

Initiation of RAFT Polymerization: Electrochemically Initiated RAFT Polymerization in Emulsion (Emulsion eRAFT), and Direct PhotoRAFT Polymerization of Liquid Crystalline Monomers*
Caroline Bray A B , Guoxin Li A , Almar Postma A , Lisa T. Strover A B , Jade Wang A and Graeme Moad A B
A B , Guoxin Li A , Almar Postma A , Lisa T. Strover A B , Jade Wang A and Graeme Moad A B
A CSIRO Manufacturing, Clayton, Vic. 3168, Australia.
B Corresponding authors. Email: caroline.bray@csiro.au; lisa.strover@csiro.au; graeme.moad@csiro.au
Australian Journal of Chemistry 74(1) 56-64 https://doi.org/10.1071/CH20260
Submitted: 28 August 2020 Accepted: 7 September 2020 Published: 2 October 2020
Journal Compilation © CSIRO 2021 Open Access CC BY-NC-ND
Abstract
We report on two important advances in radical polymerization with reversible addition–fragmentation chain transfer (RAFT polymerization). (1) Electrochemically initiated emulsion RAFT (eRAFT) polymerization provides rapid polymerization of styrene at ambient temperature. The electrolytes and mediators required for eRAFT are located in the aqueous continuous phase separate from the low-molar-mass-dispersity macroRAFT agent mediator and product in the dispersed phase. Use of a poly(N,N-dimethylacrylamide)-block-poly(butyl acrylate) amphiphilic macroRAFT agent composition means that no added surfactant is required for colloidal stability. (2) Direct photoinitiated (visible light) RAFT polymerization provides an effective route to high-purity, low-molar-mass-dispersity, side chain liquid-crystalline polymers (specifically, poly(4-biphenyl acrylate)) at high monomer conversion. Photoinitiation gives a product free from low-molar-mass initiator-derived by-products and with minimal termination. The process is compared with thermal dialkyldiazene initiation in various solvents. Numerical simulation was found to be an important tool in discriminating between the processes and in selecting optimal polymerization conditions.
Introduction
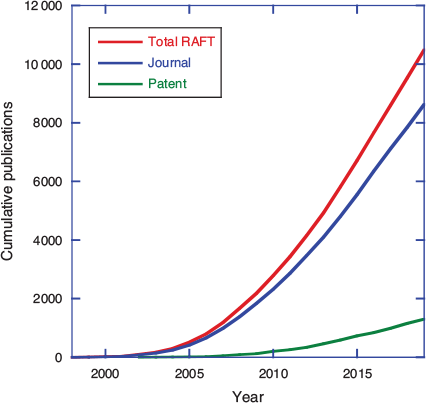
Reversible addition–fragmentation chain transfer (RAFT) polymerization, making use of thiocarbonylthio transfer agents was first reported just over 22 years ago. Thiocarbonylthio-RAFT was first disclosed in a CSIRO/DuPont patent published in January 1998.[1] A patent describing the parallel development of MADIX (RAFT with xanthate transfer agents) at Rhodia was published in December 1998.[2] The first open-literature publication also appeared in 1998.[3] RAFT arose out of a desire to achieve structural perfection in polymers (or at least to define the molar mass and end-group functionality and limit the imperfections), i.e. to invent a process for radical polymerization with the essential characteristics of living polymerization. Although an actual living radical polymerization remains a forlorn hope, RAFT polymerization has matured to be one of the most used, and arguably most effective, methods for achieving well-defined polymer structures. That first RAFT patent was, by 2005, one of the most highly cited patents in the field of chemistry and related science, and the patent literature now abounds with an ever-increasing number of RAFT-related inventions (Fig. 1). However, commercial success stories associated with RAFT polymerization are few.[4] Yet with the first RAFT patents having now reached the end of their enforceable life, we might envisage an upsurge in commercial applications.
|
For many, RAFT is now seen as a mature technology; a toolkit for producing well-defined polymers. For others, including ourselves, the technology is neither fully defined nor sufficiently well understood and is yet to reach its full potential. In the following text, we highlight two recent studies using RAFT at CSIRO on the initiation of RAFT polymerization. These are: electrochemically initiated RAFT emulsion polymerization (eRAFT), and the synthesis of low-dispersity (low Đ) side chain liquid-crystalline polymers (SCLCPs) by direct photoinitiated RAFT polymerization (no exogenous initiator). The latter work also provides an illustration of the use of numerical simulation in understanding and improving the RAFT process.
RAFT Free from Exogenous Initiators
RAFT polymerization is most commonly initiated by radicals generated from an added initiator. However, a RAFT process, wherein radicals are generated photochemically direct from the initial RAFT agent (called an iniferter), is imbedded in Otsu’s proposed mechanism for implementing living radical polymerization using the iniferter process.[5–7] Were it not for the dithiocarbamate iniferters used in Otsu’s early experiments having very low transfer coefficients (Ctr) (i.e. not being effective RAFT agents) with respect to polymerizations of the monomers then used, there would have been no RAFT left to invent 10 years later. Photoinitiated RAFT is mentioned in the first RAFT patent.[1] Publications on RAFT with direct photoinitiation appeared several years later.[8] From that time through to the current day, there has been remarkably persistent discussion on the topic of whether the dominant control mechanism in such systems is the reversible coupling equilibrium of stable radical-mediated polymerization (SRMP), the degenerate transfer process that is RAFT, or some combination of both. This despite the demonstration that when a high-Ctr RAFT agent is used, the dominant process must be RAFT (i.e. the use of photoinitiation does not turn RAFT off).[8] Only when a RAFT agent with a very low Ctr is used is the SRMP mechanism likely to dominate.[9] The early work used UV irradiation and observed the formation of at the time undefined RAFT agent-derived by-products.[8] More recent studies have made use of visible irradiation and report fewer, but still not completely defined, by-products.[10–13]
The recent upsurge in interest in direct photoinitiated RAFT and photoinduced energy or electron transfer (PET)-RAFT[14,15] can largely be linked to the desire to obtain polymers free from initiator-derived by-products (a particular concern in RAFT single-unit monomer insertion (RAFT-SUMI))[16,17] and in the synthesis of sequence-defined polymers[18,19] and the need for spatial and temporal control over the RAFT process, which is critically important in flow and high-throughput polymer synthesis.[20,21]
Electrochemically initiated RAFT polymerization, dubbed eRAFT, has recently attracted attention for similar reasons.[22–26]
Electrochemically Initiated RAFT Polymerization (eRAFT) in Emulsion
In eRAFT, initiating radicals can be generated from the RAFT agent (directly or by way of a redox mediator);[24] they may also be formed from an exogeneous electrochemically active initiator.[22,27] To date, all efforts have focussed on homogeneous solution polymerization. We hypothesized that eRAFT might be usefully extended to emulsion polymerization. We believed that the use of eRAFT polymerization in a dispersed medium might overcome some of the existing limitations to eRAFT, namely the passivation of the working electrode by radical species[22] and various side-reactions involving the RAFT agent at the working electrode.[25]
Our approach to electroactive initiation was adapted from a commonly used redox initiation system that comprises ferric sulfate hydrate (Fe2III(SO4)3.xH2O), EDTA, and sodium formaldehyde sulfoxylate (SFS) as reductant, and ammonium persulfate (APS) as oxidant and source of sulfate radical anion as initiating radicals, according to Scheme 1.

|
In the conventional system, ferric sulfate is used in very low concentration and is reduced to ferrous sulfate and recycled by the action of SFS. In our system, electrochemical reduction is used in place of the chemical reductant SFS, the Fe-EDTA complex is retained as a reduction mediator, and APS remains as the source of sulfate radical ion initiating radicals.
We decided to use a so-called ‘surfactant-free system’[28–33] that in our case comprised an amphiphilic poly(N,N-dimethyl acrylamide)-block-poly(butyl acrylate) (PDMAm-b-PBA, 2; refer to Scheme 2) macroRAFT agent. This non-ionic macroRAFT agent was chosen so as to be unlikely to be electrochemically active under the polymerization conditions (a hypothesis confirmed in the present experiments). The specific composition of macroRAFT agents, with a PDMAm block having a degree of polymerization (Xn) of ~29 (macroRAFT 2a) or 18 (macroRAFT 2b) units and a PBA block ~8 units respectively had been found by high-throughput experimentation to be most successful in mediating ambient-temperature RAFT emulsion polymerization of styrene and enabled the preparation of a dispersion of styrene monomer that appeared kinetically stable on the required timescale of several hours (see Supplementary Material). Subsequent analysis proved these to be a mixture of PDMAm and PDMAm-b-PBA macroRAFT agents. The use of hydrophilic PDMAm homopolymer macroRAFT agents directly in surfactant-free RAFT emulsion polymerization has been previously reported by Rieger et al.[34] However, the homopolymer macroRAFT agents by themselves do not allow preparation of a stable styrene monomer dispersion.

|
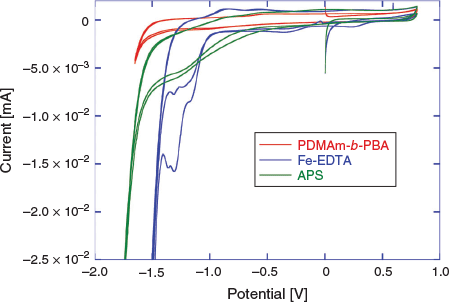
In order to determine conditions for the eRAFT emulsion polymerization, and to confirm our choice of a suitable electrochemically active initiator system, cyclic voltammetry (CV) characterization of the macroRAFT agent (2), Fe-EDTA (mediator), and APS (oxidant and radical source) was conducted (Fig. 2).
|
The CVs were conducted in aqueous solution with 0.1 M KCl supporting electrolyte (Fig. 2). According to the proposed polymerization mechanism, the amphiphilic macroRAFT agent 2 would be located at the interface between the aqueous continuous phase and the particle phase or monomer-swollen micelles, and thus would not itself be directly involved in the initiation process. Nonetheless, the CV confirmed that 2 was not subject to reduction within the potential range used, and therefore would not be degraded by electrochemical stimulus under the chosen polymerization conditions. APS could feasibly be reduced directly to initiate polymerization; however, the reduction peak observed in CV was broad and poorly defined, indicating that direct electrochemical reduction of APS would be unlikely to provide efficient (re)initiation. The Fe-EDTA complex, however, showed a strong reduction peak between –1.0 and –1.5 V corresponding to the reduction of Fe3+. Drawing on the approach used in redox-initiated RAFT, the Fe2+ species can then reduce APS, being oxidized back to Fe3+ in the process.[35,36] This mediated reduction of APS also has the advantage that the initiator is activated and radicals are generated in the continuous aqueous phase as in conventional emulsion polymerization, rather than at the electrode surface. These factors were anticipated to promote initiation of polymerization and limit passivation of the working electrode by initiator-derived radicals as observed previously in eRAFT when using exogenous radical initiators.
eRAFT emulsion polymerizations were conducted in a three-electrode bulk electrolysis cell with a glassy carbon (GC) rod counter electrode, GC rod working electrode, and an Ag/AgCl reference electrode (Fig. 3a). The emulsion compositions and polymerization data are summarized in Table 1. Polymerization proceeded very rapidly at ambient temperature (~22°C), with >99 % conversion being achieved in <120 min. Even though the product appeared as a highly viscous latex (Fig. 3b), the PDMAm-b-PBA-b-polystyrene products had low molar mass dispersity (1.20 ≤ Ð ≤ 1.25) (Fig. 3c, Table 1). The Mn as determined by gel permeation chromatography (GPC) was higher than the theoretical value (Eqn 1):

|

|
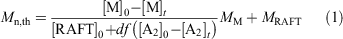
where M is the monomer, RAFT is the RAFT agent or macroRAFT agent, MM and MRAFT are the molar masses of the monomer and the RAFT agent, respectively, and the term df([A2]0 – [A2]t) is the concentration of polymer chains produced from the initiator (A2). This may indicate that the APS initiator or mediator is reacting directly with the macroRAFT agent but may also indicate imprecision in the macroRAFT concentration. In both conventional emulsion and RAFT emulsion polymerization, the initiator efficiency of persulfate is thought to be low (e.g. ~0.1),[37,38] and it is likely to be equally low in the present work. Tailing to low molar mass evident in the GPC traces (Fig. 3c) may indicate the presence of unreacted macroRAFT agent in the product but is more likely to be associated with initiator-derived chains. Dynamic light scattering (DLS) analysis shows that a monomodal particle size distribution, with Zaverage size in the range 90–120 nm and polydispersity index (PDI) ~0.2, is rapidly established and does not change substantially with polymerization time (see Supplementary Material).
Our preliminary results presented here demonstrate the potential for eRAFT emulsion polymerization to provide rapid monomer conversion and a low-dispersity product. The fact the product is a viscous mass rather than a free-flowing dispersion creates obvious limitations, particularly with respect to the desired implementation in continuous flow. Ongoing work aims to overcome this issue by optimizing the conditions for emulsion polymerization with the initial focus being the nature and concentration of the initiator components and the type of amphiphilic macroRAFT agent. Future work will also explore the application of eRAFT emulsion methodology to other monomers.
PhotoRAFT of Liquid Crystalline Monomers
Interest in SCLCPs first emerged ~40 years ago and came from a desire to combine the optical properties of liquid-crystalline materials with the mechanical properties of polymers.[39] SCLCPs show potential in various nanotechnology applications that include electro-optical materials, solar cells, and transistors.[40–42] Conventional radical polymerization can be used to prepare SCLCPs.[43–46] However, this process provides little control over molar mass dispersity (Đ) and the molecular uniformity that can be critical to many applications cannot be achieved. Methods for reversible deactivation radical polymerization (RDRP) such as atom-transfer radical polymerization (ATRP) and RAFT polymerization address this by allowing more precise control over molecular mass, microstructure, and architecture.[47–52]
In this report, we explore RAFT polymerization of 4-biphenyl acrylate. In initial experiments, a dialkyldiazene was used as the radical source. Many parameters such as the RAFT agent, solvent, initiator, monomer concentration, initiator concentration, and temperature were explored; these will not be detailed here. Conditions providing rapid polymerization, good monomer and polymer solubility, and low molar mass dispersity, made use of anisole solvent, 4-cyano-4-(((dodecylthio)carbonothioyl)thio)pentanoic acid (3) as the RAFT agent, and 2,2′-azobis(2,4-dimethylpentanenitrile) (ABPN) as initiator at 70°C (Scheme 3). These conditions provided 92 % conversion within 2 h and an apparently well-defined product with the number average molar mass obtained by GPC, Mn,GPC = 22900 and Đ = 1.06 (Table 2). However, a small shoulder was evident at higher molecular mass (Fig. 4).
|

|

|
High-molar-mass shoulders in molar mass distributions are frequently observed in high-conversion polymerization of acrylates and can be attributed to a variety of causes.[53,54] In that a high concentration of initiator had been used to ensure rapid reaction, we suspected that the shoulder might be largely attributable to dead chains formed by termination.
The fraction of living chains (L) in polymerizations with a dialkyldiazene initiator can be estimated using the expression in Eqn 2, which indicates an L value of ~0.9 for the polymerization conditions used:
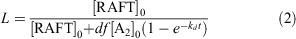
where [RAFT]0 is the initial RAFT agent concentrations and [A2]0 is the initial initiator concentration, kd is the rate coefficient for initiator decomposition (2.46 × 10−4 s−1 for ABPN at 70°C),[55] f is the initiator efficiency (assumed to be 0.7),[55] and d is the number of chains produced in radical–radical termination (1 < d < 2). Termination in polymerization of acrylates is believed to occur predominantly by combination, i.e. d = 1.0.[56]
To further understand the process, the polymerization was modelled by performing numerical simulation using PrediciTM. This enabled the molar mass distribution, including the high-molar-mass shoulder, to be reasonably predicted (Fig. 4a).
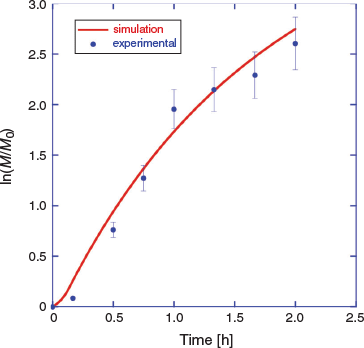
There is a short period of slow monomer conversion, attributable to initialization effects, and some curvature in the pseudo-first-order kinetic plot for longer times, associated with initiator depletion and the short half-life initiator used. These are also predicted by the PrediciTM simulation (Fig. 5).
|
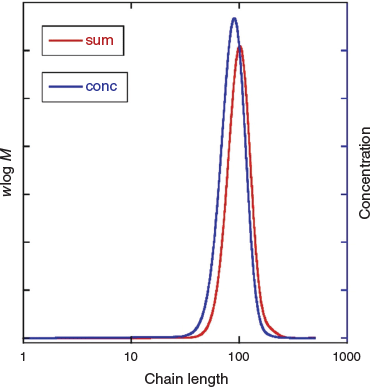
Although the high fraction of dead polymer is clearly an issue, a potentially bigger problem in the context of the intended applications of SCLCPs is the tail to lower molar mass. This tail is almost unobservable in the GPC distribution (Fig. 4a) but is immediately evident in the predicted concentration distribution (Fig. 4b). Note that for GPC distributions with refractive index detection, the intensity should be proportional to wlog M, where w is mass (weight) and M is molar mass.
Direct photoinitiation was then studied to establish if this would provide a cleaner product. Various solvents, solvent mixtures, and monomer concentrations were explored to establish optimal conditions (Table 3). Polymerizations in many solvents including DMSO that did not comprise a high proportion of anisole or DMF (e.g. DMSO, DMSO/anisole 1 : 1) were observed to form a viscous, translucent gel for monomer conversions >15 %. Interestingly, this did not seem detrimental to the breadth of the molar mass distribution or the rate of polymerization. Substantially broader molar mass distributions and lower-molar-mass product, perhaps indicative of transfer to solvent, were observed with DMF as solvent. The preferred polymerization conditions made use of anisole containing 10 % d6-DMSO to provide a lock signal facilitating in situ NMR analysis. Higher monomer (and RAFT agent) concentrations gave faster rates of polymerization. Pseudo-first-order kinetic plots and molar mass distribution for several systems are shown in Fig. 6 (50 : 50 and 90 : 10 DMSO/anisole solvent) and in Fig. S11 in the Supplementary Material (DMF, DMSO solvent).

|

|
We then modelled photoRAFT polymerization using PrediciTM (Figs 7–9). The kinetic model and the kinetic parameters used are described in detail in the Supplementary Material. We chose a rate of photodissociation of the RAFT agent to provide a similar conversion after 2 h to the thermally initiated RAFT polymerization and used the same kinetic parameters for propagation, termination, and RAFT as used in the simulation of the thermally initiated process. The concentration of propagating radicals (macroradicals) v. time is, however, quite different (Fig. 9a). As anticipated, for the case of photoinitiation the tail to low molar mass is absent. This is most apparent in the concentration distributions (compare Fig. 4b and Fig. 8) We still see a high-molar-mass shoulder, though it is substantially smaller than that observed in the thermally initiated polymerization at the same monomer conversion. It is important to note in designing the synthesis that the size of the shoulder increases with further irradiation (Fig. 7a). There is slight curvature for very high conversions as macroRAFT agent is lost through termination and the rate of initiation slows accordingly (Fig. 7b).

|
|

|
Interestingly, even though photoRAFT provides a higher-purity product (Fig. 9b), the yield of macroRAFT agent is higher in the thermally initiated experiment. With thermal initiation, the moles of dead polymer correspond to 0.5 times the moles of chains initiated, as suggested by Eqn 2. In the case of photoinitiation, for every mole of dead polymer formed, two moles of macroRAFT agent are lost and one mole of by-product (assumed to be the disulfide, (C12H25C(=S)S)2, in the kinetic model) is formed.
Nonetheless, photoRAFT is the preferred method of synthesis. To achieve highest purities, it is best not to attempt to achieve quantitative monomer conversion through prolonged irradiation. Moreover, the irradiation intensity should be as low as possible, consistent with achieving an acceptable rate of polymerization. The rate of termination, irrespective of the initiation mechanism, is proportional to the concentration of propagating radicals squared.
Conclusions
In this paper, we have provided two examples of the importance of initiation in dictating the outcome of RAFT polymerization.
Emulsion eRAFT polymerization gives rapid polymerization of styrene at ambient temperature to give low-Đ macroRAFT PS. By conducting eRAFT polymerization in emulsion, electrolytes and mediators required can be located only in the aqueous continuous phase, separate from the precursor amphiphilic macroRAFT agent and the forming macroRAFT agent product that is in the dispersed or particle phase. Use of the amphiphilic PDMAm-b-PBA-trithiocarbonate allowed ‘surfactant-free’ eRAFT polymerization in emulsion.
Direct photoinitiated RAFT polymerization of monomers with pendant mesogens (the example is 4-biphenyl acrylate) provides high-purity, low-dispersity, SCLCPs essentially free from low-molar-mass initiator-derived by-products and with minimal termination. Shortcomings in the sensitivity of conventional analysis based on GPC can be at least partially overcome through the use of numerical simulation.
Experimental
Details of materials, syntheses, and numerical simulation are provided in the Supplementary Material.
Supplementary Material
Experimental procedures and characterization details for electrochemically initiated RAFT polymerization in emulsion, high-throughput synthesis of PDMAm and PDMAm-b-PBA macroRAFT agents, high-throughput ‘surfactant-free’ emulsion polymerization using PDMAm-b-PBA macroRAFT agents, photoRAFT polymerization of liquid-crystalline monomers, and the numerical simulation of RAFT polymerization kinetics including details of the reaction scheme implemented in PrediciTM and the kinetic parameters used are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
JW thanks CSIRO Manufacturing for an Industrial Traineeship and her supervisor from Ecole Nationale Supérieure de Chimie de Rennes, Eric Le Fur. AP thanks CSIRO for a Julius Career Award. CB and LTS thank CSIRO for Research+ Postdoctoral Fellowships. CB thanks her supervisory team Richard Evans, Nino Malic, and George Simon for helpful discussion. We are grateful to Mike Horne and Bita Bayatsarmadi (CSIRO Minerals) for advice on electrochemistry, to Ben Muir, Shaun Howard, and Weidong Yang for assistance with high-throughput experimentation, to George Maurdev (CSIRO Manufacturing) for assistance with dynamic light scattering and Oliver Hutt and Simon Saubern (CSIRO Manufacturing) for helpful discussion.
References
[1] T.P. Le, G. Moad, E. Rizzardo, S.H. Thang, Polymerization with living characteristics, DuPont/CSIRO, 1998, Patent WO9801478A1.[2] P. Corpart, D. Charmot, T. Biadatti, S. Zard, D. Michelet, Block polymer synthesis by controlled radical polymerization, Rhodia Chimie, 1998, Patent WO9858974.
[3] J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo, S. H. Thang, Macromolecules 1998, 31, 5559.
| Crossref | GoogleScholarGoogle Scholar |
[4] M. Destarac, Polym. Chem. 2018, 9, 4947.
| Crossref | GoogleScholarGoogle Scholar |
[5] T. Otsu, M. Yoshida, Makromol. Chem., Rapid. Commun. 1982, 3, 127.
| Crossref | GoogleScholarGoogle Scholar |
[6] T. Otsu, M. Yoshida, A. Kuriyama, Polym. Bull. 1982, 7, 45.
| Crossref | GoogleScholarGoogle Scholar |
[7] T. Otsu, J. Polym. Sci. A Polym. Chem. 2000, 38, 2121.
| Crossref | GoogleScholarGoogle Scholar |
[8] J. F. Quinn, L. Barner, C. Barner-Kowollik, E. Rizzardo, T. P. Davis, Macromolecules 2002, 35, 7620.
| Crossref | GoogleScholarGoogle Scholar |
[9] C. P. Easterling, Y. Xia, J. Zhao, G. E. Fanucci, B. S. Sumerlin, ACS Macro Lett. 2019, 8, 1461.
| Crossref | GoogleScholarGoogle Scholar |
[10] J. Yeow, O. R. Sugita, C. Boyer, ACS Macro Lett. 2016, 5, 558.
| Crossref | GoogleScholarGoogle Scholar |
[11] T. G. McKenzie, Q. Fu, M. Uchiyama, K. Satoh, J. Xu, C. Boyer, M. Kamigaito, G. G. Qiao, Adv. Sci. 2016, 3, 1500394.
| Crossref | GoogleScholarGoogle Scholar |
[12] S. Shanmugam, J. Xu, C. Boyer, Macromol. Rapid Commun. 2017, 38, 1700143.
| Crossref | GoogleScholarGoogle Scholar | 28556363PubMed |
[13] J. Xu, S. Shanmugam, N. A. Corrigan, C. Boyer, in Controlled Radical Polymerization: Mechanisms (Eds K. Matyjaszewski, B. S. Sumerlin, N. V. Tsarevsky, J. Chiefari) 2015, Vol. 1187, pp. 247–267 (American Chemical Society: Washington, DC). [ACS Symp. Ser. 2015, 1187, 247.
[14] S. Li, G. Han, W. Zhang, Polym. Chem. 2020, 11, 1830.
| Crossref | GoogleScholarGoogle Scholar |
[15] M. D. Nothling, Q. Fu, A. Reyhani, S. Allison-Logan, K. Jung, J. Zhu, M. Kamigaito, C. Boyer, G. G. Qiao, Adv. Sci. 2020, 2001656.
| Crossref | GoogleScholarGoogle Scholar |
[16] A. Aerts, R. W. Lewis, Y. Zhou, N. Malic, G. Moad, A. Postma, Macromol. Rapid Commun. 2018, 39, 1800240.
| Crossref | GoogleScholarGoogle Scholar | 29900617PubMed |
[17] Y. Zhou, Z. Zhang, C. Reese, D. L. Patton, J. Xu, C. Boyer, A. Postma, G. Moad, Macromol. Rapid Commun. 2020, 41, 1900478.
| Crossref | GoogleScholarGoogle Scholar | 32297374PubMed |
[18] J. Xu, C. Fu, S. Shanmugam, C. J. Hawker, G. Moad, C. Boyer, Angew. Chem. Int. Ed. Engl. 2017, 56, 8376.
| Crossref | GoogleScholarGoogle Scholar | 27925363PubMed |
[19] Z. Huang, B. B. Noble, N. Corrigan, Y. Chu, K. Satoh, D. S. Thomas, C. J. Hawker, G. Moad, M. Kamigaito, M. L. Coote, C. Boyer, J. Xu, J. Am. Chem. Soc. 2018, 140, 13392.
| Crossref | GoogleScholarGoogle Scholar | 30230329PubMed |
[20] P. R. Judzewitsch, N. Corrigan, F. Trujillo, J. Xu, G. Moad, C. J. Hawker, E. H. H. Wong, C. Boyer, Macromolecules 2020, 53, 631.
| Crossref | GoogleScholarGoogle Scholar |
[21] N. Zaquen, M. Rubens, N. Corrigan, J. Xu, P. B. Zetterlund, C. Boyer, T. Junkers, Prog. Polym. Sci. 2020, 107, 101256.
| Crossref | GoogleScholarGoogle Scholar |
[22] Y. Wang, M. Fantin, S. Park, E. Gottlieb, L. Fu, K. Matyjaszewski, Macromolecules 2017, 50, 7872.
| Crossref | GoogleScholarGoogle Scholar | 29977098PubMed |
[23] Y. Wang, M. Fantin, K. Matyjaszewski, Macromol. Rapid Commun. 2018, 39, 1800221.
| Crossref | GoogleScholarGoogle Scholar | 30318665PubMed |
[24] F. Lorandi, M. Fantin, S. Shanmugam, Y. Wang, A. A. Isse, A. Gennaro, K. Matyjaszewski, Macromolecules 2019, 52, 1479.
| Crossref | GoogleScholarGoogle Scholar |
[25] L. T. Strover, A. Cantalice, J. Y. L. Lam, A. Postma, O. E. Hutt, M. D. Horne, G. Moad, ACS Macro Lett. 2019, 8, 1316.
| Crossref | GoogleScholarGoogle Scholar |
[26] W. Sang, M. Xu, Q. Yan, ACS Macro Lett. 2017, 6, 1337.
| Crossref | GoogleScholarGoogle Scholar |
[27] J. Bünsow, M. Mänz, P. Vana, D. Johannsmann, Macromol. Chem. Phys. 2010, 211, 761.
| Crossref | GoogleScholarGoogle Scholar |
[28] C. J. Ferguson, R. J. Hughes, B. T. T. Pham, B. S. Hawkett, R. G. Gilbert, A. K. Serelis, C. H. Such, Macromolecules 2002, 35, 9243.
| Crossref | GoogleScholarGoogle Scholar |
[29] C. J. Ferguson, R. J. Hughes, D. Nguyen, B. T. T. Pham, R. G. Gilbert, A. K. Serelis, C. H. Such, B. S. Hawkett, Macromolecules 2005, 38, 2191.
| Crossref | GoogleScholarGoogle Scholar |
[30] J. Zhou, H. Yao, J. Ma, Polym. Chem. 2018, 9, 2532.
| Crossref | GoogleScholarGoogle Scholar |
[31] S. J. Stace, J. Vanderspikken, S. C. Howard, G. Li, B. W. Muir, C. M. Fellows, D. J. Keddie, G. Moad, Polym. Chem. 2019, 10, 5044.
| Crossref | GoogleScholarGoogle Scholar |
[32] G. K. K. Clothier, T. R. Guimarães, M. Khan, G. Moad, S. Perrier, P. B. Zetterlund, ACS Macro Lett. 2019, 8, 989.
| Crossref | GoogleScholarGoogle Scholar |
[33] R. A. E. Richardson, T. R. Guimarães, M. Khan, G. Moad, P. B. Zetterlund, S. Perrier, Macromolecules 2020, 53, 7672.
| Crossref | GoogleScholarGoogle Scholar |
[34] J. Rieger, W. Zhang, F. o. Stoffelbach, B. Charleux, Macromolecules 2010, 43, 6302.
| Crossref | GoogleScholarGoogle Scholar |
[35] G. Kojima, M. Hisasue, Makromol. Chem. 1981, 182, 1429.
| Crossref | GoogleScholarGoogle Scholar |
[36] S. Lee, A. Rudin, J. Polym. Sci., Part A, Polym. Chem. 1992, 30, 2211.
| Crossref | GoogleScholarGoogle Scholar |
[37] K. Y. van Berkel, G. T. Russell, R. G. Gilbert, Polymer 2006, 47, 4667.
| Crossref | GoogleScholarGoogle Scholar |
[38] H. A. S. Schoonbrood, A. L. German, R. G. Gilbert, Macromolecules 1995, 28, 34.
| Crossref | GoogleScholarGoogle Scholar |
[39] T. Ganicz, W. Stańczyk, Materials 2009, 2, 95.
| Crossref | GoogleScholarGoogle Scholar |
[40] H. Khandelwal, A. P. H. J. Schenning, M. G. Debije, Adv. Energy Mater. 2017, 7, 1602209.
| Crossref | GoogleScholarGoogle Scholar |
[41] C. S. Lee, T. A. Kumar, J. H. Kim, J. H. Lee, J. S. Gwag, G.-D. Lee, S. H. Lee, J. Mater. Chem. C 2018, 6, 4243.
| Crossref | GoogleScholarGoogle Scholar |
[42] C.-S. Hsu, Prog. Polym. Sci. 1997, 22, 829.
| Crossref | GoogleScholarGoogle Scholar |
[43] H. Finkelmann, H. Ringsdorf, J. H. Wendorff, Makromol. Chem. 1978, 179, 273.
| Crossref | GoogleScholarGoogle Scholar |
[44] B. Peng, D. Johannsmann, J. Rühe, Macromolecules 1999, 32, 6759.
| Crossref | GoogleScholarGoogle Scholar |
[45] G. O. R. Alberda van Ekenstein, H. J. H. Altena, Y. Y. Tan, Eur. Polym. J. 1989, 25, 111.
| Crossref | GoogleScholarGoogle Scholar |
[46] D. Wu, B. Ni, Y. Liu, S. Chen, H. Zhang, J. Mater. Chem. A 2015, 3, 9645.
| Crossref | GoogleScholarGoogle Scholar |
[47] X.-Z. Wang, H.-L. Zhang, D.-C. Shi, J.-F. Chen, X.-Y. Wang, Q.-F. Zhou, Eur. Polym. J. 2005, 41, 933.
| Crossref | GoogleScholarGoogle Scholar |
[48] L. Angiolini, T. Benelli, L. Giorgini, F. Paris, E. Salatelli, M. P. Fontana, P. Camorani, Eur. Polym. J. 2008, 44, 3231.
| Crossref | GoogleScholarGoogle Scholar |
[49] Y. Zhu, Y. Zhou, Z. Chen, R. Lin, X. Wang, Polymer 2012, 53, 3566.
| Crossref | GoogleScholarGoogle Scholar |
[50] X. Zhang, S. Boisse, C. Bui, P.-A. Albouy, A. Brulet, M.-H. Li, J. Rieger, B. Charleux, Soft Matter 2012, 8, 1130.
| Crossref | GoogleScholarGoogle Scholar |
[51] N. I. Boiko, M. A. Bugakov, E. V. Chernikova, A. A. Piryazev, Y. I. Odarchenko, D. A. Ivanov, V. P. Shibaev, Polym. Chem. 2015, 6, 6358.
| Crossref | GoogleScholarGoogle Scholar |
[52] W. Wen, T. Huang, S. Guan, Y. Zhao, A. Chen, Macromolecules 2019, 52, 2956.
| Crossref | GoogleScholarGoogle Scholar |
[53] A. Postma, T. P. Davis, G. Li, G. Moad, M. O’Shea, Macromolecules 2006, 39, 5307.
| Crossref | GoogleScholarGoogle Scholar |
[54] Q. Yang, M. Guerre, V. Ladmiral, B. Ameduri, Polym. Chem. 2018, 9, 3388.
| Crossref | GoogleScholarGoogle Scholar |
[55] G. Moad, Prog. Polym. Sci. 2019, 88, 130.
| Crossref | GoogleScholarGoogle Scholar |
[56] T. G. Ribelli, K. F. Augustine, M. Fantin, P. Krys, R. Poli, K. Matyjaszewski, Macromolecules 2017, 50, 7920.
| Crossref | GoogleScholarGoogle Scholar |
* Dr Graeme Moad is the recipient of the Australian Academy of Science’s 2020 David Craig Medal.