

Polyion Complex Micelles for Protein Delivery*
Fan Chen A and Martina H. Stenzel A BA School of Chemistry, Centre for Advanced Macromolecular Design (CAMD), University of New South Wales, Sydney, NSW 2052, Australia.
B Corresponding author. Email: m.stenzel@unsw.edu.au

Fan Chen received his B.Sc. degree in biofunctional materials from Beijing University of Chemical Technology in 2012, followed by an M.Sc. degree in polymer science from Loughborough University in 2013. He started his Ph.D. in chemistry at the University of New South Wales under the supervision of Professor Martina Stenzel in 2014 and has recently submitted his Ph.D. thesis. His research focuses on the use of nanoparticles for cancer treatment and bioimaging techniques. |

Martina Stenzel studied chemistry at the University of Bayreuth, Germany, before completing her Ph.D. in 1999 at the Institute of Applied Macromolecular Chemistry, University of Stuttgart, Germany. She started as a post-doctoral fellow at UNSW in 1999 and is now a full professor in the School of Chemistry as well as co-director of the Centre for Advanced Macromolecular Design (CAMD). In 2019, she was elected Fellow of the Australian Academy of Science. Her research interest is focused on the synthesis of functional nanoparticles for drug delivery applications. Martina has published more than 300 peer-reviewed papers mainly on polymer and nanoparticle design. She is scientific editor of Materials Horizons and serves currently on a range of editorial boards. She is currently the chair of the National Chemistry Committee of the Australian Academy of Science. She has received a range of awards, including the 2011 Le Fèvre Memorial Prize of the Australian Academy of Science. In 2017, she received the H. G. Smith Memorial Award. |
Australian Journal of Chemistry 71(10) 768-780 https://doi.org/10.1071/CH18219
Submitted: 14 May 2018 Accepted: 22 July 2018 Published: 6 September 2018
Abstract
Proteins are ubiquitous in life and next to water, they are the most abundant compounds found in human bodies. Proteins have very specific roles in the body and depending on their function, they are for example classified as enzymes, antibodies or transport proteins. Recently, therapeutic proteins have made an impact in the drug market. However, some proteins can be subject to quick hydrolytic degradation or denaturation depending on the environment and therefore require a protective layer. A range of strategies are available to encapsulate and deliver proteins, but techniques based on polyelectrolyte complex formation stand out owing to their ease of formulation. Depending on their isoelectric point, proteins are charged and can condense with oppositely charged polymers. Using block copolymers with a neutral block and a charged block results in the formation of polyion complex (PIC) micelles when mixed with the oppositely charged protein. The neutral block stabilises the charged protein–polymer core, leading to nanoparticles. The types of micelles are also known under the names interpolyelectrolyte complex, complex coacervate core micelles, and block ionomer complexes. In this article, we discuss the formation of PIC micelles and their stability. Strategies to enhance the stability such as supercharging the protein or crosslinking the PIC micelles are discussed.
Introduction
Protein therapeutics
Protein is a vital component of our bodies built of smaller compounds known as amino acids.[1] Depending on the number and sequence of amino acid units, proteins fold into a characteristic three-dimensional conformation that determines their specific biological functions.[2] These functions cover nearly every biological process including catalysing reactions, storing ions, transporting molecules, and identifying foreigner intruders.[3] Recent studies have shown that many human diseases are related to dysfunctions of proteins, such as Alzheimer’s,[4] Parkinson’s,[5] diabetes[6] and cancer.[7] Fig. 1 shows the number, type, and target area of therapeutic proteins approved by US Food and Drug Administration (FDA) between 1 January 2011 and 31 August 2016.[8] It is clear that the pharmaceutical market for protein drugs is growing very fast and involved in a wide range of therapeutic areas to promote healthy living. This is attributed to the advantages of protein therapeutics over traditional small-molecule drugs. Proteins possess a set of complex functions and high specificity, therefore making it less likely for protein drugs to cause side effects. As our body itself produces many types of proteins for the purpose of normal biological processes, protein drugs are believed to minimise the triggering of any immune response.[9] Therefore, proteins as therapeutic agents are believed to be a promising strategy to treat these diseases and have attracted great attention.[8–10]

|
Despite these advantages, intrinsic properties of proteins, such as their large size, surface charge, and fragile tertiary structure, are the main obstacles for their clinical applications. Protein therapeutics are now mainly delivered via oral, transdermal, or parenteral means. However, some proteins can be easily digested by proteolytic enzymes and become unstable in extreme pH environments, like gastric fluid, if given orally. Transdermal delivery is currently only applicable for small molecules as the permeability of molecules larger than 500 Da in size through biological membranes is poor.[11] Proteins delivered via parenteral route are the most commonly applied pathways. However, frequent injections are required to achieve ideal therapeutic effects owing to the short half-life as they are rapidly eliminated via renal filtration.[9b] Despite some challenges, many proteins are active and do not require any additional help during administration. In fact, some proteins need to remain unaltered to elicit the desired immune response. However, most proteins benefit from a protective layer like a polymer layer that can help protect the protein and increase circulation time.
Delivery of Protein Therapeutics
To date, many efforts have been devoted to overcoming the limitations of therapeutic proteins. Among all approaches, the entrapment of proteins in nanocarriers as an efficient delivery vehicle is of great interest.[12] Nanocarriers that either chemically conjugate or physically encapsulate protein therapeutics are the two main strategies for drug delivery (Fig. 2).[13] A review by Solaro et al. discussed the various approaches and their advantages and disadvantages.[12] Among all the techniques, the formation of polyelectrolyte complexes between two oppositely charged species was presented as ‘easy to achieve’. Mixtures of charged proteins and oppositely charged proteins can easily result in the formation of large particles and can even be a tool to prepare protein-based hydrogels. Crucial for the delivery of proteins within drug carriers is, however, a well-controlled particle size, preferably below 100 nm. Key to size control is the use of block copolymers, which are composed of a neutral block and a charged polymer. The neutral block directs the self-assembly process, stabilising the resulting polyion complex (PIC) micelles against further aggregation.

|
Polyion Complex Micelles
PIC micelles were first reported by Harada and Kataoka in 1995 (Fig. 3).[14] They attempted to apply supramolecular interactions other than hydrophobic interactions to form polymeric micelles in aqueous media. Inspired by the segregation of charged molecules under charge-neutralising conditions, they mixed two oppositely charged block copolymers in aqueous solution and obtained micelles with a narrow size distribution driven by electrostatic interactions (Fig. 3). This method avoids the use of organic solvent, making PIC micelles an excellent candidate for delivering organic-solvent-sensitive therapeutic agents, such as nucleic acids[15] and proteins,[16] which are naturally occurring polyelectrolytes. Although the focus of the present review is the formation of PIC micelles with proteins, we would like to briefly mention that PIC formation using two oppositely charged block copolymers, where a charged polymer takes on the role of the protein, is a very active research field as evidenced by ref. [17] for example.[17] Moreover, condensation of other families of bioactive biopolymers into PIC micelles, such as heparin[18] and DNA,[19] is possible. PIC micelles with nucleic acids are often termed polyplexes. In particular, binding with oligonucleotides, small interfering RNA (siRNA) or DNA has been studied in depth. The main difference, though, is that these molecules are highly charged, which is in contrast to proteins, which have charged, neutral, and hydrophobic surface functionalities that affect the binding process.

|
PIC Micelles with Proteins
The design of PIC micelles with proteins including enzymes is determined by the nature of the protein, in particular the isoelectric point. A negatively charged protein is typically condensed using a positively charged block copolymer and vice versa. The binding between the polymer and the protein is usually a function of the nature of the polymer, such as the type of ionic functionality, and the polymer architecture, such as the length of the polymer. These influencing parameters need to be discussed in combination with environmental factors such as pH and ionic strength, which affect the strength of the electrostatic interactions.
Shell-Forming Polymers
The shell of the PIC micelle plays a range of roles in PIC micelles. The shell needs to stabilise the PIC micelle against aggregation and needs to ensure colloidal stability in various environments. Moreover, the shell needs to present a barrier against enzymatic attack, protecting the biomacromolecule from the environment. As usually neutral polymers are employed to form the shell, the absorption of proteins is minimised, resulting in less fouling and, therefore, often longer circulation times in the bloodstream. Preferably, the shell-forming polymer should have only limited interaction with the loaded protein in the core. Ideal shell-forming polymers are neutral and should preferably be devoid of strong H-bond donors and acceptors to avoid competition with the charged polymer for the protein binding sites. Polymers that have been used for the shell include poly(ethylene glycol) (PEG), poly(acrylamide) (PAAm),[20] poly(glyceryl methacrylate) (PGMA),[21] poly(2-hydroxyethylacrylate) (PHEA),[22] poly(N-(2-hydroxypropyl)methacrylamide) (PHPMA),[23] and poly(isopropyl oxazoline) (PiPrOx),[24] although not all polymers listed here have been used in PIC micelle formation with proteins, as some were only employed in PIC micelle formation with oppositely charged polymers. Not all polymers listed here meet the requirement of being devoid of H-bonding, but experience show that stronger ionic interactions can overcome competing forces. In fact, almost all PIC micelles with proteins discussed below were based on PEG as the stabilising block as PEG was shown not to interact with the protein and can convey high colloidal stability.
Binding Proteins Using Charged Polymers
Based on the charges of cargo therapeutics, core-forming material can be designed as cationic or anionic blocks (Fig. 4). Previously reported cationic blocks to interact with anionic proteins includes poly(l-lysine) (P(Lys),[25] poly(aminoalkyl aspartamide),[26] and poly(aminoalkyl methacrylate) (PAMA).[27] For anionic proteins, poly(acrylic acid) (PAA) and poly(aspartate) (P(Asp))[28] can be used as the ionic block for PIC micelle formation. Instead of synthetic polymers, it is possible to use charged biopolymers such as the negatively charged polymer–protein conjugate based on albumin, which can entrap positively charged proteins.[29]

|
Factors Affecting the Formation of PIC Micelles
PIC micelles are formed by mixing proteins with ionic block copolymers usually in aqueous buffer solution. In a typical experiment, the polymer is slowly titrated into the protein solution, although the opposite approach is possible, but may not always lead to the same results. The central question at this stage is what polymer protein molar ratios are ideal to obtain well-defined PIC micelles. A detailed study on the formation of PIC micelles in correlation with the amount of charged species present was carried out by van der Burgh et al.[30] The authors defined the charge ratio f+ as 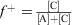 , where [C] is the concentration of cationic charges and [A] is the concentration of anionic charges. The maximum scattering intensity depicted in Fig. 5 can be found at f + = 0.5, where both charges are in equimolar amounts, whereas an excess of either charged species should result in species that are all less densely packed.[30] In reality, the ideal composition may not be at equimolar charged amounts, because in the case of proteins, the spatial distribution of surface charge may not match that of the polymers, requiring more polymer to saturate the protein. At this point, it needs to be noted that the formation of PIC micelles with proteins is different to that between two polymers. Proteins are stiff structures with limited flexibility. It is therefore up to the polymer to provide sufficient flexibility to span two charged areas in the protein and wrap around its structure. As a result, stiff polymers display lower binding strength.[31] Even highly flexible polymers need to adjust to the usually uneven distribution of charges on the protein surface,[32] leading to entropically unfavourable chain stretching.
, where [C] is the concentration of cationic charges and [A] is the concentration of anionic charges. The maximum scattering intensity depicted in Fig. 5 can be found at f + = 0.5, where both charges are in equimolar amounts, whereas an excess of either charged species should result in species that are all less densely packed.[30] In reality, the ideal composition may not be at equimolar charged amounts, because in the case of proteins, the spatial distribution of surface charge may not match that of the polymers, requiring more polymer to saturate the protein. At this point, it needs to be noted that the formation of PIC micelles with proteins is different to that between two polymers. Proteins are stiff structures with limited flexibility. It is therefore up to the polymer to provide sufficient flexibility to span two charged areas in the protein and wrap around its structure. As a result, stiff polymers display lower binding strength.[31] Even highly flexible polymers need to adjust to the usually uneven distribution of charges on the protein surface,[32] leading to entropically unfavourable chain stretching.

|
Although the structure of PIC micelles when equimolar charges are present can be imagined, excess negative or positive charges species can result in different scenarios, which are depicted in Fig. 6. Low polymer concentrations will lead to large particles as proteins will share the few available polymers, most likely forming PIC micelles with several proteins. In a second scenario, small PIC micelles are formed while free protein remains in the solution, waiting for more added polymer. Increasing the polymer concentration towards a charge-neutral point will see the formation of PIC micelles with a low size dispersity whereas an excess of polymer should create a bimodal size distribution of PIC micelles and free polymer. Alternatively, it is possible that PIC micelles might grow in size to accommodate further polymer. It is therefore conceivable that the minimum size dispersity should be found around an equimolar ratio of charged amino acids from the protein and charged polymer repeating units.

|
The second scenario depicted in Fig. 6 was indeed found by Harada et al.[16] At low polymer concentrations, here PEG-P(Asp), bimodal distributions were found indicating free lysozyme, which was used as model protein. The size corresponding to the PIC micelle did not change. This is contrast to our work, where we mixed poly(ethylene glycol) methyl ether acrylate (PEGMEA)-PAA with lysozyme and observed a decline in measured particle size, suggesting that the first model applies in this system (Fig. 6). As expected, the best size distribution was obtained at an equimolar ratio of charges. Although this can be a first approximation, equimolar ratios are not necessary and need to be identified for every system. As pointed out above, this point can be conveniently identified using the correlation between the polydispersity obtained from scattering studies and the molar ratio of polymer and protein.[33] Harada et al.[16] investigated the PIC micelles in more detail using scattering techniques. At an equimolar ratio of both charges, the PIC micelle with a hydrodynamic diameter of Dh = 47 nm was estimated to have 42 polymer chains and 36 lysozymes. Increasing the amount of PEG-P(Asp) saw an increase in particle size with the amount of polymer per particle increasing while the amount of protein slightly decreased.[34] Interestingly, the critical association concentration was found to be independent of the mixing ratio when referring to the charge concentration. Using density calculations, the authors proposed that the PEG shell increases with increasing amount of polymer, which can be explained by enhanced PEG stretching at high polymer–protein ratios.[34] There is no rule of thumb what scenario will take place as every system is based on different polymers and different size proteins. For example, PIC micelles based on the much larger glucose oxidase (GOx) led to slightly smaller sizes at higher polymer content.[33]
One important aspect to consider is that PIC micelles are dynamic structures, but reaching equilibrium can be delayed depending on the system. In theory, it should not matter if the polymer is added to the protein or vice versa. In reality, it can take several days for the equilibrium to adjust, even longer when strong polyelectrolytes are used. As a result, the two approaches, protein or polymer first, can lead to slightly different outcomes.[35] This can be understood by looking at the PIC formation process. On mixing, the negatively and positively charged polymers or proteins form random aggregates, which are determined by random collision events. This is typically a fast process and only controlled by diffusion events; therefore, complexes are formed within milliseconds. These initial structures are badly defined and do not have optimum packing parameters. In the next step, to reach equilibrium, the polymers need to rearrange themselves along the protein or by exchanging polymer with the solution. The second step can, however, be a slow process of the order of many days.[35]
When discussing PIC micelles, usually only electrostatic interactions are involved. Although these are indeed the strongest forces, other attractive forces such as H-bonding can play a role,[36] which can be sufficiently strong to bind a polymer to the protein.[37]
Effect of External Parameters
Polyion complex micelles are dynamic structures that can be assembled and disassembled depending on external parameters. Similar to traditional micelles, PIC micelles have a critical aggregate concentration (cac) where the assembled structures start to fall apart into proteins and free polymers.[34,38] The cac is influenced by the structure of the block copolymer, but also external parameters such as pH and ionic strength.
Addition of salt always leads to the disassembly of the PIC micelle. The disassembly can be a stepwise process as the addition of salt can lead to the loosening of the core structure or to rearrangement before full disassembly is observed.[39] In general, polymers with longer charged blocks display higher stability against disassembly in environments of high ionic strength.[40]
The variation in ionic strength and the coinciding assembly and disassembly was used to modulate the activity of lysozyme against Micrococcus luteus cells. The intact PIC micelle was unable to display any enzymatic activity on the cells as the shell prevented cell lysis. Addition of NaCl led to the liberation of the protein and the onset of enzymatic activity. Removal of NaCl triggered the reassembly of the PIC micelle.[41]
Similarly to increased ionic strength, the addition of surfactant can interfere with the PIC micelle formation. Addition of surfactants such as cetyltrimethylammonium bromide may not necessarily lead to disassembly, but can cause the formation of large particles and subsequent precipitation.[40]
As the PIC micelle formation is based on electrostatic interactions, the binding process can be affected by an electric field. The lysozyme activity in PIC micelles based on PEG-b-P(Asp) could be switched off and on when the solution was exposed to a threshold voltage.[42]
Effect of the Length of the Core-Forming Block and the Polymer Architecture
The length of the charged polymer, or more precisely the number of charged functional groups, should be influential in the binding event between polymer and protein. A significant amount of work on this topic has been published using charged polymers for encapsulation, but not proteins. However, the lessons learned when trying to trap polymers can be applied to proteins. The block copolymer PEG-P(Asp) with various P(Asp) block lengths was condensed with P(Lys) with 20–82 repeating units. Interestingly, the hydrodynamic radius was found to be ~24 nm independently of the length of the polymer blocks. Although the external appearance of the PIC micelles was similar, the aggregation number N and the amount of P(Lys) in the micelle changed. Block copolymers with longer charged blocks typically led to higher aggregation numbers N and higher amounts of P(Lys) in the core, which correlates with a larger core and smaller corona, but also lower PEG chain densities in the shell.[25]
The enzymatic activity of the encapsulated protein was found to be enhanced when the enzyme was wrapped into polymers as long as the substrate was small enough to be able to diffuse through the polymer shell. For example, the relative activity of lysozyme in the core of the PEG-PAA PIC micelles versus p-nitrophenyl-N-acetyl-β-chitooligosides was found to be increased compared with the free enzyme, but PIC micelles based on smaller charged polymer blocks, here poly(α,β-aspartic acid), showed higher enzymatic activity. This was explained by the position of the charged block copolymer in relation to the binding pockets with longer polymers spanning several active sites on the enzyme.[42] Contrasting results were obtained when trypsin (~23 kDa) was used instead of lysozyme (~14 kDa). Larger charged polymer blocks resulted in general in better enzymatic activity, but the activity enhancement was also dependent on the mixing ratio between polymer and protein. The identified optimum mixing ratio between polymer and protein was found to decrease with increasing molecular weight.[43]
The influence of the length of copolymers with grafted PEG chain on the formation of PIC micelles with GOx (160 kDa) was investigated.[33] Although the molecular mass of PEG-graft-poly(allyl amine) was varied between 31 and 258 kDa, the radius of gyration of the resulting PIC micelle with GOx increased from 10.8 to 12.6 nm. Detailed studies revealed that PIC micelles made from longer polymers contained fewer enzyme molecules.[33] The enzymatic activity was independent of the molecular mass as the substrate glucose was able to diffuse easily to the enzyme with PEG presenting no barrier.[33]
As PIC micelles with proteins were also prepared using graft or comb polymers, the influence of the polymer architecture needs to be discussed in terms of molar ratio of neutral and charged blocks. The negative neutral PDMAM content in the graft copolymers poly(sodium acrylate-co-sodium 2-acrylamido-2-methyl-1-propanesulfonate)-graft-poly(N,N-dimethylacrylamide) (P(NaA-coNaAMPS)-g-PDMAMx)) was varied from 0 to 75 % to investigate the binding with bovine serum albumin (BSA) below its isoelectric point, thus with BSA in a positively charged state. The absence of stabilising neutral PDMAM led to high turbidity indicative of the formation of large particles whereas PDMAM was able to stabilise the system, resulting in reduced viscosity.[44]
The presence of a single hydrophobic endgroup can strongly influence PIC micelle formation and stability. The endgroups of PEG-P(Asp)-X were varied by introducing phenyl, naphthyl, and pyrenyl terminal functionalities that were subsequently located in the core of the lysozyme-loaded PIC micelle. The resulting PIC micelles increased in size in correlation with the increased hydrophobicity of the endgroup. Hydrophobic endgroups led to a higher aggregation number and a larger amount of lysozyme in the core in addition to a higher stability in the presence of NaCl. The introduction of large aromatic groups had even more far-reaching effects as light-scattering studies suggested the formation of non-spherical particles. It was proposed that the sphere–rod transition was caused by the stretching of the P(Asp) in the PIC micelle.[45]
Discussion on block length cannot focus on the charged block alone as the shell-forming block is crucial in achieving colloidal stability.[30] If the non-charged polymer is too short, the nanoparticles can precipitate whereas very long non-charged polymer may not result in any PIC micelle formation. Longer oppositely charged polymers in the core then can help the stability of the PIC micelles.
Effect of the Structure of the Charged Functionality
Comparison of three block copolymers with similar repeating units but with different monomer structure of the ionic block revealed that not only does the length of the polymer play a role, but also the direct surroundings of the charged functionality, e.g. the pKa value. The three block copolymers PEG-block-poly(glutamic acid) (PEG-PGA), PEG-block- poly(α,β-aspartic acid) (PEG-P(Asp)) and PEG-block-poly(methacrylic acid) (PEG-PMA) were complexed with trypsin.[46] Trypsin activity was found to be dependent on the pH of the solution. Free trypsin shows only low activity at pH values below 6 as the imidazolium in the binding pocket, which is integral to the amidase activity, is protonated. The presence of negatively charged polymers can provide local buffering activity, enabling enhanced activity at low pH. Polymers with high pKa values such as PEG-PMA are more likely to be protonated and cannot stabilise the enzyme imidazolium, hampering the stabilising effect.[46]
The difference in protonation is also evident in other systems. PEG-P(Lys) has a pKa value of 9.4 whereas PEG-PAsp(DET) has two pKa values, 6.1 and 9.9, which can have a large buffering effect. The PIC micelles prepared from both polymers with Cu/Zn superoxide dismutase had similar sizes and both led to a decline in enzymatic activity of ~50 %. In this case, the advantage of PEG-PAsp(DET) can be found in its lower cytotoxicity, an attractive feature for drug delivery.[47]
Structural variations around the positive charge in cationic polymers can influence the activity of the enzyme. In general, the cationic polymers PEG-PAMA depicted in Fig. 4 decreased the ability of α-amylase to interact with the substrate p-nitrophenyl-α-d-maltoside. Addition of salt and the simultaneous disassembly of the PIC micelle restored the catalytic process. However, assembly and disassembly were dependent on the polarity of the polymer, with the most non-polar polymer, carrying a phenyl group, showing stronger binding than the other polymers.[27]
As the formation of PIC micelles is based on electrostatic interactions, the stability of PIC micelles relies on the degree of ionisation, which is governed by the pKa of the charged functionality and the surrounding pH. To make the binding process less responsive to the environment, it is possible to employ polymers that are permanently charged such as polymers with pendant sulfonate and quaternary amino groups.[48] The current literature suggests little interest in this approach for trying to trap proteins. Instead, efforts have been devoted to creating systems that respond very quickly to pH changes. Adjusting the pKa value of the polymer allows some fine-tuning of the strength of binding between polymer and protein. However, it does not present a switch that converts strong protein binding to fast protein release with minute pH changes. This can be achieved by creating a charge conversion system as was demonstrated by Lee et al.[49] The idea is here that the charged species is masked by a labile functional group carrying the opposite charge. In slightly acidic environments, this group is cleaved, liberating the oppositely charged functional group underneath.
This concept was first demonstrated on lysozyme, which was modified with citraconic anhydride creating a negatively charged protein that could revert back to its natural positively charged state on degradation of the labile amide group.[50] This idea was then applied by many researchers to trigger drug release. Chen et al. modified PEG-b-P(Lys) with four different anhydrides including succinic anhydride (SA), cis-cyclohexene-1,2-dicarboxylic anhydride (CDA), cis-aconitic anhydride (CA), and dimethylmaleic anhydride (DMMA).[51] The resulting negatively charged block copolymers containing pH-sensitive amide bonds were then employed to form PIC micelles with positively charged doxorubicin (DOX). It was found that CA- and DMMA-containing polymers showed better cytotoxicity towards HepG2 cells than the free form of DOX, whereas SA and CDA, which form stable amide linkages, resulted in PIC micelles of relatively low cytotoxicity. This concept was also applied to protein delivery. PIC micelles were obtained by synthesising PEG-b-poly[(N′-citraconyl-2-aminoethyl)aspartamide] (PEG-b-pAsp(EDA-CA)) from the reaction between the primary amine on PEG-b-poly[(2-aminoethyl)aspartamide] and citraconic anhydride.[49] As a control, a non-responsive SA system was prepared. The lysozyme containing PIC micelles underwent charge conversion at endosomal pH, pushing out the protein by repulsion (Fig. 7).

|
Strategies to Enhance PIC Micelle Stability
Supercharging of Proteins
The formation of PIC micelles relies on the presence of an abundance of charges on the surface of the protein. The charge density on the protein surface influences the binding event, with the charged polymers as proteins with high (positive or negative) zeta potential resulting in stronger polymer interactions. However, some proteins may display an isoelectric point close to physiological pH, which means that they are neither strongly negatively nor strongly positively charged. This results only in weak electrostatic interactions between the charged polymer and the protein. To address this, proteins have been chemically modified with additional charged groups to enhance binding such as by reacting available amino groups with SA, resulting in negatively charged proteins. The supercharged protein can then bind tightly to the oppositely charged polymer. The challenge here is to ensure that the protein does not lose its biological activity with excess functionalisation. This approach was studied in detail using a set of proteins – α- chymotrypsinogen, lysozyme, myoglobin, and RNase A – that are all weakly charged under physiological conditions.[52] Reaction with SA led to a strong negative charge, the intensity of which could be controlled by the amount of SA added. As a result, even originally positively charged proteins such as lysozyme displayed a strong negative charge and could be entrapped in PIC micelles based on positively charged polymers. The protein did not undergo any major structural changes during surface modification and could be formulated into tightly packed PIC micelles, with the size of the nanoparticle decreasing with increasing amount of charge on the protein. Although these PIC micelles still disassembled with the addition of NaCl, the critical concentration was noticeably higher.[52]
Supercharging of proteins can potentially alter their biological activity. This can be addressed using citraconic anhydride as was demonstrated using native immunoglobulin G (IgG), which has an almost neutral surface charge (Fig. 8). The advantage of this technique is not only the quick introduction of excess negatively charged groups on the surface of the antibody, but also the quick cleavage of this amide bond at low pH, restoring the full activity of the antibody.[26]

|
Co-Encapsulation of Charged Polymers
Addition of homopolymers that carry a similar charge to the payload can help stabilise the micelle against disassembly in an environment of high ionic strength. A study that used entrapment of different ratios of positive charged lysozyme and poly(N,N-dimethylaminoethyl methacrylate) (PDMAEMA) together into a PIC micelle revealed an enhanced stability, in particular when the homopolymer was in slight excess compared with the amount of protein. This technique also allowed the adjustment of the amount of lysozyme in the PIC micelle, which could be tailored from 5 to 15 protein molecules per nanoparticle. The challenge of this approach is the possibility that the negatively charged block copolymer interacts preferentially with the strongly charged homopolymer, expelling the protein from the core.[53] Detailed scattering studies (dynamic light scattering (DLS) and small-angle neutron scattering (SANS)) in solutions of different NaCl concentrations confirmed the enhanced stability the PIC micelles in the presence of added homopolymer.[39] This approach is successful with homopolymers, and also with oppositely charged block copolymers, which can bind proteins in addition. Lysozyme (positively charged) or α-lactalbumin (negatively charged) could therefore both be entrapped in a mixture of PAA-b-P(AAm) and poly(2-methyl vinyl pyridinium iodide)-block-poly(ethylene oxide).[35] Systems that contain two polymers with opposite charges can display a slightly different release profile when NaCl is added. Whereas a two-component PIC micelle of block copolymer and protein will ultimately lead to disassembly, a three-component system can maintain the structural integrity of the PIC micelle after protein release. The negatively charged lipase was entrapped in PIC micelles of poly(2-methyl vinyl pyridinium)-block-poly(ethylene oxide) and the anionic homopolymer PAA. Addition of salt led to lipase release, leaving behind empty PIC micelles with few structural changes (Fig. 9).[54]
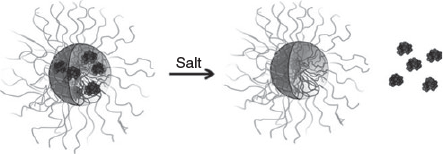
|
Crosslinking of PIC Micelles
The low stability of PIC micelles can be simply addressed by crosslinking the aggregates. The challenge here is to find a suitable crosslinker that does not affect the structural integrity of the protein. Typical crosslinkers such as glutaraldehyde[28] or genipin[55] may result in crosslinking of the cargo.
Jaturanpinyo et al.[28] crosslinked PIC micelles prepared from PEG-b-poly(α,β-aspartic acid) and trypsin, a cationic enzyme, with glutaraldehyde (Fig. 10). The resulting micelles showed enhanced resistance to disassembly at high salt concentrations while the activity of the trapped trypsin was retained.

|
Although crosslinking can permanently stabilise the PIC micelle structure, fast disassembly on an external trigger is highly desirable as it allows release of the protein on demand, making it available for its biological function. This is in particular attractive for drug delivery purposes as the therapeutic load needs to be protected in the bloodstream, but the PIC micelle needs to disassemble once the nanoparticle reaches its target. For cancer therapy, pH-responsive drug carriers are often used as it has been reported that the pH around tumour tissue is slightly acidic (~pH 6) whereas the pH of the bloodstream is 7.4.[56]
These slightly acidic cell compartments can now trigger the cleavage of a range of covalent bonds including boronic esters. Ren et al. developed a sugar/pH dual-responsive core-crosslinked PIC micelle for the delivery of either positively or negatively charged proteins.[57] The PIC micelles were initially formed from cytochrome C and a positively charged block copolymer, PEG-b-poly(l-lysine-co-ϵ-3,4-dihydroxyphenylcarboxyl-l-lysine) (PEG-b-P(Lys-co-LysCA)). Subsequently, a negatively charged block copolymer, PEG-b-poly(glutamic acid-co-glutamicamidophenylboronic acid) (PEG-b-P(Glu-co-GluPBA)) was added dropwise. The crosslinking occurred during mixing through phenylboronic acid–catechol interactions. The core-crosslinked micelles showed good stability under physiological conditions but the PIC micelle disassembled quickly on addition of excess fructose or acids (~pH 5) (Fig. 11).

|
To date, many PIC micelles have also been designed to be responsive to other biosignals, such as the redox potential of glutathione, to achieve controlled release of the cargo. It was reported that the concentration of the reduced form of glutathione is 500–1000 times higher than its oxidised form in cytoplasm and is 7 times higher near tumour tissue than normal tissue.[58] Micelles stabilised with disulfide bridges are therefore widely applied to generate redox-responsive drug carriers to deliver a range of drugs including proteins.[59] The antigen ovalbumin (OVA) and immunostimulatory CpG-DNA or the anti-inflammatory enzyme catalase (CAT) were combined with PEG-poly(l-lysine-dithiopyridine) (PEG-PLDTP) and crosslinked (Fig. 12).[59] A similar approach was employed to stabilise catalase in a PIC micelle prepared from polyethyleneimine–PEG (PEI-PEG) micelles, which were stabilised by reacting the amino groups with either a non-degradable or a redox-sensitive linker.[60]

|
A detailed study on the types of crosslinker was carried out using catalase and superoxide dismutase 1.[61] Both enzymes, separately or together, were entrapped in PEG-P(Lys) or PEG-PEI. Permanent and degradable crosslinkers such as glutaraldehyde, bis-(sulfosuccinimidyl)suberate sodium salt or 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide glycol were used. The catalytic activity was maintained although it seems that the enzymes suffered slightly under the glutaraldehyde treatment. The authors of that study acknowledged that it is likely that the crosslinking process will result in the crosslinking of not only the polymer chains, but also the proteins. To address this, the use of a slight excess of polymer was suggested to limit crosslinking with the loaded protein.[62] In all systems, bimodal distributions were observed, indicating the presence of larger PIC micelles and small particles that could be either free enzyme or single-enzyme nanoparticles. Overall, crosslinking favoured the formation of larger nanoparticles, but it was also found to have the added benefit that it reduced the toxicity of the cationic polymer and enhanced the catalytic activity of the enzyme.[62] Important though was the enhanced stability of the crosslinked systems in vivo whereas free enzyme and PIC-encapsulated enzymes seemed to succumb to degradation.[61]
Targeted PIC Delivery
As with any micelle, PIC micelles benefit from the attachment of targeting ligands. Examples include the attachment of folic acid to enhance the delivery of BSA as model drug. These micelles displayed enhanced cellular uptake on cells overexpressing folate receptors.[63] It is interesting to note that this approach has not been explored widely in the literature to increase the accumulation of protein drugs.
Application of PIC Micelles
Delivery of Therapeutic Proteins
PIC micelles have indeed been shown to be able to deliver therapeutic proteins. In one example, the protein Sprouty 1, which can act as an endogenous angiogenesis inhibitor, was delivered in a PIC micelle prepared from albumin as the charged part and a pegylated polymer as the stabilising neutral block. The aim was to create an efficient system for the treatment of breast cancer.[29]
One of the consequences of strokes is slow neurological recovery, depression, and cognitive impairment, which are thought to be caused by low levels of brain-derived neurotrophic factor (BDNF). In order to deliver this protein, the block copolymer PEG-PGA was condensed with BDNF.[36] Prior to the incubation of polymer with proteins, the authors carried out molecular simulation studies in order to understand the nature of the contact points between polymer and protein, identifying electrostatic interactions, and also H-bonding. The formulation was administered intranasally and could be accumulated in the brain.[36] Compared with free BDNF, the PIC micelle delivering BDNF was able to significantly reduce cerebral tissue loss in mice after stroke.[64]
Parkinson’s disease has been the target of PIC micelles based on catalase. PEI-PEG was used to form PIC micelles with catalase. The resulting 60–100-nm-sized PIC micelles, which the authors called nanozymes, were non-toxic, and protected the enzyme from hydrolytic degradation. The resulting nanoparticle was loaded into bone marrow macrophages as a cell-based drug carrier to make use of the receptors expressed on the surface of these cells. The uptake of the nanozymes by cells was complete within 1 h while the enzyme was slowly released again from the cell within several days.[65] Nanozyme loading did not affect α4-integrin levels of the macrophages, making these carriers potentially suitable for the treatment of Parkinson’s disease.[66] The nanozymes were shown to have enhanced uptake by murine brain microvascular endothelial cells (BMVEC), neurons and rat astrocytes compared with the free catalase.[67] This catalyse-loaded PIC system was then stabilised by crosslinking using a degradable (3,3′-dithiobis(sulfosuccinimidyl propionate) and a non-degradable (bi(sulfosuccinimidyl)suberate sodium salt) linker. The crosslinked nanozyme, which was delivered using macrophages, reduced inflammation of the neural system in mice.[60]
Reduction of oxidative stress is the key to lower brain tissue damage and to treating diseases that affect the central nervous system. The enzyme copper/zinc superoxide dismutase (CuZnSOD), which scavenges superoxide, was observed to reduce the effect of angiotensin II (AngII)-induced diseases such as hypertension and heart failure. Delivery of the enzymes in PEI-PEG micelles was observed to reduce the formation of peroxide in neurons to a greater extent than the enzyme alone.[68] The same system, but now crosslinked to stabilise the enzyme, was tested in a rat middle cerebral artery occlusion (MCAO) model. The delivered enzyme resulted in a reduction of oxidative damage. The crosslinked CuZnSOD-loaded PIC micelle was able to reduce infarct size and improve motor function in rat models.[62] To understand how the crosslinked PIC micelle was able to display this enhanced therapeutic effect, the rat brain was analysed in more detail. The nanoparticles were found to be located in the lumen of the blood vessel, but they did not cross the blood–brain barrier. The therapeutic activity of the nanoparticle was attributed to accumulation in the infarct region.[69] As the polymer used was found to display high cytotoxicity, it was later replaced by PEG-PAsp(DET), which was not only less toxic, but also displayed less accumulation in the liver and spleen.[47]
Control of Enzymatic Activity
Encapsulation of enzymes into PIC micelles creates nanoparticles that can stabilise enzymes and control catalytic activity. It is assumed that the enzyme is entrapped within the core of the nanoparticle, protected from the environment. This, however, can reduce enzymatic activity when the substrate it too big to penetrate through the polymer shell or if the polymer leads to unfolding of the protein. In general, PIC micelles seem to enhance, or at least not inhibit, the catalytic activity of enzymes such as lysozyme,[42] trypsin,[43] and GOx[33] as long as the substrate is small. In contrast, enzymatic activity can be completely hampered with large substrates.[41] In this case, a switch is required, such as the addition of salt[27] or an electric field[42] that can disassemble the PIC micelle, restoring the enzyme. There has been little focus so far on the structure of the protein once it is complexed with the charged polymer. Thermal and circular dichroism analysis of lysozyme to identify the transition from the folded to the unfolded state has revealed that the enzyme in the PIC micelle has different secondary and tertiary structures, a lower thermostability, and different unfolding and refolding behaviours.[70]
Conclusions
PIC micelles have proved to be an easy way to entrap various proteins simply by mixing polymer and protein at various molar ratios. Researchers have investigated the effect of the type of polymers on the stability of PIC micelles and the stability of the protein. As PIC micelles are inherently unstable at high ionic strength, crosslinking was investigated. Other stabilisation techniques include supercharging of proteins or the addition of more charged polymers. In conclusion, PIC micelles have been found to be able to stabilise enzymes or to safely delivery therapeutic proteins.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors would like to thank the Australian Research Council (ARC grant no. DP140100240) for funding.
References
[1] M. Granold, P. Hajieva, M. I. Toşa, F.-D. Irimie, B. Moosmann, Proc. Natl. Acad. Sci. USA 2018, 115, 41.| Crossref | GoogleScholarGoogle Scholar |
[2] N. Gregersen, P. Bross, S. Vang, J. H. Christensen, Annu. Rev. Genomics Hum. Genet. 2006, 7, 103.
| Crossref | GoogleScholarGoogle Scholar |
[3] D. Lee, O. Redfern, C. Orengo, Nat. Rev. Mol. Cell Biol. 2007, 8, 995.
| Crossref | GoogleScholarGoogle Scholar |
[4] M. Jucker, L. C. Walker, Nature 2013, 501, 45.
| Crossref | GoogleScholarGoogle Scholar |
[5] L. Breydo, J. W. Wu, V. N. Uversky, Biochim. Biophys. Acta, Mol. Basis Dis. 2012, 1822, 261.
| Crossref | GoogleScholarGoogle Scholar |
[6] G. Wilcox, Clin. Biochem. Rev. 2005, 26, 19.
[7] (a) S. Masoumi-Moghaddam, A. Amini, D. L. Morris, Cancer Metastasis Rev. 2014, 33, 695.
| Crossref | GoogleScholarGoogle Scholar |
(b) O. J. Finn, Ann. Oncol. 2012, 23, viii6.
| Crossref | GoogleScholarGoogle Scholar |
(c) C. Wu, M. Sun, L. Liu, G. W. Zhou, Gene 2003, 306, 1.
| Crossref | GoogleScholarGoogle Scholar |
[8] H. A. D. Lagassé, A. Alexaki, V. L. Simhadri, N. H. Katagiri, W. Jankowski, Z. E. Sauna, C. Kimchi-Sarfaty, F1000 Res. 2017, 6, 113.
| Crossref | GoogleScholarGoogle Scholar |
[9] (a) B. Leader, Q. J. Baca, D. E. Golan, Nat. Rev. Drug Discov. 2008, 7, 21.
| Crossref | GoogleScholarGoogle Scholar |
(b) V. P. Torchilin, A. N. Lukyanov, Drug Discov. Today 2003, 8, 259.
| Crossref | GoogleScholarGoogle Scholar |
[10] K. Fosgerau, T. Hoffmann, Drug Discov. Today 2015, 20, 122.
| Crossref | GoogleScholarGoogle Scholar |
[11] J. D. Bos, M. M. H. M. Meinardi, Exp. Dermatol. 2000, 9, 165.
| Crossref | GoogleScholarGoogle Scholar |
[12] R. Solaro, F. Chiellini, A. Battisti, Materials 2010, 3, 1928.
| Crossref | GoogleScholarGoogle Scholar |
[13] (a) D. S. Pisal, M. P. Kosloski, S. V. Balu-Iyer, J. Pharm. Sci. 2010, 99, 2557.
| Crossref | GoogleScholarGoogle Scholar |
(b) Z. Gu, A. Biswas, M. Zhao, Y. Tang, Chem. Soc. Rev. 2011, 40, 3638.
| Crossref | GoogleScholarGoogle Scholar |
[14] A. Harada, K. Kataoka, Macromolecules 1995, 28, 5294.
| Crossref | GoogleScholarGoogle Scholar |
[15] D. Wakebayashi, N. Nishiyama, K. Itaka, K. Miyata, Y. Yamasaki, A. Harada, H. Koyama, Y. Nagasaki, K. Kataoka, Biomacromolecules 2004, 5, 2128.
| Crossref | GoogleScholarGoogle Scholar |
[16] A. Harada, K. Kataoka, Macromolecules 1998, 31, 288.
| Crossref | GoogleScholarGoogle Scholar |
[17] (a) A. Harada, K. Kataoka, Polym. J. 2018, 50, 95.
| Crossref | GoogleScholarGoogle Scholar |
(b) N. Lefevre, C. A. Fustin, J. F. Gohy, Macromol. Rapid Commun. 2009, 30, 1871.
| Crossref | GoogleScholarGoogle Scholar |
[18] Y. Zhao, M. S. Lord, M. H. Stenzel, J. Mater. Chem. B 2013, 1, 1635.
| Crossref | GoogleScholarGoogle Scholar |
[19] Y. Lee, K. Kataoka, Soft Matter 2009, 5, 3810.
| Crossref | GoogleScholarGoogle Scholar |
[20] I. K. Voets, A. d. Keizer, P. d. Waard, P. M. Frederik, P. H. H. Bomans, H. Schmalz, A. Walther, S. M. King, F. A. M. Leermakers, M. A. C. Stuart, Angew. Chem. Int. Ed. 2006, 45, 6673.
| Crossref | GoogleScholarGoogle Scholar |
[21] I. K. Voets, A. de Keizer, M. A. Cohen Stuart, P. de Waard, Macromolecules 2006, 39, 5952.
| Crossref | GoogleScholarGoogle Scholar |
[22] N. Sanson, F. Bouyer, C. Gerardin, M. In, Phys. Chem. Chem. Phys. 2004, 6, 1463.
| Crossref | GoogleScholarGoogle Scholar |
[23] C. W. Scales, F. Huang, N. Li, Y. A. Vasilieva, J. Ray, A. J. Convertine, C. L. McCormick, Macromolecules 2006, 39, 6871.
| Crossref | GoogleScholarGoogle Scholar |
[24] J.-S. Park, Y. Akiyama, Y. Yamasaki, K. Kataoka, Langmuir 2007, 23, 138.
| Crossref | GoogleScholarGoogle Scholar |
[25] A. Harada, K. Kataoka, Macromolecules 2003, 36, 4995.
| Crossref | GoogleScholarGoogle Scholar |
[26] A. Kim, Y. Miura, T. Ishii, O. F. Mutaf, N. Nishiyama, H. Cabral, K. Kataoka, Biomacromolecules 2016, 17, 446.
| Crossref | GoogleScholarGoogle Scholar |
[27] K. Kuwada, T. Kurinomaru, S. Tomita, K. Shiraki, Colloid Polym. Sci. 2016, 294, 1551.
| Crossref | GoogleScholarGoogle Scholar |
[28] M. Jaturanpinyo, A. Harada, X. Yuan, K. Kataoka, Bioconjug. Chem. 2004, 15, 344.
| Crossref | GoogleScholarGoogle Scholar |
[29] Y. Y. Jiang, H. X. Lu, F. Chen, M. Callari, M. Pourgholami, D. L. Morris, M. H. Stenzel, Biomacromolecules 2016, 17, 808.
| Crossref | GoogleScholarGoogle Scholar |
[30] S. van der Burgh, A. de Keizer, M. A. Stuart, Langmuir 2004, 20, 1073.
| Crossref | GoogleScholarGoogle Scholar |
[31] M. Jonsson, P. Linse, J. Chem. Phys. 2001, 115, 10975.
| Crossref | GoogleScholarGoogle Scholar |
[32] J. M. Park, B. B. Muhoberac, P. L. Dubin, J. Xia, Macromolecules 1992, 25, 290.
| Crossref | GoogleScholarGoogle Scholar |
[33] A. Kawamura, C. Kojima, M. Iijima, A. Harada, K. Kono, J. Polym. Sci. Part A: Polym. Chem. 2008, 46, 3842.
| Crossref | GoogleScholarGoogle Scholar |
[34] A. Harada, K. Kataoka, Langmuir 1999, 15, 4208.
| Crossref | GoogleScholarGoogle Scholar |
[35] S. Lindhoud, W. Norde, M. A. Cohen Stuart, J. Phys. Chem. B 2009, 113, 5431.
| Crossref | GoogleScholarGoogle Scholar |
[36] Y. Jiang, J. M. Fay, C.‐D. Poon, N. Vinod, Y. Zhao, K. Bullock, S. Qin, D. S. Manickam, X. Yi, W. A. Banks, A. V. Kabanov, Adv. Funct. Mater. 2018, 28,
| Crossref | GoogleScholarGoogle Scholar |
[37] S. De Luca, F. Chen, P. Seal, M. H. Stenzel, S. C. Smith, Biomacromolecules 2017, 18, 3665.
| Crossref | GoogleScholarGoogle Scholar |
[38] A. V. Kabanov, T. K. Bronich, V. A. Kabanov, K. Yu, A. Eisenberg, Macromolecules 1996, 29, 6797.
| Crossref | GoogleScholarGoogle Scholar |
[39] S. Lindhoud, L. Voorhaar, R. de Vries, R. Schweins, M. A. C. Stuart, W. Norde, Langmuir 2009, 25, 11425.
| Crossref | GoogleScholarGoogle Scholar |
[40] D. Kaczmarek, J. S. Diget, B. Nyström, G. Gyulai, R. Mészáros, T. Gilányi, I. Varga, Colloids Surf. A 2017, 532, 290.
| Crossref | GoogleScholarGoogle Scholar |
[41] A. Harada, K. Kataoka, J. Am. Chem. Soc. 1999, 121, 9241.
| Crossref | GoogleScholarGoogle Scholar |
[42] A. Harada, K. Kataoka, J. Am. Chem. Soc. 2003, 125, 15306.
| Crossref | GoogleScholarGoogle Scholar |
[43] A. Kawamura, Y. Yoshioka, A. Harada, K. Kono, Biomacromolecules 2005, 6, 627.
| Crossref | GoogleScholarGoogle Scholar |
[44] M. Sotiropoulou, G. Bokias, G. Staikos, Biomacromolecules 2005, 6, 1835.
| Crossref | GoogleScholarGoogle Scholar |
[45] X. Yuan, A. Harada, Y. Yamasaki, K. Kataoka, Langmuir 2005, 21, 2668.
| Crossref | GoogleScholarGoogle Scholar |
[46] A. Harada, Y. Yoshioka, A. Kawamura, C. Kojima, K. Kono, Macromol. Biosci. 2007, 7, 339.
| Crossref | GoogleScholarGoogle Scholar |
[47] Y. Jiang, P. Arounleut, S. Rheiner, Y. Bae, A. V. Kabanov, C. Milligan, D. S. Manickam, J. Control. Release 2016, 231, 38.
| Crossref | GoogleScholarGoogle Scholar |
[48] K. Nakai, M. Nishiuchi, M. Inoue, K. Ishihara, Y. Sanada, K. Sakurai, S.-i. Yusa, Langmuir 2013, 29, 9651.
| Crossref | GoogleScholarGoogle Scholar |
[49] Y. Lee, S. Fukushima, Y. Bae, S. Hiki, T. Ishii, K. Kataoka, J. Am. Chem. Soc. 2007, 129, 5362.
| Crossref | GoogleScholarGoogle Scholar |
[50] J. K. Shetty, J. E. Kinsella, Biochem. J. 1980, 191, 269.
| Crossref | GoogleScholarGoogle Scholar |
[51] J. Chen, J. Ding, Y. Zhang, C. Xiao, X. Zhuang, X. Chen, Polym. Chem. 2015, 6, 397.
| Crossref | GoogleScholarGoogle Scholar |
[52] A. C. Obermeyer, C. E. Mills, X.-H. Dong, R. J. Flores, B. D. Olsen, Soft Matter 2016, 12, 3570.
| Crossref | GoogleScholarGoogle Scholar |
[53] S. Lindhoud, R. de Vries, W. Norde, M. A. C. Stuart, Biomacromolecules 2007, 8, 2219.
| Crossref | GoogleScholarGoogle Scholar |
[54] S. Lindhoud, R. de Vries, R. Schweins, M. A. Cohen Stuart, W. Norde, Soft Matter 2009, 5, 242.
| Crossref | GoogleScholarGoogle Scholar |
[55] F. Maggi, S. Ciccarelli, M. Diociaiuti, S. Casciardi, G. Masci, Biomacromolecules 2011, 12, 3499.
| Crossref | GoogleScholarGoogle Scholar |
[56] G. R. Martin, R. K. Jain, Cancer Res. 1994, 54, 5670.
[57] J. Ren, Y. Zhang, J. Zhang, H. Gao, G. Liu, R. Ma, Y. An, D. Kong, L. Shi, Biomacromolecules 2013, 14, 3434.
| Crossref | GoogleScholarGoogle Scholar |
[58] (a) D. P. Jones, J. L. Carlson, V. C. Mody, J. Cai, M. J. Lynn, P. Sternberg, Free Radic. Biol. Med. 2000, 28, 625.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. Russo, W. DeGraff, N. Friedman, J. B. Mitchell, Cancer Res. 1986, 46, 2845.
[59] M. J. Heffernan, N. Murthy, Ann. Biomed. Eng. 2009, 37, 1993.
| Crossref | GoogleScholarGoogle Scholar |
[60] N. L. Klyachko, M. J. Haney, Y. Zhao, D. S. Manickam, V. Mahajan, P. Suresh, S. D. Hingtgen, R. L. Mosley, H. E. Gendelman, A. V. Kabanov, E. V. Batrakova, Nanomedicine 2014, 9, 1403.
| Crossref | GoogleScholarGoogle Scholar |
[61] N. L. Klyachko, D. S. Manickam, A. M. Brynskikh, S. V. Uglanova, S. Li, S. M. Higginbotham, T. K. Bronich, E. V. Batrakova, A. V. Kabanov, Nanomedicine 2012, 8, 119.
| Crossref | GoogleScholarGoogle Scholar |
[62] D. S. Manickam, A. M. Brynskikh, J. L. Kopanic, P. L. Sorgen, N. L. Klyachko, E. V. Batrakova, T. K. Bronich, A. V. Kabanov, J. Control. Release 2012, 162, 636.
| Crossref | GoogleScholarGoogle Scholar |
[63] S. H. Kim, J. H. Jeong, C. O. Joe, T. G. Park, J. Control. Release 2005, 103, 625.
| Crossref | GoogleScholarGoogle Scholar |
[64] N. M. Harris, R. Ritzel, N. S. Mancini, Y. Jiang, X. Yi, D. S. Manickam, W. A. Banks, A. V. Kabanov, L. D. McCullough, R. Verma, Pharmacol. Biochem. Behav. 2016, 150–151, 48.
| Crossref | GoogleScholarGoogle Scholar |
[65] E. V. Batrakova, S. Li, A. D. Reynolds, R. L. Mosley, T. K. Bronich, A. V. Kabanov, H. E. Gendelman, Bioconjug. Chem. 2007, 18, 1498.
| Crossref | GoogleScholarGoogle Scholar |
[66] A. M. Brynskikh, Y. Zhao, R. L. Mosley, S. Li, M. D. Boska, N. L. Klyachko, A. V. Kabanov, H. E. Gendelman, E. V. Batrakova, Nanomedicine 2010, 5, 379.
| Crossref | GoogleScholarGoogle Scholar |
[67] M. J. Haney, Y. Zhao, S. Li, S. M. Higginbotham, S. L. Booth, H.-Y. Han, J. A. Vetro, R. L. Mosley, A. V. Kabanov, H. E. Gendelman, E. V. Batrakova, Nanomedicine 2011, 6, 1215.
| Crossref | GoogleScholarGoogle Scholar |
[68] E. G. Rosenbaugh, J. Roat, L. Gao, R.-F. Yang, D. S. Manickam, J.-X. Yin, H. D. Schultz, T. K. Bronich, E. V. Batrakova, A. V. Kabanov, I. H. Zucker, M. C. Zimmerman, Biomaterials 2010, 31, 5218.
| Crossref | GoogleScholarGoogle Scholar |
[69] Y. Jiang, A. M. Brynskikh, D. S. Manickam, A. V. Kabanov, J. Control. Release 2015, 213, 36.
| Crossref | GoogleScholarGoogle Scholar |
[70] F.-G. Wu, Y.-W. Jiang, Z. Chen, Z.-W. Yu, Langmuir 2016, 32, 3655.
| Crossref | GoogleScholarGoogle Scholar |
* Martina Stenzel is the winner of the 2017 H. G. Smith Memorial Award.