames methodology using TA100.[109] Contrary to Levin’s recently published view,[108] quantitative Ames II results for 42 with S. typhimurium TA7001-7006 cannot be compared meaningfully to predicted plate counts from the standard Ames test with S. typhimurium TA100.
Acknowledgements
The author is indebted to the many students whose work is cited in this Account, as well as to Dr Tony Bonin who contributed his valuable expertise to early Ames testing of our compounds and to the establishment of the testing facility at UNE.
References
[1] SA Glover, Anomeric Amides — Structure, Properties and Reactivity. Tetrahedron 1998, 54, 7229.| Anomeric Amides — Structure, Properties and Reactivity.Crossref | GoogleScholarGoogle Scholar |
[2] IV Alabugin, S Bresch, M Manoharan, Hybridization Trends for Main Group Elements and Expanding the Bent’s Rule Beyond Carbon: More than Electronegativity. J Phys Chem A 2014, 118, 3663.
| Hybridization Trends for Main Group Elements and Expanding the Bent’s Rule Beyond Carbon: More than Electronegativity.Crossref | GoogleScholarGoogle Scholar |
[3] HA Bent, An Appraisal of Valence-bond Structures and Hybridization in Compounds of the First-row Elements. Chem Rev 1961, 61, 275.
| An Appraisal of Valence-bond Structures and Hybridization in Compounds of the First-row Elements.Crossref | GoogleScholarGoogle Scholar |
[4] A Rauk, SA Glover, A Computational Investigation of the Stereoisomerism in Heteroatom-substituted Amides. J Org Chem 1996, 61, 2337.
| A Computational Investigation of the Stereoisomerism in Heteroatom-substituted Amides.Crossref | GoogleScholarGoogle Scholar |
[5] SA Glover, A Rauk, Conformational Stereochemistry of the HERON Amide, N-methoxy-N-dimethylaminoformamide: a Theoretical Study. J Org Chem 1999, 64, 2340.
| Conformational Stereochemistry of the HERON Amide, N-methoxy-N-dimethylaminoformamide: a Theoretical Study.Crossref | GoogleScholarGoogle Scholar |
[6] SA Glover, G Mo, A Rauk, DJ Tucker, P Turner, Structure, conformation, anomeric effects and rotational barriers in the HERON amides, N,N’-diacyl-N,N’-dialkoxyhydrazines. J Chem Soc, Perkin Trans 2 1999, 2053.
| Structure, conformation, anomeric effects and rotational barriers in the HERON amides, N,N’-diacyl-N,N’-dialkoxyhydrazines.Crossref | GoogleScholarGoogle Scholar |
[7] A-ME Gillson, SA Glover, DJ Tucker, P Turner, Crystal structures and properties of mutagenic N-acyloxy-N-alkoxyamides — ‘most pyramidal’ acyclic amides. Org Biomol Chem 2003, 1, 3430.
| Crystal structures and properties of mutagenic N-acyloxy-N-alkoxyamides — ‘most pyramidal’ acyclic amides.Crossref | GoogleScholarGoogle Scholar |
[8] Glover SA. N-Acyloxy-N-alkoxyamide – Structure, properties, reactivity and biological activity. In Richard J, editor. Advances in Physical Organic Chemistry. Vol. 42. London: Elsevier; 2008. pp. 35–123.
[9] Glover SA. N-Heteroatom-substituted hydroxamic esters. In Rappoport Z, Liebman JF, editors. The Chemistry of Hydroxylamines, Oximes and Hydroxamic, Acids, Part 2. Chichester: Wiley; 2009. pp. 839–923.
[10] SA Glover, JM White, AA Rosser, KM Digianantonio, Structures of N,N-Dialkoxyamides: Pyramidal Anomeric Amides with Low Amidicity. J Org Chem 2011, 76, 9757.
| Structures of N,N-Dialkoxyamides: Pyramidal Anomeric Amides with Low Amidicity.Crossref | GoogleScholarGoogle Scholar |
[11] SA Glover, AA Rosser, A Taherpour, BW Greatrex, Formation and HERON Reactivity of Cyclic N,N-Dialkoxyamides. Aust J Chem 2014, 67, 507.
| Formation and HERON Reactivity of Cyclic N,N-Dialkoxyamides.Crossref | GoogleScholarGoogle Scholar |
[12] SA Glover, AA Rosser, RM Spence, Studies of the Structure, Amidicity and Reactivity of N-Chlorohydroxamic Esters and N-Chloro-β,β-dialkylhydrazides: Anomeric Amides with Low Resonance Energies. Aust J Chem 2014, 67, 1344.
| Studies of the Structure, Amidicity and Reactivity of N-Chlorohydroxamic Esters and N-Chloro-β,β-dialkylhydrazides: Anomeric Amides with Low Resonance Energies.Crossref | GoogleScholarGoogle Scholar |
[13] SA Glover, AA Rosser, Heteroatom Substitution at Amide Nitrogen – Resonance Reduction and HERON Reactions of Anomeric Amides. Molecules 2018, 23, 2834.
| Heteroatom Substitution at Amide Nitrogen – Resonance Reduction and HERON Reactions of Anomeric Amides.Crossref | GoogleScholarGoogle Scholar |
[14] AM Bonin, SA Glover, GP Hammond, Reactive intermediates from the solvolysis of mutagenic O-alkyl N-acetoxybenzohydroxamates. J Chem Soc, Perkin Trans 2 1994, 1173.
| Reactive intermediates from the solvolysis of mutagenic O-alkyl N-acetoxybenzohydroxamates.Crossref | GoogleScholarGoogle Scholar |
[15] JJ Campbell, SA Glover, GP Hammond, CA Rowbottom, Evidence for the Formation of Nitrenium ions in the Acid-catalysed Solvolysis of Mutagenic N-acetoxy-N-Alkoxybenzamides. J Chem Soc, Perkin Trans 2 1991, 2067.
| Evidence for the Formation of Nitrenium ions in the Acid-catalysed Solvolysis of Mutagenic N-acetoxy-N-Alkoxybenzamides.Crossref | GoogleScholarGoogle Scholar |
[16] KL Cavanagh, SA Glover, HL Price, RR Schumacher, SN2 Substitution Reactions at the Amide Nitrogen in the Anomeric Mutagens, N-Acyloxy-N-alkoxyamides. Aust J Chem 2009, 62, 700.
| SN2 Substitution Reactions at the Amide Nitrogen in the Anomeric Mutagens, N-Acyloxy-N-alkoxyamides.Crossref | GoogleScholarGoogle Scholar |
[17] SA Glover, SN2 reactions at amide nitrogen – theoretical models for reactions of mutagenic N-acyloxy-N-alkoxyamides with bionucleophiles (OT-308CP). Arkivoc 2001, 143.
| SN2 reactions at amide nitrogen – theoretical models for reactions of mutagenic N-acyloxy-N-alkoxyamides with bionucleophiles (OT-308CP).Crossref | GoogleScholarGoogle Scholar |
[18] JJ Campbell, SA Glover, Bimolecular Reactions of Mutagenic N-(Acyloxy)-N-alkoxybenzamides with Aromatic Amines. J Chem Res (S). 1999, 23, 474.
| Bimolecular Reactions of Mutagenic N-(Acyloxy)-N-alkoxybenzamides with Aromatic Amines.Crossref | GoogleScholarGoogle Scholar |
[19] JJ Campbell, SA Glover, Bimolecular Reactions of Mutagenic N-(Acyloxy)-N-alkoxybenzamides with Aromatic Amines. J Chem Res (M). 1999, 23, 2075.
[20] JJ Campbell, SA Glover, Bimolecular Reactions of Mutagenic N-Acetoxy-N-alkoxybenzamides and N-methylaniline. J Chem Soc, Perkin Trans 2 1992, 1661.
| Bimolecular Reactions of Mutagenic N-Acetoxy-N-alkoxybenzamides and N-methylaniline.Crossref | GoogleScholarGoogle Scholar |
[21] SA Glover, G Mo, Hindered ester formation by SN2 azidation of N-acetoxy-N-alkoxyamides and N-alkoxy-N-chloroamides—Novel application of HERON rearrangements. J Chem Soc, Perkin Trans 2 2002, 1728.
| Hindered ester formation by SN2 azidation of N-acetoxy-N-alkoxyamides and N-alkoxy-N-chloroamides—Novel application of HERON rearrangements.Crossref | GoogleScholarGoogle Scholar |
[22] SA Glover, GP Hammond, AM Bonin, A Comparison of the Reactivity and Mutagenicity of N-(Benzoyloxy)-N-(benzyloxy)benzamides. J Org Chem 1998, 63, 9684.
| A Comparison of the Reactivity and Mutagenicity of N-(Benzoyloxy)-N-(benzyloxy)benzamides.Crossref | GoogleScholarGoogle Scholar |
[23] SA Glover, M Adams, Reaction of N-acyloxy-N-alkoxyamides with Biological Thiol groups. Aust J Chem 2011, 64, 443.
| Reaction of N-acyloxy-N-alkoxyamides with Biological Thiol groups.Crossref | GoogleScholarGoogle Scholar |
[24] JP Johns, A van Losenoord, C Mary, P Garcia, DS Pankhurst, AA Rosser, SA Glover, Thermal Decomposition of N-Acyloxy-N-alkoxyamides – a New HERON Reaction. Aust J Chem 2010, 63, 1717.
| Thermal Decomposition of N-Acyloxy-N-alkoxyamides – a New HERON Reaction.Crossref | GoogleScholarGoogle Scholar |
[25] Beland FA, Kadlubar FF. Metabolic activation and DNA adducts of aromatic amines and nitroaromatic hydrocarbons. In Cooper CS, Grover PL, editors. Chemical Carcinogenesis and Mutagenesis, 8 edn. Secaucus, NJ: Springer Verlag; 1990. pp. 267–325.
[26] FA Beland, FF Kadlubar, Formation and Persistence of Arylamine DNA Adducts in Vivo. Environ Health Perspect 1985, 62, 19.
| Formation and Persistence of Arylamine DNA Adducts in Vivo.Crossref | GoogleScholarGoogle Scholar |
[27] GM Blackburn, B Kellard, Chemical carcinogens - II. Chem Ind 1986, 687.
[28] D Kim, FP Guengerich, Cytochrome P450 Activation of Arylamines and Heterocyclic Amines. Annu Rev Pharmacol Toxicol 2005, 45, 27.
| Cytochrome P450 Activation of Arylamines and Heterocyclic Amines.Crossref | GoogleScholarGoogle Scholar |
[29] JA Miller, EC Miller, Some Historical Aspects of N-Aryl Carcinogens and Their Metabolic Activation. Environ Health Perspect 1983, 49, 3.
| Some Historical Aspects of N-Aryl Carcinogens and Their Metabolic Activation.Crossref | GoogleScholarGoogle Scholar |
[30] M Novak, S Rajagopal, N-arylnitrenium ions. Adv Phys Org Chem 2001, 36, 167.
| N-arylnitrenium ions.Crossref | GoogleScholarGoogle Scholar |
[31] M Novak, S Rajagopal, Correlations of Nitrenium Ion Selectivities with Quantitative Mutagenicity and Carcinogenicity of the Corresponding Amines. Chem Res Toxicol 2002, 15, 1495.
| Correlations of Nitrenium Ion Selectivities with Quantitative Mutagenicity and Carcinogenicity of the Corresponding Amines.Crossref | GoogleScholarGoogle Scholar |
[32] M Novak, MJ Kahley, E Eiger, JS Helmick, HE Peters, Reactivity and Selectivity Of Nitrenium Ions Derived from Ester Derivatives of Carcinogenic N-(4-Biphenylyl)hydroxylamine and the Corresponding Hydroxamic Acid. J Am Chem Soc 1993, 115, 9453.
| Reactivity and Selectivity Of Nitrenium Ions Derived from Ester Derivatives of Carcinogenic N-(4-Biphenylyl)hydroxylamine and the Corresponding Hydroxamic Acid.Crossref | GoogleScholarGoogle Scholar |
[33] PA Davidse, MJ Kahley, RA McClelland, M Novak, Flash Photolysis Observation and Lifetimes of 2-Fluorenyl- and 4-Biphenylylacetylnitrenium Ions in Aqueous Solution. J Am Chem Soc 1994, 116, 4513.
| Flash Photolysis Observation and Lifetimes of 2-Fluorenyl- and 4-Biphenylylacetylnitrenium Ions in Aqueous Solution.Crossref | GoogleScholarGoogle Scholar |
[34] RG Gerdes, SA Glover, JF Ten Have, CA Rowbottom, N-Acetoxy-N-alkoxyamides - a new class of nitrenium ion precursors, which are mutagenic. Tetrahedron Lett 1989, 30, 2649.
| N-Acetoxy-N-alkoxyamides - a new class of nitrenium ion precursors, which are mutagenic.Crossref | GoogleScholarGoogle Scholar |
[35] JJ Campbell, SA Glover, CA Rowbottom, Solvolysis and Mutagenesis of N-Acetoxy-N-Alkoxybenzamides — Evidence for Nitrenium Ion Formation. Tetrahedron Lett 1990, 31, 5377.
| Solvolysis and Mutagenesis of N-Acetoxy-N-Alkoxybenzamides — Evidence for Nitrenium Ion Formation.Crossref | GoogleScholarGoogle Scholar |
[36] SA Glover, AP Scott, MNDO properties of heteroatom and phenyl-substituted nitrenium ions. Tetrahedron 1989, 45, 1763.
| MNDO properties of heteroatom and phenyl-substituted nitrenium ions.Crossref | GoogleScholarGoogle Scholar |
[37] SA Glover, A Goosen, CW McCleland, JL Schoonraad, N-Alkoxy-N-acylnitrenium Ions as Possible Intermediates in Intramolecular Aromatic Substitution: Novel Formation of N-Acyl-3,4-dihydro-1H-2,1-benzoxazines and N-Acyl-4,5-dihydro-1H,3H-2,1-benzoxazepine. J Chem Soc, Perkin Trans 1 1984, 2255.
| N-Alkoxy-N-acylnitrenium Ions as Possible Intermediates in Intramolecular Aromatic Substitution: Novel Formation of N-Acyl-3,4-dihydro-1H-2,1-benzoxazines and N-Acyl-4,5-dihydro-1H,3H-2,1-benzoxazepine.Crossref | GoogleScholarGoogle Scholar |
[38] TM Banks, AM Bonin, SA Glover, AS Prakash, Mutagenicity and DNA Damage Studies of N-acyloxy-N-alkoxyamides — the Role of Electrophilic Nitrogen. Org Biomol Chem 2003, 1, 2238.
| Mutagenicity and DNA Damage Studies of N-acyloxy-N-alkoxyamides — the Role of Electrophilic Nitrogen.Crossref | GoogleScholarGoogle Scholar |
[39] Banks TM. Reactivity, Mutagenicity and DNA damage of N-Acyloxy-N-alkoxyamides. PhD thesis, University of New England, Armidale; 2003.
[40] AM Bonin, TM Banks, JJ Campbell, SA Glover, GP Hammond, AS Prakash, CA Rowbottom, Mutagenicity of Electrophilic N-acyloxy-N-alkoxyamides. Mutat Res Genet Toxicol Environ Mutagen 2001, 494, 115.
| Mutagenicity of Electrophilic N-acyloxy-N-alkoxyamides.Crossref | GoogleScholarGoogle Scholar |
[41] K Mortelmans, E Zeiger, The Ames Salmonella/microsome mutagenicity assay. Mutat Res Fundam Mol Mech Mutagen 2000, 455, 29.
| The Ames Salmonella/microsome mutagenicity assay.Crossref | GoogleScholarGoogle Scholar |
[42] TM Banks, SF Clay, SA Glover, RR Schumacher, Mutagenicity of N-acyloxy-N-alkoxyamides as an indicator of DNA intercalation Part 1: Evidence for naphthalene as a DNA intercalator. Org Biomol Chem 2016, 14, 3699.
| Mutagenicity of N-acyloxy-N-alkoxyamides as an indicator of DNA intercalation Part 1: Evidence for naphthalene as a DNA intercalator.Crossref | GoogleScholarGoogle Scholar |
[43] Hansch C, Leo AJ. Exploring QSAR, Fundamentals and Applications in Chemistry and Biology, Part I. Washington DC: American Chemical Society; 1995.
[44] AK Debnath, AJ Shusterman, RL Lopez de Compadre, C Hansch, The importance of the hydrophobic interaction in the mutagenicity of organic compounds. Mutat Res Fundam Mol Mech Mutagen 1994, 305, 63.
| The importance of the hydrophobic interaction in the mutagenicity of organic compounds.Crossref | GoogleScholarGoogle Scholar |
[45] AK Debnath, C Hansch, The importance of hydrophobicity in the mutagenicity of methanesulfonic acid esters with salmonella typhimurium TA100. Chem Res Toxicol 1993, 6, 310.
| The importance of hydrophobicity in the mutagenicity of methanesulfonic acid esters with salmonella typhimurium TA100.Crossref | GoogleScholarGoogle Scholar |
[46] AK Debnath, RL Lopez de Compadre, G Debnath, AJ Shusterman, C Hansch, Structure–activity relationship of mutagenic aromatic and heteroaromatic nitro compounds. Correlation with molecular orbital energies and hydrophobicity. J Med Chem 1991, 34, 786.
| Structure–activity relationship of mutagenic aromatic and heteroaromatic nitro compounds. Correlation with molecular orbital energies and hydrophobicity.Crossref | GoogleScholarGoogle Scholar |
[47] RLL De Compadre, AJ Shusterman, C Hansch, The Role of Hydrophobicity in the Ames Test. The Correlation of the Mutagenicity of Nitropolycyclic Hydrocarbons with Partition Coefficients and Molecular Orbital Indices. Int J Quantum Chem 1988, 34, 91.
| The Role of Hydrophobicity in the Ames Test. The Correlation of the Mutagenicity of Nitropolycyclic Hydrocarbons with Partition Coefficients and Molecular Orbital Indices.Crossref | GoogleScholarGoogle Scholar |
[48] AJ Leo, C Hansch, Role of hydrophobic effects in mechanistic QSAR. Perspect Drug Discovery Des 1999, 17, 1.
| Role of hydrophobic effects in mechanistic QSAR.Crossref | GoogleScholarGoogle Scholar |
[49] AK Ghose, A Pritchett, GM Crippen, Atomic Physicochemical Parameters for Three-Dimensional Structure Directed Quantitative Structure–Activity Relationships III: Modeling Hydrophobic Interactions. J Comput Chem 1988, 9, 80.
| Atomic Physicochemical Parameters for Three-Dimensional Structure Directed Quantitative Structure–Activity Relationships III: Modeling Hydrophobic Interactions.Crossref | GoogleScholarGoogle Scholar |
[50] LE Andrews, TM Banks, AM Bonin, SF Clay, A-ME Gillson, SA Glover, Mutagenic N-acyloxy-N-alkoxyamides: Probes for Drug–DNA Interactions. Aust J Chem 2004, 57, 377.
| Mutagenic N-acyloxy-N-alkoxyamides: Probes for Drug–DNA Interactions.Crossref | GoogleScholarGoogle Scholar |
[51] Hansch C, Leo AJ, Hoekman D. Exploring QSAR, Fundamentals and Applications in Chemistry and Biology, Part II. Washington, DC: American Chemical Society; 1995.
[52] SA Glover, RR Schumacher, The effect of hydrophobicity upon the direct mutagenicity of N-acyloxy-N-alkoxyamides – Bilinear dependence upon LogP. Mutat Res Genet Toxicol Environ Mutagen 2016, 795, 41.
| The effect of hydrophobicity upon the direct mutagenicity of N-acyloxy-N-alkoxyamides – Bilinear dependence upon LogP.Crossref | GoogleScholarGoogle Scholar |
[53] K Tuppurainen, Frontier orbital energies, hydrophobicity and steric factors as physical QSAR descriptors of molecular mutagenicity. A review with a case study: MX compounds. Chemosphere 1999, 38, 3015.
| Frontier orbital energies, hydrophobicity and steric factors as physical QSAR descriptors of molecular mutagenicity. A review with a case study: MX compounds.Crossref | GoogleScholarGoogle Scholar |
[54] BN Ames, FD Lee, WE Durston, An Improved Bacterial Test System for the Detection and Classification of Mutagens and Carcinogens. Proc Natl Acad Sci U S A 1973, 70, 782.
| An Improved Bacterial Test System for the Detection and Classification of Mutagens and Carcinogens.Crossref | GoogleScholarGoogle Scholar |
[55] CA Lipinski, F Lombardo, BW Dominy, PJ Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Delivery Rev 2001, 46, 3.
| Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings.Crossref | GoogleScholarGoogle Scholar |
[56] H Kubinyi, Quantitative structure–activity relations. 7. The bilinear model, a new model for non-linear dependence of biological activity on hydrophobic character. J Med Chem 1977, 20, 625.
| Quantitative structure–activity relations. 7. The bilinear model, a new model for non-linear dependence of biological activity on hydrophobic character.Crossref | GoogleScholarGoogle Scholar |
[57] Kubinyi H. Lipophilicity and drug activity. In Jucker E, editor. Progress in Drug Research. Vol. 23. Birkhäuser Basel; 1979. pp. 97–198.
| Crossref |
[58] H Kubinyi, OH Kehrhahn, Quantitative Structure–Activity relationships. VI. Non-linear dependence of biological activity on hydrophobic character: Calculation procedures for bilinear model. Arzneimittel-Forschung 1978, 28, 598.
[59] LE Andrews, AM Bonin, LE Fransson, A-ME Gillson, SA Glover, The Role of Steric Effects in the Direct Mutagenicity of N-Acyloxy-N-alkoxyamides. Mutat Res Genet Toxicol Environ Mutagen 2006, 605, 51.
| The Role of Steric Effects in the Direct Mutagenicity of N-Acyloxy-N-alkoxyamides.Crossref | GoogleScholarGoogle Scholar |
[60] De Kimpe N, Verhé R. Synthesis and reactivity of α-halogenated ketones. In Patai S, Rappoport Z, editors. α-Haloketones, α-Haloaldehydes and α-Haloimines. John Wiley & Sons, Inc; 1988. pp. 1–119.
[61] SA Glover, RR Schumacher, AM Bonin, LE Fransson, Steric effects on the direct mutagenicity of N-acyloxy-N-alkoxyamides – probes for drug–DNA interactions. Mutat Res Genet Toxicol Environ Mutagen 2011, 722, 32.
| Steric effects on the direct mutagenicity of N-acyloxy-N-alkoxyamides – probes for drug–DNA interactions.Crossref | GoogleScholarGoogle Scholar |
[62] Neidle S. Principles of Small Molecule-DNA Recognition. New York: Academic Press; 2008. pp. 132–203.
[63] Blackburn GM, Gait MJ, Loakes D, Williams DM. Nucleic Acids in Chemistry and Biology. Cambridge: RSC Publishing; 2006.
[64] DE Graves, LM Velea, Intercalative Binding of Small Molecules to Nucleic Acids. Curr Org Chem 2000, 4, 915.
| Intercalative Binding of Small Molecules to Nucleic Acids.Crossref | GoogleScholarGoogle Scholar |
[65] U Pindur, M Haber, K Sattler, Antitumor Active Drugs as Intercalators of Deoxyribonucleic Acid. J Chem Educ 1993, 70, 263.
| Antitumor Active Drugs as Intercalators of Deoxyribonucleic Acid.Crossref | GoogleScholarGoogle Scholar |
[66] X Qian, T-B Huang, D-z Wei, D-H Zhu, Zhu, W Yao, Interaction of naphthyl heterocycles with DNA: effects of thiono and thio groups. J Chem Soc, Perkin Trans 2 2000, 715.
| Interaction of naphthyl heterocycles with DNA: effects of thiono and thio groups.Crossref | GoogleScholarGoogle Scholar |
[67] J Sartorius, H-J Schneider, Intercalation mechanisms with ds-DNA: binding modes and energy contributions with benzene, naphthalene, quinoline and indole derivatives including some antimalarials. J Chem Soc, Perkin Trans 2 1997, 2319.
| Intercalation mechanisms with ds-DNA: binding modes and energy contributions with benzene, naphthalene, quinoline and indole derivatives including some antimalarials.Crossref | GoogleScholarGoogle Scholar |
[68] KA Stevenson, SF Yen, NC Yang, DW Boykin, WD Wilson, A substituent constant analysis of the interaction of substituted naphthalene monoimides with DNA. J Med Chem 1984, 27, 1677.
| A substituent constant analysis of the interaction of substituted naphthalene monoimides with DNA.Crossref | GoogleScholarGoogle Scholar |
[69] Y Chu, DW Hoffman, BL Iverson, A Pseudocatenane Structure Formed between DNA and a Cyclic Bisintercalator. J Am Chem Soc 2009, 131, 3499.
| A Pseudocatenane Structure Formed between DNA and a Cyclic Bisintercalator.Crossref | GoogleScholarGoogle Scholar |
[70] SF Yen, EJ Gabbay, WD Wilson, Interaction of aromatic imides with DNA. 1. Spectrophotometric and viscometric studies. Biochemistry 1982, 21, 2070.
| Interaction of aromatic imides with DNA. 1. Spectrophotometric and viscometric studies.Crossref | GoogleScholarGoogle Scholar |
[71] Wilson WD. Comprehensive Natural Product Chemistry. Oxford: Pergamon; 1999. pp. 427–476.
[72] JB Chaires, Energetics of drug-DNA interactions. Biopolymers 1997, 44, 201.
| Energetics of drug-DNA interactions.Crossref | GoogleScholarGoogle Scholar |
[73] LS Lerman, The Structure of the DNA–Acridine Complex. Proc Natl Acad Sci U S A 1963, 49, 94.
| The Structure of the DNA–Acridine Complex.Crossref | GoogleScholarGoogle Scholar |
[74] LS Lerman, Structural considerations in the interaction of DNA and acridines. J Mol Biol 1961, 3, 18.
| Structural considerations in the interaction of DNA and acridines.Crossref | GoogleScholarGoogle Scholar |
[75] Neidle S. Nucleic Acid Structure and Recognition. Oxford: Oxford University Press; 2002.
[76] BN Ames, EG Gurney, JA Miller, H Bartsch, Carcinogens as Frameshift Mutagens: Metabolites and Derivatives of 2-Acetylaminofluorene and Other Aromatic Amine Carcinogens. Proc Natl Acad Sci U S A 1972, 69, 3128.
| Carcinogens as Frameshift Mutagens: Metabolites and Derivatives of 2-Acetylaminofluorene and Other Aromatic Amine Carcinogens.Crossref | GoogleScholarGoogle Scholar |
[77] K Isono, J Yourno, Chemical Carcinogens as Frameshift Mutagens: Salmonella DNA Sequence Sensitive to Mutagenesis by Polycyclic Carcinogens. Proc Natl Acad Sci U S A 1974, 71, 1612.
| Chemical Carcinogens as Frameshift Mutagens: Salmonella DNA Sequence Sensitive to Mutagenesis by Polycyclic Carcinogens.Crossref | GoogleScholarGoogle Scholar |
[78] BN Ames, WE Durston, E Yamasaki, FD Lee, Carcinogens are Mutagens: A Simple Test System Combining Liver Homogenates for Activation and Bacteria for Detection. Proc Natl Acad Sci U S A 1973, 70, 2281.
| Carcinogens are Mutagens: A Simple Test System Combining Liver Homogenates for Activation and Bacteria for Detection.Crossref | GoogleScholarGoogle Scholar |
[79] HJ Creech, RK Preston, RM Peck, AP O’Connell, BN Ames, Antitumor and mutagenic properties of a variety of heterocyclic nitrogen and sulfur mustards. J Med Chem 1972, 15, 739.
| Antitumor and mutagenic properties of a variety of heterocyclic nitrogen and sulfur mustards.Crossref | GoogleScholarGoogle Scholar |
[80] SA Glover, RR Schumacher, Mutagenicity of N-acyloxy-N-alkoxyamides as an indicator of DNA intercalation: The role of fluorene and fluorenone substituents as DNA intercalators. Mutat Res Genet Toxicol Environ Mutagen 2021, 863–864, 503299.
| Mutagenicity of N-acyloxy-N-alkoxyamides as an indicator of DNA intercalation: The role of fluorene and fluorenone substituents as DNA intercalators.Crossref | GoogleScholarGoogle Scholar |
[81] Schumacher RR. Structural Effects upon the Mutagenicity of N-acyloxy-N-alkoxyamides. PhD thesis, University of New England, Armidale; 2011.
[82] IS Zegar, AS Prakash, RG Harvey, PR LeBreton, Stereoelectronic aspects of the intercalative binding properties of 7,12-dimethylbenz[a]anthracene metabolites with DNA. J Am Chem Soc 1985, 107, 7990.
| Stereoelectronic aspects of the intercalative binding properties of 7,12-dimethylbenz[a]anthracene metabolites with DNA.Crossref | GoogleScholarGoogle Scholar |
[83] H Zang, KS Gates, DNA Binding and Alkylation by the ‘Left Half’ of Azinomycin B. Biochemistry 2000, 39, 14968.
| DNA Binding and Alkylation by the ‘Left Half’ of Azinomycin B.Crossref | GoogleScholarGoogle Scholar |
[84] S Alcaro, RS Coleman, A Molecular Model for DNA Cross-Linking by the Antitumor Agent Azinomycin B. J Med Chem 2000, 43, 2783.
| A Molecular Model for DNA Cross-Linking by the Antitumor Agent Azinomycin B.Crossref | GoogleScholarGoogle Scholar |
[85] S Alcaro, F Ortuso, RS Coleman, DNA Cross-Linking by Azinomycin B: Monte Carlo Simulations in the Evaluation of Sequence Selectivity. J Med Chem 2002, 45, 861.
| DNA Cross-Linking by Azinomycin B: Monte Carlo Simulations in the Evaluation of Sequence Selectivity.Crossref | GoogleScholarGoogle Scholar |
[86] RS Coleman, RJ Perez, CH Burk, A Navarro, Studies on the Mechanism of Action of Azinomycin B: Definition of Regioselectivity and Sequence Selectivity of DNA Cross-Link Formation and Clarification of the Role of the Naphthoate. J Am Chem Soc 2002, 124, 13008.
| Studies on the Mechanism of Action of Azinomycin B: Definition of Regioselectivity and Sequence Selectivity of DNA Cross-Link Formation and Clarification of the Role of the Naphthoate.Crossref | GoogleScholarGoogle Scholar |
[87] RS Coleman, MT Tierney, SB Cortright, DJ Carper, Synthesis of Functional ‘Top-Half’ Partial Structures of Azinomycin A and B. J Org Chem 2007, 72, 7726.
| Synthesis of Functional ‘Top-Half’ Partial Structures of Azinomycin A and B.Crossref | GoogleScholarGoogle Scholar |
[88] MA Casely-Hayford, K Pors, CH James, LH Patterson, JA Hartley, M Searcey, Design and synthesis of a DNA-crosslinking azinomycin analogue. Org Biomol Chem 2005, 3, 3585.
| Design and synthesis of a DNA-crosslinking azinomycin analogue.Crossref | GoogleScholarGoogle Scholar |
[89] KW Bair, RL Tuttle, VC Knick, M Cory, DD McKee, [(1-Pyrenylmethyl)amino]alcohols, a new class of antitumor DNA intercalators. Discovery and initial amine side chain structure–activity studies. J Med Chem 1990, 33, 2385.
| [(1-Pyrenylmethyl)amino]alcohols, a new class of antitumor DNA intercalators. Discovery and initial amine side chain structure–activity studies.Crossref | GoogleScholarGoogle Scholar |
[90] F Secco, M Venturini, T Biver, F Sánchez, R Prado-Gotor, E Grueso, Solvent Effects on the Kinetics of the Interaction of 1-Pyrenecarboxaldehyde with Calf Thymus DNA. J Phys Chem B 2010, 114, 4686.
| Solvent Effects on the Kinetics of the Interaction of 1-Pyrenecarboxaldehyde with Calf Thymus DNA.Crossref | GoogleScholarGoogle Scholar |
[91] S Laib, A Krieg, P Häfliger, N Agorastos, DNA-intercalation on pyrene modified surface coatings. Chem Commun 2005, 5566.
| DNA-intercalation on pyrene modified surface coatings.Crossref | GoogleScholarGoogle Scholar |
[92] M Nakamura, Y Fukunaga, K Sasa, Y Ohtoshi, K Kanaori, H Hayashi, H Nakano, K Yamana, Pyrene is highly emissive when attached to the RNA duplex but not to the DNA duplex: the structural basis of this difference. Nucleic Acids Res 2005, 33, 5887.
| Pyrene is highly emissive when attached to the RNA duplex but not to the DNA duplex: the structural basis of this difference.Crossref | GoogleScholarGoogle Scholar |
[93] U Förster, C Grünewald, JW Engels, J Wachtveitl, Ultrafast Dynamics of 1-Ethynylpyrene-Modified RNA: A Photophysical Probe of Intercalation. J Phys Chem B 2010, 114, 11638.
| Ultrafast Dynamics of 1-Ethynylpyrene-Modified RNA: A Photophysical Probe of Intercalation.Crossref | GoogleScholarGoogle Scholar |
[94] F-M Chen, Binding of pyrene to DNA, base squence specificity and its implication. Nucleic Acids Res 1983, 11, 7231.
| Binding of pyrene to DNA, base squence specificity and its implication.Crossref | GoogleScholarGoogle Scholar |
[95] H-C Becker, B Nordén, DNA Binding Mode and Sequence Specificity of Piperazinylcarbonyloxyethyl Derivatives of Anthracene and Pyrene. J Am Chem Soc 1999, 121, 11947.
| DNA Binding Mode and Sequence Specificity of Piperazinylcarbonyloxyethyl Derivatives of Anthracene and Pyrene.Crossref | GoogleScholarGoogle Scholar |
[96] Z Jiang, Y Zhang, Y Yu, Z Wang, X Zhang, X Duan, S Wang, Study on Intercalations between Double-Stranded DNA and Pyrene by Single-Molecule Force Spectroscopy: Toward the Detection of Mismatch in DNA. Langmuir 2010, 26, 13773.
| Study on Intercalations between Double-Stranded DNA and Pyrene by Single-Molecule Force Spectroscopy: Toward the Detection of Mismatch in DNA.Crossref | GoogleScholarGoogle Scholar |
[97] AK Debnath, RL Lopez Compadre, AJ Shusterman, C Hansch, Quantitative Structure–Activity Relationship Investigation of the Role of Hydrophobicity in Regulating Mutagenicity in the Ames Test: 2. Mutagenicity of Aromatic and Heteroaromatic Nitro Compounds in Salmonella typhimurium TA100. Env Mol Mutagen 1992, 19, 53.
| Quantitative Structure–Activity Relationship Investigation of the Role of Hydrophobicity in Regulating Mutagenicity in the Ames Test: 2. Mutagenicity of Aromatic and Heteroaromatic Nitro Compounds in Salmonella typhimurium TA100.Crossref | GoogleScholarGoogle Scholar |
[98] M Klein, U Voigtmann, T Haack, L Erdinger, G Boche, From mutagenic to non-mutagenic nitroarenes: effect of bulky alkyl substituents on the mutagenic activity of 4-nitrobiphenyl in Salmonella typhimurium: Part I. Substituents ortho to the nitro group and in 2′-position. Mutat Res Genet Toxicol Environ Mutagen 2000, 467, 55.
| From mutagenic to non-mutagenic nitroarenes: effect of bulky alkyl substituents on the mutagenic activity of 4-nitrobiphenyl in Salmonella typhimurium: Part I. Substituents ortho to the nitro group and in 2′-position.Crossref | GoogleScholarGoogle Scholar |
[99] M Klein, L Erdinger, G Boche, From mutagenic to non-mutagenic nitroarenes: effect of bulky alkyl substituents on the mutagenic activity of nitroaromatics in Salmonella typhimurium: Part II. Substituents far away from the nitro group. Mutat Res Genet Toxicol Environ Mutagen 2000, 467, 69.
| From mutagenic to non-mutagenic nitroarenes: effect of bulky alkyl substituents on the mutagenic activity of nitroaromatics in Salmonella typhimurium: Part II. Substituents far away from the nitro group.Crossref | GoogleScholarGoogle Scholar |
[100] SH Kennedy, BD Dherange, KJ Berger, MD Levin, Skeletal editing through direct nitrogen deletion of secondary amines. Nature 2021, 593, 223.
| Skeletal editing through direct nitrogen deletion of secondary amines.Crossref | GoogleScholarGoogle Scholar |
[101] KJ Berger, JL Driscoll, M Yuan, BD Dherange, O Gutierrez, MD Levin, Direct Deamination of Primary Amines via Isodiazene Intermediates. J Am Chem Soc 2021, 143, 17366.
| Direct Deamination of Primary Amines via Isodiazene Intermediates.Crossref | GoogleScholarGoogle Scholar |
[102] SA Glover, A Rauk, JM Buccigross, JJ Campbell, GP Hammond, G Mo, LE Andrews, A-ME Gillson, The HERON reaction – Origin, Theoretical Background, and Prevalence. Can J Chem 2005, 83, 1492.
| The HERON reaction – Origin, Theoretical Background, and Prevalence.Crossref | GoogleScholarGoogle Scholar |
[103] SA Glover, G Mo, A Rauk, HERON rearrangement of N,N’-diacyl-N,N’-dialkoxyhydrazines – a theoretical and experimental study. Tetrahedron 1999, 55, 3413.
| HERON rearrangement of N,N’-diacyl-N,N’-dialkoxyhydrazines – a theoretical and experimental study.Crossref | GoogleScholarGoogle Scholar |
[104] JM Buccigross, SA Glover, Molecular orbital studies of novel N to C migrations in N,N-bisheteroatom-substituted amides—HERON rearrangements. J Chem Soc, Perkin Trans 2 1995, 595.
| Molecular orbital studies of novel N to C migrations in N,N-bisheteroatom-substituted amides—HERON rearrangements.Crossref | GoogleScholarGoogle Scholar |
[105] Glover SA. HERON Rearrangement (Heteroatom Rearrangements on Nitrogen). In O’Neil MJ, editor. Merck Index, Organic Name Reactions ONR-48, 13 edn. Whitehouse Station, N.J.: Merck & Co., Inc.; 2001.
[106] WP Unsworth, A-J Avestro, Nitrogen deletion offers fresh strategy for organic synthesis. Nature 2021, 593, 203.
| Nitrogen deletion offers fresh strategy for organic synthesis.Crossref | GoogleScholarGoogle Scholar |
[107] C Zippel, J Seibert, S Bräse, Skeletal Editing—Nitrogen Deletion of Secondary Amines by Anomeric Amide Reagents. Angew Chem Int Ed 2021, 60, 19522.
| Skeletal Editing—Nitrogen Deletion of Secondary Amines by Anomeric Amide Reagents.Crossref | GoogleScholarGoogle Scholar |
[108] Dherange BD, Yuan M, Kelly CB, Reiher CA, Grosanu C, Berger KJ, Gutierrez O, Levin MD. Direct deaminative functionalization. J Am Chem Soc 2023; 145: 17–24.
| Crossref |, S2 of Supporting Information
[109] Kamber M, Flückiger-Isler S, Engelhardt G, Jaeckh R and Zeiger E. Comparison of the Ames II and traditional Ames test responses with respect to mutagenicity, strain specificities, need for metabolism and correlation with rodent carcinogenicity. Mutagenesis 2009; 24: 359–366.
| Crossref |