

An Investigation of Five Component [3+2] Self-Assembled Cage Formation Using Amidinium···Carboxylate Hydrogen Bonds*,†
Chriso M. Thomas A B , Émer M. Foyle A B , Samuel E. Walker A and Nicholas G. White A C
A C
A Research School of Chemistry, Australian National University, Canberra, ACT 0200, Australia.
B These authors contributed equally to this work.
C Corresponding author. Email: nicholas.white@anu.edu.au
Australian Journal of Chemistry 74(11) 787-794 https://doi.org/10.1071/CH21101
Submitted: 30 April 2021 Accepted: 14 June 2021 Published: 5 July 2021
Abstract
The assembly of hydrogen bonded cages using amidinium···carboxylate hydrogen bonding interactions was investigated. A new tris-amidinium hydrogen bond donor tecton based on a tetraphenylmethane scaffold was prepared and its self-assembly with the terephthalate anion studied, and a new tricarboxylate hydrogen bond acceptor tecton was synthesised and its assembly with the 1,3-benzenebis(amidinium) hydrogen bond donor explored. In both cases, molecular modelling indicated that the formation of the cages was geometrically feasible and 1H NMR spectroscopic evidence was consistent with interactions between the components in competitive d6-DMSO solvent mixtures. DOSY NMR spectroscopy of both systems indicated that both components diffuse at the same rate as each other, and diffusion coefficients were consistent with cage formation, and with the formation of assemblies significantly larger than the individual components. An X-ray crystal structure showed that one of the assemblies did not have the desired cage structure in the solid state.
Keywords: supramolecular chemistry, cages, self–assembly, hydrogen bonding, amidinium, carboxylate, DOSY NMR, X-ray crystallography, computational modelling.
Introduction
Cage and capsule molecules and supramolecular assemblies have received significant research attention,[1] driven by possible applications in gas storage,[2] stabilisation of reactive species,[3] and catalysis.[4,5] Many of these cages are either organic cage molecules held together by covalent bonds, or metal organic cages held together by coordination bonds, however a relatively large number of cages have been assembled using non-covalent interactions such as hydrogen bonding,[6–9] while a small number have been prepared using halogen or chalcogen bonding.[10–13]
The majority of these hydrogen bonded systems have been prepared from neutral components, and contain relatively weak hydrogen bonds. However, some authors have used charge-assisted hydrogen bonds to assemble the supramolecules. Notably, in the early 2000s, Crego-Calama showed that a calix[4]arene tetra-amidinium molecule could assemble with a calix[4]arene tetra-sulfonate[14] or tetra-carboxylate[15] to give two- component capsules that were stable in methanol or water. More recently, Szumna has reported a dimeric capsule assembled from two molecules of a resorcinarene appended with four zwitterionic H-bonding groups.[16]
A subtly different approach was pioneered by Yashima and coworkers who used a crescent shaped molecule containing two amidinium motifs and assembled these around polycarboxylate anions to form five- and six-component cages.[17] Very recently, Niemeyer and coworkers have used a similar approach to form cages based on diphosphonate crescents and polyamidinium cations.[18] This kind of multicomponent approach is potentially more versatile, as several different assemblies can be assembled by varying the more easily synthesised component. Indeed, the groups of both Yashima and Niemeyer have demonstrated this by each preparing two different systems where either the readily prepared polycarboxylate (in Yashima and coworkers’ case) or polyamidinium (in Niemeyer and coworker’s case) was varied.[17,18]
Inspired by the elegant work of Yashima and coworkers,[17,19–21] Hosseini et al.,[22] and others,[23] we have investigated the use of the amidinium···carboxylate interaction in self-assembly.[24] This interaction can involve two parallel charge-assisted hydrogen bonds ( in graph set notation,[25] Fig. 1), although we note that other hydrogen bonding arrangements are also commonly observed.[26–28] These hydrogen bonds can be quite strong and survive in polar solvents or even water: indeed we have reported hydrogen bonded frameworks based on this interaction that can be formed in water,[29] and can withstand heating in water for extended periods (days).[30,31]
in graph set notation,[25] Fig. 1), although we note that other hydrogen bonding arrangements are also commonly observed.[26–28] These hydrogen bonds can be quite strong and survive in polar solvents or even water: indeed we have reported hydrogen bonded frameworks based on this interaction that can be formed in water,[29] and can withstand heating in water for extended periods (days).[30,31]
 in graph set notation[
in graph set notation[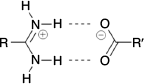
As well as investigating framework formation using the amidinium···carboxylate interaction, we have been investigating whether this interaction can be used to prepare self-assembled cages. Specifically, we envisaged designing a tris-amidinium compound with an appropriate geometry to assemble around dicarboxylate anions such as terephthalate or isophthalate.[32] Given the wide range of dicarboxylate anions available, we thought that a family of self-assembled cages of varying shapes and sizes could be prepared quite rapidly. In this work, we describe our investigation into the self-assembly of cages assembled through amidinium···carboxylate interactions.[33]
Results and Discussion
Design of Cage Systems
Before attempting to synthesise our cage systems, we used semi-empirical calculations with PM6 parameters[34] to determine if our proposed systems were geometrically feasible. We initially proposed the tris-amidinium compounds 13+ and 23+ as these appeared relatively easy to synthesise and appeared to have the correct geometry to assemble into cages with terephthalate (TP2–) and isophthalate (IP2–) anions, respectively. As can be seen in Fig. 2, the calculations suggest that the geometries of the components are suitable for cage formation.

|
Synthesis of Tectons
The tetraphenylmethane based tecton 13+ was synthesised from the known tris-alkyne 3,[35] as shown in Scheme 1. Sonogashira coupling of 3 with 3-iodobenzonitrile gave tris-nitrile 4; subsequent reaction with lithium bis(trimethylsilyl)amide in THF followed by workup with ethanolic HCl gave 1·Cl3 in good yield. As chloride anions are potentially coordinating and may interfere with the self-assembly process, we exchanged these for non-coordinating BPh4– anions in quantitative yield.

|
We attempted to prepare 23+ in an analogous manner using a Suzuki coupling of tris-bromomethyl compound 5[36] and 4-cyanophenylboronic acid in conditions similar to those reported by Kotha et al.[37] We were able to prepare the new tris-nitrile 6, albeit in relatively low yield (27 %). However, attempts to convert this into the tris-amidinium 23+ were unsuccessful with a mixture of products being observed: mass spectrometry and NMR spectroscopy indicated that some product was formed but we were not able to isolate this or drive the reaction to completion.
Given our inability to synthesise 23+, we next investigated whether it was possible to prepare the tris-carboxylate 73–. We reasoned that it would be possible to synthesise a cage from this and the bis-benzamidinium 82+, which would in effect be the ‘reverse’ of the initially proposed 22·IP3. Semi-empirical geometry optimisations suggested that this reverse cage, 83·72, was geometrically feasible (Fig. S21, Supplementary Material). The reaction of 5 with 4-methoxycarbonylphenylboronic acid gave the tris-ester 9 in modest yield (26 %), which was subsequently hydrolysed to give the tris-carboxylic acid 73H in good yield (81 %). This was then converted into the soluble tetrabutylammonium (TBA) salt, TBA3·7, using TBA·OH (Scheme 2).

|
Investigation of Cage Formation in Solution
13+/TP2–
We initially investigated the formation of cages from 13+ and TP2– anions using 1H NMR spectroscopy. Mixing solutions of 1·(BPh4)3 and TBA2·TP in d6-DMSO resulted in only small shifts (< 0.05 ppm) of the resonances of 13+, although significant peak broadening was observed (Fig. S15, Supplementary Material). The small shifts are perhaps not unexpected given the distance between the amidinium/carboxylate groups and the nearest C–H proton. Importantly, DOSY NMR spectroscopy shows that 13+ and TP2– diffuse at the same rate, consistent with them forming a supramolecular assembly (Fig. 3 and Fig. S16, Supplementary Material). These experiments indicated a diffusion coefficient of 0.75 × 10−10 m2 s−1, consistent with a species having a solvodynamic radius of 15 Å. The calculated structure of 12·TP3 (Fig. 2) is clearly non-spherical, but has approximate dimensions of 40 × 18 Å so a calculated radius of 15 Å (i.e. diameter of 30 Å) is consistent with cage formation.[38] Notably, this value is significantly larger than that recorded for 1·(BPh4)3 in the absence of the TP2– anion (diffusion coefficient for 1·(BPh4)3 = 1.2 × 10−10 m2 s−1, solvodynamic radius = 9.5 Å, Fig. S17, Supplementary Material). We attempted to grow single crystals to enable us to gain information about the solid state structure of the assembly using X-ray crystallography, but despite numerous attempts we were unable to obtain X-ray quality crystals.

|
82+/73–
We initially studied cage formation from the tricarboxylate 73– and bis-amidinium compound 82+ using 1H NMR spectroscopy in d6-DMSO. As shown in Fig. 4, significant shifts for the C–H peaks of both compounds are observed when solutions of the compounds are mixed. Downfield shifts of ~0.15 ppm are observed for both the peak adjacent to the carboxylate group in 73– and the peak between the amidinium groups in 82+, while smaller upfield shifts are observed for the other amidinium proton resonances. Significant broadening of the resonances corresponding to the amidinium N–H groups of 82+ is observed upon mixing with 73– such that these peaks cannot be resolved. DOSY NMR shows that the peaks corresponding to both 82+ and 73– diffuse at the same rate, with a diffusion coefficient of 1.0 × 10−10 m2 s−1 (Fig. S18, Supplementary Material), corresponding to a solvodynamic radius of 11 Å, which is again consistent with cage formation (approximate dimensions: 26 × 18 Å). The solvodynamic radius of the mixture of 73– and 82+ is significantly larger than that of TBA3·7 (diffusion coefficient = 1.5 × 10−10 m2 s−1, solvodynamic radius = 7.4 Å, Fig. S19, Supplementary Material).

|
Crystal Structure of 83·72
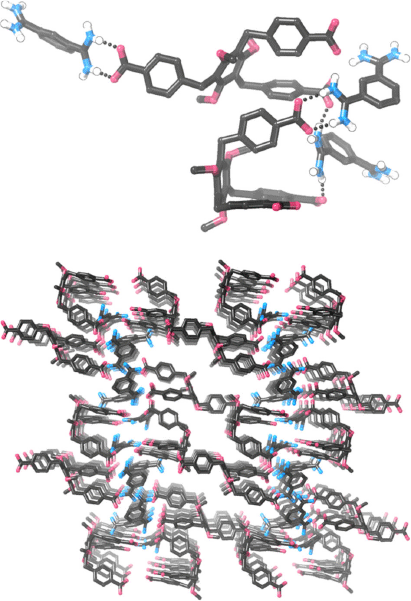
We carried out numerous experiments to try and obtain single crystals of either 12·TP3 or 83·72. These systems were resistant to crystallisation but we were eventually able to obtain very small single crystals of 83·72. To minimise the chance of obtaining crystals of relatively insoluble TBA·BPh4, which would be a possible by-product during formation of 83·72, we mixed TBA3·7 and the bis-amidinium molecule 8·(BPh4)2 in acetone, which precipitated 83·72 free from either TBA+ or BPh4– ions (as revealed by 1H NMR spectroscopy). After trialling several solvent/anti-solvent conditions, we were able to obtain crystals by diffusing pentane vapour into a DMSO solution of this compound. Crystals were small and weakly diffracting, but with the use of synchrotron radiation it was possible to obtain low quality data and determine the structural connectivity. Despite numerous attempts, it was not possible to resolve the apparently disordered hexyloxy chains of 73+, and so PLATON-SQUEEZE[39] was used to include these, as well as areas that appear to correspond to disordered solvent molecules, in the model. While detailed inferences about bond lengths/angles are not appropriate given the relatively poor quality of the data, it is clear that in the crystalline state, 83·72 does not exist as a hydrogen bonded cage (Fig. 5). A variety of hydrogen bonding arrangements are seen, including the desired ‘paired’  hydrogen bonding arrangement as well as others. It is notable that of the two molecules of 73– in the asymmetric unit, the expected (and desired) scaffolding[40] of the 1,3,5-trialkoxybenzene motif has not occurred and this leads to one of the carboxylate groups pointing in the opposite direction to the others, precluding cage formation. We were also able to obtain crystals by diffusing pentane vapour into a DMSO/methanol solution of 83·72; in this case crystals again required synchrotron radiation, but data were of even lower quality. While a stable model could not be constructed, the molecular connectivity seems very similar to that shown in Fig. 5 (i.e. the crystals appear to be isostructural, but not isomorphous).
hydrogen bonding arrangement as well as others. It is notable that of the two molecules of 73– in the asymmetric unit, the expected (and desired) scaffolding[40] of the 1,3,5-trialkoxybenzene motif has not occurred and this leads to one of the carboxylate groups pointing in the opposite direction to the others, precluding cage formation. We were also able to obtain crystals by diffusing pentane vapour into a DMSO/methanol solution of 83·72; in this case crystals again required synchrotron radiation, but data were of even lower quality. While a stable model could not be constructed, the molecular connectivity seems very similar to that shown in Fig. 5 (i.e. the crystals appear to be isostructural, but not isomorphous).
|
Discussion
DOSY NMR results are consistent with formation of the targeted self-assembled cages for both 12·TP3 and 83·72. That is, the radii of the assemblies calculated from DOSY NMR experiments match well with the modelled cage structures, which are the smallest non-strained structures we can envisage that obey the principle of maximal site occupancy.[41] While we cannot rule out the formation of small amounts of other species that are in equilibrium with the cages, the observed diffusion coefficients, and the fact that the peaks for both amidinium and carboxylate species diffuse at the same rate, are both consistent with cage assembly. We attempted to further probe the solution assembly of these systems by electrospray ionisation mass spectrometry, but did not obtain conclusive evidence for the cages, or any other obvious aggregate. A similar finding was observed by Niemeyer and coworkers who studied somewhat related hydrogen bonded cages.[18,42]
It is noteworthy that these cages assemble in the competitive and highly polar solvent d6-DMSO. Indeed, we found that the cages showed very poor solubility in all but the most polar solvents. This is in contrast to the multi-component systems reported by the groups of Yashima[17] and Niemeyer,[18] which were prepared in CDCl3 or 4:1 CDCl3/CD3OD, respectively. We note that both of these systems contained N-substituted amidinium groups, where each amidinium nitrogen atom contained one hydrogen atom and one alkyl substituent, where our systems contain no solubilising substituents at the amidinium nitrogen atoms (Fig. 6). While there are synthetic challenges associated with introducing alkyl substituents, the ability to use less polar solvents to assemble the cages is clearly advantageous. We designed 73– to include three hexyloxy chains attached to the central benzene ring to aid solubility, but this was clearly not sufficient.

|
X-Ray crystallography of single crystals of 83·72 did not show the expected cage structure, which we attribute to crystallisation favouring a polymeric structure containing relatively little solvent in preference to a porous cage that would be difficult to pack closely. We note that we have previously attempted to form self-assembled hexagonal macrocycles from simple benzenebis-amidinium and benzenedicarboxylate components, but in that case observed little evidence for hexagon formation.[28] It is interesting that in this related system we do appear to be able to form relatively well defined self-assembled structures. We attribute this to the smaller number of components required to self-assemble as well as their higher charge.
Conclusions
A new tris-amidinium and a new tricarboxylate hydrogen bonding tecton were synthesised and their assembly with an appropriate dicarboxylate or bis-amidinium to form [3+2] self-assembled cages was investigated. While the low solubility of the cages meant we were unable to study this process in solvents other than DMSO, 1H and DOSY NMR spectroscopy data in this solvent were consistent with the formation of the target cages.
Experimental
General Remarks
The tetraphenyl tris-alkyne 3,[35] the tris(hexyloxy)benzene tris-bromomethyl compound 5,[36] bis-amidinium 8·(BPh4)2,[28] TBA2·TP,[43] and TBA2·IP[28] were prepared as previously described. Dry THF was distilled over sodium before use. Other chemicals were brought from commercial suppliers and used as received. NMR spectra were recorded on Bruker Avance 400 spectrometers and are referenced to the residual solvent signal.[44] Electrospray ionisation mass spectrometry data were acquired on a Micromass Waters ZMD spectrometer.
Tetraphenyl Tris-Nitrile 4
A solution of 3-iodobenzonitrile (0.18 g, 0.77 mmol), Pd(PPh3)4 (0.015 g, 0.013 mmol), and CuI (0.0025 g, 0.013 mmol) in triethylamine (11 mL) was cooled to 0°C. A solution of the tris-alkyne 3 (0.095 g, 0.22 mmol) in THF (6 mL) was added dropwise, and then the reaction was warmed to room temperature and stirred under N2 overnight. The solvent was removed under reduced pressure and the resulting solid was washed with methanol (10 mL). The remaining residue was dissolved in dichloromethane (50 mL), washed with water (30 mL), and dried (MgSO4). The solvent was removed under reduced pressure and purified by column chromatography (eluent: 1:1 dichloromethane/pet. spirits) to give 4 as a yellow powder. Yield: 0.13 g (0.18 mmol, 82 %).
δH (400 MHz, CDCl3) 7.79 (s, 3H), 7.72 (d, J 8.2, 3H), 7.61 (d, J 8.2, 3H), 7.44–7.48 (m, 9H), 7.22 (d, J 8.0, 6H), 7.09 (d, J 8.6, 2H), 6.83 (d, J 8.6, 2H), 3.81 (s, 3H). δC (101 MHz, CDCl3) 158.0, 147.0, 140.3, 137.7, 137.4, 132.1, 131.2, 131.1, 130.9, 130.0, 125.5, 120.8, 113.3, 93.8, 90.5, 88.1, 64.4, 55.4 (1 peak not detected/overlapping). HR ESI-MS (+ve) m/z 748.2385; calcd for [C53H31N3O·Na]+ 748.2365 Da.
Tetraphenyl Tris-Amidinium 1·Cl3
A solution of 4 (0.050 g, 0.069 mmol) in dry THF (10 mL) was cooled to –78°C; under a nitrogen atmosphere a solution of LiHMDS in THF (1.0 M, 0.31 mL, 0.31 mmol) was added dropwise. The reaction mixture was allowed to warm to room temperature and stirred overnight under a nitrogen atmosphere. The resulting golden-yellow solution was cooled to 0°C and ethanolic HCl (prepared by cautiously adding 1 mL of acetyl chloride to 10 mL of ethanol) was added and stirred for 15 min. The resulting suspension was taken to dryness under reduced pressure and the solid suspended in water (10 mL). It was centrifuged for 45 min, the supernatant decanted, and the solid thoroughly air-dried to give 1·Cl3 as a yellow powder. Yield: 0.040 g (0.046 mmol, 67 %).
δH (400 MHz, d6-DMSO) 9.40 (br. s, 12 H), 8.00 (s, 3H), 7.84–7.89 (m, 6H), 7.69 (dd, J 7.6, 7.4, 3H), 7.57 (d, J 7.8, 6H), 7.23 (d, J 7.8, 6H), 7.06 (d, J 8.2, 2H), 6.94 (d, J 8.2, 2H), 3.75 (s, 3H). δC (101 MHz, d6-DMSO) 165.6, 158.2, 147.6, 137.5, 136.7, 132.1, 131.8, 131.4, 130.2, 129.6, 129.0, 123.5, 120.3, 114.2, 91.1, 88.8, 64.5, 55.7 (1 peak not detected/overlapping). HR ESI-MS (+ve) m/z 259.7830; calcd for [C53H43N6O]3+ (13+) 259.7833 Da.
Tetraphenyl Tris-Amidinium 1·(BPh4)3
A solution of NaBPh4 (0.019 g, 0.056 mmol) in water (2 mL) was mixed with a suspension of 1·Cl3 (0.015 g, 0.016 mmol) in water (3 mL) and the suspension sonicated for 30 min. The resulting solid was isolated by filtration, washed with water (2 × 10 mL), and dried under vacuum to give 1·(BPh4)3 as a pale orange powder in quantitative yield. Yield: (0.028 g, 0.016 mmol, 100 %).
δH (400 MHz, d6-DMSO) 9.26 (br. s, 12 H), 7.97 (s, 3H), 7.88 (d, J 7.8, 3H), 7.83 (d, J 7.9, 3H), 7.67 (dd, J 7.9, 7.8, 3H), 7.57 (d, J 8.0, 6H), 7.23 (d, J 8.0, 6H), 7.17 (br. s, 12H), 7.06 (d, J 8.5, 2H), 6.89–6.94 (m, 14H), 6.79 (dd, J 7.2, 7.2, 6 H), 3.75 (s, 3H). δC (101 MHz, d6-DMSO) 165.3, 163.1–164.6 (m), 158.0, 147.4, 137.3, 136.6, 136.0, 131.9, 131.6, 131.2, 130.1, 129.6, 128.8, 125.8, 123.3, 122.0, 120.1, 114.0, 90.0, 88.6, 64.4, 55.6. ESI MS (+ve) m/z 259.7; calcd for [C53H43N6O]3+ (13+) 259.8 Da.
Tris(hexyloxy)benzene Tris-Nitrile 6
To a solution of 5 (0.400 g, 0.608 mmol, 1 equiv.) in THF (70 mL) was added 4-cyanophenylboronic acid (0.352 g, 2.40 mmol, 3.91 equiv.) and K2CO3 (aq) (2.0 M, 9.4 mL), and the resultant pale yellow solution was deoxygenated with bubbling N2 for 20 min. After this time, Pd(PPh3)4 (0.103 g, 0.094 mmol, 0.154 equiv.) was added and the solution was heated to 75°C for 48 h under N2, during which time the solution turned black. After this, the solution was cooled to room temperature and water (100 mL) was added. This was then extracted with ethyl acetate (3 × 100 mL), and the resultant organic layer was washed with water (100 mL) and brine (100 mL), and then dried (MgSO4). The solution was then concentrated under vacuum to leave the crude product as a black oily residue, which was purified via column chromatography (4:1 pet. spirits/EtOAc) to give the product as a colourless oil. Yield 0.119 g (0.164 mmol, 27 %).
δH (400 MHz, CDCl3) 7.53 (d, J 8.3, 6H), 7.23 (d, J 8.3, 6H), 4.04 (s, 6H), 3.52 (t, J 6.6, 6H), 1.54–1.58 (m, 6H), 1.18–1.24 (m, 18H), 0.83 (t, J 6.9, 9H). δC (101 MHz, CDCl3) 156.9, 147.1, 132.2, 128.9, 123.1, 119.1, 109.9, 74.4, 31.6, 30.9, 30.2, 25.7, 22.6, 14.1. HR ESI MS (+ve) m/z 724.4472; calcd for [C48H57N3O3·H]+ 724.4478 Da.
Tris(hexyloxy)benzene Tris-Ester 9
To a solution of 5 (0.496 g, 0.734 mmol, 1 equiv.) in THF (87 mL) was added 4-methoxycarbonylphenylboronic acid (0.531 g, 2.97 mmol, 4.0 equiv.) and K2CO3 (aq) (2.0 M, 11 mL) to produce a pale yellow solution. The solution was deoxygenated with bubbling N2 for 20 min and then Pd(PPh3)4 (0.131 g, 0.109 mmol, 0.149 equiv.) was added. The solution was heated at 75°C for 48 h under N2, during which time it turned black. The mixture was cooled to room temperature and water (100 mL) was added. The solution was extracted with ethyl acetate (3 × 75 mL), and the combined organic layers were washed with water (75 mL) and brine (75 mL), and then dried (MgSO4). The solution was concentrated under vacuum to give the crude product as a black oily solid, which was purified by column chromatography (gradient: 5–10 % EtOAc in pet. spirits) to give the product as a colourless oil. Yield 0.156 g (0.189 mmol, 26 %).
δH (400 NMR, CDCl3) 7.91 (d, J 8.2, 6H), 7.21 (d, J 8.2, 6H), 4.06 (s, 6H), 3.89 (s, 9H), 3.51 (t, J 6.6, 6H), 1.54 dt, J 6.6, 6.6, 6H), 1.08–1.25 (m, 18H), 0.80 (t, J 7.0, 9H). δC (101 MHz, CDCl3) 167.3, 156.8, 147.3, 129.7, 128.2, 127.9, 123.5, 74.2, 52.1, 31.7, 30.9, 30.2, 25.7, 22.7, 14.1. HR ESI MS (+ve) m/z 823.4792; calcd for [C51H66O9·H]+ 823.4785 Da.
Tris(hexyloxy)benzene Tris-Carboxylic Acid 73H
NaOH (aq) (2.0 M, 2 mL) was added to a solution of 9 (0.110 g, 0.133 mmol) in methanol (14 mL) and THF (15 mL). The yellow solution was then refluxed for 24 h under N2, during which time it turned a reddish colour. After cooling to room temperature the solution was concentrated under vacuum until the volume of solvent was reduced by approximately half. Concentrated HCl (aq) was then added dropwise until no more solid formed (~1 mL). The resulting white powder was isolated via filtration and washed with water (3 × 10 mL) and diethyl ether (1 × 10 mL) and dried under vacuum to give 73H. Yield 0.082 g (0.105 mmol, 81 %).
δH (400 MHz, d6-DMSO) 12.70 (br. s, 3H), 7.82 (d, J 8.2, 6H), 7.20 (d, J 8.2, 6H,), 4.03 (s, 6H), 3.54 (t, J 6.7, 6H), 1.47 (dt, J 6.7, 6.7, 6H), 1.04–1.17 (m, 18H), 0.74 (t, J 6.9, 9H). δC (101 MHz, d6-DMSO) 167.3, 156.3, 146.6, 129.3, 128.3, 127.9, 123.0, 73.5, 30.9, 30.1, 29.4, 24.9, 22.0, 13.8. HR ESI MS (–ve) m/z 779.4156; calcd for [C48H68O9]– 779.4159 Da.
Tris(hexyloxy)benzene Tris-Carboxylate TBA3·7
The tricarboxylic acid 73H (0.060 g, 0.077 mmol, 1 equiv.) was suspended in ethanol (5 mL) and a solution of TBA·OH in methanol (1.0 M, 0.23 mL, 0.23 mmol, 3.0 equiv.) was added causing the solid to dissolve. The resultant solution was stirred at room temperature for 1 h under N2. The solution was then concentrated under vacuum to give the product as a pale yellow oil. Yield 0.116 g (0.077 mmol, 100 %).
δH (400 NMR,CDCl3) 7.84 (d, J 7.9, 6H), 7.05 (d, J 7.9, 6H), 3.93 (s, 6H), 3.56 (t, J 6.4, 6H), 3.13–3.24 (m, 24H), 1.53–1.58 (m, 6H), 1.43–1.45 (m, 24H), 1.20–1.28 (m, 42H) 0.79–0.84 (m, 45H). δC (101 MHz, CDCl3) 171.9, 156.5, 142.9, 137.3, 129.4, 127.3, 124.2, 100.2, 74.1, 58.7, 31.9, 30.5, 25.8, 24.1, 22.7, 19.8, 14.2, 13.8. HR ESI MS (–ve) m/z 509.8429; calcd for [C64H93O9N]2– (TBA·72–) 509.8420 Da.
X-Ray Crystallography
Data were collected on the MX2 beamline[45] at the Australian Synchrotron at 100 K. Raw frame data (including data reduction, interframe scaling, and unit cell refinement) were processed using XDS.[46] The structure was solved using Superflip[47] and refined using full-matrix least-squares on F2 within the Crystals suite.[48] All non-hydrogen atoms were refined with anisotropic displacement parameters. A thermal ellipsoid plot and crystallographic data table are included in the Supplementary Material and the data in CIF format have been uploaded to the Cambridge Structural Database (CCDC: 2080276).
Despite numerous attempts to grow high quality crystals, only small and weakly diffracting crystals could be obtained. It was not possible to resolve the hexyloxy chains, and so these were represented with an oxygen and carbon atom with the remaining electron density included in the model using PLATON-SQUEEZE.[39] It was felt that this was a more honest representation of the structure than ‘constructing’ hexyl chains at arbitrary locations using copious amounts of crystallographic restraints. While the resulting structural model is of relatively low quality, it unambiguously allows the determination of the structural connectivity.
Calculations
Gas phase energy minimisations of 12·TP3 and 22·IP3 were conducted using PM6 parameters[34] within Spartan.[49] The gas phase energy minimisation of 83·72 is described in the Supplementary Material. Atomic coordinates for all three structures are provided in the Supplementary Material.
Supplementary Material
1H and 13C NMR spectra for new compounds as well as details of studies of cage self–assembly, X-ray crystallography, and computational modelling experiments are available on the Journal’s website.
Data Availability Statement
The data that support this study are available in the article and accompanying online Supplementary Material.
Conflicts of Interest
The authors declare no conflicts of interest.
Declaration of Funding
The authors thank the Australian Research Council (Australian Government RTP Scholarship to EMF and DE170100200 to NGW) and the ANU Summer Scholars Program (SEW) for funding.
Acknowledgements
The authors thank Tom Anglim Lagones and Esther van Praag for preliminary experiments on related systems, Prof. Christian Doonan for providing an initial sample of 3, and Chris Blake and Dr Daniel Preston for assistance with DOSY NMR spectroscopy. Parts of this work were conducted using the MX2 Beamline of the Australian Synchrotron.
References
[1] M. Yoshizawa, J. K. Klosterman, M. Fujita, Angew. Chem. Int. Ed. 2009, 48, 3418.| Crossref | GoogleScholarGoogle Scholar |
[2] T. Hasell, A. I. Cooper, Nat. Rev. Mater. 2016, 1, 16053.
| Crossref | GoogleScholarGoogle Scholar |
[3] A. Galan, P. Ballester, Chem. Soc. Rev. 2016, 45, 1720.
| Crossref | GoogleScholarGoogle Scholar | 26797259PubMed |
[4] V. Mouarrawis, R. Plessius, J. I. van der Vlugt, J. N. H. Reek, Front Chem. 2018, 6, 623.
| Crossref | GoogleScholarGoogle Scholar | 30622940PubMed |
[5] Y. Xue, X. Hang, J. Ding, B. Li, R. Zhu, H. Pang, Q. Xu, Coord. Chem. Rev. 2021, 430, 213656.
| Crossref | GoogleScholarGoogle Scholar |
[6] L. R. MacGillivray, J. L. Atwood, Nature 1997, 389, 469.
| Crossref | GoogleScholarGoogle Scholar |
[7] T. Heinz, D. M. Rudkevich, J. Rebek, Nature 1998, 394, 764.
| Crossref | GoogleScholarGoogle Scholar |
[8] Y. Liu, C. Hu, A. Comotti, M. D. Ward, Science 2011, 333, 436.
| Crossref | GoogleScholarGoogle Scholar | 21778396PubMed |
[9] L. Adriaenssens, P. Ballester, Chem. Soc. Rev. 2013, 42, 3261.
| Crossref | GoogleScholarGoogle Scholar | 23321897PubMed |
[10] O. Dumele, N. Trapp, F. Diederich, Angew. Chem. Int. Ed. 2015, 54, 12339.
| Crossref | GoogleScholarGoogle Scholar |
[11] L. Turunen, U. Warzok, R. Puttreddy, N. K. Beyeh, C. A. Schalley, K. Rissanen, Angew. Chem. Int. Ed. 2016, 55, 14033.
| Crossref | GoogleScholarGoogle Scholar |
[12] L.-J. Riwar, N. Trapp, K. Root, R. Zenobi, F. Diederich, Angew. Chem. Int. Ed. 2018, 57, 17259.
| Crossref | GoogleScholarGoogle Scholar |
[13] Y.-J. Zhu, Y. Gao, M.-M. Tang, J. Rebek, Y. Yu, Chem. Commun. 2021, 57, 1543.
| Crossref | GoogleScholarGoogle Scholar |
[14] F. Corbellini, R. Fiammengo, P. Timmerman, M. Crego-Calama, K. Versluis, A. J. R. Heck, I. Luyten, D. N. Reinhoudt, J. Am. Chem. Soc. 2002, 124, 6569.
| Crossref | GoogleScholarGoogle Scholar | 12047176PubMed |
[15] F. Corbellini, L. Di Costanzo, M. Crego-Calama, S. Geremia, D. N. Reinhoudt, J. Am. Chem. Soc. 2003, 125, 9946.
| Crossref | GoogleScholarGoogle Scholar | 12914457PubMed |
[16] B. Kuberski, A. Szumna, Chem. Commun. 2009, 1959.
| Crossref | GoogleScholarGoogle Scholar |
[17] H. Katagiri, Y. Tanaka, Y. Furusho, E. Yashima, Angew. Chem. Int. Ed. 2007, 46, 2435.
| Crossref | GoogleScholarGoogle Scholar |
[18] M. Kohlhaas, M. Zähres, C. Mayer, M. Engeser, C. Merten, J. Niemeyer, Chem. Commun. 2019, 55, 3298.
| Crossref | GoogleScholarGoogle Scholar |
[19] Y. Tanaka, H. Katagiri, Y. Furusho, E. Yashima, Angew. Chem. Int. Ed. 2005, 44, 3867.
| Crossref | GoogleScholarGoogle Scholar |
[20] M. Ikeda, Y. Tanaka, T. Hasegawa, Y. Furusho, E. Yashima, J. Am. Chem. Soc. 2006, 128, 6806.
| Crossref | GoogleScholarGoogle Scholar | 16719458PubMed |
[21] Y. Nakatani, Y. Furusho, E. Yashima, Angew. Chem. Int. Ed. 2010, 49, 5463.
| Crossref | GoogleScholarGoogle Scholar |
[22] M. W. Hosseini, R. Ruppert, P. Schaeffer, A. De Cian, N. Kyritsakas, J. Fischer, J. Chem. Soc. Chem. Commun. 1994, 2135.
| Crossref | GoogleScholarGoogle Scholar |
[23] T. Kusukawa, K. Matsumoto, H. Nakamura, W. Iizuka, K. Toyama, S. Takeshita, Org. Biomol. Chem. 2013, 11, 3692.
| Crossref | GoogleScholarGoogle Scholar | 23625021PubMed |
[24] N. G. White, Dalton Trans. 2019, 48, 7062.
| Crossref | GoogleScholarGoogle Scholar | 30667427PubMed |
[25] J. Bernstein, R. E. Davis, L. Shimoni, N.-L. Chang, Angew. Chem. Int. Ed. Engl. 1995, 34, 1555.
| Crossref | GoogleScholarGoogle Scholar |
[26] N. Kamali, M. Aljohani, P. McArdle, A. Erxleben, Cryst. Growth Des. 2015, 15, 3905.
| Crossref | GoogleScholarGoogle Scholar |
[27] L. Pop, N. D. Hadade, A. van der Lee, M. Barboiu, I. Grosu, Y.-M. Legrand, Cryst. Growth Des. 2016, 16, 3271.
| Crossref | GoogleScholarGoogle Scholar |
[28] M. Thomas, T. Anglim Lagones, M. Judd, M. Morshedi, M. L. O’Mara, N. G. White, Chem. Asian J. 2017, 12, 1587.
| Crossref | GoogleScholarGoogle Scholar | 28544634PubMed |
[29] M. Morshedi, M. Thomas, A. Tarzia, C. J. Doonan, N. G. White, Chem. Sci. 2017, 8, 3019.
| Crossref | GoogleScholarGoogle Scholar | 28451369PubMed |
[30] S. A. Boer, M. Morshedi, A. Tarzia, C. J. Doonan, N. G. White, Chem. – Eur. J. 2019, 25, 10006.
| Crossref | GoogleScholarGoogle Scholar | 31267583PubMed |
[31] J. Nicks, S. A. Boer, N. G. White, J. A. Foster, Chem. Sci. 2021, 12, 3322.
| Crossref | GoogleScholarGoogle Scholar | 34164102PubMed |
[32] For a review of the supramolecular chemistry of these species in an anion recognition context, see: S. M. Butler, K. A. Jolliffe, Org. Biomol. Chem. 2020, 18, 8236.
| Crossref | GoogleScholarGoogle Scholar | 33001119PubMed |
[33] As noted by a referee, carboxylates are relatively basic in DMSO (e.g. pKa of benzoic acid = 11.1 in DMSO; F. G. Bordwell, Acc. Chem. Res. 1988, 21, 456–463) and it is conceivable that some proton transfer from the amidinium to carboxylate groups could be envisaged. While in this study, the N–H peaks broadened upon addition of a carboxylate such that they could not be well resolved, other authors (e.g. refs [19] and [23]) have shown convincingly that the amidinium···carboxylate form dominates in similar systems, and indeed that mixing benzamidines and benzoic acids in DMSO results in proton transfer to give the amidinium···carboxylate form.
[34] J. J. P. Stewart, J. Mol. Model. 2007, 13, 1173.
| Crossref | GoogleScholarGoogle Scholar |
[35] A. Avellaneda, P. Valente, A. Burgun, J. D. Evans, A. W. Markwell-Heys, D. Rankine, D. J. Nielsen, M. R. Hill, C. J. Sumby, C. J. Doonan, Angew. Chem. Int. Ed. 2013, 52, 3746.
| Crossref | GoogleScholarGoogle Scholar |
[36] É. M. Foyle, N. G. White, CrystEngComm 2020, 22, 2526.
| Crossref | GoogleScholarGoogle Scholar |
[37] S. Kotha, K. Mandal, K. K. Arora, V. R. Pedireddi, Adv. Synth. Catal. 2005, 347, 1215.
| Crossref | GoogleScholarGoogle Scholar |
[38] We note that there are considerable difficulties with determining solvodynamic radii for non-spherical species. As well as using the Stokes–Einstein equation, we have calculated the diffusion values for the supramolecular assemblies and for 13+ or 73– alone, relative to TBA+ or BPh4– as internal references. In each case, the increase in size calculated using this method is the same within 10 % as the value calculated using the Stokes–Einstein equation (see Supplementary Material for more details).
[39] A. L. Spek, Acta Crystallogr. 2015, C71, 9.
[40] X. Wang, F. Hof, Beilstein J. Org. Chem. 2012, 8, 1.
| Crossref | GoogleScholarGoogle Scholar | 22423267PubMed |
[41] R. Krämer, J.-M. Lehn, A. Marquis-Rigault, Proc. Natl. Acad. Sci. USA 1993, 90, 5394.
| Crossref | GoogleScholarGoogle Scholar | 11607405PubMed |
[42] Niemeyer and coworkers (ref. [18]) suggested that the failure to obtain mass spectrometric evidence for related self–assembled cages may be due to either the dissociation of the assemblies at the very low concentrations used for mass spectrometry, or to protonation/deprotonation of the neutral capsules during the ESI process destabilising the hydrogen bonds and resulting in fragmentation. We suggest similar processes are at play in our system.
[43] A. J. Lowe, F. M. Pfeffer, Chem. Commun. 2008, 1871.
| Crossref | GoogleScholarGoogle Scholar |
[44] H. E. Gottlieb, V. Kotlyar, A. Nudelman, J. Org. Chem. 1997, 62, 7512.
| Crossref | GoogleScholarGoogle Scholar | 11671879PubMed |
[45] D. Aragao, J. Aishima, H. Cherukuvada, R. Clarken, M. Clift, N. P. Cowieson, D. J. Ericsson, C. L. Gee, S. Macedo, N. Mudie, S. Panjikar, J. R. Price, A. Riboldi-Tunnicliffe, R. Rostan, R. Williamson, T. T. Caradoc-Davies, J. Synchrotron Radiat. 2018, 25, 885.
| Crossref | GoogleScholarGoogle Scholar | 29714201PubMed |
[46] W. Kabsch, J. Appl. Cryst. 1993, 26, 795.
| Crossref | GoogleScholarGoogle Scholar |
[47] L. Palatinus, G. Chapuis, J. Appl. Cryst. 2007, 40, 786.
| Crossref | GoogleScholarGoogle Scholar |
[48] P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout, D. J. Watkin, J. Appl. Cryst. 2003, 36, 1487.
| Crossref | GoogleScholarGoogle Scholar |
[49] Spartan 18 2018 (Wavefunction Inc.: Irvine, CA).
* Nicholas G. White is the joint winner of the 2020 Rennie Memorial Medal.
† A previous version of this manuscript has been deposited on the Chemrxiv server, https://doi.org/ 10.26434/chemrxiv.14515845.