

Revisiting Staudinger and Ruzicka’s altered pyrethrolone: the cyclopentadienone dimers derived from pyrethrin I, cinerin I and jasmolin I
Oliver E. Hutt A , Jamie A. Freemont A , Stella Kyi A , Stuart W. Littler A , Ross P. McGeary B , Peter J. Duggan A C , John Tsanaktsidis A , Helen F. Cole D , Maurice G. Kerr D , Elizabeth H. Krenske B and John H. Ryan A *
A C , John Tsanaktsidis A , Helen F. Cole D , Maurice G. Kerr D , Elizabeth H. Krenske B and John H. Ryan A *
A
B
C
D
Abstract
The reactions of pyrethrin I, cinerin I and jasmolin I with sodium hydroxide in ethanol afforded an approximately 1:1 ratio of two respective cyclopentadienone dimers, isolated in good yield. A combination of one-dimensional and two-dimensional NMR spectroscopic studies allowed determination of the structure and stereochemistry of the dimers. The dimers are formed by cycloaddition reactions of the less substituted alkene of the cyclopentadienone, and by regioisomeric endo transition states. Density functional theory calculations were in accord with the experimental findings showing the products formed by ambimodal transition states. One such transition state led to two initial products with the less stable product undergoing facile conversion to the other more stable, experimentally observed product. These studies clarify the structures of the altered pyrethrolone reported by Staudinger and Ruzicka in 1924.
Keywords: altered pyrethrolone, biologically active molecules, cinerin I, cyclopentadienone, density functional calculations, dimerisation, electrocyclic reactions, elimination, jasmolin I, pyrethrin I.
Introduction
The Australian pyrethrum industry, based in Tasmania, has grown to be a major player in the natural insecticide market.1 The industry contributes both to the agricultural sector through the growing of the pyrethrum daisy Tanacetum cinerariifolium and to the chemical sector through the production of commercial-grade refined pyrethrum extracts that in turn are the basis of a wide range of products. The active ingredients in the refined pyrethrum extracts, known as pyrethrins, are a series of six insecticidal compounds (Fig. 1).2,3 The pyrethrins continue to be a source of inspiration for the development of synthetic analogues referred to as pyrethroids.3 Despite the availability of a large array of synthetic analogues, the pyrethrins continue to find use in a range of human, pet and household applications where the very limited persistence and low mammalian toxicity of the natural compounds offers some advantage.3

The elucidation of the structures of the pyrethrins is intimately linked to the development of organic chemistry through the early to mid-20th century.2,3 The insecticidal principle was first recognised by Fujitani and Yamamoto as containing an ester residue,4–6 then by Staudinger and Ruzicka as comprising two compounds, designated as pyrethrin I and pyrethrin II (with correct molecular formula for pyrethrin I (1a) and pyrethrin II (2a), however assigned as isomeric structures with different positioning of the side chain double bonds).7 Subsequently, minor constituents were discovered, cinerin I (1b) and cinerin II (2b) by LaForge and Barthel,8–10 and jasmolin I (1c) and jasmolin II (2c) by Godin.11 The determination of the absolute configuration of the pyrethrins involved significant endeavour, with the stereochemistry of the carboxyl residues first elucidated by Crombie and Inouye,12,13 and that of the alkoxy residues by Katsuda.14
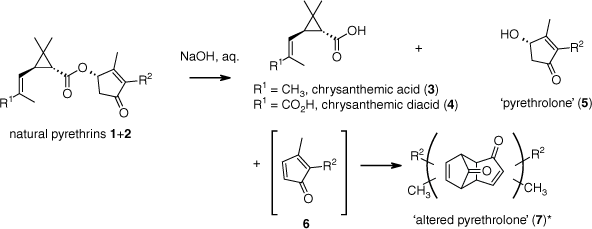
During the early studies of the chemistry of the pyrethrins, saponification reactions were performed (Scheme 1).2,15–17 Chrysanthemic acid (3) and chrysanthemic diacid (4) were recovered, representing the carboxyl residues of the pyrethrins. However, the alcohols (thought to be a single alcohol pyrethrolone (5)) were not obtained; instead, what was isolated was a material identified as altered pyrethrolone (7).2,15–17 It became accepted that hydrolysis of the natural pyrethrins is accompanied by competing elimination (E1cB) processes leading to cyclopentadienones 6, which are destabilised and highly reactive owing in part to their antiaromatic character, and undergo rapid Diels–Alder dimerisation.2 A range of 2,3-disubstituted cyclopentadienones are known to exist as cyclopentadienone dimers.18 Therefore, it has been suggested that the altered pyrethrolone (7) is a mixture of cyclopentadienone dimers (Scheme 1).2
Hydrolysis of the natural pyrethrins; regio- and stereo-chemistries (in 7) are not defined.2,15–17
Although the processes involved in formation of the naturally derived cyclopentadienone dimers 7 are generally understood,2,19–22 the structures and stereochemistry of the dimers 7 have not been reported in the peer-reviewed chemical literature. The most closely related system studied is an ester of allethrolone (8), a synthetic analogue of the pyrethrin-derived alcohols 5.19 Saponification of phthalate ester 9 produced two cyclopentadienone dimers, assigned as regioisomeric exo cycloadducts, with one depicted as regioisomer 10 (Scheme 2).19

In contrast to the above results described by LaForge,19 other cyclopentadienone dimerisation reactions display a strong preference for the kinetically favoured endo cycloadducts.23–27 For example, 2-bromocyclopentadienone (11) dimerises to produce the endo cycloadduct 12 (Scheme 3).28 The endo stereochemistry of 12 was elegantly confirmed by [2 + 2]-photocycloaddition of the diene 12 to give cage cyclobutane 13, which could only be achieved for the endo adduct. Then Favorskii ring-contraction of diketone 13 produced cubane-1,4-dicarboxylic acid 14, as part of the first synthesis of cubane (Scheme 3).28

Given the ambiguities in the literature, we sought to fully characterise the cyclopentadienone dimers derived from the natural pyrethrins. A preliminary study of the cyclopentadienone dimers from pyrethrin I (1a) was communicated at a pyrethrum symposium,29 and herein, we report the full study of the dimers resulting from pyrethrin I (1a), cinerin I (1b) and jasmolin I (1c).
Results and discussion
The base-promoted elimination of the natural pyrethrins 1 + 2 would lead to three cyclopentadienones 6, which in turn could lead to six Diels–Alder cycloaddition reactions, each of which could produce a number of regio- and stereo-isomers. We reasoned that separation of such complex mixtures of dimers could be difficult, whereas separation of the products from base-promoted elimination of a single natural pyrethrin would be relatively straightforward. Additionally, we only needed access to one series of the natural pyrethrins to produce all three cyclopentadienones 6 and, therefore, we chose to isolate pyrethrin I (1a), cinerin I (1b) and jasmolin I (1c) from the natural source.
Isolation of pyrethrin I (1a), cinerin I (1b) and jasmolin I (1c)
A sample of pyrethrin I (1a) was obtained by repeated silica gel chromatography of a sample of refined pyrethrum extract. The first pass enabled separation of the fast-eluting 1a–c from 2a to c and the second pass enabled separation of major component 1a from minor components 1b and 1c.
Although cinerin I (1b) and jasmolin I (1c) could be separated from the mixture on an analytical scale using normal or reverse-phase chromatography, preparative-scale chromatographic isolation was hampered by the similar retention times of all three components, compounded by the relatively minor amounts of these components compared with pyrethrin I (1a) (see Table 1, sample A).
| Sample | 1a | 2a | 1b | 2b | 1c | 2c | Other | |
|---|---|---|---|---|---|---|---|---|
| A | 49.9 | 29.5 | 5.5 | 4.6 | 3.4 | 1.9 | 5.2 | |
| B | 13.5 B | 8.0 A, B | 11.3 B | 9.2 A, B | 24.4 A | 18.2 | 15.4 | |
| C | 3.3 B | 1.7 A, B | 13.8 B | 10.8 A, B | 29.3 A | 21.8 A | 19.3 |
Sample A: refined pyrethrum extract prior to heat treatment. Sample B: after heating the extract at 230°C for 8 min. Sample C: after heating the extract at 230°C for 15 min. See Supplementary material for representative traces. Other refers to all other significant peaks.
It was thought that if pyrethrin I (1a) could be removed from the mixture of the three components, then it would be easier to separate the minor components chromatographically. We found that pyrethrin I/II (1a/2a) could be largely removed by heating samples of the pyrethrum extract containing the six esters at 230°C (Table 1, samples B and C). However, on inspection of the UPLC chromatograms of the heated samples, peak shoulders for the cinerin I/II (1b/2b) and jasmolin I/II (1c/2c) peaks were detected, indicating the formation of closely related impurities that would be very difficult to remove from the desired compounds.
The fast degradation of pyrethrin I/II (1a/2a) at such elevated temperatures was thought to be due to free radical reactions of the diene sidechain of 1a/2a. To test this hypothesis, we investigated the effect of adding a free radical initiator to a mixture of pyrethrin I (1a), cinerin I (1b) and jasmolin I (1c), readily obtained by column chromatography of the pyrethrum extract on silica gel. Addition of minor amounts of azobis(isobutyronitrile) (AIBN) to the mixture in toluene at 85°C resulted in a slight reduction in the levels of pyrethrin I (1a). Addition of further quantities of AIBN (up to ~0.50 mol equiv.) resulted in complete reaction of pyrethrin I (1a). The levels of cinerin I (1b) and jasmolin I (1c) did not appear to be affected by this process. The pyrethrin I (1a)–AIBN derived products could be readily separated by chromatography on silica gel, affording a mixture of cinerin I (1b) and jasmolin I (1c). This mixture of 1b and 1c was readily separated by preparative reverse-phase HPLC, which allowed isolation of cinerin I (1b) and jasmolin I (1c) in gram quantities with purity of greater than 95% by UPLC (Scheme 4).

Hydrolysis of pyrethrin I (1a), cinerin I (1b) and jasmolin I (1c)
A sample of pyrethrin I (1a) was exposed to sodium hydroxide in ethanol and after consumption of the starting material, the reaction mixture was worked up, yielding a mixture of two major products. The products were readily separated by flash column chromatography on silica, resulting in 30 and 38% yields of the respective faster-eluting and slower-eluting products (Table 2; percentage yields calculated assuming the products are the expected dimers). The treatment of cinerin I (1b) and jasmolin I (1c) under the same conditions also each afforded two products, isolated in similar yields, with the slower-eluting product isolated in slightly higher yield than the faster-eluting product (Table 2).
Structural elucidation of the cyclopentadienone dimers
The products from the hydrolysis of pyrethrin I (1a) were shown to be endo cyclopentadienone dimers. For both products, the high-resolution electrospray ionisation mass spectrum (positive ion mode) contained a peak at m/z 343.1657 assigned to the [M + Na+] ion, supporting a molecular formula of C22H24O2. The infrared spectrum of the higher RF product showed strong absorption bands at νmax 1775 and 1692 cm−1 assigned to bridged ketone and enone carbonyl groups respectively, whereas for the lower RF product, the corresponding bands were observed at νmax 1775 and 1695 cm−1. The 13C NMR spectrum of the higher RF product contained resonances at δ 204.7 and 201.3 ppm, assigned to the two carbonyl carbons, and at δ 16.2 and 15.8 ppm assigned to the two methyl groups, whereas for the lower RF product, the corresponding resonances occurred at δ 205.2, 201.1, 17.3 and 15.8 ppm. For the higher RF product, the two methyl groups exhibited resonances at δ 1.95 and 1.65 ppm in the 1H NMR spectrum, whereas for the lower RF product, these resonances occurred at δ 2.05 and 1.62 ppm. For both products, the 1H and 13C NMR spectra contained two distinct sets of signals assigned to the two diene side chains.
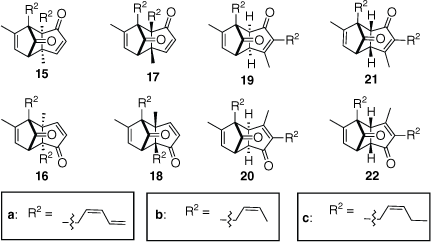
The cyclopentadienone 6a derived from pyrethrin I (1a) could dimerise by addition of the diene of one molecule to either double bond of the other, by either endo or exo transition states and by two regioisomeric orientations, so, in principle, eight isomers could be formed (Fig. 2). Analysis of the NMR spectra of the isolated dimers confirmed cycloaddition to the unsubstituted alkene of the cyclopentadienone moiety of 6a. In the 1H NMR spectra of the higher RF dimer and lower RF dimer, the respective methine signals at δH 3.14 and 2.64 ppm, and δH 3.09 and ~2.85 ppm are assigned to the bridgehead protons H3a and H7a (see Fig. 3). If cycloaddition had occurred at the substituted alkene of cyclopentadienone 6a, these signals would not be apparent and instead additional signals due to two olefinic protons would be present. This analysis allows exclusion of structures 15a–18a.
The structural formulae of the eight potential cyclopentadienone dimers derived from pyrethrin I (1a), cinerin I (1b) or jasmolin I (1c).
1H–1H COSY (bold) and NOE correlations (arrows) for higher RF dimer 21a and lower RF dimer 22a (COSY, correlation spectroscopy).
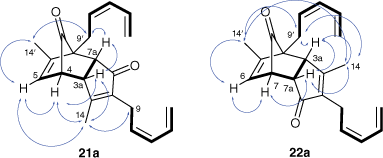
A range of NMR experiments allowed full assignment of all 1H and 13C resonances (see Supplementary materials) and assignment of the higher RF dimer as 21a and lower RF dimer as 22a. As shown in Fig. 3, the 1H–1H correlation spectroscopy (1H–1H COSY) spectrum of the higher RF dimer indicated the presence of the partial structures depicted in bold. Next, the relative stereochemistry of the higher RF dimer was characterised by a nuclear Overhauser enhancement (nOe) spectroscopy (NOESY) experiment, which showed nOe correlations between the signals due to the following protons: H3a with H7a, H14 and H4, H4 with H3a, H14 and H5, H5 with H4, H14′ with H9′ and H5, and H9′ with H7a. In particular, the nOe correlation between the signals due to H5 and H14 could only be achieved with the depicted regio- and stereo-chemistry of 21a. For the alternative stereoisomeric exo structure 19a, H5 and H14 would be too distant to exhibit a nOe correlation. For the lower RF dimer, the 1H–1H COSY spectrum again allowed assignment of partial structures depicted in bold. The NOESY spectrum showed several correlations consistent with the assigned structure. In particular, the correlation between the signals of H14 and H14′ is only possible for the depicted regio- and stereo-chemistry of 22a.
The cyclopentadienone dimers derived from cinerin I (1b) and jasmolin I (1c) were assigned in an analogous fashion, with the higher RF dimers being assigned structures 21b and 21c respectively and the lower RF dimers being assigned structures 22b and 22c respectively. Diagnostically, the 1H and 13C NMR resonances for the protons and carbons of the cyclic core were very similar within the series of the higher or the lower RF dimers. For example, for the higher RF dimers derived from 1a, 1b and 1c, respective resonances at δH 5.68, 5.67 and 5.67 ppm were assigned to the ring vinyl proton H5 of 21a, 21b and 21c. Similarly, for the lower RF dimers from 1a, 1b and 1c, the respective resonances at δH 5.92, 5.91 and 5.91 ppm were assigned to the ring vinyl proton H6 of 22a, 22b and 22c.
A proposed pathway for the formation of the respective cyclopentadienone dimers 21a–c and 22a–c from the natural pyrethrins 1a–c is shown in Scheme 5. Thus, reaction of natural pyrethrins (1a–c) with sodium hydroxide could result in ester hydrolysis to give the sodium salt of chrysanthemic acid (3) and pyrethrolones 23a–c. The latter could undergo E1cB reaction to give the transient cyclopentadienones 6a–c (path a). Alternatively, the cyclopentadienones 6a–c could form directly from E1cB elimination of chrysanthemic acid (3) from natural pyrethrins 1a–c (path b).19,22 Diels–Alder dimerisation of the cyclopentadienones 6a–c would then occur by endo transition state TS-A leading to compounds 21a–c or by regioisomeric endo transition state TS-B leading to compound 22a–c. The comparable yields obtained for the regioisomeric endo dimers imply that the regioisomeric endo transition states TS-A and TS-B are of similar energy.

Density functional theory calculations were performed to explore the endo and exo selectivities of the Diels–Alder dimerisations (Fig. 4). The calculations modelled the cyclopentadienones 6a–c as the methyl derivative 6d (R2 = Me) and were performed with B3LYP-D3(BJ)/6-311+G(d,p) using the solvent model based on solute electron density (SMD) of ethanol. In agreement with the experiment, the two endo transition states (TSs, labelled as 21d-TS and 22d-TS in Fig. 4) are strongly favoured, lying 10–12 kcal mol−1 (1 kcal mol–1 = 4.186 kJ mol–1) lower in energy than the corresponding exo transition states 19d-TS and 20d-TS. The two endo TSs differ in energy by 1 kcal mol−1, with 22d-TS being favoured, consistent with the small selectivity for 22 relative to 21 observed in experiments. The two endo cycloadditions have very small barriers (3–4 kcal mol−1), consistent with the antiaromaticity of the reacting cyclopentadienones, and lead to cycloadducts that are 22 kcal mol−1 more stable than the reactants.
Calculated transition structures for Diels–Alder dimerisations of cyclopentadienone 6d (R2 = Me) leading to 19d, 20d, 21d and 22d, calculated with B3LYP-D3(BJ)/6-311+G(d,p) in SMD implicit ethanol. The standard atom colouring scheme has been used (red is O). ΔG‡, change in Gibbs energy of activation; ΔEdist, change in energy of distortion; ΔEint, change in energy of interaction. Distances (dashed lines) in Ångstroms, energies in kilocalories per mole.
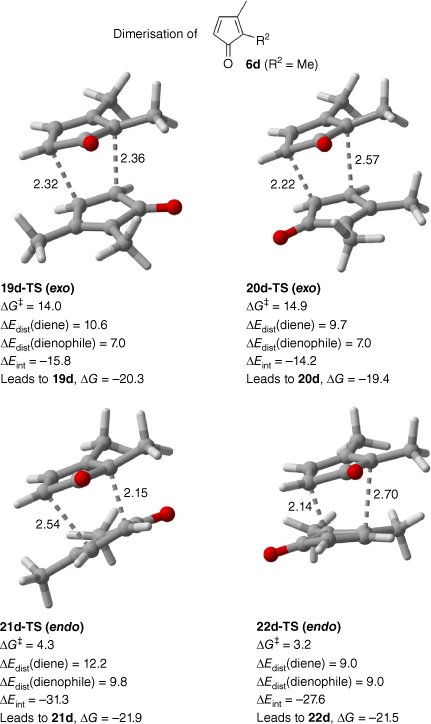
A distortion–interaction analysis30 was performed to explore the origin of the endo selectivity (Fig. 4). The diene and dienophile were found to undergo a similar amount of distortion, or even a few kilocalories per mole more distortion, when forming the endo TSs compared with the exo TSs, but the interaction between the diene and dienophile is much stronger (by 13–15 kcal mol−1) in the endo TSs than the exo TSs, leading overall to the substantial endo preference. This is likely attributable to the stronger π–π interactions that occur between the cycloaddends in the endo TSs.
Of note, the two endo TSs are ambimodal.31 Each gives rise to two alternative cycloaddition pathways that branch out after the TS (i.e. [4 + 2] and [2 + 4] pathways) on a bifurcating energy surface. The forming bonds drawn in Fig. 4 depict one of the two possible pathways for each TS. In the case of 22d-TS, the [4 + 2] and [2 + 4] pathways both give rise to the same cycloadduct by virtue of the C2 symmetry of the TS. In the case of 21d-TS, the two pathways give different products initially, as illustrated in Fig. 5. One pathway gives rise to 21d whereas the other gives rise to the less stable cycloadduct 18d. A facile Cope rearrangement of 18d over a small barrier of (0.5 kcal mol−1 relative to the starting cycloaddends) then leads to 21d. The mechanism for the Cope rearrangement of 18 to afford 21 is shown in Fig. 5. A similar type of interconversion between isomeric Diels–Alder dimers of the parent cyclopentadienone has previously been described by Caramella and coworkers.32

Conclusions
The cyclopentadienone dimers produced on hydrolysis of the natural pyrethrins pyrethrin I (1a), cinerin I (1b) and jasmolin I (1c) were isolated and the structures determined. In each case, isomeric products were isolated in ~1:1 ratio, a similar overall result to that obtained for the allethrolone esters.19 However, the products were shown, by 2-D NMR spectroscopy experiments, to be regioisomeric endo cyclopentadienone dimers 21 and 22, rather than exo cyclopentadienone dimers as previously proposed for the allethrolone esters.19 Density functional theory calculations confirmed the experimental findings, and a study of the ambimodal endo TSs revealed that one of the TSs is associated with a bifurcating energy surface with two distinct initial products, with the less stable product converting to the more stable product by a facile Cope rearrangement. Given that in this work, the side chain of the cyclopentadienone does not seem to have a significant influence on the product distribution, the results of this study serve to raise doubt about the original assignments of the dimers derived from the allethrolone ester 10.19 The present work also serves to clarify the structures of the altered (verändert) pyrethrolone first reported by Staudinger and Ruzicka in 1924.7
A study of the dimerisation of cyclopentadienones from allethrolone esters is warranted to confirm endo stereochemistry of the cycloadducts. For the natural pyrethrins, a study of this process with at least one of pyrethrin II, cinerin II or jasmolin II (2a–c) would be useful to check any influence the acryloyl moiety, embedded within the pyrethric acid side of these natural pyrethrins, may have on the outcome of the reactions. Furthermore, characterisation of the products from the reaction of pyrethrin I (1a) with AIBN may provide insight into the nature of this free radical process. With the exception of a recent study on autoxidation processes,33 the present work appears to be the only example of free radical chemistry of pyrethrins. Finally, future work could explore the question as to whether the isolated dimers represent previously undiscovered natural products. The only known natural products that appear to form by cyclopentadienone Diels–Alder dimerisation processes are guaiane dimers, including vielanin A and xylopidimers A–C and D, isolated from the roots of Xylopia vielana.23,34
Experimental
General experimental
All reaction flasks were oven-dried, and all reactions were carried out under an atmosphere of N2. All solvents used were HPLC grade. NMR spectra were recorded on a Bruker Av400 spectrometer. 1H NMR and 13C NMR were recorded at 400 and 100 MHz respectively. 1H NMR chemical shifts are reported in ppm with the internal chloroform signal at 7.26 ppm. The data are reported as s, singlet; d, doublet; t, triplet; m, multiplet or unresolved; brs, broad singlet; obs, obscured; coupling constant(s) in hertz, integration. 13C NMR chemical shifts are reported in ppm with the internal chloroform signal at 77.0 ppm. Infra-red spectra were recorded on a Thermo Scientific Nicolet 6700 Fourier transform (FT)-IR. High-resolution mass spectra (HRMS) were recorded on a Waters Q-TOF II, employing electrospray ionisation (ES) with 35-eV cone voltage, lock spray and sodium iodide as a reference sample. Analyses were performed on a Waters Acquity UPLC with 50- × 2.1-mm BEH C18 column using 55% ACN/H2O mobile phase at 0.4 mL min–1 flow rate and detection by photo diode array detector scanning 190–400 nm. Data acquisition and processing used Waters Empower software and representative chromatograms were extracted at 223 nm. Prep-HPLC separations were performed on a Waters HPLC using a 300- × 40-mm Deltaprep C18 column, a mobile phase of 50 or 55% acetonitrile/water, a flow rate of 80 mL min–1, and detection by a Waters 490E programmable multi wavelength detector set at 230 nm.
Isolation of pyrethrin I (1a)
A refined extract of natural pyrethrins (79% natural pyrethrins by HPLC, courtesy of Botanical Resources Australia Pty Ltd) was subjected to dry column vacuum chromatography (silica, gradient elution, pentane then ethyl acetate/petroleum spirits 1:23 v/v, then ethyl acetate/petroleum spirits 1:4 v/v). Fractions containing the faster-eluting natural pyrethrins 1a–c, free from the slower-eluting natural pyrethrins 2a–c, were concentrated under vacuum and subjected to column chromatography (silica gel, ethyl acetate/petroleum spirits 1:24, then 1:12 v/v). Fractions enriched in the slower-eluting pyrethrin I (1a) were combined and concentrated to afford pyrethrin I (1a) of 90% purity by UPLC analysis. The 13C NMR spectrum matched that reported in the literature.35 Other mixed fractions were combined ready for treatment with AIBN (see below).
Isolation of cinerin I (1b) and jasmolin I (1c)
To a round-bottomed flask was added a ~90:9:1 mixture of pyrethin I (1a), cinerin I (1b) and jasmolin I (1c, 11.5 g, ~35 mmol pyrethrin I) and toluene (50 mL). The flask was heated under nitrogen at 85°C for 15 min. A blast shield was installed, then AIBN (caution: special precautions should be taken when dealing with AIBN; please refer to safety data sheets) (2.8 g, 17.5 mmol) was dissolved in toluene (~25 mL) and added slowly dropwise, by a syringe fitted with a wide-bore needle, to the reaction flask. The reaction was stirred for 22 h at 85°C. TLC analysis (ethyl acetate/petroleum spirits 1:4 v/v) showed a characteristic new spot on the baseline due to the products from pyrethrin I (1a). The reaction mixture was cooled and then filtered through a short column of silica, followed by elution with ethyl acetate/petroleum spirits (1:9 v/v, 200 mL) and ethyl acetate/petroleum spirits (1:4 v/v, 200 mL). The fractions containing the faster-eluting components were combined and concentrated to give a mixture of cinerin I (1b) and jasmolin I (1c) (1.3 g, 1b:1c 94:6 v/v). This material was subjected to preparative HPLC, which yielded cinerin I (1b, 1.15 g) and jasmolin I (1c, 0.162 g) in 99.9 and 99.5% purity respectively as assessed by UPLC analysis. The 13C NMR of 1b and 1c matched that reported in the literature.35
Representative procedure for hydrolysis of the natural pyrethrins: 3,6-dimethyl-2,7-di((Z)-penta-2,4-dien-1-yl)-3α,4,7,7α-tetrahydro-1H-4,7-methanoindene-1,8-dione 21a29 and 3,5-dimethyl-2,4-di((Z)-penta-2,4-dien-1-yl)-3α,4,7,7α-tetrahydro-1H-4,7-methanoindene-1,8-dione 22a29
To a round-bottomed flask was added pyrethrin 1 (1a, 0.232 g, 90% purity, 0.64 mmol of 1a) and ethanol (2 mL). Freshly powdered sodium hydroxide (0.050 g, 1.2 mmol) was added, and the reaction mixture was stirred at room temperature for 30 min. At this time, TLC analysis (heptane/ethyl acetate 4:1 v/v) indicated that the starting material had been consumed. The reaction mixture was diluted with ethyl acetate (20 mL) and washed with water (10 mL) and brine (10 mL). The organic layer was then dried with magnesium sulfate, filtered and the solvent removed under reduced pressure. The resulting oily yellow residue was subjected to chromatography on silica gel (gradient: dichloromethane, then 0.5% methanol in dichloromethane, then 1% acetone in dichloromethane, then 5% acetone in dichloromethane). The fractions containing the component (Rf 0.3, heptane/ethyl acetate 4:1 v/v) were evaporated under reduced pressure to give title compound 21a (0.031 g, 30%) as an oil. νmax (KBr) 1775, 1692, 1639 cm−1. δH (400 MHz, CDCl3) 6.92 (dtd, J 16.8, 10.2, 1.1, 1H, H12′), 6.72 (dtd, J 16.7, 10.2, 1.1, 1H, H12), 6.14 (t, J 11.0, 1H, H11′), 5.97 (t, J 10.9, 1H, H11), 5.68 (m, 1H, H5), 5.63 (m, 1H, H10′), 5.14–5.25 (m, 5H, H13, H13′, H10), 3.22 (bt, J 5.2, 1H, H3a), 3.14 (t, J 4.2, 1H, H4), 3.06 (dd, J 15.1, 7.8, 1H, H9a), 2.99 (dd, J 14.8, 8.2, 1H, H9b), 2.90 (dd, J 15.2, 7.3, 1H, H9′a), 2.81 (dd, J 15.2, 7.3, 1H, H9′b), 2.64 (d, J 6.0, 1H, H7a), 1.95 (s, 3H, H14), 1.65 (d, J 1.6, 3H, H14′). δC (100 MHz, CDCl3) 204.7 (C1/C8), 201.3 (C1/C8), 167.3 (C3), 145.7 (C6), 143.0 (C2), 132.2 (C12′), 131.9 (C11′), 131.7 (C12), 129.9 (C11), 127.4 (C10), 121.0 (C5), 118.0 (C13/C13′), 117.9 (C13/C13′), 59.2 (C7), 49.5 (C3a), 45.3 (C4), 45.1 (C7a), 24.0 (C9′), 21.8 (C9), 16.2 (H14), 15.8 (H14′). HRMS m/z (Q-TOF ES+) Calcd for C22H24O2Na: 343.1674; found: 343.1657. Concentration of the fractions containing the component (RF 0.2, heptane/ethyl acetate 4:1 v/v) afforded the title compound 22a (0.039 g, 38%) as an oil. νmax (KBr) 1775, 1695, 1653 cm−1. δH (400 MHz, CDCl3) 6.72 (m, 2H, H12, H12′), 6.16 (t, J 11.0, 1H, H11), 5.98 (t, J 10.7, 1H, H11′), 5.92 (m, 1H, H6), 5.60 (m, 1H, H10), 5.15–5.30 (m, 5H, H10′, H13, H13′), 3.28 (dd, J 4.7, 3.7, 1H, H7), 3.09 (m, 1H, H3a), 3.04 (m, 2H, H9′), 2.83–2.87 (m, 3H, H7a, H9), 2.05 (s, 3H, H14), 1.62 (d, J 1.6, 3H, H14′). δC (100 MHz, CDCl3) 205.2 (C1/C8), 201.1 (C1/C8), 167.5 (C3), 146.1 (C5), 140.9 (C2), 131.6 (C12), 131.4 (C12′), 131.3 (C11), 129.9 (C11′), 127.3 (C10′), 127.0 (C10), 123.2 (C6), 118.5 (C13), 118.1 (C13′), 58.9 (C4), 49.1 (C7), 45.6 (C3a), 45.2 (C7a), 24.3 (C9), 21.7 (C9′), 17.3 (C14), 15.8 (C14′). HRMS m/z (Q-TOF ES+) Calcd for C22H24O2Na: 343.1674; found: 343.1657.
2,7-Di((Z)-but-2-en-1-yl)-3,6-dimethyl-3α,4,7,7α-tetrahydro-1H-4,7-methanoindene-1,8-dione 21b and 2,4-di((Z)-but-2-en-1-yl)-3,5-dimethyl-3α,4,7,7α-tetrahydro-1H-4,7-methanoindene-1,8-dione 22b
A sample of cinerin I (1b) (0.120 g, 0.37 mmol) was treated according to the representative procedure. After chromatography, concentration of the fractions containing the faster-eluting component (RF 0.7, heptane/ethyl acetate 4:1 v/v) afforded the title compound 21b (0.014 g, 26%) as a colourless oil. νmax (KBr) 1776, 1692, 1638 cm−1. δH (400 MHz, CDCl3) 5.67 (brs, 1H, H5), 5.51–5.61 (m, 2H, H10′, H11′), 5.39–5.47 (m, 1H, H11), 5.12–5.18 (m, 1H, H10), 3.20 (t, J 5.1, 1H, H3a), 3.13 (t, J 4.0, 1H, H4), 2.91 (dd, J 14.7, 7.2, 1H, H9a), 2.85 (dd, J 14.8, 7.5, 1H, H9b), 2.74 (dd, J 15.3, 7.0, 1H, H9a′), 2.68 (dd, J 15.5, 6.4, 1H, H9b′), 2.62 (d, J 6.0, 1H, H7a), 1.94 (s, 3H, H13), 1.74 (d, J 6.2, 3H, H12′), 1.68 (obscured d, 3H, H12), 1.66 (d, J 1.4, 3H, H13′). δC (100 MHz, CDCl3) 205.0 (C1/C8), 201.7 (C1/C8), 166.8 (C3), 146.4 (C6/C2), 143.1 (C6/C2), 126.8 (C10′), 126.1 (C10), 125.4 (C11′), 124.8 (C11), 120.9 (C5), 59.3 (C7), 49.6 (C4), 45.2 (two signals coinciding: C3a, C7a), 23.1 (C9′), 21.0 (C9), 16.1 (C13), 15.7 (C13), 12.9 (C12′), 12.8 (C12). HRMS m/z (Q-TOF ES+) Calcd for C20H24O2Na: 319.1674, found: 319.1664. Concentration of the fractions containing the slower-eluting component (RF 0.6, heptane/ethyl acetate 4:1 v/v) afforded the title compound 22b (0.017 g, 31%) as a colourless oil. νmax (KBr) 1778, 1698, 1635 cm−1. δH (400 MHz, CDCl3) 5.91 (dd, J 3.2, 1.5, 1H, H6), 5.61 (m, 1H, H11′), 5.53 (m, 1H, H10′), 5.45 (m, 1H, H11), 5.14 (m, 1H, H10), 3.26 (t, J 4.2, 1H, H7), 3.07 (d, J 6.0, 1H, H3a), 2.94 (dd, J 14.9, 6.8, 1H, H9a), 2.87 (obscured dd, 1H, H9b), 2.84 (t, J 5.6, 1H, H7a), 2.68 (m, 2H, H9′), 2.05 (s, 3H, H13), 1.72 (d, J 6.4, 3H, H12′), 1.67 (d, J 6.9, 3H, H12), 1.63 (d, J 1.5, 3H, H13′). δC (100 MHz, CDCl3) 205.6 (C1/C8), 201.5 (C1/C8), 167 (C3), 146.7 (C5), 141.1 (C2), 126.2 (C11′), 126.1 (C10), 125.5 (C10′), 124.9 (C11), 123.0 (C6), 59.1 (C4), 49.1 (C7), 45.6 (C3a), 45.2 (C7a), 23.4 (C9′), 20.9 (C9), 17.1 (C13), 15.7 (C13′), 13.1 (C12′), 12.8 (C12). HRMS m/z (Q-TOF ES+) Calcd for C20H24O2Na: 319.1674; found: 319.1651.
3,6-Dimethyl-2,7-di((Z)-pent-2-en-1-yl)-3α,4,7,7α-tetrahydro-1H-4,7-methanoindene-1,8-dione 21c and 3,5-dimethyl-2,4-di((Z)-pent-2-en-1-yl)-3α,4,7,7α-tetrahydro-1H-4,7-methanoindene-1,8-dione 22c
A sample of jasmolin I (1c) (0.120 g, 0.36 mmol) was treated according to the representative procedure. After chromatography, concentration of the fractions containing the faster-eluting component (RF 0.9, 1% methanol in dichloromethane) afforded the title compound 21c (0.014 g, 24%) as a colourless oil. νmax (KBr) 1775, 1692, 1638 cm−1. δH (400 MHz, CDCl3) 5.67 (m, 1H, H5), 5.50 (m, 2H, H10′, H11′), 5.35 (m, 1H, H11), 5.09 (m, 1H, H10), 3.20 (t, J 5.1, 1H, H3a), 3.13 (t, J 4.0, 1H, H4), 2.90 (dd, J 15.0, 7.4, 1H, H9a), 2.84 (1H, dd, J 14.9, 7.7, H9b), 2.74 (dd, J 15.5, 5.8, 1H, H9a‘), 2.67 (dd, J 15.8, 5.1, 1H, H9b‘), 2.62 (d, J 6, 1H, H7a), 2.22 (m, 2H, H12′), 2.11 (m, 2H, H12), 1.95 (s, H14), 1.65 (d, J 1.6, 3H, H14′), 0.98 (two coincidental t, J 7.8, 6H). δC (100 MHz, CDCl3) 205.0 (C1/C8), 201.7 (C1/C8), 166.8 (C3), 146.4 (C6), 143.1 (C2), 134.5 (C10′), 132.5 (C10), 124.5 (C11′), 123.8 (C11), 120.9 (C5), 59.3 (C7), 49.6 (C4), 45.3 (C3a), 45.2 (C7a), 23.4 (C9′), 21.3 (C9), 20.6 (C12), 20.5 (C12′), 16.0 (C14), 15.8 (C14′), 14.1 (C13), 14.0 (C13′). HRMS m/z (Q-TOF ES+) Calcd for C22H28O2Na requires 347.1987; found: 347.1989. Concentration of the fractions containing the slower-eluting component (RF 0.5, 1% methanol in dichloromethane) afforded the title compound 22c (0.016 g, 27%) as a colourless oil. νmax (KBr) 1778, 1698, 1634 cm−1. δH (400 MHz, CDCl3) 5.91 (dd, J 3.3, 1.6, 1H, H6), 5.35 (m, 1H, H11), 5.49 (m, H10, 2H, H10′), 5.07 (m, 1H, H11′), 3.25 (dd, J 4.7, 3.7, 1H, H7), 3.08 (d, J 6.0, 1H, H3a), 2.93 (dd, J 15.1, 6.5, 1H, H9a), 2.87 (obscured dd, 1H, H9b), 2.83 (t, J 5.6, 1H, H7a), 2.68 (m, 2H, H9′), 2.14 (m, 4H, H12, H12′), 2.04 (s, 3H, H14), 1.60 (d, J 1.6 Hz, 3H, H14′), 1.02 (t, J 7.5, 3H, H13), 0.97 (t, J 7.5, 3H, H13′). δC (100 MHz, CDCl3) 205.6 (C1/C8), 201.5 (C1/C8), 167.2 (C3), 146.7 (C5), 141.1 (C2), 133.9 (C11′), 132.6 (C10), 124.4 (C10′), 123.8 (C11), 123.0 (C6), 59.0 (C4), 49.1 (C7), 45.6 (C3a), 45.2 (C7a), 23.7 (C9′), 21.2 (C9), 20.8 (C12′), 20.5 (C12), 17.0 (C14), 15.8 (C14′), 14.1 (C13), 13.9 (C13′). HRMS m/z (Q-TOF ES+) Calcd for C22H28O2Na requires 347.1987; found: 347.1981.
Density functional theory calculations
Calculations were performed using Gaussian 16 (rev. C.01, see https://gaussian.com/gaussian16/).36 Geometries were optimised37–41 with B3LYP-D3(BJ)/6-311+G(d,p) using the SMD42 of ethanol. Distortion–interaction analyses of each TS partner were performed at the same level of theory. Intrinsic reaction coordinate calculations43,44 were performed on all TSs, which allowed the reactant and product structures connected to each TS along minimum-energy pathways to be identified. Gibbs free energies are reported at a standard state of 298.15 K and 1 mol L–1.
Supplementary material
Numbering schemes, 1-D and 2-D NMR spectra and HPLC chromatograms of all new compounds; UPLC traces of representative samples of refined pyrethrum extract before and after heat treatment; computed geometries and energies of species calculated with density functional theory: supplementary material is available online.
Data availability
The data that support this study are available in the article and accompanying online supplementary material. Further details can be shared on reasonable request to the corresponding author.
Declaration of funding
The CSIRO research was partly funded by Horticulture Australia Ltd (Project Number PY09004), the Commonwealth of Australia DIISR Enterprise Connect ‘Researchers in Business’ Program and Botanical Resources Australia Pty Ltd. We thank the Australian Research Council for funding (DP180103047 to E. H. Krenske).
Acknowledgements
The authors thank Dr Roger Mulder, Dr Jo Cosgriff and Carl Braybrook for their assistance with collection of NMR and MS data. Computational resources were provided by the Australian National Computational Infrastructure through the National Computational Merit Allocation Scheme.
References
1 Fist AJ, Chung B. Pyrethrum and poppy: two Tasmanian industries of world significance. Acta Hortic 2015; 1073: 85-90.
| Crossref | Google Scholar |
3 Krief A. Pyrethroid insecticides. Arkivoc 2021; Part I: 48-54.
| Google Scholar |
4 Fujitani J. Chemistry and pharmacology of insect powder. Arch Exp Pathol Pharmakol 1909; 61: 47-75.
| Crossref | Google Scholar |
5 Yamamoto R. The insecticidal principle in Chrysanthemum cinerariaefolium. Part I. J Chem Soc Jpn 1919; 40: 126.
| Google Scholar |
6 Yamamoto R. The insecticidal principle in Chrysanthemum cinerariaefolium. Parts II and III. On the constitution of the pyrethronic acid. J Chem Soc Jpn 1923; 44: 311.
| Google Scholar |
7 Staudinger H, Ruzicka L. Insektentötende Stoffe. X. Helv Chim Acta 1924; 7: 448-458 [In German].
| Crossref | Google Scholar |
8 LaForge FB, Barthel WF. Constituents of pyrethrum flowers. XVI. Heterogeneous nature of pyrethrolone. J Org Chem 1944; 09: 242-249.
| Crossref | Google Scholar |
9 LaForge FB, Barthel WF. Constituents of pyrethrum flowers. XVII. The isolation of five pyrethrolone semicarbazones. J Org Chem 1945; 10: 106-113.
| Crossref | Google Scholar |
10 LaForge FB, Barthel WF. Constituents of pyrethrum flowers. XVII. The structure and isomerism of pyrethrolone and cinerolone. J Org Chem 1945; 10: 114-120.
| Crossref | Google Scholar |
11 Godin PJ, Sleeman RJ, Snarey M, Thain EM. The jasmolins, new insecticidally active constituents of Chrysanthemum cinerariaefolium VIS. J Chem Soc C 1966; 332-334.
| Crossref | Google Scholar |
12 Crombie L, Harper SH. The chrysanthemumcarboxylic acids. Part VI. The configurations of the chrysanthemic acids. J Chem Soc 1954; 470.
| Crossref | Google Scholar |
13 Inouye Y, Takeshita Y, Ohno M. Studies on synthetic pyrethroids: part V. Synthesis of geometrical isomers of chrysanthemum dicarboxylic acid. Bull Agric Chem Soc Jpn 1955; 19: 193-199.
| Crossref | Google Scholar |
14 Katsuda Y, Chikamoto T, Inouye Y. The absolute configuration of naturally derived pyrethrolone and cinerolone. Bull Agric Chem Soc Jpn 1958; 22: 427-428.
| Crossref | Google Scholar |
15 Staudinger H, Ruzicka L. Insektentotende Stoffe. I. Über Isolierung und Konstitution des wirksamen Teiles des dalmatinischen Insektenpulvers. Helv Chim Acta 1924; 7: 177-201 [In German].
| Crossref | Google Scholar |
16 Staudinger H, Ruzicka L. Insektentotende Stoffe. II. Zur Konstitution der Chrysanthemum-monocarbonsäure und -dicarbonsäure. Helv Chim Acta 1924; 7: 201-211 [In German].
| Crossref | Google Scholar |
17 Staudinger H, Ruzicka L. Insektentotende Stoffe. III. Konstitution des Pyrethrolons. Helv Chim Acta 1924; 7: 212-235 [In German].
| Crossref | Google Scholar |
18 Ogliaruso MA, Romanelli MG, Becker EI. Chemistry of cyclopentadienones. Chem Rev 1965; 65: 261-367.
| Crossref | Google Scholar |
19 LaForge FB, Green N, Schechter MS. Dimerized cyclopentadienones from esters of allethrolone. J Am Chem Soc 1952; 74: 5392-5394.
| Crossref | Google Scholar |
20 Allen CFH, VanAllen JA. A carbonyl bridged compound in which the bridge ring is saturated. J Org Chem 1955; 20: 323-327.
| Crossref | Google Scholar |
21 Crombie L, Elliot M, Harper SH. Experiments on the synthesis of the pyrethrins. Part III. Synthesis of dihydrocinerin I and tetrahydropyrethrin I; a study of the action of N-bromosuccinimide on 3-methyl-2-n-alkyl (and alkenyl) cyclopent-2-en-1-ones. J Chem Soc 1950; 971-978.
| Crossref | Google Scholar |
22 Elliott M, Harper SH, Kazi MA. Experiments on the synthesis of the pyrethrins. XIV. Rethrins and the cyclopentadienone related to 3-methylcyclopent-2-enone. J Sci Food Agric 1967; 18: 167-171.
| Crossref | Google Scholar |
23 Guo Y, Xie Y, Wu G, Cheng T, Zhu S, Yan S, Jin H, Zhang W. Xylopidimers A–E, five new guaiane dimers with various carbon skeletons from the roots of Xylopia vielana. ACS Omega 2019; 4: 2047-2052.
| Crossref | Google Scholar | PubMed |
24 Kong X, Yang S, Yu F, Vasamsetty L, Liu J, Liu S, Liu X, Fang X. Cyclopentadienone formation from β,γ-unsaturated cyclopentenones and its application in Diels–Alder reactions. J Org Chem 2018; 83: 8953-8961.
| Crossref | Google Scholar | PubMed |
25 Harmata M, Barnes CL, Brackley J, Bohnert G, Kirchhoefer P, Kürti L, Rashatasakhon P. Generation of cyclopentadienones from 2-bromocyclopentenones. J Org Chem 2001; 66: 5232-5236.
| Crossref | Google Scholar | PubMed |
26 Fuchs B, Pasternak M, Scharf G. The dimer of 2,3-dimethyl-3,4-(o,o’-biphenylene)cyclopentadienone: thermal and photochemical transformations. J Chem Soc, Chem Commun 1976; 53-54.
| Crossref | Google Scholar |
27 DePuy CH, Isaks M, Eilers KL, Morris GF. The formation of cyclopentadienones. J. Org. Chem 1964; 29: 3503-3507.
| Crossref | Google Scholar |
28 Eaton PE, Cole TW. The cubane system. J Am Chem Soc 1964; 86: 962-964.
| Crossref | Google Scholar |
29 Hutt OE, Freemont JA, Littler S, Duggan PJ, Tsanaktsidis J, Cole H, Kerr M, Ryan JH. Staudinger and Ruzicka’s altered pyrethrolone: the cyclopentadienone dimers derived from pyrethrin I. Acta Hortic 2015; 1073: 181-190.
| Crossref | Google Scholar |
30 Bickelhaupt FM, Houk KN. Analysing reaction rates with the distortion/interaction–activation strain model. Angew Chem Int Ed 2017; 56: 10070-10086.
| Crossref | Google Scholar | PubMed |
31 McLeod D, Thøgersen MK, Jessen NI, Jørgensen KA, Jamieson CS, Xue X-S, Houk KN, Liu F, Hoffmann R. Expanding the frontiers of higher-order cycloadditions. Acc Chem Res 2019; 52: 3488-3501.
| Crossref | Google Scholar | PubMed |
32 Quadrelli P, Romano S, Toma L, Caramella P. A bispericyclic transition structure allows for efficient relief of antiaromaticity enhancing reactivity and endo selectivity in the dimerization of the fleeting cyclopentadienone. J Org Chem 2003; 68: 6035-6038.
| Crossref | Google Scholar | PubMed |
33 Freemont JA, Littler SW, Hutt OE, Mauger S, Meyer AG, Winkler DA, Kerr MG, Ryan JH, Cole HF, Duggan PJ. Molecular makers for pyrethrin autoxidation in stored pyrethrum crop: analysis and structure determination. J Agric Food Chem 2016; 64: 7134-7141.
| Crossref | Google Scholar | PubMed |
34 Kamperdick C, Phuong NM, Sung TV, Adam G. Guaiane dimers from Xylopia vielana. Phytochem 2001; 56: 335-340.
| Crossref | Google Scholar | PubMed |
35 Crombie L, Pattenden G, Simmonds DJ. Carbon-13 magnetic resonance spectra of natural pyrethrins and related compounds. J Chem Soc Perkin Trans 1 1975; 1500-1502.
| Google Scholar | PubMed |
36 Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ. Gaussian 16, revision C.01. Wallingford, CT, USA: Gaussian, Inc.; 2019.
37 Lee C, Yang W, Parr RG. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 1988; 37: 785-789.
| Crossref | Google Scholar | PubMed |
38 Becke AD. Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 1993; 98: 5648-5652.
| Crossref | Google Scholar |
39 Becke AD. A mixture of Hartree–Fock and local density-functional theories. J Chem Phys 1993; 98: 1372-1377.
| Crossref | Google Scholar |
40 Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys Chem 1994; 98: 11623-11627.
| Crossref | Google Scholar |
41 Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H–Pu. J Chem Phys 2010; 132: 154104.
| Crossref | Google Scholar | PubMed |
42 Marenich AV, Cramer CJ, Truhlar DG. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 2009; 113: 6378-6396.
| Crossref | Google Scholar | PubMed |
43 Gonzalez C, Schlegel HB. An improved algorithm for reaction path following. J Chem Phys 1989; 90: 2154-2161.
| Crossref | Google Scholar |
44 Gonzalez C, Schlegel HB. Reaction path following mass-weighted internal coordinates. J Phys Chem 1990; 94: 5523-5527.
| Crossref | Google Scholar |