

Progress Towards Direct Hydrogen Peroxide Fuel Cells (DHPFCs) as an Energy Storage Concept*
Ciaran J. McDonnell-Worth A B and Douglas R. MacFarlane A
A B and Douglas R. MacFarlane A
A Faculty of Science, Monash University, Scenic Boulevard and Wellington Road, Clayton, Vic. 3800, Australia.
B Corresponding author. Email: ciaran.mcdonnell-worth@monash.edu

Ciaran McDonnell-Worth is a Research Fellow in the MacFarlane group at Monash University. He is a B.Sc. (Hons) graduate and obtained his Ph.D. degree from Monash University. His research interests are in electrogenerated fuels and clean energy storage. |

Professor Doug MacFarlane is an Australian Laureate Fellow and leader of the Energy Program in the Australian Centre for Electromaterials Science. He is one of the pioneers in the field of ionic liquids and his group along with collaborators in Australia and worldwide has published more than 650 papers and 30 patents. Professor MacFarlane is a B.Sc. (Hons) graduate of Victoria University of Wellington and received his Ph.D. degree from Purdue University. He was appointed Professor of Chemistry at Monash University in 1995. He was elected to the Australian Academy of Science in 2007 and in 2018 was awarded the Academy’s Craig Medal for achievements in chemistry He was elected to the Australian Academy of Technological Sciences and Engineering in 2009. He is an International Fellow of the Queen’s University Belfast, a Visiting Professor of the Chinese Academy of Sciences, and the Huangshan Distinguished Visiting Professor at HuFei University of Technology. |
Australian Journal of Chemistry 71(10) 781-788 https://doi.org/10.1071/CH18328
Submitted: 9 July 2018 Accepted: 9 August 2018 Published: 7 September 2018
Abstract
This review introduces the concept of direct H2O2 fuel cells and discusses the merits of these systems in comparison with other ‘clean-energy’ fuels. Through electrochemical methods, H2O2 fuel can be generated from environmentally benign energy sources such as wind and solar. It also produces only water and oxygen when it is utilised in a direct H2O2 fuel cell, making it a fully reversible system. The electrochemical methods for H2O2 production are discussed here as well as the recent research aimed at increasing the efficiency and power of direct H2O2 fuel cells.
Introduction
Phasing out fossil fuels as an energy storage system is an inevitability as sources of oil become harder to access both physically and politically.[1] The influence of carbon dioxide and other greenhouse gases on weather patterns and the environment has also become a serious cause for concern.[2] The amount of renewable energy generated by wind, solar, hydro, and geothermal sources is on the rise and will likely begin to eclipse fossil fuels in the future. Unfortunately, many of these renewable energy sources are either intermittent or limited to certain geographic regions, and as such cannot fully replicate the ubiquity afforded by petrol, coal or natural gas. To sustain the same modes of energy transportation and utilisation that our society is comfortable with today, the energy produced by renewable sources must be stored in efficient and inexpensive media. Electrochemically generated chemical fuels are ideal for this purpose as they can be produced on demand with renewable energies and stored and transported when those energy sources are unavailable.
Rather than being burned in conventional combustion engines, chemical fuels can also be used in fuel cells to convert their stored chemical potential energy directly into electrical energy. In general, a fuel cell is composed of two compartments, one containing an anode on which the fuel is electrochemically oxidised and the other containing a cathode on which an oxidant (usually O2) is reduced. Separating these two segments is a membrane that allows charge to pass through the fuel cell but keeps the fuel and the oxidant from coming in direct contact with each other, or the opposite electrodes. The power that is generated by the fuel cell is a result of the difference in the electrochemical potential of the oxidation of the fuel and the reduction of the oxidant.
The fuel most famously associated with clean energy is undoubtedly H2, which has garnered media attention for its use in real-world applications[3] as well as its long pedigree of intense scientific research.[4–6] H2 can be generated electrochemically by the water-splitting process in which an electrochemical cell is used to oxidise water while simultaneously reducing protons to H2. This gas can be collected and later utilised as a fuel.
Although it ticks many of the boxes that are desirable in a ‘green’ fuel (its products when it is burned are only water and energy), H2 has several drawbacks that are not trivial to overcome. One of these is the storage of H2, which is largely a problem of both cost and efficiency. H2 storage has long been a problem for its utilisation as a fuel, as its energy density by volume at atmospheric pressure is far below that which would be practicable in a vehicle or even a stationary fuel storage facility. In order for it to be viable, H2 gas must either be compressed to ~70 MPa pressure, cryogenically stored as a liquid below 21 K or be physically or chemically captured in other materials.[7] In terms of compression, storage at higher pressures allows more fuel to be stored per unit volume, but also increases the amount of external equipment required to maintain that pressure safely.[8,9] This raises the cost of any system incorporating pressurised H2 gas and also increases its weight. There is also a certain amount of energy required to pressurise the H2 in the first place, all of which lowers the efficiency of these types of storage systems. Other options are physiochemical storage (using materials such as a metal–organic frameworks)[10–13] or chemical storage (such as in metal hydrides)[14,15] to adsorb or chemically bind H2. These materials are certainly safer than pressurised or liquid storage of H2, but usually necessitate energy losses such as heating to release H2 from the storage material during operation[16] and may suffer from impurities that lower their performance over time.[17]
One particularly interesting alternative fuel to H2 that has garnered some attention in the past decade is H2O2. H2O2 is well known as an oxidant and is used in several chemical and industrial processes that make use of this property.[18–23] It has even been used as the oxidant in other types of fuel cells where it has been shown to perform even better than O2 in some cases.[24–27] However, H2O2 is quite unique when applied to fuel cells because it can be both oxidised and reduced, with each process occurring at a different electrochemical potential (NHE = normal hydrogen electrode):
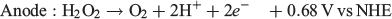
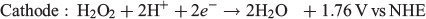
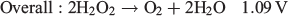
This opens up the possibility of using H2O2 as the fuel and the oxidant in the same fuel cell with a maximum theoretical potential of 1.09 V, which is reasonably close to other fuel cells such as those using methanol/air (1.21 V) and H2/air (1.23 V). The major advantage that an H2O2 fuel cell has over H2 is that H2O2 can be present in an aqueous solution at room temperature, thus requiring no pressurisation or additional storage media. Given this potential use as an energy storage medium, we review in the present paper recent progress in the production of H2O2 and its use in fuel cells.
Energy Density of H2O2
The energy density of an H2O2 fuel cell can be estimated and will ultimately depend on the concentration of H2O2 that can be safely used. At a conservative concentration of 7 wt-% in water, the theoretical energy density can be calculated from the Gibbs free energy of the state of the system before and after fuel cell operation. The total energy generated by the simultaneous reduction and oxidation of 2 mol of H2O2 to 1 mol of O2 and 2 mol of H2O is −234 kJ mol−1.[28] For a concentration of 70 wt-% of H2O2 in water (0.89 kg H2O2 L−1), this is equivalent to 3.1 MJ per litre of 70 wt-% H2O2. By comparison, a tank of H2(g) at 70 MPa at room temperature can produce 6.8 MJ L−1 (theoretical) when operating in a fuel cell. However, an estimated 20 % of that energy is used to compress the H2(g) in the first place,[7] which lowers the maximum practical energy to ~5.4 MJ L−1. Although this statistic may favour the use of compressed H2(g), it is not only the volume of the fuel that needs to be considered in the practical application of these systems. As mentioned above, pressurisation inevitably requires additional and specialised equipment for compressing the gas[13] and to prevent leakage and relieve pressure in case of faults.[9] In total, the gravimetric capacity of compressed H2(g) is ~13 wt-%,[17] meaning that most of the weight of the system is taken up by the storage vessel. Given that the storage of H2O2 is relatively simple compared with H2, the scale of application of the technology can be large or small with little change to the storage mechanism.
Sustainable Methods for Hydrogen Peroxide Production
Hydrogen peroxide is known as a ‘green’ oxidant because it only produces water and oxygen when it decomposes. As such, there is a much interest in using it for advanced oxidation processes such as the decolourisation of waste dye[20,29–31] and as the oxidant in chemical reactions.[19,22,32–37] Large-scale H2O2 production is almost exclusively performed via the anthraquinone auto-oxidation process in which O2 is reduced by 2-alkyl-anthrahydroquinone. First, H2 (g) is produced (usually via steam reformation) and used to hydrogenate the quinone over a Pd catalyst. The hydrogenated quinone must then be filtered, and then oxidised with air to form H2O2, which is subsequently extracted and distilled into an aqueous phase. This method is unsuitable in the context of renewable fuels primarily because of its reliance on hydrogen gas, which, when produced by steam reformation, is inherently non-renewable and ultimately energy inefficient. The direct electrochemical methods of production discussed below therefore provide a sustainable route for H2O2.
The Pd catalyst must also be roasted to clean it of organics and some toxic by-products must be treated or disposed of, which is energy-intensive and adds to the cost of the process.[38] The process is also only economically viable when it is performed on a large scale.
One of the benefits of electrochemical generation of H2O2 is that the devices used to produce the fuel can be constructed on a smaller or larger scale depending on their specific use. Much interest has been shown in designing electrocatalytic H2O2 batch reactors[39] for on-site industrial facilities that make use of advanced oxidation processes (AOPs),[33,40,41] pulp bleaching[18] or waste management[42] by H2O2. It may even be possible to downsize an electrochemical H2O2 production unit to the size that would fit into a household garage so that a steady supply of fuel could be generated and stored in homes.
There are two pathways available for electrochemical H2O2 production: O2 reduction and water oxidation. Both pathways may yield H2O2 in conjunction with other products, such as water in the case of O2 reduction and O2 in the case of water oxidation, depending on the specificity of the production method. H2O2 may also be produced by the direct reaction of H2(g) and O2(g);[43] however, a sustainable source of H2(g) is necessary for this process. In the following sections, we explore these processes in more detail.
Electrochemical H2O2 Generation
Oxygen Reduction
Electrochemical O2 reduction to H2O2 is a two-electron process and takes place according to the following redox reaction (E0 is the standard electrode potential):
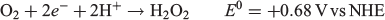
A wide variety of electrochemical catalysts have been found to reduce O2 to H2O2 with a greater or lesser degree of specificity, including TiO2 particles,[44] platinum,[45–47] manganese oxides,[48,49] iron oxides,[50] metal alloys such as Pd/Au[51] and Pt/Hg,[52] copper complexes,[53] cobalt oxides[54–57] and boron- or nitrogen-doped carbon materials.[58–60] The design of the electrocatalyst is important, as the competing four-electron O2 reduction to H2O:[61–63]
can cause efficiency losses. Bonakdapour et al. found that for non-noble metal catalysts, the amount of H2O2 produced from oxygen reduction varied greatly depending on the catalyst loading on the surface of a rotating ring disc electrode.[62] In fact, by altering the catalyst loading from 20 to 800 µg cm−2, the fraction of H2O2 observed at the ring from an Fe–N–C catalyst fell from 95 to as low as 5 %. This was attributed to the lifetime of H2O2 in the bulk of the catalyst being shortened either by further oxidation to water or by a chemical reaction with itself to produce O2 and water. By reducing the thickness of the electrode, most H2O2 was produced at the surface and was released to the electrolyte near the electrode before being further oxidised on the electrode.
In terms of bulk electrochemical production of H2O2 from O2, the surface area of a catalyst per unit area is a major limiting factor in production rates. Higher surface area means a higher number of catalytically active sites available for O2 reduction at any one time. Some materials such as porous carbon have been used for O2 reduction to H2O2 and have good activity, but highly varying faradaic efficiency.[64–69] Liu et al. have reported very good H2O2 production rates from O2 reduction, as high as 2249.4 mg L−1 h−1 using hierarchically porous carbon in a solution at pH 1.[70] These materials have a surface area of ~2000 m2 g−1, which was considered to be one of the major contributing factors to their high activity. The faradaic efficiency of these materials, however, varied greatly with changes to the electrolyte pH. The highest efficiency of 85–90 % was achieved in pH 1 electrolytes and the efficiency decreased with increasing pH, most likely owing to the propensity for H2O2 to disproportionate at higher pH.
In terms of pH, the two-electron reduction of O2 to H2O2 has been found to be highly favoured on Ag(111) in acidic electrolytes when the overpotential applied is kept below a certain threshold.[71] As higher overpotentials were applied, the rate of complete four-electron water oxidation increased until only O2 evolution was observed. In alkaline environments, there was very little H2O2 produced at all overpotentials applied. This behaviour was hypothesised to be highly dependent on the interaction between the catalytic surface and the intermediate reactants and spectating species. As such, certain catalysts are able to perform the two-electron O2 reduction only in alkaline environments.[57] Air pressure also has a significant effect on H2O2 generation from O2 reduction, with much higher rates seen at 3 MPa compared with atmospheric pressure.[72] This was attributed to the higher solubility of O2 in the electrolyte.
Photocatalytic H2O2 production has also been observed in water saturated with O2. Graphitic carbon nitride,[73] TiO2, zinc oxide,[74,75] and Co and Ru complex[76,77] catalysts have shown highly selective (in some cases greater than 90 %) two-electron O2 reduction to H2O2 simply by irradiation with sunlight or even sonication, reaching H2O2 concentrations of up to 350 µM in 1 h.
Water Oxidation
Water oxidation to H2O2 has been studied less thoroughly than O2 reduction but nonetheless shows promise as an efficient electrochemical production method. The process was observed by Izgorodin et al.[78] in an ionic liquid-based electrolyte using a manganese oxide catalyst. The presence of free amine in the alkaline electrolyte was proposed to solvate the H2O2 by hydrogen bonding, which limits the reaction to a two-electron oxidation. Using chronoamperometry, the potential of the working electrode was kept at 0.59 V versus Ag/AgCl. H2O2 was produced with a faradaic efficiency of 77 %. Further work elaborated on this by testing other cation and anion pairs for the electrolyte, including secondary ammonium cations (diethylammonium) and longer-chain alkylammonium cations (hexylammonium) as well as larger anions such as methanesulfonate and ethanesulfonate.[79] Although it was found that none of these produced more H2O2 than butylammonium sulfate (BAS), the efficiency of the process was improved by identifying the necessary applied potential at which H2O2 is produced but not further oxidised to O2(g). It was also found that H2O2 production from water oxidation occurs in a limited pH range starting at approximately pH 9.0 and reaching a maximum at pH 10.5. Photo-assisted electrocatalysed water oxidation to H2O2 has also been demonstrated on WO3/BiVO4 catalysts in aqueous electrolytes.[80]
By performing water oxidation to generate H2O2 rather than reducing O2, an interesting synergy can be exploited. Ando et al.[81] proposed a dual system in which H2O2 is produced simultaneously with H2 according to the reaction shown below:
This system was hypothesised to be more cost-effective as two products of value would be produced: H2 as a fuel and H2O2 as an industrial oxidant.
Following the same reasoning, it may be possible to combine both of the H2O2 production methods outlined above into one system where H2O2 is produced at the cathode (O2 reduction to H2O2) and anode (H2O oxidation to H2O2) simultaneously as shown below:
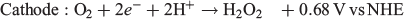
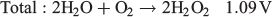
Given that water oxidation to H2O2 only occurs in alkaline electrolytes and O2 reduction catalysts for H2O2 production generally perform better in acidic environments, there is much work to be done on catalysts to make this process viable.
Hydrogen Peroxide in Fuel Cells
In general, fuel cells generate power because of the difference in electrochemical potential between the oxidative and reductive reactions spontaneously occurring on the electrodes. The power generated by a fuel cell is determined by the product of the current being drawn and the potential difference between the two electrodes at that current. As a larger current is drawn, that potential difference is reduced and as such each fuel cell will have a current at which the power is at a maximum. Therefore, this maximum power point is strongly influenced by the open circuit potential of the cell (which is the cell potential at which no current is being drawn from the cell) and the limiting current density (which is the total current density that can be drawn when the electrodes are short-circuited). Although the open circuit potential (OCP) is limited by thermodynamics, increases in the limiting current density can increase the maximum achievable power density of the fuel cell as well. This requires improvement in the electrochemical catalysts that are used for both the oxidation and reduction processes.
These components of an H2O2 fuel cell are particularly important when it comes to achieving OCP close to thermodynamic potential (1.09 V). Unfortunately, this value is difficult to achieve in a real cell owing to significant overpotentials associated with both reactions. This has been demonstrated by numerous studies, in particular by those who have attempted to incorporate H2O2 as an oxidant into other types of fuel cells and semi-fuel cells.[24–26,82,83] One of the problems associated with using H2O2 as a reductant or an oxidant is that on many catalysts, it is possible for H2O2 to be reduced and oxidised on the same surface[84–86] (Fig. 1), effectively causing the decomposition of the H2O2 and generating a ‘mixed’ potential on the electrode.

|
Jing et al.[87] investigated this phenomenon by measuring the OCPs of H2O2 on various noble metals in an electrochemical cell with electrolytes containing H2O2. OCP is defined here as the potential applied to the electrode of interest at which there is zero current in the cell compared with an external reference electrode. The interpretation given by Jing et al. was that rather than defining the oxidation and reduction potentials of H2O2, it was more useful to observe the degree to which the OCP trended towards the theoretical oxidation and reduction potentials. For example, in both acidic and alkaline media, they found that on Pt, Au, Pd, and glassy carbon (GC) electrodes, the OCP tended to be closer to the standard theoretical oxidation potential and concluded that this represented a mixed potential where both H2O2 reduction to H2O and H2O2 oxidation to O2 were taking place on the same electrode, but with a greater propensity towards the oxidation reaction. As these two processes are taking place simultaneously on the same electrode, the total current being recorded will be the sum of both processes. This is problematic for fuel cells as not only is the maximum power of the cell diminished, but some fuel will be lost owing to unproductive disproportionation of the H2O2, which lowers its overall fuel efficiency. An ideal catalyst is therefore one that has a strong selectivity towards one of the reactions and not the other.
H2O2 Electrocatalysts
Several studies have looked at reduction and oxidation catalysts for H2O2 for varying purposes such as fuel cells or H2O2 detectors.[88–90] In alkaline environments, perovskite[91,92] and cobalt-based catalysts (particularly those based on Co3O4) have shown excellent H2O2 reduction properties. Cathodes incorporating spinel-structured Co3O4 or copper–cobalt nanoparticles were found to exhibit high activity towards H2O2 reduction in 3 M NaOH.[93,94] The H2O2 reduction current remained steady over the course of 30 min of chronoamperometry, indicating that the Co3O4 cathode is reasonably stable in alkaline electrolytes. In work performed by Zhihao et al., three-dimensional gold–cobalt oxide cathodes were synthesised[95] and showed large reduction current peaks at −0.28 V versus Ag/AgCl in highly alkaline 3 M KOH solutions. Catalysts based around the dye Prussian blue have also been found to reduce H2O2 with high faradaic efficiency in neutral to slightly acidic electrolytes.[96]
H2O2 oxidation catalysts for the anodic electrode have not been studied nearly as thoroughly in the context of fuel cells as the reduction catalysts, as the latter reaction is thought to be the major cause of energy loss through high overpotentials. Metals such as nickel or gold are often used as anodes in direct hydrogen peroxide fuel cells (DHPFCs) except in the case of some two-compartment fuel cell studies where the cathode and the anode can be the same material. For example, Yang et al. constructed a DHPFC[97] in which the cathode and the anode were both Pd particles electrodeposited onto carbon fibre cloth as discussed further below. Recently, cheaper alternatives to the Pd cathodes such as Ni and Co catalysts deposited on TiC nanowires[98] and nickel ferric ferrocyanide nanoparticles[99] showing similar or higher power densities have been demonstrated in two-compartment fuel cells.
Direct Hydrogen Peroxide Fuel Cell Designs
Two-Compartment Fuel Cells
The first DHPFC was designed by Hasegawa et al.[100] using two electrolytes, solutions of H2SO4 and NaOH containing H2O2, brought into contact with each other by a microfluidic flow cell. This cell achieves power generation by the reduction and oxidation of H2O2 in the acidic and alkaline electrolytes respectively.
As the electrolytes are not separated by a membrane, the charge is balanced by the formation of a Na2SO4 solution at the point of mixing. The maximum power density for this cell was found to be 23 mW cm−2 at 300 mV and 76 mA cm−2. As no membrane is required for this cell design, the authors proposed that the cost and electrical resistivity are reduced, making it superior in that respect to H2 or methanol fuel cells. However, in a practical application of the technology, this cell is limited by both the scalability of a microchannel design and, critically, by the consumption of the components of the electrolyte other than the fuel and the oxidant. Subsequent two-compartment fuel cell designs have included a membrane separating the acidic and the alkaline electrolytes. Fig. 2 describes a possible arrangement of such a two-compartment DHPFC.

|
One of the major sources of inefficiency in a fuel cell is the membrane that separates the anodic and cathodic chambers. Membranes introduce resistance into the cell that results in loss of power density. They also complicate cell construction and limit its size, shape, and the distance between electrodes. Some fuel cells can function without a membrane and these are usually based around the use of the laminar flow of electrolytes through microchannels. An example of this is given by Hasegawa et al.[100] as described above. However, as the two different electrolytes are in contact with each other, there is usually going to be some degree of crossover of ions due to concentration gradients. The microchannel design also limits the size and geometry of the cell if large-scale designs are necessary.
Despite the drawbacks of fuel cells containing membranes, they have been included in the majority of designs of two-compartment DHPFCs since 2005. The focus of the research into these types of fuel cells has been on the catalytic materials used for the cathode and anode, increasing the selectivity for H2O2 oxidation and reduction and improving the limiting current densities. Sanli et al. developed a two-compartment cell that utilised a Ni-carbon paper anode and a Pt cathode.[101] Nickel was used because, in previous DHPFCs,[102] it was found to have the lowest rate of unproductive H2O2 decomposition compared with other metals such as Au, Ag, Pt, and Pd.
One-Compartment DHPFCs
The unique operation of the H2O2 fuel cell also allows the construction of a different membraneless design called a ‘single-compartment DHPFC’ (Fig. 3). Here, a single electrolyte is used in conjunction with two different catalysts. An open-circuit potential is produced based on the selectivity that the catalysts have towards the oxidation and reduction of H2O2, rather than the pH difference at the surface of the cathode and anode. Yamazaki et al.[102] were the first to design a one-electrolyte, one-compartment DHPFC using metals such as Ag, Au, Pt, Pd, and Ni as catalysts. In that study, an aqueous 1 M NaOH solution containing 0.3 M H2O2 was used as the fuel into which two different metals were immersed, yielding an OCP of ~100 mV. This OCP is obviously much lower than the theoretical 1.09 V and this was ascribed to the problem of mixed potentials that was discussed above. In particular, the high overpotential at the cathode was suggested to be the major contributor to the poor performance of the cell. By modifying the surface of the Ag metal with nanostructured Ag–Pb alloys, the total voltage produced by the cell was improved from 100 to 150 mV.[103] Interestingly, the introduction of small amounts of Pb to the alloy (Ag : P 7 : 3 and Ag : Pb 9 : 1) gave higher OCPs and limiting current densities than just Ag nanoparticles, or when the Pb ratio was too high (Ag : Pb 6 : 4). These experiments were all conducted in 1 M NaOH electrolyte; however, H2O2 is known to be unstable in most alkaline environments. An exception to this is alkaline ammonium-based solutions containing free amines, which were discussed previously for their use in low-overpotential water oxidation.[78,79]

|
Yamada et al. have used several metal-based complexes as H2O2 reduction catalysts for single-compartment fuel cells that make use of acidic electrolytes as the fuel medium. Complexes such as protonated iron–phthalocyanine,[104] Fe3[{CoIII(CN)6}2],[105] and pyrazine-bridged Fe[MC(CN)4] (MC = Pt2+ and Pd2+)[106] have been used as H2O2 reduction catalysts with great success. Fuel cells using these electrodes have achieved very high OCPs of ~0.8 V along with high limiting current densities up to 15 mA cm−2 for total power densities up to 4 mW cm−2. Yamada et al. found that the presence of Sc3+ ions in the electrolyte solution inhibited the disproportionation of H2O2 and extended the time that a stable output potential could be maintained while a current was being produced by the cell. The stabilising effect was attributed to the trapping of HO2• produced from the H2O2 by the Fe ions. Instead of acting as the initiator for further reaction, the Sc3+ trapped the radical as Sc3+-bound O2•–. Although stabilisation of the H2O2 solution may be important for improving the efficiency of these types of fuel cells, another important aspect to consider is the need to regenerate or treat the electrolyte between cycles of electrochemical H2O2 production and fuel cell operation. Given that the O2•– radical is bound to the Sc3+ ions, the electrolyte may need to be regenerated or may begin to lose effectiveness after several cycles of operation.
Prussian blue, another cyano-complex, has been used as a cathode catalyst[107] to produce reasonably high OCPs in acidic one-compartment H2O2 fuel cells with a maximum power density of 1.5 mW cm−2.
Conclusions
H2O2 is a viable alternative to other ‘green’ fuels as it can be produced electrochemically from renewable feedstocks. Compared with H2 gas, it has a reasonably similar energy density per volume and can be stored without pressurisation or solid-state storage materials. Owing to its unique properties as both an oxidant and reductant, the H2O2 fuel cell is flexible in its design as either a two-compartment device with a membrane or a one-compartment device with only one electrolyte. Further work is needed to bring these types of fuel cells to practical application, including improving the maximum power density and investigating the long-term cyclability of a complete H2O2 fuel production and utilisation system. This can be done through investigating new catalysts that reduce the overpotential needed for both H2O2 oxidation and reduction and also have good stability in high concentrations of H2O2. Given that H2O2 can be produced electrochemically by both O2 reduction and water oxidation, an extremely efficient H2O2 generation system may be designed that produces both the fuel and oxidant simultaneously with no loss of energy to unneeded reactions. This, in combination with the H2O2 fuel cell, creates a reversible fuel cell in which the electrogenerated H2O2 can be stored and transported to be utilised in a fuel cell at a later stage. To make this practical, both the electrolyte and catalysts must be stable for long-term cyclability. There is also a need to increase the concentration of the electrogenerated H2O2 solutions either during production or by post-processing. As H2O2 is an oxidant, in high concentrations, this fuel poses some immediate safety concerns if accidentally exposed to the environment. However, when exposed to heat and light, H2O2 quickly decomposes to water and oxygen, eliminating long-term environmental hazards. Despite these concerns, there is considerable potential in this H2O2 system as an energy storage solution and it certainly deserves further investigation and development.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
This research did not receive any specific funding.
References
[1] S. Shafiee, E. Topal, Energy Policy 2009, 37, 181.| Crossref | GoogleScholarGoogle Scholar |
[2] T. Zielinski, in Impact of Climate Changes on Marine Environments (Eds J. M. Węsławski, K. Kuliński, T. Zielinski) 2015, Ch. 1, 1–148 (Springer: Berlin).
[3] N. V. Patel, Foreign Policy 2015, 213, 28.
[4] J. Tollefson, Nature 2010, 464, 1262.
| Crossref | GoogleScholarGoogle Scholar |
[5] B. G. Ram, K. K. Pant, in Hydrogen Fuel (Ed. B. G. Ram) 2008, Ch. 1, pp. 2–32 (CRC Press: Boca Raton, FL).
[6] A. Szyszka, Int. J. Hydrogen Energy 1998, 23, 849.
| Crossref | GoogleScholarGoogle Scholar |
[7] S. Sharma, S. K. Ghoshal, Renew. Sustain. Energy Rev. 2015, 43, 1151.
| Crossref | GoogleScholarGoogle Scholar |
[8] R. Ortiz Cebolla, B. Acosta, P. Moretto, N. Frischauf, F. Harskamp, C. Bonato, D. Baraldi, Int. J. Hydrogen Energy 2014, 39, 6261.
| Crossref | GoogleScholarGoogle Scholar |
[9] M. Royle, D. Willoughby, Process Saf. Environ. Prot. 2011, 89, 452.
| Crossref | GoogleScholarGoogle Scholar |
[10] L. Ma, D. J. Mihalcik, W. Lin, J. Am. Chem. Soc. 2009, 131, 4610.
| Crossref | GoogleScholarGoogle Scholar |
[11] I. A. Ibarra, S. Yang, X. Lin, A. J. Blake, P. J. Rizkallah, H. Nowell, D. R. Allan, N. R. Champness, P. Hubberstey, M. Schroder, Chem. Commun. 2011, 8304.
| Crossref | GoogleScholarGoogle Scholar |
[12] Y. Yan, S. Yang, A. J. Blake, M. Schröder, Acc. Chem. Res. 2014, 47, 296.
| Crossref | GoogleScholarGoogle Scholar |
[13] M. Lototskyy, V. A. Yartys, J. Alloys Compd. 2015, 645, S365.
| Crossref | GoogleScholarGoogle Scholar |
[14] F. Schuth, B. Bogdanovic, M. Felderhoff, Chem. Commun. 2004, 2249.
| Crossref | GoogleScholarGoogle Scholar |
[15] C. M. Rangel, V. R. Fernandes, Y. Slavkov, L. Bozukov, Int. J. Hydrogen Energy 2009, 34, 4587.
| Crossref | GoogleScholarGoogle Scholar |
[16] I. P. Jain, Int. J. Hydrogen Energy 2009, 34, 7368.
| Crossref | GoogleScholarGoogle Scholar |
[17] S. Niaz, T. Manzoor, A. H. Pandith, Renew. Sustain. Energy Rev. 2015, 50, 457.
| Crossref | GoogleScholarGoogle Scholar |
[18] U. Suess Hans, Pulp Bleaching Today 2010 (De Gruyter: Berlin).
[19] K. Sato, R. Noyori, Science 1998, 281, 1646.
| Crossref | GoogleScholarGoogle Scholar |
[20] A. Aleboyeh, H. Aleboyeh, Y. Moussa, Dyes Pigments 2003, 57, 67.
| Crossref | GoogleScholarGoogle Scholar |
[21] P. Blach, Z. Böstrom, S. Franceschi-Messant, A. Lattes, E. Perez, I. Rico-Lattes, Tetrahedron 2010, 66, 7124.
| Crossref | GoogleScholarGoogle Scholar |
[22] H. Egami, T. Oguma, T. Katsuki, J. Am. Chem. Soc. 2010, 132, 5886.
| Crossref | GoogleScholarGoogle Scholar |
[23] L. Cui, S. Furuhashi, Y. Tachikawa, N. Tada, T. Miura, A. Itoh, Tetrahedron Lett. 2013, 54, 162.
| Crossref | GoogleScholarGoogle Scholar |
[24] N. A. Choudhury, R. K. Raman, S. Sampath, A. K. Shukla, J. Power Sources 2005, 143, 1.
| Crossref | GoogleScholarGoogle Scholar |
[25] S. J. Lao, H. Y. Qin, L. Q. Ye, B. H. Liu, Z. P. Li, J. Power Sources 2010, 195, 4135.
| Crossref | GoogleScholarGoogle Scholar |
[26] G. Agladze, P. Nikoleishvili, V. Kveselava, G. Tsurtsumia, G. Gorelishvili, D. Gogoli, I. Kakhniashvili, J. Power Sources 2012, 218, 46.
| Crossref | GoogleScholarGoogle Scholar |
[27] D. M. F. Santos, P. G. Saturnino, R. F. M. Lobo, C. A. C. Sequeira, J. Power Sources 2012, 208, 131.
| Crossref | GoogleScholarGoogle Scholar |
[28] R. S. Disselkamp, Int. J. Hydrogen Energy 2010, 35, 1049.
| Crossref | GoogleScholarGoogle Scholar |
[29] S. Haji, B. Benstaali, N. Al-Bastaki, Chem. Eng. J. 2011, 168, 134.
| Crossref | GoogleScholarGoogle Scholar |
[30] V. M. Daskalaki, E. S. Timotheatou, A. Katsaounis, D. Kalderis, Desalination 2011, 274, 200.
| Crossref | GoogleScholarGoogle Scholar |
[31] S. Yang, P. Wang, X. Yang, L. Shan, W. Zhang, X. Shao, R. Niu, J. Hazard. Mater. 2010, 179, 552.
| Crossref | GoogleScholarGoogle Scholar |
[32] D. Sloboda-Rozner, P. L. Alsters, R. Neumann, J. Am. Chem. Soc. 2003, 125, 5280.
| Crossref | GoogleScholarGoogle Scholar |
[33] K.-P. Ho, K.-Y. Wong, T. H. Chan, Tetrahedron 2006, 62, 6650.
| Crossref | GoogleScholarGoogle Scholar |
[34] Y. Xu, N. R. B. J. Khaw, Z. Li, Green Chem. 2009, 11, 2047.
| Crossref | GoogleScholarGoogle Scholar |
[35] P. Jin, Z. Zhao, Z. Dai, D. Wei, M. Tang, X. Wang, Catal. Today 2011, 175, 619.
| Crossref | GoogleScholarGoogle Scholar |
[36] J. Kim, S. Jung, S. Park, S. Park, Tetrahedron Lett. 2011, 52, 2866.
| Crossref | GoogleScholarGoogle Scholar |
[37] F. Nikbakht, A. Heydari, Tetrahedron Lett. 2014, 55, 2513.
| Crossref | GoogleScholarGoogle Scholar |
[38] Q. Chen, J. Clean. Prod. 2006, 14, 708.
| Crossref | GoogleScholarGoogle Scholar |
[39] M. Giomo, A. Buso, P. Fier, G. Sandonà, B. Boye, G. Farnia, Electrochim. Acta 2008, 54, 808.
| Crossref | GoogleScholarGoogle Scholar |
[40] G. Fioroni, F. Fringuelli, F. Pizzo, L. Vaccaro, Green Chem. 2003, 5, 425.
| Crossref | GoogleScholarGoogle Scholar |
[41] G. B. Payne, J. Am. Chem. Soc. 1959, 81, 4901.
| Crossref | GoogleScholarGoogle Scholar |
[42] I. Oller, S. Malato, J. A. Sánchez-Pérez, Sci. Total Environ. 2011, 409, 4141.
| Crossref | GoogleScholarGoogle Scholar |
[43] I. Yamanaka, T. Onisawa, T. Hashimoto, T. Murayama, ChemSusChem 2011, 4, 494.
| Crossref | GoogleScholarGoogle Scholar |
[44] M. A. Ghanem, A. M. Al-Mayouf, M. N. Shaddad, F. Marken, Electrochim. Acta 2015, 174, 557.
| Crossref | GoogleScholarGoogle Scholar |
[45] A. von Weber, E. T. Baxter, H. S. White, S. L. Anderson, J. Phys. Chem. C 2015, 119, 11160.
| Crossref | GoogleScholarGoogle Scholar |
[46] M. Gara, E. Laborda, P. Holdway, A. Crossley, C. J. V. Jones, R. G. Compton, Phys. Chem. Chem. Phys. 2013, 15, 19487.
| Crossref | GoogleScholarGoogle Scholar |
[47] A. Ohma, K. Fushinobu, K. Okazaki, Electrochim. Acta 2010, 55, 8829.
| Crossref | GoogleScholarGoogle Scholar |
[48] M. Gennari, D. Brazzolotto, J. Pécaut, M. V. Cherrier, C. J. Pollock, S. DeBeer, M. Retegan, D. A. Pantazis, F. Neese, M. Rouzières, R. Clérac, C. Duboc, J. Am. Chem. Soc. 2015, 137, 8644.
| Crossref | GoogleScholarGoogle Scholar |
[49] L. R. Aveiro, A. G. M. da Silva, V. S. Antonin, E. G. Candido, L. S. Parreira, R. S. Geonmonond, I. C. de Freitas, M. R. V. Lanza, P. H. C. Camargo, M. C. Santos, Electrochim. Acta 2018, 268, 101.
| Crossref | GoogleScholarGoogle Scholar |
[50] W. R. P. Barros, Q. Wei, G. Zhang, S. Sun, M. R. V. Lanza, A. C. Tavares, Electrochim. Acta 2015, 162, 263.
| Crossref | GoogleScholarGoogle Scholar |
[51] J. S. Jirkovský, I. Panas, E. Ahlberg, M. Halasa, S. Romani, D. J. Schiffrin, J. Am. Chem. Soc. 2011, 133, 19432.
| Crossref | GoogleScholarGoogle Scholar |
[52] S. Siahrostami, A. Verdaguer-Casadevall, M. Karamad, D. Deiana, P. Malacrida, B. Wickman, M. Escudero-Escribano, E. A. Paoli, R. Frydendal, T. W. Hansen, I. Chorkendorff, I. E. L. Stephens, J. Rossmeisl, Nat. Mater. 2013, 12, 1137.
| Crossref | GoogleScholarGoogle Scholar |
[53] S. Kakuda, R. L. Peterson, K. Ohkubo, K. D. Karlin, S. Fukuzumi, J. Am. Chem. Soc. 2013, 135, 6513.
| Crossref | GoogleScholarGoogle Scholar |
[54] M. Campos, W. Siriwatcharapiboon, R. J. Potter, S. L. Horswell, Catal. Today 2013, 202, 135.
| Crossref | GoogleScholarGoogle Scholar |
[55] I. Yamanaka, R. Ichihashi, T. Iwasaki, N. Nishimura, T. Murayama, W. Ueda, S. Takenaka, Electrochim. Acta 2013, 108, 321.
| Crossref | GoogleScholarGoogle Scholar |
[56] K. Mase, K. Ohkubo, S. Fukuzumi, J. Am. Chem. Soc. 2013, 135, 2800.
| Crossref | GoogleScholarGoogle Scholar |
[57] G. Göransson, E. Ahlberg, Electrochim. Acta 2014, 146, 638.
| Crossref | GoogleScholarGoogle Scholar |
[58] S. Chen, Z. Chen, S. Siahrostami, D. Higgins, D. Nordlund, D. Sokaras, T. R. Kim, Y. Liu, X. Yan, E. Nilsson, R. Sinclair, J. K. Nørskov, T. F. Jaramillo, Z. Bao, J. Am. Chem. Soc. 2018, 140, 7851.
| Crossref | GoogleScholarGoogle Scholar |
[59] Y. Sun, I. Sinev, W. Ju, A. Bergmann, S. Dresp, S. Kühl, C. Spöri, H. Schmies, H. Wang, D. Bernsmeier, B. Paul, R. Schmack, R. Kraehnert, B. Roldan Cuenya, P. Strasser, ACS Catal. 2018, 8, 2844.
| Crossref | GoogleScholarGoogle Scholar |
[60] S. Kabir, K. Artyushkova, A. Serov, P. Atanassov, ACS Appl. Mater. Interfaces 2018, 10, 11623.
| Crossref | GoogleScholarGoogle Scholar |
[61] C. Di Bari, S. Shleev, A. L. De Lacey, M. Pita, Bioelectrochemistry 2016, 107, 30.
| Crossref | GoogleScholarGoogle Scholar |
[62] A. Bonakdarpour, M. Lefevre, R. Yang, F. Jaouen, T. Dahn, J.-P. Dodelet, J. R. Dahn, Electrochem. Solid-State Lett. 2008, 11, B105.
| Crossref | GoogleScholarGoogle Scholar |
[63] V. G. Khomenko, K. V. Lykhnytskyi, V. Z. Barsukov, Electrochim. Acta 2013, 104, 391.
| Crossref | GoogleScholarGoogle Scholar |
[64] T.-P. Fellinger, F. Hasché, P. Strasser, M. Antonietti, J. Am. Chem. Soc. 2012, 134, 4072.
| Crossref | GoogleScholarGoogle Scholar |
[65] T. Murayama, I. Yamanaka, J. Phys. Chem. C 2011, 115, 5792.
| Crossref | GoogleScholarGoogle Scholar |
[66] I. Yamanaka, T. Murayama, Angew. Chem. Int. Ed. 2008, 47, 1900.
| Crossref | GoogleScholarGoogle Scholar |
[67] V. Čolić, S. Yang, Z. Révay, I. E. L. Stephens, I. Chorkendorff, Electrochim. Acta 2018, 272, 192.
| Crossref | GoogleScholarGoogle Scholar |
[68] F. Yu, Y. Chen, H. Ma, New J. Chem. 2018, 42, 4485.
| Crossref | GoogleScholarGoogle Scholar |
[69] V. L. Kornienko, G. A. Kolyagin, G. V. Kornienko, V. A. Parfenov, I. V. Ponomarenko, Russ. J. Electrochem. 2018, 54, 258.
| Crossref | GoogleScholarGoogle Scholar |
[70] Y. Liu, X. Quan, X. Fan, H. Wang, S. Chen, Angew. Chem. Int. Ed. 2015, 54, 6837.
| Crossref | GoogleScholarGoogle Scholar |
[71] B. B. Blizanac, P. N. Ross, N. M. Markovic, Electrochim. Acta 2007, 52, 2264.
| Crossref | GoogleScholarGoogle Scholar |
[72] J. F. Pérez, A. Galia, M. A. Rodrigo, J. Llanos, S. Sabatino, C. Sáez, B. Schiavo, O. Scialdone, Electrochim. Acta 2017, 248, 169.
| Crossref | GoogleScholarGoogle Scholar |
[73] Y. Shiraishi, S. Kanazawa, Y. Kofuji, H. Sakamoto, S. Ichikawa, S. Tanaka, T. Hirai, Angew. Chem. Int. Ed. 2014, 53, 13454.
| Crossref | GoogleScholarGoogle Scholar |
[74] C. Kormann, D. W. Bahnemann, M. R. Hoffmann, Environ. Sci. Technol. 1988, 22, 798.
| Crossref | GoogleScholarGoogle Scholar |
[75] N. Her, J.-S. Park, Y. Yoon, Chem. Eng. J. 2011, 166, 184.
| Crossref | GoogleScholarGoogle Scholar |
[76] K. Mase, M. Yoneda, Y. Yamada, S. Fukuzumi, ACS Energy Lett. 2016, 1, 913.
| Crossref | GoogleScholarGoogle Scholar |
[77] Y. Isaka, K. Oyama, Y. Yamada, T. Suenobu, S. Fukuzumi, Catal. Sci. Technol. 2016, 6, 681.
| Crossref | GoogleScholarGoogle Scholar |
[78] A. Izgorodin, E. Izgorodin, D. R. MacFarlane, Energy Environ. Sci. 2012, 5, 9496.
| Crossref | GoogleScholarGoogle Scholar |
[79] C. McDonnell-Worth, D. R. MacFarlane, RSC Adv. 2014, 4, 30551.
| Crossref | GoogleScholarGoogle Scholar |
[80] K. Fuku, K. Sayama, Chem. Commun. 2016, 5406.
| Crossref | GoogleScholarGoogle Scholar |
[81] Y. Ando, T. Tanaka, Int. J. Hydrogen Energy 2004, 29, 1349.
| Crossref | GoogleScholarGoogle Scholar |
[82] N. Luo, G. H. Miley, K.-J. Kim, R. Burton, X. Huang, J. Power Sources 2008, 185, 685.
| Crossref | GoogleScholarGoogle Scholar |
[83] R. R. Bessette, J. M. Cichon, D. W. Dischert, E. G. Dow, J. Power Sources 1999, 80, 248.
| Crossref | GoogleScholarGoogle Scholar |
[84] X. Li, D. Heryadi, A. A. Gewirth, Langmuir 2005, 21, 9251.
| Crossref | GoogleScholarGoogle Scholar |
[85] I. Katsounaros, W. B. Schneider, J. C. Meier, U. Benedikt, P. U. Biedermann, A. A. Auer, K. J. J. Mayrhofer, Phys. Chem. Chem. Phys. 2012, 14, 7384.
| Crossref | GoogleScholarGoogle Scholar |
[86] R. J. Bowen, H. B. Urbach, J. H. Harrison, Nature 1967, 213, 592.
| Crossref | GoogleScholarGoogle Scholar |
[87] X. Jing, D. Cao, Y. Liu, G. Wang, J. Yin, Q. Wen, Y. Gao, J. Electroanal. Chem. 2011, 658, 46.
| Crossref | GoogleScholarGoogle Scholar |
[88] L. Shi, X. Niu, T. Liu, H. Zhao, M. Lan, Microchim. Acta 2015, 182, 2485.
| Crossref | GoogleScholarGoogle Scholar |
[89] Z. Yin, J. Wu, Z. Yang, Biosens. Bioelectron. 2011, 26, 1970.
| Crossref | GoogleScholarGoogle Scholar |
[90] C. Debiemme-Chouvy, Biosens. Bioelectron. 2010, 25, 2454.
| Crossref | GoogleScholarGoogle Scholar |
[91] G. Wang, Y. Bao, Y. Tian, J. Xia, D. Cao, J. Power Sources 2010, 195, 6463.
| Crossref | GoogleScholarGoogle Scholar |
[92] T. Poux, A. Bonnefont, A. Ryabova, G. Kerangueven, G. A. Tsirlina, E. R. Savinova, Phys. Chem. Chem. Phys. 2014, 16, 13595.
| Crossref | GoogleScholarGoogle Scholar |
[93] D. Cao, J. Chao, L. Sun, G. Wang, J. Power Sources 2008, 179, 87.
| Crossref | GoogleScholarGoogle Scholar |
[94] N. C. D. Nath, T. Debnath, E.-K. Kim, M. A. Ali Shaikh, J.-J. Lee, Electrochim. Acta 2018, 273, 474.
| Crossref | GoogleScholarGoogle Scholar |
[95] Z. Li, Y. He, X. Ke, L. Gan, J. Zhao, G. Cui, G. Wu, J. Power Sources 2015, 294, 136.
| Crossref | GoogleScholarGoogle Scholar |
[96] R. Araminaitė, R. Garjonytė, A. Malinauskas, J. Solid State Electrochem. 2010, 14, 149.
| Crossref | GoogleScholarGoogle Scholar |
[97] F. Yang, K. Cheng, X. Liu, S. Chang, J. Yin, C. Du, L. Du, G. Wang, D. Cao, J. Power Sources 2012, 217, 569.
| Crossref | GoogleScholarGoogle Scholar |
[98] X. Wang, K. Ye, H. Zhang, X. Ma, K. Zhu, K. Cheng, G. Wang, D. Cao, Int. J. Hydrogen Energy 2017, 42, 15044.
| Crossref | GoogleScholarGoogle Scholar |
[99] X. Xiao, F. Yang, K. Cheng, X. Wang, H. Zhang, K. Ye, G. Wang, D. Cao, Int. J. Hydrogen Energy 2017, 42, 22856.
| Crossref | GoogleScholarGoogle Scholar |
[100] S. Hasegawa, K. Shimotani, K. Kishi, H. Watanabe, Electrochem. Solid-State Lett. 2005, 8, A119.
| Crossref | GoogleScholarGoogle Scholar |
[101] A. E. Sanli, A. Aytaç, Int. J. Hydrogen Energy 2011, 36, 869.
| Crossref | GoogleScholarGoogle Scholar |
[102] S.-i. Yamazaki, Z. Siroma, H. Senoh, T. Ioroi, N. Fujiwara, K. Yasuda, J. Power Sources 2008, 178, 20.
| Crossref | GoogleScholarGoogle Scholar |
[103] Y. Yamada, Y. Fukunishi, S.-i. Yamazaki, S. Fukuzumi, Chem. Commun. 2010, 7334.
| Crossref | GoogleScholarGoogle Scholar |
[104] Y. Yamada, S. Yoshida, T. Honda, S. Fukuzumi, Energy Environ. Sci. 2011, 4, 2822.
| Crossref | GoogleScholarGoogle Scholar |
[105] Y. Yamada, M. Yoneda, S. Fukuzumi, Chem. – Eur. J. 2013, 19, 11733.
| Crossref | GoogleScholarGoogle Scholar |
[106] Y. Yamada, M. Yoneda, S. Fukuzumi, Inorg. Chem. 2014, 53, 1272.
| Crossref | GoogleScholarGoogle Scholar |
[107] S. A. Mousavi Shaegh, N.-T. Nguyen, S. M. Mousavi Ehteshami, S. H. Chan, Energy Environ. Sci. 2012, 5, 8225.
| Crossref | GoogleScholarGoogle Scholar |
* Douglas R. MacFarlane was awarded the 2018 David Craig Medal of the Australian Academy of Science.