

On-Water Reactivity and Regioselectivity of Quinones in C–N Coupling with Amines: Experimental and Theoretical Study
Maximiliano Martínez-Cifuentes A C , Graciela Clavijo-Allancan A , Carolina Di Vaggio-Conejeros A , Boris Weiss-López B and Ramiro Araya-Maturana A CA Departamento de Química Orgánica y Fisicoquímica, Facultad de Ciencias Químicas Y Farmacéuticas, Universidad de Chile, Casilla 233, Santiago 1, Chile.
B Departamento de Química, Facultad de Ciencias, Universidad de Chile, Casilla 653, Santiago, Chile.
C Corresponding authors. Email: mmartinez@ug.uchile.cl; raraya@ciq.uchile.cl
Australian Journal of Chemistry 67(2) 217-224 https://doi.org/10.1071/CH13355
Submitted: 6 July 2013 Accepted: 10 September 2013 Published: 11 October 2013
Abstract
A study about the oxidative coupling of some representative carbo- and heterocyclic non-symmetrical quinones with aryl- and alkylamines, was carried out comparing dichloromethane and water as reaction mediums. We found that the on-water reactions gave better or, at worst, the same results as a conventional organic medium like dichloromethane. Descriptors derived from conceptual density functional theory and approaches of electrostatic nature, such as the molecular electrostatic potential, were used to explain the observed chemical reactivity and regioselectivity. Further, the on-water conditions were used to obtain 24 new aminoquinones with potential biological activity.
Introduction
Quinone derivatives are considered a privileged skeleton in medicinal chemistry as anticancer,[1] antifungal,[2,3] and anti-parasitic[4] drugs. Quinones also have relevance in industrial applications such as dyes,[5] and in the biodegradation of priority pollutants.[6] Among quinones, aminoquinone derivatives are remarkable, based on the potential biological applications they posses. The aminoquinone motif[7] is a component of the molecular framework of numerous natural products, such as kinamycins,[8] caulibugulones,[9] griffithazanone A,[10] cyclosmenospongine,[11] and others, and they have been used as reactive intermediates for the synthesis of many biologically relevant compounds.[12,13]
The synthesis of aminoquinones has been achieved, mainly, through oxidative coupling of amines with quinones.[14] Significant attention has been devoted to improve the efficiency of this reaction using Lewis acids like Ce(iii),[15] Cu(ii),[16] or Au(iii)[17] salts. Nevertheless, the use of heavy metals and volatile organic compounds (VOCs) as solvents present serious environmental impact, and reducing their use is a central issue in green chemistry.[18] In view of the latter, new methodologies without catalysts, using water as reaction medium have been developed in recent years.[19,20] In addition to its positive environmental properties, water presents unique reactivity and selectivity that cannot be achieved by conventional organic solvents.[21,22]
On the other hand, several descriptors derived from conceptual density functional theory (DFT)[23] and approaches of pure electrostatic nature, such as molecular electrostatic potentials,[24] have been used to assess chemical reactivity.
In this paper we report on the reactions between 1,4-quinone derivatives: 2-methyl-1,4-benzoquinone I, 8,8-dimethylnaphthalene-l,4,5(8H)-trione II, 1,3-dimethyl-5,8-dioxo-5,8-dihydroisoquinoline-4-carboxylic acid methyl ester III (used as quinone-model compounds), with N-phenylpiperazine IVa and aniline Va (used as amine-model compounds). Additionally, several derivatives of IVa and Va with different substituents were investigated to measure the scope of this reaction (Fig. 1).
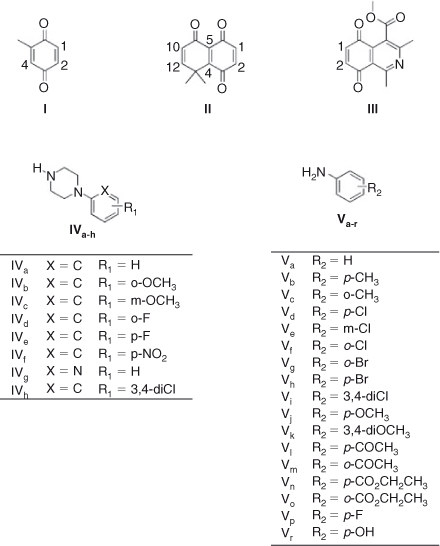
|
Quinones I and II have been used before to obtain bioactive natural and synthetics products.[25,26] Also, isoquinoline quinone III has been used as a substrate in oxidative coupling reactions with amines to produce anti-tumour quinones.[27]
The two goals of this work were: first, to contribute to the understanding of how water affects the reactivity and selectivity of quinones in C–N coupling reactions with amines, and second, to study a non-catalytic eco-friendly methodology, for the regioselective oxidative coupling reaction of amines with quinones. The results are explained using DFT calculations. Global and local reactivity indexes and molecular electrostatic potential (MEP) are used to discuss the changes in reactivity and regioselectivity observed experimentally.
Theoretical Background
The energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is indicative of the chemical reactivity of a molecule. Using Koopman’s theorem for closed-shell compounds, the hardness, η, and the electronic chemical potential, μ, can be defined as η = (I –A)/2 and μ = –(I+A)/2, where I and A are the ionization potential and electron affinity respectively. Parr et al.[28] have defined a descriptor to quantify the global electrophilic power as an electrophilicity index (ω): ω = μ2/2η.
Global reactivity indexes such as μ and η have been approximated in terms of the one electron energies of the frontier molecular orbitals HOMO and LUMO, ϵH and ϵL, using the expression μ ≈ (ϵH+ϵL)/2 and η ≈ (ϵL–ϵH), from the ground state of the molecules.[29] The local electrophilicity,[30] ωk, condensed to atom k, was obtained by projecting the global quantity onto the atomic centre k in the molecule, by using the condensed electrophilic Fukui function,[31] fk+ according to ωk = ω fk+.
In addition to the electrophilicity index, several other approaches have been proposed for the quantitative assessment of nucleophilicity. Since electrophilicity and nucleophilicity are inversely proportional, it has been suggested that nucleophilicity, N, can be considered as the multiplicative inverse of the electrophilicity index (Eqn 1).[32] Recently, new equations have been proposed for nucleophilicity. One of them uses the HOMO energy of the nucleophile and calculates the difference with respect to the HOMO of tetracyanoethylene (TCE) (Eqn 2). TCE is used as reference because it presents the lowest HOMO among a great number of molecules.[33] Other methods to calculate the nucleophilicity based on the electrodonating power concept (ω-)[34] have defined the nucleophilicity as the inverse of electrodonating power (ω-) and they have proposed two equations to calculate it (Eqns 3 and 4), describing the latter as the best[34b]:

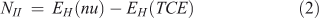

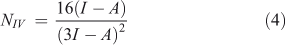
where the ionization potential (I) was approximated to EHOMO and the electroaffinity (A) was approximated to ELUMO.
On the other hand, the molecular electrostatic potential (MEP) is related to the electron density and also is a very useful descriptor of reactivity for electrophilicity and nucleophilicity.[35,36] To visualize the reactive sites of the studied quinones, we calculated their MEP.
The efficiency of functionals employing different basis sets, in predicting electronic properties including reactivity indexes of some organic molecules has been systematically evaluated. There being no significant change between the different levels of theory,[37] our calculations were carried out at the B3LYP/6–31G(d,p) level of theory.
Results and Discussion
The addition of aniline to quinone I, through an in-water, on-water domino process starting from toluhydroquinone has been reported. In this work, only the mono-addition product was obtained.[38] In contrast, the products obtained from the reaction of quinone I and p-benzoquinone with a series of anilines in the two previous methods were only bis-addition products.[39] Thus, we carried out the reaction of quinone I and II with aniline (Va) and additionally tested the reaction with 1-phenylpiperazine (IVa), commonly found within molecules with biological activities.[40]
The reaction between I and IVa in equimolar ratio, using dichloromethane as solvent, yielded two regioisomers, VI and VII in 60 : 40 ratio, and 67 % yield. A modification of the reaction conditions, using water as reaction milieu, changes the results only slightly, obtaining a 55 : 45 ratio and 66 % yield. Surprisingly, when aniline was used as nucleophile in dichloromethane no reaction was observed, but when water was used as solvent, three products were obtained X,VIII, and XI, in a ratio of 70 : 20 : 10 and 87 % overall yield, taking into account that 50 % of the starting quinone acts as an oxidant of the amine hydroquinone intermediates of the reaction. Considering a stepwise reaction for the formation of the bis addition products, we can postulate a regioselectivity of near 90 % for the first addition (Scheme 1; Tables 1 and 2).

|

|

|
When 8,8-dimethyl-8H-naphthalen-1,4,5-trione II, a more electrophilic quinone, reacts with both amines, a different behaviour is observed. The reaction of quinone II with phenylpiperazine in dichloromethane as solvent generates only the single regioisomer XII in 73 % yield. When the reaction is carried out on-water the regioselectivity remains unaltered, and the yield decreases slightly. Using aniline a rather different behaviour is observed; when dichloromethane is used as solvent a mixture of regioisomers XIV and XV is obtained, in ratio 70 : 30 and 40 % total yield, lower compared with that obtained with phenylpiperazine (Scheme 2; Tables 3 and 4) .
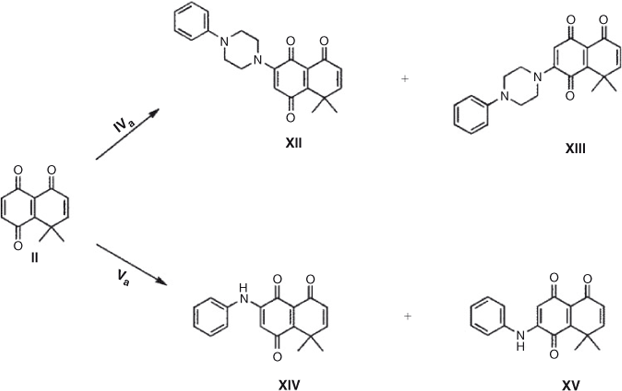
|

|

|
On the other hand, the totally regio-controlled reactions of III with aniline derivatives have been reported using CeCl3·7H2O in ethanol at room temperature, to give an aminoquinone with anti-tumor activity.[27] To test for this reaction in water, we chose some previously used aniline derivatives. The products of this reaction were also obtained with a totally regiocontrolled outcome. Although with slightly lower yields, this simple change of conditions ensured products free of potentially toxic metals, such as cerium(iii) (Scheme 3; Table 5).
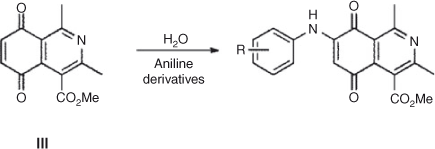
|
|
Analysis of Global and Local Properties
We calculated the global and local electrophilicities of quinones I, II, III, IX, and X, and the global nucleophilicity of amines IVa and Va. Also, to study solvation effects in the reactivity of these molecules, we calculated their complexes with water molecules. Different methodologies have been used to include water solvation in theoretical studies of reactivity, either in implicit and/or explicit forms. Recently, five different methods to study the inclusion of solvent effects on reactivity properties have been reported: 1) polarizable continuum model (PCM), an implicit form; 2) using 499 point charges (PC), an explicit form, 3) using 2 explicit water molecules and 497 point charges representing the same number of water molecules, 4) 40 explicit water molecules and 459 point charges, and 5) 2 explicit water molecules in vacuum; it was found that only the continuum model (PCM) was incapable of obtaining a significant change in the energies of the HOMO and LUMO orbitals, with respect to the values obtained in gas phase. Furthermore, the differences in the values of the reactivity indexes among the explicit methodologies are minimal. Therefore, the coordination of each basic centre in a studied molecule with a water molecule in vacuum is a reasonable choice to obtain reliable results for reactivity indexes.[41]
The global and local electrophilicities of quinone I and the corresponding water complexes are presented in Table 6. We found that global electrophilicity of this quinone (ω = 3.67) increased their value remarkably when solvated with water (ω = 4.36), which explains the differences observed when the reaction with aniline was carried out on-water instead of dichloromethane. In both cases, solvated or not with water, local electrophilicity showed the same correlation, with C(2) being the most electrophilic carbon (ωk = 0.40 not solvated, and ωk = 0.47 solvated) and C(4) being the least (ωk = 0.30 not solvated, and ωk = 0.32 solvated). This result is in agreement with the experimental products ratio (Chart 1).

|

|
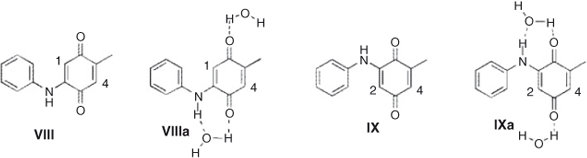
Since the reaction of quinone I with aniline gave a second addition product, we also calculated the properties of the mono addition product and its water complexes. First, the potential energy surface along dihedrals α and β were carried out for molecules VIII and IX. The minimum energy conformations of both molecules are presented in Fig. 2.

|
We found that water solvation also increased the reactivity of mono addition products. The calculated local electrophilicity of both VIII and IX showed that position 4 was largely more reactive than 1 and 2, which explains why only X and XI were obtained as products (Chart 2; Table 7).
|

|
The interaction of water molecules with quinone II has been considered in two different coordination modes: two water molecules, one with carbonyls 1 and 2, and the other with carbonyl 3 (mode a, Chart 3), or three water molecules, one at each carbonyl group, 1, 2, and 3 (mode b). Depending on the coordination mode with water, the global electrophilicity showed different behaviour. Unsolvated quinone shows a global electrophilicity of 4.43 (Table 8).

|

|
Local electrophilicity showed that C(1) (ωk = 0.45) is more reactive than C(2) (ωk = 0.30), which agrees with the experimental regioselectivity in dichloromethane. C(4) and C(5) also have a high electrophilicity; nevertheless it has been proposed that the gem-dimethyl group generates a steric effect which blocks the reactivity of these carbons for Diels–Alder reactions.[42] This is in accordance with the results for oxidative addition reactions.
In coordination mode a, the global electrophilicity of the solvated quinone was slightly reduced, from 4.43 to 4.33, and the local electrophilicity on C(1) and C(2) showed a larger difference (0.44 vs 0.22) than in the non-solvated case (0.45 vs 0.30). On the other hand, in coordination mode b, the global electrophilicity increased their value from 4.43 to 4.87, and the local electrophilicity difference of C(1) and C(2) remains the same, 0.51 and 0.36. The results from coordination mode a are in agreement with the increase in the observed regioselectivity when the reaction with aniline was performed in water instead of dichloromethane. On the other hand, coordination mode b correctly predicts the reactivity increase when the reaction was performed in water. The coordination energies of modes a and b are very similar (ΔE = 0.12 kcal mol–1; 1 kcal mol–1 = 4.186 kJ mol–1); in this way, both modes contribute to explain the observed increase in reactivity and regioselectivity (Chart 3).
Reactivity indices of isoquinoline quinone III and the complex with two water molecules were also calculated. We found that water solvation increases the electrophilicity of the molecule from 3.78 to 4.33. The local electrophilicity difference between carbon 8 and 10 also increase slightly. These results are in agreement with the experimental observations, which showed that the effect of water was almost equivalent to use of Ce(iii) salts as catalyst in ethanol, as previously reported (Chart 4; Table 9).[27]

|

|
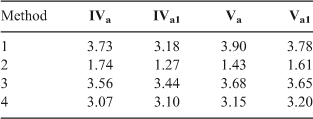
We also calculated the nucleophilicity of amines IVa and Va and their water complexes IVa1 and Va1, through methods 1 to 4 mentioned in the theoretical background section (Table 10). From this data we observe that only method 2 reproduces correctly the reactivity increase of aniline IV when solvated with water; it also predicts correctly that phenylpiperazine V is more reactive than aniline IV without the water solvation, which agrees with the experimental behaviour observed in dichloromethane.
|
Molecular Electrostatic Potential
To generate the MEP the colour-coded values were projected onto the 0.02 a.u. iso-potential energy surface. The red colour indicates negative sites of the molecule, showing possible positions for electrophilic attack, while the blue colour indicates positive sites, potentially suitable for nucleophilic attacks. We calculated MEP for quinones I, II, III, VIII, IX and their water complexes. As expected, quinone I showed two electron-rich sites on the oxygens and three electron-deficient sites on C(1), C(2), and C(4). The MEP of complex Ia showed that C(1) and C(2) increased their electron-deficient character, while in C(4) it almost disappears. The MEP of aminoquinones IX and X and their water complexes showed that C(4) is more electron-deficient than C(1), which is in agreement with the local electrophilicities calculated before and also with the experimental results (Fig. 3).

|
Quinone II showed three electron-rich sites on the carbonyl oxygen atoms. The two neighbouring carbonyl oxygens generate a strong negative region around that side of the molecule. On the other hand, the carbonyl oxygen closest to the gem-dimethyl group exhibited a less electron-rich site; besides it is distorted due to steric effects of the gem-dimethyl moiety. This molecule also showed electron-deficient sites on C(1) and C(2). Complex IIa showed that the electron density of the water molecule coordinated to oxygen at the carbonyl between C(2) and C(4) is blocking C(2). This stereo-electronic hindrance, along with the variation of the local electrophilicity in C(1) and C(2), can explain the increase in regioselectivity of the reaction with aniline. The complex IIb showed an electron density distribution similar to II(Fig. 4).

|
Isoquinoline quinone III exhibited four electron-deficient sites, localized on two carbonyl carbons, C(1) and C(2), being these last positions where oxidative addition can occur. The water complex, IIIa, showed an increase in the electron-deficient character of C(1), but it disappears from C(2) due to the hindrance induced by coordination of water with the neighbouring carbonyl. These results showed that both the local reactivity difference and the stereo-electronic effects are involved in the high regioselectivity at C(1) displayed by the reaction (Fig. 5).
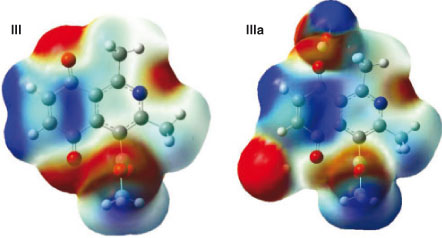
|
Use of On-Water Methodology to Obtain Substituted Aminoquinones Derived from Quinone II
Quinone II was used as a scaffold to produce biologically active compounds. Because of the remarkable results, the on-water conditions were used to produce a series of aniline and N-phenylpiperazine derivatives. In order to evaluate the effect of the electronic nature of the substituents on the reactivity and regioselectivity of these reactions, they were carried out with a series of substituted amines (Chart 5; Table 11).

|

|
The results show that the on-water reaction tolerated the different nature of the substituents on the aromatic ring of the amine. Better yields are obtained with the stronger electron-donor substituents, which increased the nucleophilicity of the aromatic amines. However, strong electron-releasing groups also gave the corresponding products, although with lower yields. For phenylpiperazines, the substituent did not directly affect the nucleophilicity of the nitrogen, but differences were also detected with the modification of that substituent.
Conclusions
In summary, we found that water can increase both reactivity and regioselectivity in the C–N coupling of amines with quinones. For the cases of this study, the reactions on-water gave better or, at worst, the same results as a conventional organic medium, like dichloromethane. The global and local electrophilicities of quinones were in agreement with the experimental reactivity. Nucleophilicity of amines were calculated using four different methods, where only the one based on the electrodonating power provided results according to the experimental reactivity. MEP of the quinones allowed us to visualize changes on electron density generated by water solvation, which helps to explain changes of reactivity with the same tendency of electrophilicity calculations. MEP also showed that stereo-electronic hindrance was an important factor to explain certain results. Further, we extended the on-water reaction to aniline and N-phenylpiperazine derivatives with quinone II with remarkable results, regardless of the nature of the amine substituents. The results reported here may motivate similar studies on a wider range of organic reactions, where green media not only give results similar to conventional ones but also improved yields and selectivities.
Synthetic General Procedure
The reaction of quinone I with amine IVa was carried out using condition B. The reactions of quinones I and II with amines IVa and Va were carried out using conditions A and B, both described below. The reactions of quinone II with amines IVb to IVg and Vb to Vn were carried out using conditions B. The synthesis of compounds II,[43] VIII-IX, XI,[44] and XVI-XIX[27] have been previously described.
-
Dissolving reactants in dichloromethane and stirring at room temperature overnight.
-
Using water as the reaction medium and stirring at room temperature overnight.
Computational Details
Calculations were carried out using DFT with the B3LYP functional,[45,46] together with the standard 6–31G(d,p) basis set. No imaginary frequencies were found at the optimized molecular geometries, which indicate that they are real minima of the potential energy surface. All calculations were carried out using the Gaussian 03[47] program package, running in a Microsystem cluster of blades. Calculations of global and local DFT-defined chemical reactivity descriptors were carried out using Fukui2 program.[48]
Supplementary Material
1H-RMN, 13C-RMN, IR, HRMS data and melting points for new compounds, as well as Cartesian coordinates and energies for the optimized structure calculated are available on the Journal’s website.
Acknowledgements
This work was supported by FONDECYT grant 1110176, and Anillo ACT-1107. M.M-C thanks CONICYT for PhD fellowship No. 21090023 and Beca Chile No. 75120034.
References
[1] P. R. Dandawate, A. C. Vyas, S. B. Padhye, M. W. Singh, J. B. Baruah, Mini Rev. Med. Chem. 2010, 10, 436.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXosFCntb8%3D&md5=9ca8f457be283808a2a071b689677a04CAS | 20370705PubMed |
[2] M. M. M. Santos, N. Faria, J. Iley, S. J. Coles, M. B. Hursthouse, M. L. Martins, R. Moreira, Bioorg. Med. Chem. Lett. 2010, 20, 193.
| Crossref | GoogleScholarGoogle Scholar |
[3] L. Mendoza, R. Araya-Maturana, W. Cardona, T. Delgado-Castro, C. García, C. Lagos, M. Cotoras, J. Agric. Food Chem. 2005, 53, 10080.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1Gju73O&md5=1548e1eba309ceffb26e4c8a7d87ae71CAS | 16366698PubMed |
[4] E. N. da Silva, R. F. S. Menna-Barreto, M. C. F. R. Pinto, R. S. F. Silva, D. V. Teixeira, M. C. B. V. de Souza, C. A. De Simone, S. L. De Castro, V. F. Ferreira, A. V. Pinto, Eur. J. Med. Chem. 2008, 43, 1774.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXptlOlsrY%3D&md5=8b333f94ee3185f53b3110d6c17a7282CAS | 18045742PubMed |
[5] E. S. B. Ferreira, A. N. Hulme, H. McNab, A. Quye, Chem. Soc. Rev. 2004, 33, 329.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmtVWgtbc%3D&md5=7edc723f662e22bff8690b451783efefCAS |
[6] F. J. Cervantes, T. Duong-Dag, A. D. L. Akkermans, G. Lettinga, J. A. Field, Water Sci. Technol. 2003, 48, 9.
| 1:CAS:528:DC%2BD3sXpvFemsrg%3D&md5=039405759f2889d89e489ff1321ec22fCAS | 14640194PubMed |
[7] M. Arshadi, M. Ghiaci, Appl. Catal. A Gen. 2011, 399, 75.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXmtVOqu7Y%3D&md5=3bb3b74d0068de412a845ea9002fb904CAS |
[8] S. Omura, A. Nakagawa, H. Yamada, T. Hata, A. Furusaki, T. Watanabe, Chem. Pharm. Bull. (Tokyo) 1973, 21, 931.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3sXkslyms78%3D&md5=3909fe44de4b42ab5b02c81f846ffa40CAS | 4727361PubMed |
[9] D. J. Milanowski, K. R. Gustafson, J. A. Kelley, J. B. McMahon, J. Nat. Prod. 2004, 67, 70.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXmtVak&md5=94d9e4926f63ebd0ca11f9c184acfc0fCAS | 14738389PubMed |
[10] Y. J. Zhang, M. Kong, R. Y. Chen, D. Q. Yu, J. Nat. Prod. 1999, 62, 1050.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjvFGhurY%3D&md5=d54d9a55cff2a543b2da168b25b800b4CAS | 10425141PubMed |
[11] N. K. Utkina, V. A. Denisenko, O. V. Scholokova, M. V. Virovaya, N. G. Prokofèva, Tetrahedron Lett. 2003, 44, 101.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XpsVensrY%3D&md5=f808a68e88685122990a36a0790f2b1aCAS |
[12] Y. Kita, H. Tohma, M. Inagaki, K. Hatanaka, T. Yakura, J. Am. Chem. Soc. 1992, 114, 2175.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38Xht12gt7w%3D&md5=a903436c6a4a09e270f814587ca8463aCAS |
[13] C.-P. Chuang, A.-I. Tsai, Tetrahedron 2007, 63, 11911.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1SmtrnK&md5=3234f02ab33846385d9fd508ed1a3108CAS |
[14] (a) J. W. Macleod, R. H. Thomson, J. Org. Chem. 1960, 25, 36.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF3cXotVGguw%3D%3D&md5=0308583991ad13589e4de9ca4ec8a1e0CAS |
(b) C. A. Panetta, P. W. J. Fan, R. Fattah, J. C. Greever, Z. L. He, C. L. Hussey, D. Z. Sha, L. D. Wescott, J. Org. Chem. 1999, 64, 2919.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. K. Machocho, T. Win, S. Grinberg, S. Bittner, Tetrahedron Lett. 2003, 44, 5531.
| Crossref | GoogleScholarGoogle Scholar |
[15] (a) Y. T. J. Pratt, J. Org. Chem. 1962, 27, 3905.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF3sXjt1ah&md5=760c2469ce6de6ded1099dcf7d118a2fCAS |
(b) M. Aguilar-Martinez, G. Cuevas, M. Jimenez-Estrada, I. Gonzalez, B. Lotina-Hennsen, N. Macias-Ruvalcaba, J. Org. Chem. 1999, 64, 3684.
| Crossref | GoogleScholarGoogle Scholar |
[16] C. S. Lisboa, V. G. Santos, B. G. Vaz, N. C. de Lucas, M. N. Eberlin, S. J. Garde, J. Org. Chem. 2011, 76, 5264.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXnt1ansrc%3D&md5=b71369f33d45e9d87372c470b5d9eabaCAS |
[17] C. H. Jiang, S. Z. Wang, Synlett 2009, 1099.
| 1:CAS:528:DC%2BD1MXmtF2msLk%3D&md5=ed45de9978cec72e3c3d24baede66af1CAS |
[18] P. Tundo, A. Perosa, F. Zecchini, Methods and Reagents for Green Chemistry: An Introduction 2007 (Wiley-VCH: Weinhein).
[19] J. S. Yadav, B. V. S. Reddy, T. Swamy, K. S. Shankar, Monatsh. Chem. 2008, 139, 1317.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlais7fK&md5=579ed73ce6f1add13ab63a5e36b66674CAS |
[20] P. Norcott, C. Spielman, C. S. P. McErlean, Green Chem. 2012, 14, 605.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XjtFymsbY%3D&md5=83d24a32142e8b5560e39e9082006a1eCAS |
[21] (a) N. Azizi, M. S. Saidi, Org. Lett. 2005, 7, 3649.
| 1:CAS:528:DC%2BD2MXmsFGgtbg%3D&md5=3a3e52c0deed093e03ad21a1482de61dCAS | 16092841PubMed |
(b) N. Azizi, F. Aryanasab, L. Torkiyan, A. Ziyaei, M. R. Saidi, J. Org. Chem. 2006, 71, 3634.
| Crossref | GoogleScholarGoogle Scholar |
[22] (a) G. L. Khatik, R. Kumar, A. K. Chakraborti, Org. Lett. 2006, 8, 2433.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xkt1Ogsbc%3D&md5=7e37a97572cf630f9374fc8cd32815efCAS | 16706544PubMed |
(b) J. S. Yadav, T. Swamy, B. V. S. Reddy, R. Krishna, J. Mol. Catal. Chem. 2007, 274, 116.
| Crossref | GoogleScholarGoogle Scholar |
[23] (a) P. K. Chattaraj, S. Giri, S. Duley, Chem. Rev. 2011, 111, PR43.
| Crossref | GoogleScholarGoogle Scholar | 21306180PubMed |
(b) S. Pan, M. Sola, P. K. Chattaraj, J. Phys. Chem. A 2013, 117, 1843.
| Crossref | GoogleScholarGoogle Scholar |
[24] (a) P. Politzer, J. S. Murray, Theor. Chem. Acc. 2002, 108, 134.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XnsFSgs7g%3D&md5=98c16fc59e946cea31f6bb4ae1be5358CAS |
(b) O. E. Kasende, J. T. Muya, L. Broeckaert, G. Maes, P. Geerlings, J. Phys. Chem. A 2012, 116, 8608.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. Mathew, C. H. Suresh, Organometallics 2011, 30, 1438.
| Crossref | GoogleScholarGoogle Scholar |
[25] (a) M. Yogo, C. Ito, H. Furukawa, Chem. Pharm. Bull. (Tokyo) 1991, 39, 328.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXkslWqt70%3D&md5=1be173b19927fb1cad86ca7e530489cbCAS |
(b) H. J. Knölker, K. R. Reddy, Heterocycles 2003, 60, 1049.
| Crossref | GoogleScholarGoogle Scholar |
(c) P. H. Bernardo, C. L. L. Chai, M. Le Guen, G. D. Smith, P. Waring, Bioorg. Med. Chem. Lett. 2007, 17, 82.
| Crossref | GoogleScholarGoogle Scholar |
[26] (a) R. Araya-Maturana, W. Cardona, B. K. Cassels, T. Delgado-Castro, J. Ferreira, D. Miranda, M. Pavani, H. Pessoa-Mahana, J. Soto-Delgado, B. Weiss-López, Bioorg. Med. Chem. 2006, 14, 4664.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XltVGqtbg%3D&md5=09cee77e53e8529be3b33749ad50ce8dCAS | 16504517PubMed |
(b) L. Mendoza, R. Araya-Maturana, W. Cardona, T. Delgado-Castro, C. García, C. Lagos, M. Cotoras, J. Agric. Food Chem. 2005, 53, 10080.
| Crossref | GoogleScholarGoogle Scholar |
[27] J. A. Valderrama, J. A. Ibacache, V. Arancibia, J. Rodriguez, C. Theoduloz, Bioorg. Med. Chem. 2009, 17, 2894.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjvV2hsL8%3D&md5=1fd867a984026e91a8a08e4346ae85d4CAS | 19269832PubMed |
[28] R. G. Parr, L. V. Szentpaly, S. J. Liu, J. Am. Chem. Soc. 1999, 121, 1922.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXhtFagt7o%3D&md5=47231e31f76563fd22dc1288a85c7e38CAS |
[29] R. G. Parr, W. Yang, Density Functional Theory of Atoms and Molecules 1989 (Oxford University Press: New York, NY).
[30] L. R. Domingo, M. J. Aurell, P. Pérez, R. Contreras, Tetrahedron 2002, 58, 4417.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xjsl2itr8%3D&md5=42430ac1ae7a6b830bc19167e550f9ebCAS |
[31] R. Contreras, P. Fuentealba, M. Galván, P. Pérez, Chem. Phys. Lett. 1999, 304, 405.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXislWlt70%3D&md5=e714fc3eebd8c17a2e167eea5d9da255CAS |
[32] (a) P. K. Chattaraj, B. Maiti, J. Phys. Chem. A 2001, 105, 169.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXos1WqurY%3D&md5=76fb8453a44ea7772c0128f8279c0f23CAS |
(b) P. Jaramillo, P. Perez, R. Contreras, W. Tiznado, P. Fuentealba, J. Phys. Chem. A 2006, 110, 8181.
| Crossref | GoogleScholarGoogle Scholar |
(c) F. De Vleeschouwer, V. Van Speybroeck, M. Waroquier, P. Geerlings, F. De Proft, Org. Lett. 2007, 9, 2721.
| Crossref | GoogleScholarGoogle Scholar |
[33] (a) R. Contreras, J. Andres, V. S. Safont, P. Campodonico, J. G. Santos, J. Phys. Chem. A 2003, 107, 5588.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXkvVOqsLg%3D&md5=6b61b7539b0b0fc09319f607d9d32a64CAS |
(b) L. R. Domingo, E. Chamorro, P. Pérez, J. Org. Chem. 2008, 73, 4615.
| Crossref | GoogleScholarGoogle Scholar |
[34] (a) J. L. Gazquez, A. Cedillo, A. Vela, J. Phys. Chem. A 2007, 111, 1966.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhslOrsbg%3D&md5=4837f87d826795bb03134b3e09d697c8CAS | 17305319PubMed |
(b) S. Pratihar, S. Roy, J. Org. Chem. 2010, 75, 4957.
| Crossref | GoogleScholarGoogle Scholar |
[35] F. J. Luque, J. M. Lopez, M. Orozco, Theor. Chem. Acc. 2000, 103, 343.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXjsV2htLY%3D&md5=92055c5bd1a3e11723293ec72950b984CAS |
[36] N. Okulik, A. H. Jubert, Internet Electron. J. Mol. Des. 2005, 4, 17.
| 1:CAS:528:DC%2BD2MXitVyhu7o%3D&md5=62576877935ac471918d5521139aecccCAS |
[37] R. Vijayaraj, V. Subramanian, P. K. Chattaraj, J. Chem. Theory Comput. 2009, 5, 2744.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFOitbjE&md5=cee19a71813ee26168708909887c888fCAS |
[38] P. Norcott, C. Spielman, C. S. P. McErlean, Green Chem. 2012, 14, 605.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XjtFymsbY%3D&md5=83d24a32142e8b5560e39e9082006a1eCAS |
[39] (a) L. C. A. Barbosa, U. A. Pereira, C. R. A. Maltha, R. R. Teixeira, V. N. M. Valente, J. R. O. Ferreira, L. V. Costa-Lotufo, M. O. Moraes, C. Pessoa, Molecules 2010, 15, 5629.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVGnsLfJ&md5=f831969f4e23e84f3e5b9fdbe457e892CAS |
(b) J. S. Yadav, B. V. Subba Reddy, T. Swamy, K. S. Shankar, Monatsh. Chem. 2008, 139, 1317.
| Crossref | GoogleScholarGoogle Scholar |
[40] (a) M. Staszewski, K. Walczynski, Med. Chem. Res. 2013, 22, 1287.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xot12htrk%3D&md5=e0d8b619f45b9727fd732b5c64f558e1CAS |
(b) A. Ahmadi, M. Khalili, A. Nafarie, A. Yazdani, B. Nahri-Niknafs, Mini Rev. Med. Chem. 2012, 12, 1282.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. Handzlik, M. Bajda, M. Zygmunt, D. Maciag, M. Dybala, M. Bednarski, B. Filipek, B. Malawska, K. K. Kononowicz, Bioorg. Med. Chem. 2012, 20, 2290.
| Crossref | GoogleScholarGoogle Scholar |
[41] G. M. A. Junqueira, L. C. Rocha, V. T. Cotta, E. T. César, Chem. Phys. Lett. 2012, 538, 54.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XnslKhtb4%3D&md5=2d0346bbbf7f1e4a1754b9e75db73cefCAS |
[42] J. A. Valderrama, R. Araya-Maturana, F. J. Zuloaga, J. Chem. Soc. Perkin Trans. 1993, 1, 1103.
[43] R. Araya-Maturana, B. K. Cassels, T. Delgado-Castro, J. A. Valderrama, B. E. Weiss-López, Tetrahedron 1999, 55, 637.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXpvVGmug%3D%3D&md5=815f2811daef02ec9bdbc70b0c7cf323CAS |
[44] S. C. Srivastava, U. Hornemann, Angew. Chem. 1976, 88, 87.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE28XovFOmug%3D%3D&md5=d2229e33b95910e18db0a204e8cf7d4fCAS |
[45] A. D. Becke, J. Chem. Phys. 1993, 98, 5648.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXisVWgtrw%3D&md5=f6cf3fb1eb0aa0bd90e09e70f7fe63aeCAS |
[46] P. J. Stephens, F. J. Devlin, C. F. Chahalowsky, M. J. Frisch, J. Phys. Chem. 1994, 98, 11623.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXmvVSitbY%3D&md5=e1e5e9babf6e7231adf55e499646050fCAS |
[47] M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, N. Rega, P. Salvador, J. J. Dannenberg, D. K. Malick, A. D. Rabuck, K. Rahavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. AlLaham, C. Y. Peng, A. Nanayakkara, M. Challa Combe, P. Gill, B. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head Gordon, E. S. Replogle, J. A. Pople, Gaussian03, Revision E. 01 2004 (Gaussian, Inc.: Wallingford, CT).
[48] E. Chamorro, P. Perez, J. Chem. Phys. 2005, 123, 114107.
| Crossref | GoogleScholarGoogle Scholar | 16392551PubMed |