

Decorating Semiconductor Silver-Tetracyanoquinodimethane Nanowires with Silver Nanoparticles from Ionic Liquids
Chuan Zhao A B , Changlong Xiao A , Hubert M. Chan A and Xunyu Lu AA School of Chemistry, The University of New South Wales, Sydney, NSW 2052, Australia.
B Corresponding author. Email: chuan.zhao@unsw.edu.au
Australian Journal of Chemistry 67(2) 213-216 https://doi.org/10.1071/CH13393
Submitted: 25 July 2013 Accepted: 9 September 2013 Published: 25 September 2013
Abstract
Hybrid semiconducting silver-tetracyanoquinodimethane (AgTCNQ) nanowires decorated with Ag nanoparticles have been synthesized at room temperature in the ionic liquid 1-butyl-3-methylimidazolium tetrafluoroborate. Hydroquinone was applied to reduce Ag+ and TCNQ to silver nanoparticles, and TCNQ–, respectively, under ambient conditions. AgTCNQ nanowires were formed via spontaneous electrolysis between Ag metal nanoparticles and TCNQ, and reaction between Ag+ and TCNQ–. Microscopic, spectroscopic, and X-ray characterizations all confirmed the formation of crystalline Ag nanoparticle–AgTCNQ nanowire hybrid structures. The ionic liquid was used as a reaction medium, but also as a stabilizing (or blocking) agent to control the nucleation and growth rate of AgTCNQ wires.
Introduction
Downscaling the size of flash and dynamic random access memory is becoming more and more challenging as the physical limits of the storage mechanism are approached.[1–4] Charge transfer metal-7,7,8,8-tetracyanoquinodimethane (TCNQ) complexes have drawn considerable interest owing to their novel electrical, optical, and/or magnetic properties. In particular, the discovery of electrically and optically bistable behaviour and memory effects of both AgTCNQ and CuTCNQ has sparked considerable interest in AgTCNQ and CuTCNQ non-volatile-based memory devices.[5–10] Complementary metal oxide semiconductor devices fabricated from these TCNQ materials have been downscaled in size to an area of 0.25 μm.[9] A range of techniques have therefore been introduced, including vacuum vapour deposition of TCNQ onto metal surfaces at low temperature[11] and thermal vapour deposition of TCNQ on Ag at 150–180°C.[12] Most published ‘wet’ methods for AgTCNQ preparation consist of a reaction between dissolved TCNQ in acetonitrile and metallic Ag, where metallic Ag is spontaneously oxidized into Ag+ and TCNQ is reduced into TCNQ– radical anions before precipitating together, resulting in a bluish-black film at the Ag surface.[7,13] Recent contributions from this laboratory have also demonstrated the application of room temperature ionic liquids for the preparation of AgTCNQ by electrocrystallization[14,15] and photocrystallization[16] methods to achieve several novel nanostructures.
An interesting recent trend in nanomaterial research is to design and synthesize metal–semiconductor hybrid nanostructures, which could effectively combine the properties of both materials into one system. Decoration of organic semiconductor nanowires with metal nanoparticles has been proven to be an attractive route to organic–inorganic hybrid nanomaterials.[17,18] The rational design of such heterostructures can in principle lead to novel and advantageous functionalities such as increased conductivity, due to the influence of the metallic component, and enhanced charge separation, due to the energy band alignment, which may be tailored to fit a specific application. The advantages of such metal–semiconductor hybrid structures have been demonstrated in various applications such as photocatalysts for solar energy conversion with enhanced efficiencies and field-effect transistors with high mobility and current on/off ratio.[18,19] Pearson et al. reported CuTCNQ microrods decorated with gold nanoparticles via a galvanic replacement reaction in aqueous solution.[20,21] Recently, Xiao et al. reported the reaction between Ag nanoparticles and TCNQ microparticles in aqueous solution which led to the formation of hybrid Ag nanoparticle-decorated AgTCNQ nanowire materials.[6] Importantly, the as-fabricated hybrid nanostructures showed good performance in non-volatile memory devices with multiple write-read-erase-read cycles in air.
In this paper, we describe a one-pot solution route for the synthesis of AgTCNQ nanowires in ionic liquids containing dissolved TCNQ and Ag salts. The AgTCNQ nanowires were characterized by a range of microscopic and spectroscopic techniques. Interestingly, we found the AgTCNQ nanowires were uniformly decorated with Ag nanoparticles. To the best of our knowledge, this study represents the first example of the use of ionic liquids for the fabrication of metal nanoparticle–semiconductor nanowire hybrid materials.
Results and Discussion
The ionic liquid 1-butyl-3-methylimidazolium tetrafluoroborate [BMIM][BF4] was chosen for use in this study because of the much higher solubility of TCNQ, AgBF4, and hydroquinone (H2Q) than that found in other ionic liquids tested. It is known that semiconducting metal–TCNQ materials such as AgTCNQ and CuTCNQ can be prepared in acetonitrile via the so-called ‘spontaneous electrolysis’ method where TCNQ dissolved in acetonitrile reacts with metallic Ag. In this reaction, metallic Ag is oxidized to the Ag+ cation and TCNQ is reduced to the TCNQ– radical anions to give AgTCNQ at the metal surface. Recently, we also have shown that AgTCNQ can be fabricated from ionic liquids through an electrocrystallization method where Ag+ was electrochemically reduced and the electrodeposited Ag nanoparticles on the electrode surface react with TCNQ dissolved in [BMIM][BF4] to form AgTCNQ.[15]
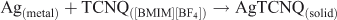
In the study, chemical reduction of Ag+ cation in ionic liquid [BMIM][BF4] was achieved by using H2Q as a reducing agent. In a control experiment, equimolar amounts of Ag+ and H2Q were mixed in [BMIM][BF4] and stirred for 1 h to yield a black solution. After repetitive washing with ethanol, centrifugation, and drying under vacuum, a black powder was obtained. Transmission electron microscopy (TEM) studies confirm that Ag nanoparticles of diameter ~20 nm were formed (Fig. 1a). This result is in good agreement with studies described previously under similar reaction conditions.[22]

|
In another control experiment, equimolar amounts of (13 μM) TCNQ and H2Q were mixed in [BMIM][BF4] for 1 h. The ultraviolet-visible (UV-Vis) spectrum (Fig. 1) exhibits a strong adsorption band with λmax = 399 nm, which resembles that reported in [BMIM][BF4] and can be assigned to the well understood 1Agf →1B3u transition for TCNQ.[16] The absorbance over the wavelength range of 700–800 nm, which is not observed for neutral TCNQ, is attributed to the absorbance from TCNQ– and is assigned to the TCNQ– 2B2g → 2B1u transition,[23,24] suggesting TCNQ is also reduced by H2Q to TCNQ– in [BMIM][BF4]. Thus, the presence of TCNQ– enables another possible route to form AgTCNQ, via the following reaction scheme:

Fig. 2b shows that AgTCNQ nanowires with diameters of 50 to 200 nm and lengths of >1 μm are formed. Energy dispersive X-ray (EDX) analysis of the nanowires shows that the samples contain Ag, C, and N, confirming AgTCNQ formation. TEM images obtained at higher magnifications (Figs 2c, d) reveal that evenly distributed crystalline Ag nanoparticles with diameters of around 10 nm are formed on the surface of AgTCNQ, and are confirmed to be Ag by elemental analysis.

|
Raman spectroscopy is a frequently used method to detect the vibrational modes of different chemical bonds between metal atoms and TCNQ molecules in metal–TCNQ complexes. Thus, we applied Raman spectroscopy in this study to confirm the formation of AgTCNQ. Fig. 3a shows the four characteristic principle vibration modes at 1205 cm–1 (C=CH bending), 1382 cm–1 (C–CN wing stretching), 1603 cm–1 (C=C stretching), and 2210 cm–1 (C–N stretching) confirming that the nanowires are AgTCNQ.[25] When compared with the Raman spectrum of neutral TCNQ (Fig. 3b), the two principle vibration modes of TCNQ at 1455 cm–1 (C–CN wing stretching) and 2227 cm–1 (C–N stretching) are actually red-shifted by 73 and 17 cm–1 respectively in AgTCNQ, which is consistent with that obtained from other metal–TCNQ compounds, and can be attributed to the charge transfer between atomic Ag and free TCNQ, in which TCNQ is reduced to TCNQ– to form AgTCNQ.[26]

|
Powder X-ray diffraction (XRD) has been used to distinguish the phase of as-prepared AgTCNQ. The two forms of AgTCNQ materials, so called phase I and II, have been reported.[13] Fig. 4 displays the XRD pattern of the as-prepared AgTCNQ nanoparticles, which confirms the formed AgTCNQ are crystalline in nature and matches well with the characteristic peaks of the reported phase II AgTCNQ with an orthorhombic structure, where the Ag atom is coordinated to four N atoms in a distorted tetrahedral environment and TCNQ molecules stack face-to-face.[25] The Ag diffraction peaks observed at 38.26 and 44.49° further confirm the presence of Ag nanoparticles. Infrared (IR) spectra recorded for the hybrid AgTCNQ materials confirm the formation of AgTCNQ (Fig. 5a). The three strong bands for the C≡N stretching mode of TCNQ molecules are observed at 2202, 2185, and 2170 cm–1, respectively. A strong band at 1504 cm–1 corresponding to the C=C stretching mode of TCNQ phenyl rings, and a weak adsorption peak at 822 cm–1 attributed to the C–H bending mode are all indicative of reduced TCNQ, TCNQ– anion radical, rather than [TCNQ]2–, [TCNQ‐TCNQ]2– dimer, or neutral TCNQ, which is in good accordance with the characteristic IR data reported for phase II AgTCNQ.[25]

|

|
All data obtained confirm the formation of Ag nanoparticle–AgTCNQ nanowire hybrid structures. The detailed formation mechanism is still under investigation but can generally be attributed to two possible effects. First, the slow diffusion rate of Ag+, TCNQ, Ag nanoparticles, and TCNQ– in [BMIM][BF4] media, owing to the high viscosity of the ionic liquid (120 cP at 25°C) which slows down the nucleation and growth of AgTCNQ. The effect is supported by the observation that attempts to synthesize AgTCNQ material from non-viscous media, whilst maintaining all other synthetic conditions, resulted in smooth surfaced nanowires with no decoration of Ag nanoparticles. The second possible effect as reported in in previous studies, is that Ag nanoparticles prepared from ionic liquids often contain an adsorbed ionic liquid layer.[22] The adsorbed [BMIM][BF4] layer on the Ag nanoparticle surface can act as a blocking agent that slows down the nucleation and growth of AgTCNQ. This is confirmed by IR spectra of the Ag nanoparticles–AgTCNQ nanowires (Fig. 4a). In comparison to the IR spectra of pure [BMIM][BF4] (Fig. 5b), the presence of characteristic peaks of [BMIM][BF4] between 2800 to 3000 cm–1, originated from the butyl chain attached to the imidazolium ring, and at 3122 and 3163 cm–1, attributed to the antisymmetric and symmetric C–H vibrational mode of the imidazolium ring,[27] all suggest a [BMIM][BF4] layer is adsorbed onto the surface of the hybrid materials. As the sample has already been washed repetitively with acetonitrile and ethanol before the spectroscopic measurements, it is concluded that this adsorption is relatively strong.
Conclusion
In summary, we have prepared AgTCNQ nanowires using ionic liquids, which can potentially be applied in non-volatile memory devices and catalysis. Ionic liquids play multiple roles in the synthesis and represent a novel class of media for fabrication of functional hybrid nanomaterials, which could be easily extended to other metal–TCNQ charge transfer complexes. It is noteworthy that ionic liquids themselves are designable solvents and could be tailored to specific applications. The approach thus opens opportunities for fabrication of a range of novel inorganic–organic, conductor/semiconductor nanostructures with tunable physical properties.
Experimental
1-Butyl-3-methylimidazolium tetrafluoroborate ([BMIM][BF4], 98 %) was purchased from Merck. 7,7,8,8-Tetracyanoquinodimethane (TCNQ, 98 %), silver tetrafluoroborate (AgBF4), and hydroquinone were purchased from Sigma. Absolute ethanol from Aldrich was used as provided.
In a typical synthesis, 8 mL [BMIM][BF4] solution containing equal amounts (5 mM) of TCNQ and AgBF4 was prepared at room temperature in an ultrasonic bath, yielding a yellow-green coloured solution. Silver-TCNQ nanoparticles were grown at room temperature by adding 1 mL of freshly prepared reducing reagent, 40 mM H2Q, dissolved in [BMIM][BF4]. The mixture was stirred for 1 h to yield a dark blue solution. Ethanol was then added to the specimen tube to reduce the viscosity and the AgTCNQ material was collected by centrifugation after washing three times with acetonitrile and ethanol, and finally drying under vacuum.
Electronic spectra were obtained with a Varian Cary 5 UV-Vis spectrophotometer (Varian OS2 software) using a 1 cm path length cell under benchtop laboratory conditions. Powder XRD patterns of synthesized AgTCNQ were collected at 40 kV and 25 mA using a Phillips 1729 X-ray diffractrometer with Cu Kα radiation in the range from 2 to 60° 2θ in steps of 0.02° with a counting time of 2 s step–1. Infrared spectra were obtained with a Bruker IFS Equinox Fourier transform IR system equipped with a Golden Gate single bounce diamond Micro-ATR (attenuated total reflectance) cell. Raman spectra were collected with a Renishaw RM 2000 Raman spectrograph and microscope using a laser strength of 18 mW at a wavelength of 780 nm. TEM measurements were undertaken with a Philips CM 200 field emission gun transmission electron microscope. AgTCNQ samples for TEM imaging were prepared by suspending blue AgTCNQ solid in ethanol for 24 h to obtain a very diluted suspension. A drop of the suspension was then drop casted onto a copper grid electrode and dried in air.
Acknowledgement
This study was financed by an ARC Discovery Grant (DP110102569).
References
[1] G. Atwood, Science 2008, 321, 210.| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXoslOhsLw%3D&md5=96a9ad58e3117f1a255a169f641a1c51CAS | 18621659PubMed |
[2] R. Bez, A. Pirovano, Mater. Sci. Semicond. Process. 2004, 7, 349.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXpvVelu7c%3D&md5=0c79162140d4ce78caab5898eac7d6d8CAS |
[3] R. Müller, S. De Jonge, K. Myny, D. J. Wouters, J. Genoe, P. Heremans, Appl. Phys. Lett. 2006, 89, 223501.
| Crossref | GoogleScholarGoogle Scholar |
[4] T. Oyamada, H. Tanaka, K. Matsushige, H. Sasabe, C. Adachi, Appl. Phys. Lett. 2003, 83, 1252.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXmtFOksbg%3D&md5=9407b2fdbed18aa3d8b0088ed88b91e6CAS |
[5] H. B. Liu, Q. Zhao, Y. L. Li, Y. Liu, F. S. Lu, J. P. Zhuang, S. Wang, L. Jiang, D. B. Zhu, D. P. Yu, L. F. Chi, J. Am. Chem. Soc. 2005, 127, 1120.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXitlGmuw%3D%3D&md5=4de1a624c8a33a04a4374b29ec8f219bCAS |
[6] J. C. Xiao, Z. Y. Yin, Y. C. Wu, J. Guo, Y. H. Cheng, H. Li, Y. Z. Huang, Q. Zhang, J. Ma, F. Boey, H. Zhang, Q. C. Zhang, Small 2011, 7, 1242.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXltlelu78%3D&md5=2c27e69453279be580490ba0160dc399CAS |
[7] Y. L. Liu, Z. Y. Ji, Q. X. Tang, L. Jiang, H. X. Li, M. He, W. P. Hu, D. Q. Zhang, L. Jiang, X. K. Wang, C. Wang, Y. Q. Liu, D. B. Zhu, Adv. Mater. 2005, 17, 2953.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xlt1WmsQ%3D%3D&md5=397f9b305e8d29eab2d1b4b4440aa2efCAS |
[8] R. Müller, J. Genoe, P. Heremans, Appl. Phys. Lett. 2006, 88, 24205.
[9] R. Müller, S. Jonge, K. Myny, D. J. Wouters, J. Genoe, P. Heremans, Solid-State Electron. 2006, 50, 601.
| Crossref | GoogleScholarGoogle Scholar |
[10] K. Xiao, J. Tao, Z. W. Pan, A. A. Puretzky, I. N. Ivanov, S. J. Pennycook, D. B. Geohegan, Angew. Chem. Int. Ed. 2007, 46, 2650.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXksVWrtLc%3D&md5=0d1e8e3393ffe19a0b9a53506a19b5dbCAS |
[11] E. I. Kamitsos, W. M. Risen, Solid State Commun. 1983, 45, 165.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3sXpvFCksA%3D%3D&md5=99aeeba729f23f1da20d6f3329636893CAS |
[12] K. Xiao, J. Tao, A. A. Puretzky, I. N. Ivanov, S. T. Retterer, S. J. Pennycook, D. B. Geohegan, Adv. Funct. Mater. 2008, 18, 3043.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlWnt7nE&md5=837d0095c0971ad1d7b72f3222b3d5cfCAS |
[13] R. A. Heintz, H. H. Zhao, O. Y. Xiang, G. Grandinetti, J. Cowen, K. R. Dunbar, Inorg. Chem. 1999, 38, 144.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXnvFWrtLk%3D&md5=682d01216c4f7155deb043dafe694ea2CAS |
[14] H. Wang, X. H. Qu, J. X. Lu, A. M. Bond, C. Zhao, CrystEngComm 2011, 13, 4762.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXptVWlt7s%3D&md5=9c13b0b428991a3d13528b0204ee48ceCAS |
[15] C. Zhao, D. R. MacFarlane, A. M. Bon, J. Am. Chem. Soc. 2009, 131, 16195.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1Okur3P&md5=3cd37bda5e2bff1f84dbc80a9e8d9370CAS | 19831410PubMed |
[16] C. Zhao, A. M. Bond, J. Am. Chem. Soc. 2009, 131, 4279.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXivF2qtLw%3D&md5=b6083f714fbc06900837047a008fc44dCAS | 19317503PubMed |
[17] A. L. Briseno, S. C. B. Mannsfeld, E. Formo, Y. J. Xiong, X. M. Lu, Z. N. Bao, S. A. Jenekhe, Y. N. Xia, J. Mater. Chem. 2008, 18, 5395.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlaqs7jJ&md5=e3150e10cdefb6dda386810a616358a7CAS |
[18] I. Jen-La Plante, S. E. Habas, B. D. Yuhas, D. J. Gargas, T. Mokari, Chem. Mater. 2009, 21, 3662.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXot1ejtbg%3D&md5=7337f249290061630e76cd1f49c32575CAS |
[19] D. Duonghong, E. Borgarello, M. Gratzel, J. Am. Chem. Soc. 1981, 103, 4685.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXks1yltrw%3D&md5=b94a390a63b79446083a119e7aff3916CAS |
[20] A. Pearson, A. P. O’Mullane, S. K. Bhargava, V. Bansal, Inorg. Chem. 2012, 51, 8791.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtFagurnJ&md5=50c18375430e3789d2ef5b0ae6f1dcd7CAS | 22853734PubMed |
[21] A. Pearson, A. P. O’Mullane, V. Bansal, S. K. Bhargava, Inorg. Chem. 2011, 50, 1705.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXosV2gtQ%3D%3D&md5=cbf2047a66a696438c908f8e87e645bdCAS | 21247089PubMed |
[22] S. M. Zhang, C. L. Zhang, J. W. Zhang, Z. J. Zhang, H. X. Dang, Z. S. Wu, W. M. Liu, Acta Phys.-Chim. Sin. 2004, 20, 554.
| 1:CAS:528:DC%2BD2cXks1Klt7Y%3D&md5=48d72db62ca08515cad48050c2292116CAS |
[23] S. G. Liu, Y. Q. Liu, P. J. Wu, D. B. Zhu, H. Tian, K. C. Chen, Thin Solid Films 1996, 289, 300.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXntFOitA%3D%3D&md5=ef0a86c0c1c44feacd0b850c835f7b03CAS |
[24] D. A. Lowitz, J. Chem. Phys. 1967, 46, 4698.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF2sXksVymu7o%3D&md5=3537287a5ebbf31ea1b54a1a48aefdf3CAS |
[25] S. A. O’Kane, R. Clérac, H. Zhao, X. Ouyang, J. R. Galán-Mascarós, R. Heintz, K. R. Dunbar, J. Solid State Chem. 2000, 152, 159.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXksFyit7g%3D&md5=2cf10104558b05c215a5fd0bd007f765CAS |
[26] B. Lewis, J. C. Anderson, Nucleation and Growth of Thin Films 1987 (Academic Press: New York, NY).
[27] Y. Jeon, J. Sung, C. Seo, H. Lim, H. Cheong, M. Kang, B. Moon, Y. Ouchi, D. Kim, J. Phys. Chem. B 2008, 112, 4735.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXjslahu78%3D&md5=8f0fe337b68ad1b82aa7cb53f9cd729eCAS | 18366215PubMed |