

Preliminary Investigation of a Novel 18F Radiopharmaceutical for Imaging CB2 Receptors in a SOD Mouse Model
Giancarlo Pascali A B C E , Daniele Panetta C , Mariarosaria De Simone C D , Silvia Burchielli C , Valentina Lucchesi D , Elena Sanguinetti C , Simone Zanoni B , Patricia Iozzo C , Giuseppe Saccomanni D , Clementina Manera D and Piero A. Salvadori C
A B C E , Daniele Panetta C , Mariarosaria De Simone C D , Silvia Burchielli C , Valentina Lucchesi D , Elena Sanguinetti C , Simone Zanoni B , Patricia Iozzo C , Giuseppe Saccomanni D , Clementina Manera D and Piero A. Salvadori C
A Nuclear Medicine Department, Prince of Wales Hospital, Barker Street, Randwick, NSW 2031, Australia.
B School of Chemistry and Biological Resources Imaging Laboratory, University of New South Wales, High Street, Kensington, NSW 2052, Australia.
C Institute of Clinical Physiology, National Research Council (CNR), Via G. Moruzzi, Pisa 56124, Italy.
D Department of Pharmacy, University of Pisa, Via Bonanno Pisano, Pisa 56126, Italy.
E Corresponding author. Email: g.pascali@unsw.edu.au
Australian Journal of Chemistry 74(6) 443-452 https://doi.org/10.1071/CH20247
Submitted: 12 August 2020 Accepted: 21 December 2020 Published: 1 February 2021
Journal Compilation © CSIRO 2021 Open Access CC BY-NC-ND
Abstract
We successfully radiolabelled a novel prospective cannabinoid type 2 receptor ligand with 18F and tested its biodistribution in animal models by positron emission tomography (PET)/computed tomography (CT) imaging. The radiolabelling process was conducted on an alkyl mesylate fragment of the main naphthyridine core, using highly efficient microfluidic technology. No preliminary protection was needed, and the product was purified by semi-prep HPLC and SPE formulation, allowing the desired diastereomeric mixture to be obtained in 29 % radiochemical yield and > 95 % radiochemically pure. SOD1G93A mice were used as model of overexpression of CB2 receptors; PET imaging revealed a significant increase of the tracer distribution volume in the brain of symptomatic subjects compared with the asymptomatic ones.
Introduction
The cannabinoid (CB) receptor system is involved in numerous physiological processes and its functioning is mediated by two main subtype families, CB1 and CB2. Both receptor types are substrates for the same set of natural cannabinoid molecules, but they exert different effects, mostly linked to their tissue localization. In fact, while CB1 is mostly localized in the central nervous system, thus modulating behaviour and feelings, CB2 is primarily expressed peripherally and involved in the inflammation cascade. For this reason, and due to their action, CB2 agonists have been assessed for their use as antinociceptives with reduced central effects. However, it has been reported that CB2 receptors are expressed in the brain during early neuroinflammatory processes, particularly in pre-activated microglia, and CB2 agonists have been tested for reducing neuronal degeneration and improving cognitive functional scores.[1] Therefore, due to the low natural abundance of CB2 in the healthy brain, it has been suggested to use selective CB2 tracers to identify the neuroinflammation phenomena that are linked to several neurodegenerative diseases, such as Parkinson’s disease (PD), Alzheimer’s disease (AD), and amyotrophic lateral sclerosis (ALS), in order to provide a specific tool for diagnosis and therapeutic follow-up.
Following this lead, we initiated a research program aimed at designing and testing novel positron emission tomography (PET) radiopharmaceuticals for imaging the CB2 receptor population in genetically modified animal models mimicking ALS.
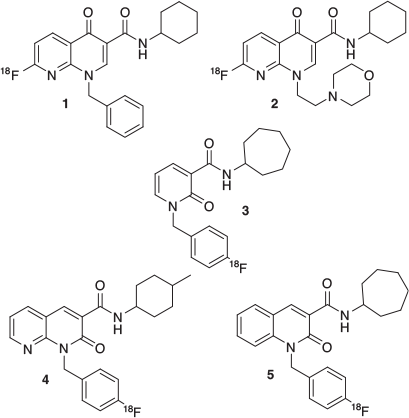
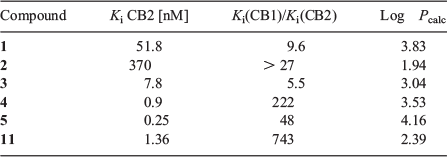
We have described the synthesis and pharmacology tests of a series of 1,8-naphthyridin-, quinolin- and pyridin- one-3-carboxamides, with high potency and selectivity for CB2 receptors (Fig. 1); many of these compounds had a fluorine atom, and interesting properties as prospective radiotracers (Table 1).[2–5] Our initial studies[6] on radiofluorinating these scaffolds revealed the instability of the carbon–fluorine bond in the ortho position of the pyridine-like nitrogen of 1 and 2, as we noticed extensive defluorination.[7,8] In successive studies, we have investigated structures 3, 4, and 5, bearing a more stable fluorobenzyl moiety,[9] but the synthesis of the radiolabelling precursor, as well as the radiofluorination process itself, proved to be quite challenging. We have therefore directed our attention to structures bearing a fluoroalkyl moiety, forecasting easier radiochemical synthesis and good stability.
|
|
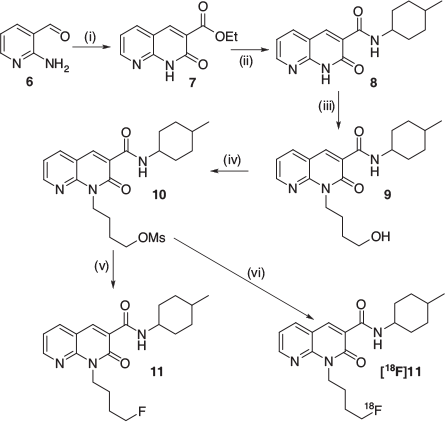
Among the candidates of the molecular lead developed, compound 11 showed good potency and CB2 selectivity, Log Pcalc in the 1–3 range, and reliable synthesis (Fig. 2).[3] In particular, the synthetic route adopted involved the production of the mesylate intermediate 10, that represents a suitable radiofluorination precursor. In this work we will describe the production of [18F]11 and its biodistribution in an ALS mouse model, based on a genetically modified superoxide dismutase 1 (SOD1).
|
Results and Discussion
Radiolabelling Optimization
The radiofluorination of 10 was performed using an Advion NanoTek microfluidic system. The use of this apparatus allowed to streamline the optimization of reaction conditions while minimizing chemicals, radioactivity, and time employed.[10] The F-18 complex was prepared as previously described[6] using DMSO as reconstituting solvent, and it was loaded in the storage loop of Pump 3 (P3), while the mesylate precursor was dissolved in DMSO (11.8 mg mL−1) and loaded in the storage loop of Pump 1 (P1); a 15.6 µL flow microreactor was used for all the experiments. In these experimental sets, the starting activity was 5.8 GBq, a bolus of 15–20 µL of fluorinating complex was used in each reaction, and delivered activity ranged from 120 to 40 MBq. Reduction of radioactive concentration was due to decay over 2.5 h, as needed for the reaction optimization process. Reaction parameters were varied in the process as follows: reaction temperature: 110–190°C, reactants residence time: 9.4–93.8 s, and mass of precursor: 178.5–2380 µg. The crude reaction mixtures were analyzed by both radio-HPLC and radio-TLC, reporting similar overall trends but radiochemical conversion (RCC)[11,12] calculated from TLC was higher than from HPLC.
Graphs from the optimization experiments are shown in Fig. 3. From these results, it was possible to identify the optimized reaction conditions for the radiolabelling. In particular, we found that temperature had the main effect on RCC, which increased at higher temperature, and reached its highest value at 190°C. Residence time had no influence at 190°C, while at 170°C a longer residence time (i.e. lower flow rates) provided a higher RCC. The effect of precursor mass variation in each bolus reaction was tested at 170°C and did not influence RCC, except a slight decrease at the lowest value of 178.5 µg.

|
Production of [18F]11
The advantage of using a microfluidic system for both optimization and production is the capability to easily scale up the process without needing to refresh the hardware moving on to higher activities.[13,14] Following the optimization tests above, it was decided to use a temperature of 190°C and a residence time of 23.4 s, given by a 1:1 mixing ratio of 20 µL min−1 flow of the radiofluorination and precursor solutions. In the preclinical production runs, a starting activity of 34.4 ± 6 GBq was used, and such radiofluoride solution was pre-concentrated, dried, and reconstituted as previously reported;[6] for this process, 0.7 mL of DMSO were used as reconstituting solvent, to provide the radiofluorinating mixture. A volume of 200 µL for both solutions was used; the concentration of the precursor ranged 1.2 to 1.3 mg mL−1 such that, when used in the production radiolabelling run, provided a total bolus reaction mass of 240–260 µg. Using larger bolus volumes was not expected to negatively affect the RCC, given the positive bolus-effect reported for this microfluidic approach.[15]
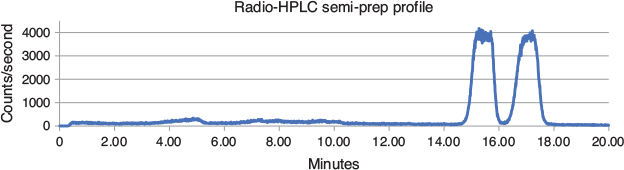
The hardware utilised in the production was modified as previously described[16] to allow reaction, purification and formulation to be controlled by the same software (Fig. 4). In detail, the crude product from the flow-chemistry system was directed towards the storage loop of a manual HPLC injector, connected to an external pump and a semi-prep column. The outlet of the chromatographic column was monitored by a customized radioactivity probe;[17] this allowed detection of the product fraction, that was collected separately for water dilution (Fig. 5). This diluted fraction was then passed through a C-18 SPE cartridge, which retained the organic product. The SPE was eluted with EtOH and the eluate evaporated and reconstituted with a 10 % ethanolic injectable saline. In a typical production run, 845 ± 393 MBq of [18F]11 was available for use, that was > 95 % radiochemically pure and with a molar activity (AM) of 25 ± 14 MBq nmol−1 at the end of synthesis (EOS; Fig. 6). This production process was repeated in 16 runs. A limited number of runs (4) had losses of radioactivity in the initial step of radiofluorination complex preparation (i.e. fluoride pre-concentration, drying, and reconstitution), possibly due to aerosol formation. No failures were recorded in the flow radiolabelling step and successive purification and formulation.

|
|

|
The semi-prep chromatographic separation highlighted the diastereoisomerism of 11. In fact, in the typical purification run, two radioactive peaks of similar heights were detected, and they were due to the presence of cis and trans conformers at the methylcyclohexyl substituent. The precursor 10 was used as a diastereoisomeric mixture and no attempt was made to purifying this precursor or synthesize the diastereoisomerically pure compound, as the biological assay for 11 was tested with such a mixture.
The overall process consisted of two phases, namely a) radiofluorinating the mixture preparation and b) tracer production. On average, phase a) lasted 40 min, while phase b) lasted 50 min; however, due to concurrent animal and scanner preparation, phase b) was not always started right after phase a). This condition, added to the reported variability in recovering the radiofluorinating mixture,[18] made it difficult to evaluate the radiochemical yield (RCY) of the process for each production run.[11,12] In this case, we evaluated this parameter by assessing the total radioactive content of the bolus reaction; this value was obtained performing a new sample radiofluorination reaction at EOS and measuring it for radioactivity content. By this method, we obtained a RCY of 29 ± 12 %, which is lower than the RCC, but still allowed the scan of up to three subjects per production run during imaging sessions.
Animal Models
The ALS mouse animal model was developed by introducing a mutation in the antioxidant enzyme Cu/Zn superoxide dismutase 1 (SOD1) gene, which has been linked to the cause of ~20 % of familial ALS. Expression of a mutant human SOD1 transgene in mice (i.e. SOD1G93A mice) results in altered motor neuron functioning, thus inducing symptoms mimicking human ALS, making this model the most widely used for studies on this disease.[19] Neuron degeneration and death is accompanied by microglia activation, which represents a hallmark of neuroinflammation. In particular, microglia priming, i.e. before phagocytosis is exerted, is associated with an upregulation of CB2 receptors,[20] and this is also readily apparent in SOD1G93A mice.[21] In this work we were interested in evaluating the ability of [18F]11, a prospective CB2 receptor ligand, to highlight the differential uptake in an animal model featuring ALS versus wild-type subjects.
SOD1G93A mice develop evident symptoms at 12–16 weeks, represented by progressive motor deficits (tremor and shaking in one or more limbs) leading to paralysis.[22] In our study, we monitored the mice for occurrence of locomotor symptoms, and scanned both asymptomatic (n = 11) and symptomatic (n = 11) SOD1G93A to assess the potential diagnostic value of CB2 imaging. We also studied [18F]11 in CD1 (n = 3) and C57BL (n = 4) mouse strains, as reference models for healthy subjects. In addition, blocking experiments with commercially available CB2 receptor agonist (AM630) were performed on five SOD1G93A animals, three of which were clearly symptomatic, while the others (121 days old) were studied just before symptoms onset.
Binding Assessment by In Vitro Autoradiography
In order to qualitatively assess the binding of [18F]11 in the tissues of interest, we performed an in vitro autoradiography assay using brain slices obtained from CD1 and SOD1 mice, which included the primary motor cortex (MC) and cerebellum (C). The slices were incubated with a solution of [18F]11 and then exposed onto a phosphor plate for radioactive imaging. Pre-blocking studies were also conducted by incubating contiguous slices with the CB2 ligand AM630 before radiopharmaceutical incubation, to assess the level of specificity of [18F]11 for the target.
Radioactive distribution was obtained by laser scan of the phosphor plates, recorded in digital light units (DLU) and corrected for regions of interest (ROIs). No calibration was performed on the phosphor imager at this time. MC was identified in the slices and ROIs were drawn manually. The specificity of [18F]11 uptake was calculated by Eqn 1:
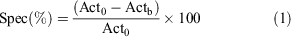
where Act0 and Actb are the DLU mm−2 values for unblocked and blocked slices. Applying the calculation, we obtained an average specific binding in the MC in both CD1 (31 ± 15 %) and SOD1 animals (21 ± 10 %). Peculiarly, only the SOD1 animals showed a distinctive asymmetry in uptake, which revealed a repeatable higher specific uptake in the right hemisphere versus the controlateral (24 ± 12 % versus 17 ± 11 %). In the cerebellum, a complete lack of specificity was observed (−13.3 ± 10.3 %).
From this data, it is possible to hypothesize that [18F]11 has a specific binding in the brain, in particular for MC areas, that could be involved in the movement disorders observed in SOD1 subjects; likewise, the observed left–right difference might be related to the asymmetric appearance of movement disorders, although we cannot make any inference on this point as symptoms were not recorded following this parameter during animal monitoring.
PET/CT Imaging
PET dynamic imaging and CT of all animals were carried out separately by using a YAP-(S)PET II microPET and a XALT high resolution scanner, respectively, as explained in the Experimental section. PET field of view (FOV) included the brain and the heart.
ROIs were manually drawn on three organs: cardiac left ventricular cavity (LV), whole brain (WB), and gallbladder (GB). The LV was used to evaluate blood compartment and the related input function, WB represented the target organ, while GB showed a peculiar high accumulation, possibly related to excretion. Time activity curves (TAC) were evaluated from the raw Bq mL−1 data, corrected for injected dose and animal weight; examples of TAC are reported in Fig. 7.

|
The data obtained showed considerable variability (e.g. Fig. 7a) that did not allow a significantly low P value to be reached in any group comparison. Average values for each time-point in each group revealed a delayed washout from the WB region (Fig. 7b) when compared with LV TACs (Fig. 7c). Gallbladder accumulation of CB2 radiolabelled tracers has been reported in previous studies,[23] and we have noticed this phenomenon also in our experiments (Fig. 7d).
A comparison between early and late frames of the WB TACs showed trend differences between study groups. While in the first 5 min CD1 and C57BL groups (i.e. control subjects) showed an immediate washout, in both unblocked groups of SOD1 mice we observed a noticeable uptake; on the other hand, the SOD1 group receiving a pre-blocking dose of competitor was characterized by markedly lower standardized uptake values (SUV) in these same early frames (Fig. 8a). This differential uptake among groups, in particular with the competitor population, disappeared in the later time points (Fig. 8b), showing a full washout from the brain (Fig. 9).

|

|
Therefore, we analyzed the data using a Logan plot to assess group differences in tissue distribution volume ratio (DVR).
Logan Analysis of Dynamic Uptake
A Logan plot is a graphical method for the quantification of PET imaging data developed for reversibly binding radiotracers with rapid dissociation constants and efflux from tissue. Moreover, this method concerns tracers whose kinetics can be described in terms of first-order, constant-coefficient, and linear differential equations. In its original version, the arterial plasma tracer concentration represents the input function,[24] but blood sampling can be avoided using the non-receptor-containing region as reference.[25] In our case, we used the LV data as proxy for the input function to estimate DVR, which is defined as Eqn 2:
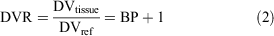
where DVtissue and DVref are the distribution volume in the target tissue and in the reference one, and BP is the binding potential.[26] Following this definition, higher values of DVR reflect an increased expression of the binding in the tissue of interest. In the Logan plot, a linear trend is reached after a certain time (t > t*), in which the slope represents the DVR, according to Eqn 3:

where Cref(t) and Ctissue(t) are the tracer concentration in the reference tissue (LV) and in the target tissue (WB), respectively. The term including the average tissue-to-plasma efflux constant (q) was omitted because the ratio of Ctissue(t) over Cref(t) is assumed to be reasonably constant.[25]
The optimal value of t* was chosen by visual inspection of all the Logan plots and set to use at t = x min (i.e. the last 7 time points). The selection of earlier or later time points (namely 5 to 10) yielded consistent results with average percentage difference of DVRs less than 4 %.
Finally, the least square estimation of DVR was weighted using the inverse of the variance of the PET measurement, calculated as Eqn 4:[27]

where C(tk) is the acquired mean value of the tracer activity over the kth relative time scan interval Δtk. The Logan analysis was implemented in MATLAB R2020a (The MathWorks, Natick, MA, USA), using the cumtrapz.m function to perform the cumulative trapezoidal numerical integrations shown in Eqn 3.
DVR findings were compared to assess binding differences in the target tissue between groups, assuming a P value < 0.05 as significant; in particular the higher the DVR, the higher the binding in the target tissue. The DVR was similar between the healthy groups, CD1 and C57BL, and the asymptomatic SOD1 mice. However, the C57BL group featured a higher similarity with the transgenic group (P = 0.1651, Fig. 10a), whereas the difference seems more pronounced with the CD1 group (P = 0.0528), in which significance was almost achieved. Interestingly, the SOD1G93A mice have a (C57BL/6 × SJL)F1 strain of origin,[28] and this finding may point out the importance of preferring the C57BL strain as control model.

|
A significant difference in DVRs was instead found between asymptomatic and symptomatic SOD1 groups (P = 0.0033) (Fig. 10b). This finding may suggest that an increased expression of CB2 receptors occurs during disease progression in this animal model.
The DVR from the group in which a competitor was used was intermediate between the asymptomatic and symptomatic group, and significantly different only from the asymptomatic SOD1 group (P = 0.0440) and not from the symptomatic one (P = 0.5061); a potential explanation is that the competition test was performed on overtly symptomatic (n = 3) or late asymptomatic (n = 2) subjects, which were age matched, thus potentially already presenting the biochemical hallmark of disease progression.
Conclusion
This work describes the radiolabelling and the preliminary in vivo biodistribution of a prospective PET radiopharmaceutical for imaging the CB2 receptor, based on a naphthyridine core and labelled with 18F on a side alkyl chain bearing a mesylate group. The radiolabelling process, as performed via microfluidics, semi-prep HPLC purification, and SPE formulation, allowed the desired product to be obtained in 29 % RCY with an AM = 24 MBq nmol−1 EOS. In vitro autoradiography and competition tests were used to preliminarily explore radiotracer specificity on the motor cortex and cerebellum. Even if autoradiography did not reveal strong specificity of the tracer, the biodistribution of the radiolabelled product was studied using PET and CT imaging in Superoxide Dismutase 1 gene model mice (SOD1G93A), and in CD1 and C57BL mice as healthy controls. SOD1 mice were divided into asymptomatic and symptomatic groups. Our investigation focussed on assessing the radioactive uptake in brain regions, and revealed quick washout and non-significant differences of SUV. For these reasons, a Logan plot analysis was conducted to estimate differences in the distribution volume ratio (DVR) in the brain between healthy, and asymptomatic and symptomatic SOD1 groups as a possible indicator of early specific extraction of the tracer in a highly reversible mode. This analysis revealed a significant difference between asymptomatic and symptomatic SOD1 animals, whereas the symptomatic SOD1 had a noticeably higher DVR. Even if not conclusive, in particular in regard with tracer specificity, the reported outcome seems to align with the research hypothesis that an increased CB2 receptor expression occurs in the symptomatic subjects, and may support the potential use of [18F]11 as a PET imaging tracer for CB2 receptors in the brain. Further studies would be required to fully understand the whole-body distribution of such a molecule, its metabolism, and the inter-species uptake differences.
Experimental
Radiochemistry
General
All chemicals and solvents were purchased from Sigma–Aldrich and used without further purification. The high-purity grade solvents were vented through a soda lime/molecular sieves trap upon use. Micro-SPE cartridges MP-1 were purchased from ORTG (USA). [18F]fluoride was produced at a PETtrace cyclotron (GE Healthcare, USA) by proton bombardment (Ep = 16.7 MeV, 5–15 min at 20–25 µA) of a 1.3 mL [18O]water (enrichment > 98 %) silver target. Preparative radio-HPLC were run using isocratic conditions (60/40 = CH3CN/H2O at 3 mL min−1) using a standalone Waters 515 pump, a Rheodyne 7125 manual injector, a Phenomenex Synergi Fusion-RP 80A (4 µm, 250 × 10 mm) column, while radioactive peaks were monitored by a prototype CsI scintillator detector (Tyndall, Ireland).[17] Analytical radio-HPLCs were obtained using a Delta 600 pump system (Waters, USA) equipped with a Gabi Star flow-through gamma detector (Raytest, Germany) connected in series to a 996 Photo Diode Array (PDA) UV detector (Waters, USA) on a Phenomenex Synergi Hydro-RP 80A (4 µm, 150 × 4.6 mm) using 65/35 = CH3CN/H2O. TLC analyses were performed using silica plates and 100 % EtOAc as eluent and acquired using a Cyclone PLUS (Perkin–Elmer, USA).
Radiosynthesis was performed using an Advion microfluidic system equipped with a 15.6 µL internal volume microreactor. The apparatus was customized for performing automatic HPLC peak collection and SPE formulation of the collected peak.[16]
Production of [18F]11
A 1.2 ± 1 mg mL−1 solution of 10 in dry DMSO was prepared from 10-fold concentrated stock solution; this precursor was charged into the storage loop of Pump 1 (P1). The dry fluorination complex was prepared as previously reported,[6] employing 15 ± 1 mg mL−1 of K222 in CH3CN and 80 µL of a 50 mg mL−1 solution of K2CO3 in H2O. The dried residue was reconstituted with 0.7 mL of DMSO and this solution was charged into the storage loop of Pump 3 (P3). In order to prime the flow system, three ‘dummy’ reactions were performed by delivering into the microreactor 30, 10, and 10 µL at 20 µL min–1 for each channel at 150, 170, and 190°C respectively. The production was performed by delivering 200 µL from P1 and P3 at 20 µL min−1 into the microreactor heated at 190°C; the reaction mixture was directed towards the 0.5 mL loop of a manual injector valve. The isocratic elution conditions used allowed collection of the radioactive product as a double peak (two conformers) at ~15 and 17 min.
The collected fractions were diluted with water, passed on a C18 Waters Sep-Pak C18 Plus Light and the product was eluted with 1.5 mL of EtOH. The obtained ethanolic solution was dried and the residue reconstituted with a suitable volume (0.5–2 mL) of 10 % ethanolic saline and sterile filtered. The product was ready for animal injection (518–1380 MBq) with a RCY of 29 ± 12 % (from the starting fluoride complex solution). The whole production process took around 50 min from the start of bolus delivering to obtaining the injectable solution. The product was > 95 % radiochemically pure by both HPLC and TLC and AM was 25 ± 14 MBq nmol−1 at EOS.
Autoradiographic Assessment
Animals were housed in light and temperature controlled conditions (12 h light: 12 h dark; 22 ± 2°C) and given ad libitum water and food. All procedures were performed using protocols approved by the Institutional Animal Care and Use Committee. At sacrifice, all mice were deeply anesthetized and brains were promptly removed, washed in PBS 1X (pH = 7.4) and then individually included in an OCT matrix. After freezing with dry ice, samples were stored at –80°C until sectioning in 20 μm thick coronal sections by a cryostat.
The obtained tissue sections of the primary motor cortex, red nucleus-midbrain, and cerebellum were collected and placed on polylysine-coated glass slides. Each area was studied in duplicate. Tissue sections were heat-fixed for 5 min at 50°C with a heating chamber and then pre-blocked or not with 100 μL of antagonist saline solution (AM630, 15 μM) for 45 min. Pre-blocked sections were washed three times with phosphate buffer 1X (pH = 7.0) before incubation. Brain tissue slices were incubated for 1 h with 100 μL of [18F]11 (0.8 MBq mL−1). During the incubation time at room temperature, the tissue sections were placed in a humidified black box to reduce evaporation. The excess of radioactivity was washed three times by phosphate buffer 1X. The sections were then dried and exposed to a phosphor plate for 1 min, before being scanned using a Cyclone Plus. The intensity of ex vivo auto-radiographic images was measured using Optiquant software (Perkin Elmer, USA) by manually drawing ROIs, and the raw data were evaluated using Microsoft Excel.
Study Populations
A total of n = 34 mice (The Jackson Laboratories, Bar Harbor, Maine) were employed for the study. Specifically, we studied three animal groups: CD1 (n = 3), C57BL (n = 4), and SOD1G93A (n = 27). Within the SOD1 group, mice were classified as follow: n = 11 as asymptomatic (age = 72–114 days), n = 11 as symptomatic (age = 137–175), and n = 5 employed in pre-blocking experiments with AM630. All mice were housed under 12-h light/12-h dark cycles and controlled room temperature (22°C), with ad libitum access to food and fresh water. Health status and locomotor abilities were regularly monitored, and underwent PET/CT imaging as required: abnormally reduced activity, hindlimb suspension, beam walk, limb tremor, or shaking were used to classify SOD1 mice as symptomatic. At the end of in vivo procedures, all animals were killed. The experimental protocol was conducted in accordance with the D.L.116/92 implementation of European Economic Community directive 609/86 regarding the protection of animals used for experimental and other scientific purposes.
PET and CT Imaging Protocol and Analysis
Prior to imaging, anaesthesia was induced by xylazine and ketamine and maintained by gas inhalation of isoflurane. The tail veins were accessed and catheterized for tracer administration. CT imaging was performed for anatomical co-localization of PET images using the XALT scanner,[29] with animals in a supine position, with the following settings: 35 kV, 1 mm Al filtration, 211 views over 200° (short-scan), 0.27 mAs per view, 80 s total scan time. Images were reconstructed with a custom cone-beam FBP algorithm on 3D matrices of 480 × 480 × 720 voxels, with an isotropic voxel size of 84 micron. Upon completion of the CT scan, animals were transferred on the same carbon fibre bed to the PET scanner (YAP(S)PET II, I.S.E. s.r.l., Vecchiano, Italy). Animals received an i.v. injection of 3–17 MBq (median: 7.7 MBq) of tracer depending on their weight in 100–200 μL dilution volume, followed by 100 μL of saline. A 60 min dynamic PET scan of the brain region was performed immediately after injection. PET images were reconstructed using an iterative OSEM algorithm, as previously described,[30,31] on a 3D matrix size of 128 × 128 × 27 slices (voxel size: 0.375 × 0.375 × 1.5 mm3). The dynamic protocol was set up with the following time frames: 9 × 60 s; 5 × 120 s; and 8 × 300 s. After the dynamic scan, a whole-body multi-bed position static PET scan was performed, with the total duration of 15 min (5 min per bed position). All images were corrected for random coincidences, dead time, decay correction, and calibrated in kBq cc−1. ROI drawing was performed on Amide software (http://amide.sourceforge.net/), using a cylindrical volume for LV (1.5 × 1.5 × 3 mm, drawn from first 2 frames), an ellipsoid for WB (4 × 7 × 3 mm, drawn from last 2 frames), and an ellipsoid for GB (2 × 3 × 2 mm, drawn from last 2 frames). Time activity curves (TAC) were then derived on each animal, and standardized uptake value (SUV) curves were calculated by correcting for injected dose and animal weight.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors thank the Australian National Imaging Facility for support. These results were obtained within the project Positron Emission Tomography and Lateral Amyotrophic Sclerosis (PETALS II) funded by the Fondazione Italiana di Ricerca per la Sclerosi Laterale Amiotrofica (ARISLA).
References
[1] S. Yamagishi, Y. Iga, M. Nakamura, C. Takizawa, D. Fukumoto, T. Kakiuchi, S. Nishiyama, H. Ohba, H. Tsukada, K. Sato, Y. Ouchi, J. Neuroinflammation 2019, 16, 208.| Crossref | GoogleScholarGoogle Scholar | 31707986PubMed |
[2] C. Manera, G. Saccomanni, B. Adinolfi, V. Benetti, A. Ligresti, M. G. Cascio, T. Tuccinardi, V. Lucchesi, A. Martinelli, P. Nieri, E. Masini, V. Di Marzo, P. L. Ferrarini, J. Med. Chem. 2009, 52, 3644.
| Crossref | GoogleScholarGoogle Scholar | 19435366PubMed |
[3] V. Lucchesi, D. P. Hurst, D. M. Shore, S. Bertini, B. M. Ehrmann, M. Allarà, L. Lawrence, A. Ligresti, F. Minutolo, G. Saccomanni, H. Sharir, M. Macchia, V. Di Marzo, M. E. Abood, P. H. Reggio, C. Manera, J. Med. Chem. 2014, 57, 8777.
| Crossref | GoogleScholarGoogle Scholar | 25272206PubMed |
[4] C. Manera, V. Benetti, M. P. Castelli, T. Cavallini, S. Lazzarotti, F. Pibiri, G. Saccomanni, T. Tuccinardi, A. Vannacci, A. Martinelli, P. L. Ferrarini, J. Med. Chem. 2006, 49, 5947.
| Crossref | GoogleScholarGoogle Scholar | 17004710PubMed |
[5] C. Manera, G. Saccomanni, A. M. Malfitano, S. Bertini, F. Castelli, C. Laezza, A. Ligresti, V. Lucchesi, T. Tuccinardi, F. Rizzolio, M. Bifulco, V. Di Marzo, A. Giordano, M. MacChia, A. Martinelli, Eur. J. Med. Chem. 2012, 52, 284.
| Crossref | GoogleScholarGoogle Scholar | 22483967PubMed |
[6] G. Pascali, G. Mazzone, G. Saccomanni, C. Manera, P. A. Salvadori, Nucl. Med. Biol. 2010, 37, 547.
| Crossref | GoogleScholarGoogle Scholar | 20610159PubMed |
[7] G. Pascali, D. Panetta, S. Burchielli, M. De Simone, E. Sanguinetti, V. Lucchesi, S. Del Carlo, G. Saccomanni, C. Manera, M. Macchia, P. A. Salvadori, J. Label. Compd. Radiopharm. 2013, 56, P-213.
[8] G. Pascali, D. Panetta, S. Del Carlo, G. Saccomanni, C. Manera, M. Macchia, P. A. Salvadori, J. Nucl. Med. 2012, 53, 1632.
[9] G. Saccomanni, G. Pascali, S. Del Carlo, D. Panetta, M. De Simone, S. Bertini, S. Burchielli, M. Digiacomo, M. Macchia, C. Manera, P. A. Salvadori, Bioorg. Med. Chem. Lett. 2015, 25, 2532.
| Crossref | GoogleScholarGoogle Scholar | 25956416PubMed |
[10] G. Pascali, L. Matesic, T. L. Collier, N. Wyatt, B. H. Fraser, T. Q. Pham, P. A. Salvadori, I. Greguric, Nat. Protoc. 2014, 9, 2017.
| Crossref | GoogleScholarGoogle Scholar | 25079426PubMed |
[11] M. M. Herth, S. Ametamey, D. Antuganov, A. Bauman, M. Berndt, A. F. Brooks, G. Bormans, Y. S. Choe, N. Gillings, U. O. Häfeli, M. L. James, K. Kopka, V. Kramer, R. Krasikova, J. Madsen, L. Mu, B. Neumaier, M. Piel, F. Rösch, T. Ross, R. Schibli, P. J. H. Scott, V. Shalgunov, N. Vasdev, W. Wadsak, B. M. Zeglis, Nucl. Med. Biol. 2021, 93, 19.
| Crossref | GoogleScholarGoogle Scholar |
[12] H. H. Coenen, A. D. Gee, M. Adam, G. Antoni, C. S. Cutler, Y. Fujibayashi, J. M. Jeong, R. H. Mach, T. L. Mindt, V. W. Pike, A. D. Windhorst, Nucl. Med. Biol. 2017, 55, v.
| Crossref | GoogleScholarGoogle Scholar | 29074076PubMed |
[13] L. Matesic, I. Greguric, G. Pascali, Aust. J. Chem. 2018, 71, 811.
| Crossref | GoogleScholarGoogle Scholar |
[14] L. Matesic, A. Kallinen, I. Greguric, G. Pascali, Nucl. Med. Biol. 2017, 52, 24.
| Crossref | GoogleScholarGoogle Scholar | 28599133PubMed |
[15] J. Ungersboeck, S. Richter, L. Collier, M. Mitterhauser, G. Karanikas, R. Lanzenberger, R. Dudczak, W. Wadsak, Nucl. Med. Biol. 2012, 39, 1087.
| Crossref | GoogleScholarGoogle Scholar | 22633218PubMed |
[16] G. Pascali, A. Berton, M. DeSimone, N. Wyatt, L. Matesic, I. Greguric, P. A. Salvadori, Appl. Radiat. Isot. 2014, 84, 40.
| Crossref | GoogleScholarGoogle Scholar | 24296158PubMed |
[17] V. Arima, G. Pascali, O. Lade, H. R. Kretschmer, I. Bernsdorf, V. Hammond, P. Watts, F. De Leonardis, M. D. Tarn, N. Pamme, B. Z. Cvetkovic, P. S. Dittrich, N. Vasovic, R. Duane, A. Jaksic, A. Zacheo, A. Zizzari, L. Marra, E. Perrone, P. A. Salvadori, R. Rinaldi, Lab Chip 2013, 13, 2328.
| Crossref | GoogleScholarGoogle Scholar | 23639996PubMed |
[18] J. Ungersboeck, C. Philippe, L.-K. Mien, D. Haeusler, K. Shanab, R. Lanzenberger, H. Spreitzer, B. K. Keppler, R. Dudczak, K. Kletter, M. Mitterhauser, W. Wadsak, Nucl. Med. Biol. 2011, 38, 427.
| Crossref | GoogleScholarGoogle Scholar | 21492791PubMed |
[19] K. E. Lewis, A. L. Rasmussen, W. Bennett, A. King, A. K. West, R. S. Chung, M. Chuah, J. Neuroinflammation 2014, 11, 55.
| Crossref | GoogleScholarGoogle Scholar | 24655927PubMed |
[20] J. Ashton, M. Glass, Curr. Neuropharmacol. 2007, 5, 73.
| Crossref | GoogleScholarGoogle Scholar | 18615177PubMed |
[21] E. A. Horne, N. Stella, Future Lipidol. 2008, 3, 435.
| Crossref | GoogleScholarGoogle Scholar |
[22] M. Leitner, S. Menzies, C. Lutz, Working with ALS Mice: Guidelines for Preclinical Testing & Colony Management 2009. Available at: http://jackson.jax.org/rs/444-BUH-304/images/Working_with_ALS_Mice.pdf
[23] R. Ahmad, M. Koole, N. Evens, K. Serdons, A. Verbruggen, G. Bormans, K. Van Laere, Mol. Imaging Biol. 2013, 15, 384.
| Crossref | GoogleScholarGoogle Scholar | 23508466PubMed |
[24] J. Logan, J. S. Fowler, N. D. Volkow, A. P. Wolf, S. L. Dewey, D. J. Schlyer, R. R. MacGregor, R. Hitzemann, B. Bendriem, S. John Gatley, D. R. Christman, J. Cereb. Blood Flow Metab. 1990, 10, 740.
| Crossref | GoogleScholarGoogle Scholar | 2384545PubMed |
[25] J. Logan, J. S. Fowler, N. D. Volkow, G. J. Wang, Y. S. Ding, D. L. Alexoff, J. Cereb. Blood Flow Metab. 1996, 16, 834.
| Crossref | GoogleScholarGoogle Scholar | 8784228PubMed |
[26] M. A. Mintun, M. E. Raichle, M. R. Kilbourn, G. F. Wooten, M. J. Welch, Ann. Neurol. 1984, 15, 217.
| Crossref | GoogleScholarGoogle Scholar | 6609679PubMed |
[27] B. M. Mazoyer, R. H. Huesman, T. F. Budinger, B. L. Knittel, J. Comput. Assist. Tomogr. 1986, 10, 645.
| Crossref | GoogleScholarGoogle Scholar | 3488337PubMed |
[28] 002300 - B6SJL-Tg(SOD1*G93A)<dl>1Gur/J. Available at https://www.jax.org/strain/002300, n.d.
[29] D. Panetta, N. Belcari, A. Del Guerra, A. Bartolomei, P. A. Salvadori, Phys. Med. 2012, 28, 166.
| Crossref | GoogleScholarGoogle Scholar | 21501966PubMed |
[30] T. Liistro, L. Guiducci, S. Burchielli, D. Panetta, N. Belcari, S. Pardini, A. Del Guerra, P. A. Salvadori, P. Iozzo, J. Cereb. Blood Flow Metab. 2010, 30, 895.
| Crossref | GoogleScholarGoogle Scholar | 20179723PubMed |
[31] S. Moehrs, M. Defrise, N. Belcari, A. Del Guerra, A. Bartoli, S. Fabbri, G. Zanetti, Phys. Med. Biol. 2008, 53, 6925.
| Crossref | GoogleScholarGoogle Scholar | 19001696PubMed |