

Spectroelectrochemistry: A Powerful Tool for Studying Fundamental Properties and Emerging Applications of Solid-State Materials Including Metal–Organic Frameworks
Deanna M. D’Alessandro A C and Pavel M. Usov A B
A C and Pavel M. Usov A B
A School of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia.
B Department of Chemistry, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo, 152-8550 Japan.
C Corresponding author. Email: deanna.dalessandro@sydney.edu.au

Deanna obtained her BSc in chemistry, physics and mathematics from James Cook University followed by PhD research with Em/Prof. Richard Keene which received the 2006 RACI Cornforth Medal and a 2007 IUPAC Prize for Young Chemists. Following postdoctoral work with Prof. Jeff Long at UC Berkeley (2007–2009) as the Dow Chemical Company Fellow (American-Australian Association) and an 1851 Fellow, she built her independent research exploring emergent electronic phenomena in framework materials. She has held a L’Oréal Australia for Women in Science Fellowship, ARC QEII and Future Fellowships, and was the recipient of the 2017 LeFévre Medal (Australian Academy of Science). |

Pavel completed his BSc in chemistry at the University of Adelaide (2010) followed by Honours Class I (2011) and PhD research (2015) in the development of spectroelectrochemical methods for framework materials at the University of Sydney with A/Prof. Deanna D’Alessandro. Following a postdoctoral position with A/Prof. Amanda Morris at Virginia Tech in the USA in the area of electrocatalytic framework materials, he received a Japan Society for the Promotion of Science (JSPS) Fellowship at the Tokyo Institute of Technology, Japan, to work with Prof. Masaki Kawano. Pavel is currently a Specially Appointed Assistant Professor in the same department. |
Australian Journal of Chemistry 74(2) 77-93 https://doi.org/10.1071/CH20301
Submitted: 9 October 2020 Accepted: 30 November 2020 Published: 22 January 2021
Journal Compilation © CSIRO 2021 Open Access CC BY
Abstract
Spectroelectrochemistry (SEC) encompasses a broad suite of electroanalytical techniques where electrochemistry is coupled with various spectroscopic methods. This powerful and versatile array of methods is characterised as in situ, where a fundamental property is measured in real time as the redox state is varied through an applied voltage. SEC has a long and rich history and has proved highly valuable for discerning mechanistic aspects of redox reactions that underpin the function of biological, chemical, and physical systems in the solid and solution states, as well as in thin films and even in single molecules. This perspective article highlights the state of the art in solid-state SEC (ultraviolet–visible–near-infrared, infrared, Raman, photoluminescence, electron paramagnetic resonance, and X-ray absorption spectroscopy) relevant to interrogating solid state materials, particularly those in the burgeoning field of metal–organic frameworks (MOFs). Emphasis is on developments in the field over the past 10 years and prospects for application of SEC techniques to probing fundamental aspects of MOFs and MOF-derived materials, along with their emerging applications in next-generation technologies for energy storage and transformation. Along with informing the already expert practitioner of SEC, this article provides some guidance for researchers interested in entering the field.
Introduction
Electroanalytical methods such as voltammetry techniques have been the primary tool for interrogation of redox-active compounds with a well-established theoretical framework to interpret the data; nevertheless, the nature of the electrogenerated species can be difficult to elucidate from electrochemical techniques alone. The integration of electrochemistry with different spectroscopic techniques has thus been actively pursued to gain information in ‘real time’ regarding the effects of a redox transformation, including the mechanism and kinetics of a reaction, by probing both the substrate and products. Spectroelectrochemistry (SEC) has a long and rich history across the scientific fields from chemistry to biology and physics since its first description in 1964;[1,2] the historical context has been elegantly reviewed by Dunsch[3] and Kuwana.[2] SEC techniques underpin many areas of industrial importance in the pharmaceutical, food, clinical, environmental, and catalysis fields, and SEC has been an important technique in the development of chemical compounds such as inks and dyestuffs that have been prized industrially for centuries.[4,5] In contemporary research, SEC techniques have enormous utility for addressing materials properties, such as energy storage and transformation, that underpin solutions to global scientific and technological challenges.[6]
SEC techniques have been successfully applied to interrogate a wide range of electroactive compounds, with over 5000 literature reports on the topic, including most prominently in the areas of polynuclear metal complexes, conducting polymers, carbon nanostructures (e.g. fullerenes, carbon nanotubes), metalloproteins, and electrocatalysts, among others. They are particularly suitable for the in situ detection and characterisation of short-lived species generated during redox transformations, allowing derivation of redox mechanisms. The current trends in the development of SEC methodologies include miniaturisation and standardisation of experimental set-ups with the aim of reducing costs, increasing throughput, and improving the accessibility of the techniques to a wider community of researchers.
Common spectroscopic techniques used in SEC are represented by readily accessible spectroscopic instrumentation including ultraviolet–visible–near-infrared (UV-Vis-NIR),[7,8] infrared (IR),[9,10] Raman,[11,12] photoluminescence,[13–15] and electron paramagnetic resonance (EPR),[16,17] with nuclear magnetic resonance (NMR),[18,19] X-ray absorption (XAS),[20–22] X-ray diffraction,[23,24] and atomic emission SEC[25] among other relatively more specialised methods.
A serious advantage of SEC techniques compared with electrochemistry alone for elucidating redox processes is that the spectroscopic technique chosen can help to separate the faradaic and non-faradaic contributions to the current signal.[26] This is because the spectral signal is relatively more sensitive than that from electrochemistry, and is not affected by a high capacitive current. This underscores the usefulness of SEC for materials such as metal complexes, polymers, perovskites, and metal–organic frameworks (MOFs), which are typically semiconducting and exhibit high non-faradaic currents in DC electrochemistry that can impede the analysis of redox processes of interest. It should be noted that AC voltammetry provides a powerful addition to any suite of electroanalytical techniques for such capacitive materials.[27]
The limitations to consider for SEC include the restrictions on the choice of electrode materials, solvents, and supporting electrolytes, which are bounded by the spectroscopic method. Moreover, the experiments are typically performed under steady-state conditions, in which reaction equilibria must be carefully considered.
This perspective focuses on the utility of SEC techniques for the burgeoning field of solid-state materials, particularly MOFs, which have been implicated in next-generation technologies for energy storage and transformation (e.g. batteries, supercapacitors, fuel cells, electrocatalysts, electrochromic devices). From this contemporary viewpoint, key concepts and selected contributions over the past 10 years are highlighted. The reader is referred to excellent reviews in the field including those on catalytic applications of MOFs,[28] SEC in the solution state,[29,30] and SEC of adsorbed molecular species relevant to surface-enhanced Raman scattering (SERS), as well as SEC of polymeric thin films for electronics and spintronics,[31] sol-gel films,[32] membranes, sensors,[26] self-assembled monolayers,[33] and even single molecules at an electrochemical interface.[34] In the field of MOF research, which is a focus herein, SEC techniques have received limited attention with only in situ powder X-ray diffraction (PXRD),[35,36] XAS,[37] UV-Vis-NIR, and EPR spectroscopy[38–45] measurements reported to date in the literature. As the number of redox-active frameworks keeps expanding, however, the demand for practical and robust SEC methodologies will continue to grow. In this regard, a goal of the present review is to motivate researchers to consider the prospects for SEC techniques in offering new fundamental insights, as well as providing a basis for emerging applications relevant to addressing global scientific and technological challenges.[6]
General Experimental Considerations for SEC: From the Novice to More Expert User
For researchers interested in entering the field, several cells and integrated instruments for SEC have been commercialised and are now actively marketed by companies such as Metrohm, Gamry Instruments, Biologic, Spectroelectrochemistry Partners, and Pine Research, among others. These apparatus offer a straightforward entry-point to the field, with options to purchase either SEC cells for integration with existing instruments, or complete SEC systems. For researchers with access to spectroscopic instruments in the laboratory or further afield at synchrotron facilities, many SEC cell designs have now been published and provide an excellent starting point for a custom-built cell. The advantage here is that the cell can be adapted to a specific electrochemical problem and spectroscopic set-up. The advent of quite economical screen-printed electrodes (based on many electrode materials) along with 3-D printing offers further prospects for the future in terms of the design and manufacture of SEC cells.
The central component of the experimental SEC cell design is a three-electrode assembly consisting of working, auxiliary, and reference electrodes, with a similar configuration to that used for voltammetry experiments; the cell is modified to enable the spectroscopic detection of the redox-active species (two-electrode configurations have also been utilised).[46–48] One of the most important design parameters is the thickness of the diffusion layer around the working electrode, which determines the electrochemical responsiveness of the cell. The fastest response time is achieved with the smallest diffusion layer; however, this can compromise the spectroscopic signal intensity. In order to overcome this limitation, working electrodes with large surface areas such as indium–tin oxide (ITO)-coated glass slides or Pt gauze are typically employed. During the SEC experiment, the spectral changes are monitored as a function of potential, which is generally applied in a stepwise fashion. The occurrence of isosbestic points in the SEC data indicates the reversibly of electrochemical transformations and the absence of side reactions that indicate a chemical transformation or decomposition process. Furthermore, SEC experiments may require low-temperature conditions owing to instability of the electrochemically generated species and as such, low-temperature attachments are often incorporated into the cell designs.
The criteria for optimising SEC cells are different from those of electrochemical cells alone, requiring consideration of the specific spectroscopic technique. An important consideration is the response time: for electrochemistry, measurements are typically of the order of seconds to minutes, whereas a formally reversible process in electrochemistry (i.e. defined by the Nernst equation as having a peak-to-peak current ratio of 1.0 and a peak-to-peak voltage difference of 59 mV for a one-electron process) may become less reversible under the relatively longer timescale conditions of SEC. In the latter case, greater time is needed to establish equilibrium of an electrogenerated product on changing the potential. The need to establish steady-state conditions is solved in part by the use of a thin-layer cell, whereby there is a thin layer of electrolyte either containing a homogeneous analyte or the electrolyte alone, minimising solvent absorption. This issue is not always a problem as it depends on the sensitivity of the spectroscopic technique to changes in the spectral response as a function of the applied potential, and the possibility of using signal averaging, or phase-sensitive detection. The advent of optically transparent electrodes (OTEs) at the inception of the field were instrumental to the development of transmission cells, the most commonly adopted OTEs being ITO along with semitransparent meshes such as Pt and Au gauzes, among others. Reflectance mode aids in extending the range of materials to opaque solids[1] and the advent of fibre optics has further facilitated the alignment of the source with both the electrode and the sample.[29]
Bidimensional SEC has also been described, simultaneously combining transmission measurements normal to an electrode surface with measurements in a parallel configuration.[26] This technique enables information about the surface charge transfer processes and those in the diffusion layer to be deconvoluted, providing a more complete picture of the mechanism of an electrode reaction, such as for example the evolution of a solid electrocatalyst in concert with the production of catalytic products in the solution or gas phases.
Application of SEC Methods to Electroactive Solids Including MOFs
UV-Vis-NIR Spectroscopy
The UV-Vis-NIR region of the electromagnetic spectrum for materials constructed from transition metals and/or organic chromophores contains bands corresponding to the absorption of light due to the excitation of electrons from one energy level to another. The spectral properties of a species are governed by its electronic structure, which can significantly vary between different redox states. As such, UV-Vis-NIR spectroscopy has been extensively used in SEC techniques because it can provide insights into the electronic structures of an electroactive material in different redox states.
UV-Vis-NIR spectroscopy is typically performed in transmittance mode where the light passes through the sample while its absorption spectrum is recorded. Transmission SEC experiments have not been limited to solution-state measurements and a wide variety of insoluble materials such as polymers and inorganic oxides have been investigated using this technique. In these cases, the materials are typically deposited as a thin film onto the transparent conductive substrate.[49–51] The main advantage of this type of measurement is that the molar extinction coefficient (ϵ) can be straightforwardly derived using the Beer–Lambert law. For SEC experiments in transmittance configuration, the optically transparent thin-layer electrochemical (OTTLE) cell has been extensively utilised.[52,53] The main feature of this cell is a cuvette with a short path length (<1 mm) that serves as the working electrode compartment. This arrangement results in a very short equilibration time and reduces effects of diffusion. The working electrode must be sufficiently transparent and therefore typically consists of noble metal microgrids or ITO-coated slides. Another important consideration in the design of the OTTLE cell is the window material, which determines its effective spectroscopic range. Numerous variations of the cell design have been reported in the literature that are typically custom-made for a specific application and include additional features such as gas seals and variable-temperature attachments.[54]
UV-Vis-NIR SEC has proved particularly useful over the decades for interrogating electrochromism, that is, the visible colour variation accompanying a change in the redox state of a system. Electrochromism may result in a shift in the energy of a transition (colour) and/or a change in the intensity (for example, colouration versus bleaching). The phenomenon is important for displays and smart windows that are relevant to building energy systems, particularly considering that half of solar power is delivered in the NIR region (14300 cm−1 (700 nm) to 3300 cm−1 (3000 nm)). Materials capable of transmitting or blocking solar irradiation are important, with prominent examples including inorganic materials such as Prussian blue and WO3, the latter being used in electrochromic windows for Boeing’s Dreamliner aeroplanes.[55] Conductive polymers such as polyaniline and polythiophene have also received close attention in this regard.[56,57] Oxidation of these polymers results in the appearance of polaron bands that are characteristic of their conductive properties.
Owing to the versatility of transmission SEC, a wide range of discrete organic and inorganic compounds have been investigated using this technique. A notable class of materials where UV-Vis-NIR SEC has proved particularly useful is multinuclear coordination complexes. The prime example of this class is the Creutz–Taube ion, [(Ru(NH3)5)2(μ-pyrazine)]5+, which consists of two ruthenium centres (in a mixed-valence state) bridged by pyrazine.[58] Owing to the delocalised nature of the bridging ligand, the formal charge on each ruthenium centre is +2.5. The Creutz–Taube ion represents a subclass of coordination complexes capable of generating a mixed-valence state that can give rise to intervalence charge transfer (IVCT) bands in the Vis-NIR region of the electromagnetic spectrum. As the mixed-valence state is often unstable and can disproportionate into fully oxidised and fully reduced states, its analysis can be challenging. As a result, UV-Vis-NIR SEC has been used extensively for the in situ analysis of IVCT bands.[59,60] The determination of band parameters such as peak height and width at half-height can provide important information regarding the degree of electronic delocalisation between the metal centres. UV-Vis-NIR SEC techniques have helped to establish structure–property relationships in mixed-valence complexes and broaden understanding of the nature of electron transfer in these systems.
Several coordination frameworks have been analysed using transmission SEC with a strong focus on the Prussian blue family.[61,62] Prussian blue itself, KFeIII[FeII(CN)6], is known to possess two additional redox states, fully oxidised Berlin green, FeIII[FeIII(CN)6], and fully reduced Prussian white, K2FeII[FeII(CN)6], each having different spectroscopic properties. The SEC experiments demonstrated that this material is capable of reversible switching between the states.
The electrochromic properties of MOFs have also been of significant interest, particularly those materials that possess both coloured and fully bleached states, these being potentially relevant in the field of electrochromic ‘smart windows’. A common experimental strategy involves UV-Vis SEC transmission measurements on either a semitransparent mechanically immobilised MOF powder, such as that reported by Lu and coworkers for the tris(4-(1H-1,2,4-triazol-1-yl)phenyl)amine (TTPA) redox-active ligand in MOFs such as [MnCl2(TTPA)(DMF)] and [CuCl2(TTPA)·1.5DMF],[63] or a transparent MOF film. In the latter case, Dincă and coworkers prepared thin films of a series of MOFs containing redox-active ligands, [Zn(DPZNIR)] (DPZNI = N,N′-di-(3,5-dimethylpyrazolato)-1,4,5,8-naphthalenecarboxydiimide; R = H, SEt, and NHEt).[43] They showed that the films undergo reversible spectroscopic switching on application of a cathodic potential due to reduction of the DPZNI ligands. At present, the difficulty of depositing transparent MOF films limits their implementation in electrochromic devices; however, continual advancements are being made in this area.[64] A later report from the same group on the [M2(NDISA)(H2O)2] (M = Mg2+, Ni2+) frameworks (where H4NDISA = N,N′-bis(3-carboxy-4-hydroxyphenyl)-1,4,5,8-naphthalenetetradicarboximide), shown in Fig. 1, revealed that the mesopores could facilitate ion and electrolyte diffusion during electrochromic cycling.[65] Here, the NDISA ligand undergoes reversible formation of its radical anion and dianion states owing to the central electroactive NDI core.

|
The redox-active naphthalenetetradicarboximide core has also been exploited to develop more robust zirconium-based MOFs, a particularly challenging goal given that d0 Zr4+ (and d10 Zn2+), which are typically used in MOFs, do not mediate strong charge transfer.[66] Ott, Cohen, and coworkers developed a UiO-type MOF, [Zr(dcphOH-NDI)] based on electroactive 4,4′-[1,4-naphthalene-bis(ethyne-2,1-diyl)]-dibenzoate (dcphOH-NDI), which was grown as a thin film on fluorine-doped tin oxide (FTO).[66] A surface-assembled monolayer (SAM) of dcphOH-NDI was first grown on FTO, followed by immersion of the SAM@FTO into the bulk synthesis mixture. SEC data were subsequently used to calculate the kinetics of electron transport through the films by monitoring the absorbance at 475 nm on SEC reduction of NDI in the MOF to NDI•−. The diffusion coefficient was calculated from the Cottrell equation, indicating that mass transport and electron hopping within the MOF films were fast. By changing the size of the counter cation in the supporting electrolyte, the authors also demonstrated a concomitant variation in the diffusion coefficient.
Transmission UV-Vis-NIR has also been possible using MOFs covalently bound to an optically transparent ITO substrate as the working electrode, in order to improve surface robustness. Crystalline films of redox-active 9,10-phenylanthracene (H4L1 = 5′,5′′′′-anthracene-9,10-diyl)bis(([1,1′:3′,1′′-terphenyl]-4,4′′-dicarboxylic acid) in MFM-186 (MFM = Manchester Framework Material) and tetraphenylethylene (H8L2 = 4′,4′′′,4′′′′′,4′′′′′′′-(ethene-1,1,2,2-tetrayl)tetrakis(([1,1′-biphenyl]-3,5-dicarboxylic acid) in MFM-180 (also known as PCN-922) were prepared, and the absorption spectra measured as a function of time.[67] As shown in Fig. 2 for MFM-186, the SEC spectra on oxidation were consistent with the formation of a ligand-centred radical of the redox-active core.

|
It is noteworthy that SEC on components of MOFs, which can be solubilised to enable straightforward transmission SEC measurements, have also proved very instructive in the field. A highlighted case here is the report of the NH2-MIL-125(Ti) [Ti8O8(OH)4(2OC-NH2-C6H3-CO2)6] MOF, which has attracted significant attention owing to its relevance in the field of photocatalysis for solar fuel production.[68] On reduction, soluble clusters of Ti8O8(O2C2(CH3)3)16 generated a new band in the NIR region that was attributed to Ti3+/Ti4+ mixed valency (Fig. 3). This result provided strong evidence for the ligand-to-metal-charge transfer (LMCT) character of the NH2-MIL-125(Ti) photoexcited state, enabling important mechanistic insights into the function of the photocatalyst.

|
Owing to the exquisite colours of porphyrinic chromophores, UV-Vis SEC has proved particularly valuable for obtaining information on the species involved in electrocatalytic mechanisms of porphyrin-based MOFs. Morris and coworkers used a transmission approach to obtain SEC data for a MOF based on CoIIITCPP (where TCPP = TCPP-H2 = 4,4′,4″,4‴-(porphyrin-5,10,15,20-tetrayl)tetrabenzoate) where the MOF was grown solvothermally as a thin film on a SAM of the porphyrin linker adsorbed onto an FTO substrate (Fig. 4a, b).[69] SEC analysis showed that on reduction, the cobalt ions underwent reversible reduction processes between their CoIII/CoII/CoI states, which were exploited to electrocatalytically reduce CCl4. The diffusion coefficient for ambipolar electron and cation transport through the film was calculated with the aid of SEC data.

|
Porphyrinic MOFs have also been implicated in electrocatalytic schemes for CO2 reduction to fuels, SEC having been employed to gain insight into the evolution of redox states in these systems. The Fe-based MOF-525 [Zr6O4(OH)4(H2TCPP)3] developed by Farha, Hupp, and coworkers was deposited by electrophoretic deposition onto a transparent FTO working electrode for transmission SEC studies (Fig. 4c–e).[70] Here, the FeIII/FeII couple of the porphyrin was identified as being central to the electrocatalytic conversion of CO2 to CO and H2. Farha, Hupp, and coworkers also developed a Co-based MOF, [Al2(OH)2CoTCPP], which was grown in a microwave reactor onto an FTO electrode functionalised with a metal oxide thin film.[71] SEC of the thin-film MOF confirmed the CoII/CoI reduction process that occurred concomitantly with CO production from CO2 with high product selectivity.
Reflectance spectroscopy provides an alternative approach for measuring UV-Vis-NIR spectra and is particularly suitable for analysing solid samples; however, its integration with SEC has been more challenging and has been less extensively explored than transmission spectroscopy. The reflectance data are generally represented as a Kubelka–Munk function, F(R)[1] where R is the reflectance of the sample, from which it is more difficult to derive the molar extinction coefficient,[72] although single-crystal absorption spectroscopy can be used for this purpose.[73]
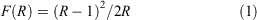
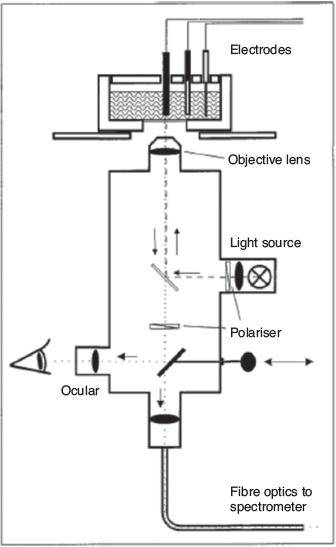
Pioneering work in designing reflectance UV-Vis-NIR SEC techniques was carried out by Scholz and coworkers (Fig. 5).[74,75] The key feature of the design was a modified optical microscope that allowed focusing of the light from the source onto the sample, which was immobilised onto a paraffin-impregnated graphite working electrode. The spectrometer measured the diffuse reflectance from the sample. Using this experimental set-up, the authors were able to detect the spectral changes associated with the oxidation of several octacyanomolybdate- and octacyanotungstate-based materials with fairly high resolution and good signal-to-noise ratio.[75] Despite being a powerful SEC technique, the set-up required multiple customised components (microscope, spectrometer) in addition to the electrochemical cell and was therefore difficult to replicate by other researchers. Nevertheless, it provided a valuable foundation for the development of simpler and more accessible reflectance UV-Vis-NIR SEC techniques.
|
Recently, we reported a robust SEC cell for diffuse reflectance UV-Vis-NIR spectroelectrochemical measurements (Fig. 6).[44] The first-generation cell design contained a small central compartment that minimised the required volume for the supporting electrolyte. The transparent ITO-coated quartz slide (25 × 25 × 0.15 mm) utilised as a working electrode allowed spectral collection from the electrode–sample interface. This compartment was connected to two side-arms used to harbour high surface area Pt coil counter and Ag wire reference electrodes. The powdered material was mechanically immobilised onto the slide with a spatula with a thin strip of Teflon tape (~1 mm) placed across it in order to prevent powder detachment. Strips of Cu tape with conducting adhesive were used to connect the working electrode to the electrode leads from the potentiostat. The SEC cell was compatible with a wide variety of solvents, both aqueous and organic. The spectral measurements were performed using a commercial Harrick Omni Diff Probe attachment for a conventional spectrophotometer, which reduced the requirements for customised components. An O-ring was placed between the central compartment and the working electrode to create a seal when the Omni Diff Probe was placed on top. Owing to this feature, the cell could be used continuously in the SEC experiments without significant evaporation of electrolyte. The first-generation SEC cell has been successfully used to characterise a wide variety of redox-active frameworks. The limitations of the cell design included the lack of air-free and low-temperature capabilities, which precluded characterisation of short-lived species, as well as use of high-cost ITO-coated quartz slides. Nevertheless, this cell has proved particularly important for observing the spectroscopic features of MOFs that are otherwise challenging to obtain via ex situ chemical oxidation or reduction. A wide range of MOFs have been interrogated,[5,41,73,76–83] including the recent observation and mechanistic interpretation of through-space electron transfer in MOFs incorporating cofacially aligned electroactive ligands such as thiazolothiazoles.[73,77,79] A closely related cofacial MOF incorporating tetrathiafulvalene (TTF) ligands, [Cd2(Py2TTF)2(bpdc)2] (Py2TTF = 2,6-bis(4′-pyridyl)-tetrathiafulvalene; bpdc2– = 4,4′-biphenyldicarboxylate) demonstrated the same through-space charge transfer mechanism (Fig. 7).[84] SEC proved instrumental in characterising the mechanism and quantifying the extent of charge transfer between the ligands.

|

|
A second-generation SEC cell was subsequently developed to enable lower than ambient room temperature operation.[85] The cell consisted of a glass body, which contained a central cylindrical compartment that harboured the working electrode surrounded by an outer section (Fig. 6c). Two side-arms were connected to the central section and served as counter and reference electrode compartments separated by the glass frits.
Another variation of reflectance spectroscopy uses attenuated total reflection (ATR) for probing the sample. As the light beam passes over the sample multiple times, ATR can offer increased sensitivity compared with conventional diffuse reflectance measurements. Several reports regarding the development of ATR-based SEC techniques have appeared in the literature.[86–88] The major challenge encountered with this type of instrumentation is the optical alignment of the spectrometer and the SEC cell; however, the advent of optical fibre technologies has significantly simplified this process. The modern ATR SEC experimental set-up generally consists of an optical fibre serving as the waveguide leading to the electrochemical cell. This method has successfully been applied to spectroelectrochemical characterisation of polymeric films as well as detection of analyte species adsorbed from solution onto the working electrode.[89] ATR SEC is most suitable for sensing low concentrations of redox-active compounds in solution or solid matrices as well as characterisation of surface-confined redox processes.
Infrared SEC
Owing to the sensitivity of molecular vibrations to the structural and electronic properties of a molecule, the development of IR SEC techniques has also received considerable attention.[10,90,91] Owing to the very short timescales (10−11−10−13 s) of vibrational transitions, the IR spectra can resolve very fast processes such as intramolecular electron transfer.[92] The instrumentation for IR spectroscopy is similar to that for UV-Vis-NIR spectroscopy in both transmission and reflectance modes, which means that they often utilise similar cell designs. The OTTLE cell, modified with IR-transparent windows such CaF2, has been routinely employed for characterisation of redox processes in coordination complexes, which often undergo intramolecular charge transfer such as metal-to-ligand charge transfer (MLCT), LMCT, and IVCT transitions.[93,94] In certain cases, especially when observing lower-energy stretches, SEC experiments are conducted in deuterated solvents in order to improve the transparency of the cell.[95] Furthermore, time-resolved IR SEC used to investigate dynamic process associated with redox transformations is potentially a powerful tool for the interrogation of charge transport properties in semiconducting or conducting systems.
A different experimental configuration commonly used for IR SEC employs ATR, which is particularly suitable for solid-state samples.[96,97] In this configuration, the beam impinges on an IR-transparent material at a specific angle, causing it to undergo multiple internal reflections, which increase its interaction with the sample. For this purpose, germanium or silicon crystals are typically employed that also serve as working electrodes (often coated with a gold film) in the SEC experiments. The main advantage of the ATR configuration is that the redox transformation takes place at the interface between the ATR crystal and the sample. The small penetration of the IR beam into the sample (~1 µm) allows rapid electrochemical conversion and corresponding spectroscopic detection. One series of redox-active systems extensively characterised using IR-ATR SEC are the oxo-centred trinuclear ruthenium clusters bridged by ligands such as pyrazine and 4,4′-bipyridine to form dimers. Multiple elegant reports detailing the SEC investigations of these systems were published by Kubiak and coworkers.[98,99] The dimers have several stable redox states including mixed-valence states, and coordinated carbon monoxide molecules serve as IR probes, thus making these systems particularly suitable for SEC measurements. From these experiments, the authors were able to determine the rates of electron transfer between the clusters. In addition, they investigated the effects of ligand substitution on the ruthenium clusters on the degree of electronic communication, which laid the groundwork for the establishment of structure–function relationships in the delocalised mixed-valence systems. This example demonstrates that IR SEC can be a powerful tool for assessing electron transfer properties in extended redox-active systems; however, its full potential has seldom been exploited in MOFs.
Recently, the application of surface-enhanced IR absorption (SERIA) was reported for a phthalocyanine-based MOF by Dong and Feng using ATR mode.[100] The 2D MOF consisted of a copper-phthalocyaninato ligand and zinc-bis(hydroxy) complex (ZnO4) as the linkage, namely PcCu-O8-Zn. Here, the MOF was deposited as a film onto an Au surface (acting as the IR signal amplifier and working electrode). SERIA was used to probe the reaction process and the nature of the electrocatalytic sites for the MOF that was drop-cast onto the electrode surface. Highly selective CO2 conversion to CO and H2 was achieved (i.e. syngas formation), with SERIA indicating that the CO formed was bound to the CuN4 and CuO4 centres of the MOF. Coupled with SECXAS (see the X-Ray Spectroscopies section below), the in situ SEC techniques enabled intimate insights into the electrocatalytic mechanism.
An important application of IR SEC involves probing structural changes occurring during electron transfer reactions. Although not strictly classified as MOFs, metal hexacyanometallates of the Prussian blue family including cobalt and copper hexacyanoferrate prepared as thin films on a gold working electrode have been investigated by polarisation modulation IR SEC (in reflectance mode).[101] Here, coordination of a metal ion to either the C- or N-terminus of the CN− bridging ligand can be identified from IR by the distinct frequency of the T1u mode of the cyanide stretching mode ν(C≡N), the position revealing the coordination and symmetry around the CN− bridging ligand. SEC analysis provided unambiguous evidence for the oxidation of N-coordinated Co2+ to Co3+ in K2CoII[FeII(CN)6] as the first electrochemical oxidation process, followed by partial oxidation of the C-coordinated Fe2+ to Fe3+.
A significant recent application of IR SEC has involved following the evolution of adsorbed species during the electrocatalytic evolution of an oxide-derived Cu/Carbon (OD Cu/C) catalyst, which was formed by the carbonisation of the MOF HKUST-1, [Cu3(BTC)2] (BTC = benzene-1,3,5-tricarboxylate) (Fig. 8).[102] Strikingly, the active electrocatalytic centres are highly dispersed Cu nanoparticles that are formed within the carbon matrix, and promote the electrochemical reduction of CO2 to C1 and C2 liquid fuels including methanol and ethanol at fairly low overpotentials. The production of the C2-derived C2H5OH was particularly noteworthy. IR-active vibrations corresponding to the formation of formate ions (HCOO−) and alcohols (O–H) were detected, along with CO (bridge-bonded and linear-bonded). For ethanol formation, a C–C bond was hypothesised to be formed between surface-bound C1 oxygenates; although further work is required, these observations suggest that IR SEC can be a particularly powerful technique for interrogating electrocatalytic mechanisms.

|
Raman SEC
The vibrational properties of a material can also be measured using Raman spectroscopy, which, in contrast to IR spectroscopy, utilises light (UV-Vis-NIR) scattering. A small amount of the energy of the incident beam is absorbed by the sample as vibrational energy, which decreases the frequency of the scattered light − an effect known as the Raman shift.[103] Another major difference is that Raman spectroscopy generally excites symmetric vibrational modes (no change in dipole moment) whereas IR spectroscopy excites asymmetric modes (change in dipole moment), making these techniques complementary to each other. Raman SEC, just like its IR counterpart, has been extensively used for investigating spectroscopic changes associated with redox transformations.[104–106] The SEC experiments are typically performed either in transmittance mode using a variation of the OTTLE cell[107] or reflectance mode in conjunction with a confocal microscope.[108] The added advantage of Raman spectroscopy is an increase in band intensities if the excitation wavelength coincides with an absorption band of the material in a phenomenon known as resonance enhancement.[109] As a result, it can be considerably more sensitive compared with IR spectroscopy. Because resonance enhancement depends on the electronic properties of the material, resonance-Raman spectra can provide information on electronic structure. The major drawback of this effect is that it can also cause fluorescence emission, which is generally much higher in intensity and therefore can preclude the detection of Raman bands.
A confocal Raman SEC technique was recently reported for MOFs containing donor and acceptor functionalities, namely [(Zn(DMF))2(TTFTC)(DPNI)] (DMF = N,N′-dimethylformamide, TTFTC = tetrathiafulvalene tetracarboxylate, DPNI = N,N′-di(4-pyridyl)-1,4,5,8-naphthalenetetracarboxydiimide).[38,78] Such donor–acceptor MOFs have been inspired by the historically rich field of ‘organic metals’ such as the archetypal tetrathiafulvalene–tetracyanoquinodimethane (TTF–TCNQ), which demonstrates near-metallic conductivity.[110] As shown in Fig. 9, the cell consisted of a widely available and economical screen-printed electrode to which the MOF was immobilised onto the working electrode.[78] A droplet of electrolyte completed the electrochemical cell. Although reductive SEC was not possible using this cell design, application of an oxidising potential led to an increase in the intensity of vibrations associated with TTFTC•+ and DPNI0 on their increased formation, and a corresponding decrease in vibrations associated with TTFTC and DPNI•−.

|
Lee and coworkers recently reported the application of Raman SEC to probe the structural evolution of a MOF-derived catalyst based on the zeolitic imidazolate framework ZIF-67, [Co(Meim)2] (Meim = 2-methylimidazolate) (Fig. 10).[111] Earlier work demonstrated that the MOF acts as a precursor to generate on electrochemical oxidation[112] cobalt oxide species that are responsible for the electrocatalytic oxygen evolution reaction (OER), which is important for energy-related electrocatalysis such as water splitting. By probing the evolution of the Raman-active stretching modes, the electrocatalytic mechanism was ascribed to the formation of α-Co(OH)2 and β-Co(OH)2 from the MOF, these oxides retaining some porosity. The work demonstrates the utility of Raman SEC for probing both catalytic mechanisms and the structural evolution of a solid catalyst in real time.

|
Another method for increasing the sensitivity of Raman spectroscopy is using SERS due to the phenomenon of plasmon resonance when electromagnetic radiation of the incident Raman light beam interacts with a metallic surface.[113] The effect arises most commonly when the analyte is adsorbed onto a metallic surface.[113,114] The spectral enhancement occurs when a potential is applied to the surface, with an enhancement factor of up to 1015, allowing the detection of single molecules.[115] The experimental set-up of SERS can be straightforwardly modified for SEC experiments where the working electrode is used to induce surface enhancement.[98] Despite these benefits, Raman SEC has not been applied to date for the investigation of redox-active MOFs. There is anecdotal evidence to suggest that SERS effects do arise from the surfaces of MOFs themselves; however, the reproducibility of the effect is an issue. SERS effects are known to occur for microstructures containing a periodic structure; the preparation of well-defined MOF surfaces or single crystals may be particularly instructive to probe these effects in the future. Although the abovementioned examples have involved confocal Raman instruments, at the time of writing, companies such as Metrohm have developed micro-Raman SEC instruments that may help to grow the community of researchers employing the technique in the future.
EPR SEC
When electrochemical processes are accompanied by changes in the spin state of a material and/or its components, EPR spectroscopy is useful for the characterisation of redox transformations because it provides information regarding the nature and the environment of unpaired electrons that are present in concentrations as low as 10−14 M.[116] Because electron spin also interacts with the nuclear spins, EPR spectroscopy can help to determine the extent of electron delocalisation throughout the molecule. The design requirements for EPR SEC cells include microwave transparency (for which quartz tubes or flat cells are well suited) as well as low-dielectric-constant solvents such as dichloromethane or tetrahydrofuran. Moreover, in order to improve signal-to-noise ratio, SEC experiments are typically conducted at low temperatures to reduce the rotational motion of molecules and stabilise the electrogenerated species. Because EPR spectroscopy is only capable of detecting unpaired electrons, it is particularly suitable for interrogating redox-induced diamagnetic–paramagnetic transitions. As a result, EPR SEC has been extensively used to analyse electrochemical generation of organic radicals and changes in the magnetic properties of coordination complexes as well as structural changes of the active sites in metalloproteins.[117,118]
The utility of the EPR SEC technique can be further expanded by performing other spectroscopic measurements (UV-Vis-NIR) in parallel. This approach was pioneered by Dunsch and coworkers, who were the first to design the EPR/UV-Vis-NIR SEC cell for characterising conductive polymers such as polyaniline and polypyrrole.[119–121] The design of the cell needed to account for the requirements of both EPR and UV-Vis-NIR spectroscopies, with the SEC experiments performed on a polymer film that was electrodeposited onto an ITO-coated glass substrate. The spectroscopic measurements were carried out simultaneously inside the EPR cavity, which allowed the authors to observe the appearance of the signals corresponding first to polarons, and then to bipolarons on application of oxidative potentials.[121] This study provided important insights into the nature of the dramatic conductivity increase in conjugated organic polymers on doping with oxidants. In recent years, the repertoire of materials analysed by EPR/UV-Vis-NIR SEC has been expanded to include other conductive polymers,[122,123] and discrete organic and coordination compounds[124,125] as well as carbon-based materials such as fullerenes and carbon nanotubes.[126,127] Combinations of two or more spectroscopy techniques with electrochemistry can yield a plethora of novel techniques capable of unravelling complex redox transformations, and their development remains an ongoing challenge.
The EPR SEC cell design shown in Fig. 11 is used for organic free radicals in fluid and frozen media and is compatible with conventional commercial spectrometers (X-, Q-, and W-bands) with a standard sample tube or thin-layer tube with variable temperature (depending on the electrolyte solvent freezing temperature).[128] In the MOF [(Zn(DMF))2(TTFTC)(DPNI)], where an inherent degree of charge transfer occurs between the donor and acceptor in the as-synthesised state, reduction leads to an increase in the formation of TTFTC0 from TTFTC•+ and DPNI•− from DPNI0.[38] The corresponding EPR signal can be resolved into two underlying contributions due to the organic radicals of the TTFTC•+ and DPNI•−.[38] EPR SEC using pulsed EPR techniques such as Electron Nuclear Double Resonance (ENDOR) has also been applied to study MOFs incorporating the redox-active tris[4-(pyridin-4-yl)phenyl]amine (NPy3) ligand, such as [Zn(NPy3)(NO2)2·xMeOH·xDMF], although it must be noted that in this example, the oxidised MOF was generated in the EPR SEC cell, which was quench-cooled to 20 K to perform the measurement.[81]

|
Photoluminescence SEC
For compounds that yield good fluorescence, photoluminescence SEC is instructive, with many applications in the solution state, particularly for very sensitive and selective biosensors,[13] optical devices, and electrochemiluminescence studies. Applications to solid materials such as MOFs are comparatively scarce. Here, the cells are generally more complex than those used in UV-Vis-NIR SEC as both the fluorescence quantum yield and the amount of radiation from the excitation source determine the fluorescence intensity.[129] To date, the technique has been applied to follow the quenching of the fluorescence signal in the abovementioned [Zn(NPy3)(NO2)2·xMeOH·xDMF] MOF on oxidation and formation of the radical cation form of the NPy3 ligand.[81]
X-Ray Spectroscopies
X-Ray absorption spectroscopy, encompassing both near-edge structure (XANES) and extended fine structure (EXFAS), has proved very fruitful for determining coordination number, geometry, and redox state using synchrotron-based radiation sources.[20,130] XANES and EXAFS SEC has been successfully applied to discern the active site and mechanistic details of MOF-based catalysts, particularly those based on earth-abundant metals such as iron to replace the noble metals. These materials are relevant to the oxygen reduction reaction (ORR), which is a critical component of practical fuel cells relevant to electrochemical energy storage and conversion devices.
Mukerjee, Jia, and coworkers investigated a MOF-derived Fe–N–C catalyst with high activity for ORR.[131] As shown in Fig. 12, the ZIF known as ZIF-8, [Zn(Meim)2], was used as a host for an iron phenanthroline complex that was prepared using mechanochemical grinding coupled with heat treatment to give a porous carbonaceous material containing metallic iron. In combination with other analytical methods, Fe K-edge XANES of the FePhenMOF–ArNH3 catalyst identified the active site as being a non-planar Fe2+ Fe–N4 moiety embedded in a distorted carbon matrix, which undergoes oxidation to an in-plane Fe3+ Fe–N4 moiety. Exceptional ORR activity was obtained owing to the highly porous carbon matrix, the findings being relevant to practical H2/air fuel cells. The XAS SEC cell shown in Fig. 13a[132] was employed in this work, with the electrocatalyst embedded in a Nafion matrix that was immobilised on carbon cloth. Although the catalytic species here are not crystalline MOFs, the coordination framework does play an integral role in structurally immobilising the catalytically active centres. Mukerjee and coworkers subsequently released a critical review outlining the advantages and disadvantages of XAS SEC for interrogating solid materials.[133]

|

|
SEC XAS has also been adopted, in concert with the SEC IR method SERIA, to elucidate the mechanistic transformation in the aforementioned 2D MOF PcCu-O8-Zn.[100] The valence state and coordination number of the metal centres in the MOF were determined using Zn K-edge XANES and Cu K-edge EXFAS (Fig. 13b–d). From these measurements, the redox states and geometries of ZnII and CuII were determined to be maintained throughout the electrocatalytic process, with no metals or metallic clusters forming. Thus, the Zn4O centres were identified as being active for the CO2 reduction reaction (CO2RR), while the CuN4 sites were active for the hydrogen evolution reaction (HER) via a synergistic reaction pathway for CO2/H2 (i.e. syngas) production with very high selectivity.
Summary and Outlook
This review article has highlighted the prospects for application of SEC techniques in the burgeoning field of MOFs; the methodologies presented are also applicable to other electroactive solids at the forefront of materials for energy storage and conversion such as graphenes, carbonaceous materials, battery materials, electrochromics, and perovskites.[134] As the number of potential MOFs featuring a wide range of physical and chemical properties is near infinite, the opportunities for the rational design and fine-tuning of desired molecular materials abound. Elucidation of the intricate interplay between the framework structure and the electron migration becomes crucial for realisation of the full potential of redox-active MOFs, and in this regard, SEC techniques have enormous future potential.
To reap the benefits of SEC techniques for solids, fabrication of thin films is highly advantageous to enable transmission measurements on optically transparent electrodes, and techniques including synthesis on a SAM or metal oxide have been noted in the aforementioned examples, along with electrophoretic deposition. A key requirement for transmission measurements is uniformly dispersed thin crystalline films to reduce backscattering issues and facilitate charge and mass transport. Reflectance SEC offers an alternative to transmission methods; again, surface roughness, which may impact signal-to-noise quality, is an issue. Miniaturisation (to enable the use of small MOF and electrolyte quantities), ultrafast detection, and sensitivity improvements all rank as important parameters to consider in future experimental designs. Moreover, the control of temperature and the redox potential window (of the solid and the electrolyte) are critical considerations, particularly where electrogenerated species may be fleeting, and there is a need to capture high-quality data efficiently. Surmounting these issues would enable the cell to be more easily replicated and accessible to the wider scientific community.
It is interesting to note that SEC methods to gain insights into electrocatalytic mechanisms of MOFs have been very fruitful over the past 5 years, with the mechanisms for charge transport (e.g. the role of redox hopping) and catalytic processes being important goals.[135] Multispectroelectrochemistry may prove particularly useful here for the simultaneous detection of MOF structure evolution (i.e. the catalyst), and reaction product formation in the electrolyte or gas phases.
Some interesting prospects also exist for new, currently uncharted solid-state SEC techniques. For example, although enormous advances have been made in NMR measurements coupled with electrochemistry, the challenge of developing a cell for solid-state measurements remains.[29] Such experiments are particularly difficult owing to the external magnetic field and the dependence on the electromagnetic environment of the material containing nuclei with non-zero nuclear spins. Recently, Richter proposed the application of a ‘pouch’ SEC cell for solid-state NMR, which was employed to quantitatively monitor an evolved reaction product (CO2) from the electrocatalytic oxidation of ethanol.[136] An important future prospect is the monitoring, in real time, of material evolution itself, providing a comprehensive picture of structural effects coupled with the substrate redox processes and changes in the reactant and product composition.
Another interesting prospect is the exploitation of surface-enhanced effects in IR and Raman spectroscopies to amplify signals. This phenomenon is attributed to the interaction between light and the periodic arrays of crystalline ordered nanostructures in MOFs and perovskites giving rise to surface plasmon resonance effects.[114] Although SERS can be challenging and difficult to reproduce, well-ordered thin films of crystalline solids such as MOFs may prove particularly instructive.
Finally, it is important to acknowledge the companies that have sought to improve the accessibility of SEC techniques to researchers through the marketplace. To date, the majority of these techniques are geared towards analytes in the solution phase, but there is a strong scope to develop readily accessible solid-state SEC cells. Given the expanding interests in the application of SEC techniques to address materials that are relevant to challenges in energy and the environment,[6] this may be a profitable goal.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
The authors gratefully acknowledge the Australian Research Council for their support (FT170100283).
References
[1] T. Kuwana, R. K. Darlington, D. W. Leedy, Anal. Chem. 1964, 36, 2023.| Crossref | GoogleScholarGoogle Scholar |
[2] T. Kuwana, W. R. Heineman, Acc. Chem. Res. 1976, 9, 241.
| Crossref | GoogleScholarGoogle Scholar |
[3] L. Dunsch, J. Solid State Electrochem. 2011, 15, 1631.
| Crossref | GoogleScholarGoogle Scholar |
[4] W. Kaim, G. K. Lahiri, Coord. Chem. Rev. 2019, 393, 1.
| Crossref | GoogleScholarGoogle Scholar |
[5] R. Murase, B. W. Ding, Q. Y. Gu, D. M. D’Alessandro, Philos. Trans.- Royal Soc., Math. Phys. Eng. Sci. 2019, 377, 20180226.
| Crossref | GoogleScholarGoogle Scholar |
[6] https://www.un.org/sustainabledevelopment/sustainable-development-goals/.
[7] K. R. Idzik, P. Rapta, P. J. Cywinski, R. Beckert, L. Dunsch, Electrochim. Acta 2010, 55, 4858.
| Crossref | GoogleScholarGoogle Scholar |
[8] U. Schröder, F. Scholz, Inorg. Chem. 2000, 39, 1006.
| Crossref | GoogleScholarGoogle Scholar | 12526381PubMed |
[9] A. Neckel, Microchim. Acta 1987, 93, 263.
| Crossref | GoogleScholarGoogle Scholar |
[10] K. Ashley, S. Pons, Chem. Rev. 1988, 88, 673.
| Crossref | GoogleScholarGoogle Scholar |
[11] M. Kalbac, L. Kavan, M. Zukalová, L. Dunsch, Chem. – Eur. J. 2006, 12, 4451.
| Crossref | GoogleScholarGoogle Scholar | 16552794PubMed |
[12] T. Itoh, R. L. McCreery, J. Am. Chem. Soc. 2002, 124, 10894.
| Crossref | GoogleScholarGoogle Scholar | 12207545PubMed |
[13] P. Audebert, F. Miomandre, Chem. Sci. 2013, 4, 575.
| Crossref | GoogleScholarGoogle Scholar |
[14] T. S. Pinyayev, C. J. Seliskar, W. R. Heineman, Anal. Chem. 2010, 82, 9743.
| Crossref | GoogleScholarGoogle Scholar | 21053915PubMed |
[15] M. Dias, P. Hudhomme, E. Levillain, L. Perrin, Y. Sahin, F.-X. Sauvage, et al. Electrochem. Commun. 2004, 6, 325.
| Crossref | GoogleScholarGoogle Scholar |
[16] R. T. Boeré, A. M. Bond, T. Chivers, S. W. Feldberg, T. L. Roemmele, Inorg. Chem. 2007, 46, 5596.
| Crossref | GoogleScholarGoogle Scholar | 17552513PubMed |
[17] T. Sixt, J. Fiedler, W. Kaim, Inorg. Chem. Commun. 2000, 3, 80.
| Crossref | GoogleScholarGoogle Scholar |
[18] R. D. Webster, Anal. Chem. 2004, 76, 1603.
| Crossref | GoogleScholarGoogle Scholar | 15018557PubMed |
[19] U. Bussy, P. Giraudeau, V. Silvestre, T. Jaunet-Lahary, V. Ferchaud-Roucher, M. Krempf, et al. Anal. Bioanal. Chem. 2013, 405, 5817.
| Crossref | GoogleScholarGoogle Scholar | 23673569PubMed |
[20] L. R. Sharpe, W. R. Heineman, R. C. Elder, Chem. Rev. 1990, 90, 705.
| Crossref | GoogleScholarGoogle Scholar |
[21] I. B. Polovov, V. A. Volkovich, J. M. Charnock, B. Kralj, R. G. Lewin, H. Kinoshita, et al. Inorg. Chem. 2008, 47, 7474.
| Crossref | GoogleScholarGoogle Scholar | 18665589PubMed |
[22] N. R. S. Farley, S. J. Gurman, A. R. Hillman, Electrochem. Commun. 1999, 1, 449.
| Crossref | GoogleScholarGoogle Scholar |
[23] M. Fleischmann, A. Oliver, J. Robinson, Electrochim. Acta 1986, 31, 899.
| Crossref | GoogleScholarGoogle Scholar |
[24] M. G. Samant, M. F. Toney, G. L. Borges, L. Blum, O. R. Melroy, Surf. Sci. 1988, 193, L29.
| Crossref | GoogleScholarGoogle Scholar |
[25] K. Ogle, Corrosion 2019, 75, 1398.
| Crossref | GoogleScholarGoogle Scholar |
[26] J. Garoz-Ruiz, J. V. Perales-Rondon, A. Heras, A. Colina, Electroanalysis 2019, 31, 1254.
| Crossref | GoogleScholarGoogle Scholar |
[27] A. M. Bond, N. W. Duffy, S.-X. Guo, J. Zhang, D. Elton, Anal. Chem. 2005, 77, 186A.
| Crossref | GoogleScholarGoogle Scholar | 15889488PubMed |
[28] T. E. Rosser, E. Reisner, ACS Catal. 2017, 7, 3131.
| Crossref | GoogleScholarGoogle Scholar |
[29] J. J. A. Lozeman, P. Fuhrer, W. Olthuis, M. Odijk, Analyst 2020, 145, 2482.
| Crossref | GoogleScholarGoogle Scholar |
[30] W. Kaim, J. Fiedler, Chem. Soc. Rev. 2009, 38, 3373.
| Crossref | GoogleScholarGoogle Scholar | 20449056PubMed |
[31] P. Bujak, I. Kulszewicz-Bajer, M. Zagorska, V. Maurel, I. Wielgus, A. Pron, Chem. Soc. Rev. 2013, 42, 8895.
| Crossref | GoogleScholarGoogle Scholar | 24030727PubMed |
[32] A. K. Surca, G. Drazic, M. Mihelcic, J. Sol-Gel Sci. Technol. 2020, 95, 587.
[33] O. Aleveque, C. Gautier, E. Levillain, Curr. Opin. Electrochem. 2019, 15, 34.
| Crossref | GoogleScholarGoogle Scholar |
[34] R. Hao, Z. Y. Peng, B. Zhang, ACS Omega 2020, 5, 89.
| Crossref | GoogleScholarGoogle Scholar | 31956755PubMed |
[35] G. Ferey, F. Millange, M. Morcrette, C. Serre, M. L. Doublet, J. M. Greneche, et al. Angew. Chem. Int. Ed. 2007, 46, 3259.
| Crossref | GoogleScholarGoogle Scholar |
[36] G. de Combarieu, M. Morcrette, F. Millange, N. Guillou, J. Cabana, C. P. Grey, et al. Chem. Mater. 2009, 21, 1602.
| Crossref | GoogleScholarGoogle Scholar |
[37] Gd. Combarieu, S. Hamelet, F. Millange, M. Morcrette, J.-M. Tarascon, G. Férey, et al. Electrochem. Commun. 2009, 11, 1881.
| Crossref | GoogleScholarGoogle Scholar |
[38] C. F. Leong, B. Chan, T. B. Faust, D. M. D’Alessandro, Chem. Sci. 2014, 5, 4724.
| Crossref | GoogleScholarGoogle Scholar |
[39] C. F. Leong, B. Chan, T. B. Faust, P. Turner, D. M. D’Alessandro, Inorg. Chem. 2013, 52, 14246.
| Crossref | GoogleScholarGoogle Scholar | 24283401PubMed |
[40] C. F. Leong, T. B. Faust, P. Turner, P. M. Usov, C. J. Kepert, R. Babarao, et al. Dalton Trans. 2013, 9831.
| Crossref | GoogleScholarGoogle Scholar | 23519323PubMed |
[41] C. Hua, D. M. A. D’Alessandro, CrystEngComm 2014, 16, 6331.
| Crossref | GoogleScholarGoogle Scholar |
[42] C. Hua, P. Turner, D. M. D’Alessandro, Dalton Trans. 2013, 6310.
| Crossref | GoogleScholarGoogle Scholar | 23518920PubMed |
[43] C. R. Wade, M. Li, M. Dincă, Angew. Chem. 2013, 125, 13619.
| Crossref | GoogleScholarGoogle Scholar |
[44] P. M. Usov, C. Fabian, D. M. D’Alessandro, Chem. Commun. 2012, 48, 3945.
| Crossref | GoogleScholarGoogle Scholar |
[45] F. J. Rizzuto, T. B. Faust, B. Chan, C. Hua, D. M. D’Alessandro, C. J. Kepert, Chem. – Eur. J. 2014, 20, 17597.
| Crossref | GoogleScholarGoogle Scholar | 25346539PubMed |
[46] L. Leon, J. D. Mozo, Trends Analyt. Chem. 2018, 102, 147.
| Crossref | GoogleScholarGoogle Scholar |
[47] F. W. Richey, B. Dyatkin, Y. Gogotsi, Y. A. Elabd, J. Am. Chem. Soc. 2013, 135, 12818.
| Crossref | GoogleScholarGoogle Scholar | 23915377PubMed |
[48] D. G. Sanderson, L. B. Anderson, Anal. Chem. 1985, 57, 2388.
| Crossref | GoogleScholarGoogle Scholar |
[49] W. A. Gazotti, G. Casalbore-Miceli, S. Mitzakoff, A. Geri, M. C. Gallazzi, M. A. De Paoli, Electrochim. Acta 1999, 44, 1965.
| Crossref | GoogleScholarGoogle Scholar |
[50] R. J. Mortimer, Chem. Soc. Rev. 1997, 26, 147.
| Crossref | GoogleScholarGoogle Scholar |
[51] M. Mastragostino, A. M. Marinangeli, A. Corradini, S. Giacobbe, Synth. Met. 1989, 28, 501.
| Crossref | GoogleScholarGoogle Scholar |
[52] I. Piljac, M. Tkalcec, B. Grabaric, Anal. Chem. 1975, 47, 1369.
| Crossref | GoogleScholarGoogle Scholar |
[53] W. R. Heineman, J. Chem. Educ. 1983, 60, 305.
| Crossref | GoogleScholarGoogle Scholar |
[54] W. A. Nevin, A. B. P. Lever, Anal. Chem. 1988, 60, 727.
| Crossref | GoogleScholarGoogle Scholar |
[55] R. Holze, J. Solid State Electrochem. 2004, 8, 982.
| Crossref | GoogleScholarGoogle Scholar |
[56] G. Zotti, G. Schiavon, Synth. Met. 1989, 31, 347.
| Crossref | GoogleScholarGoogle Scholar |
[57] E. M. Genies, M. Lapkowski, J. Electroanal. Chem. Interfacial Electrochem. 1987, 220, 67.
| Crossref | GoogleScholarGoogle Scholar |
[58] C. Creutz, H. Taube, J. Am. Chem. Soc. 1973, 95, 1086.
| Crossref | GoogleScholarGoogle Scholar |
[59] D. M. D’Alessandro, F. R. Keene, Chem. Soc. Rev. 2006, 35, 424.
| Crossref | GoogleScholarGoogle Scholar | 16636726PubMed |
[60] D. M. D’Alessandro, F. R. Keene, Chem. Rev. 2006, 106, 2270.
| Crossref | GoogleScholarGoogle Scholar | 16771450PubMed |
[61] K. Itaya, K. Shibayama, H. Akahoshi, S. Toshima, J. Appl. Phys. 1982, 53, 804.
| Crossref | GoogleScholarGoogle Scholar |
[62] D. Ellis, M. Eckhoff, V. D. Neff, J. Phys. Chem. 1981, 85, 1225.
| Crossref | GoogleScholarGoogle Scholar |
[63] C. M. Ngue, Y. H. Liu, Y. S. Wen, M. K. Leung, C. W. Chiu, K. L. Lu, Dalton Trans. 2019, 10122.
| Crossref | GoogleScholarGoogle Scholar | 31180414PubMed |
[64] I. Hod, W. Bury, D. M. Karlin, P. Deria, C.-W. Kung, M. J. Katz, et al. Adv. Mater. 2014, 26, 6295.
| Crossref | GoogleScholarGoogle Scholar | 25070374PubMed |
[65] K. AlKaabi, R. Wade Casey, M. Dincă, Chem 2016, 1, 264.
| Crossref | GoogleScholarGoogle Scholar |
[66] B. A. Johnson, A. Bhunia, H. H. Fei, S. M. Cohen, S. Ott, J. Am. Chem. Soc. 2018, 140, 2985.
| Crossref | GoogleScholarGoogle Scholar | 29421875PubMed |
[67] T. Mitra, F. Moreau, A. Nevin, C. U. Perotto, A. Summerfield, E. S. Davies, et al. Chem. Sci. 2018, 9, 6572.
| Crossref | GoogleScholarGoogle Scholar | 30310589PubMed |
[68] J. G. Santaclara, M. A. Nasalevich, S. Castellanos, W. H. Evers, F. C. M. Spoor, K. Rock, et al. ChemSusChem 2016, 9, 388.
| Crossref | GoogleScholarGoogle Scholar | 26871265PubMed |
[69] S. R. Ahrenholtz, C. C. Epley, A. J. Morris, J. Am. Chem. Soc. 2014, 136, 2464.
| Crossref | GoogleScholarGoogle Scholar | 24437480PubMed |
[70] I. Hod, M. D. Sampson, P. Deria, C. P. Kubiak, O. K. Farha, J. T. Hupp, ACS Catal. 2015, 5, 6302.
| Crossref | GoogleScholarGoogle Scholar |
[71] N. Kornienko, Y. B. Zhao, C. S. Kiley, C. H. Zhu, D. Kim, S. Lin, et al. J. Am. Chem. Soc. 2015, 137, 14129.
| Crossref | GoogleScholarGoogle Scholar | 26509213PubMed |
[72] P. Kubelka, J. Opt. Soc. Am. 1948, 38, 448.
| Crossref | GoogleScholarGoogle Scholar | 18916891PubMed |
[73] P. W. Doheny, J. K. Clegg, F. Tuna, D. Collison, C. J. Kepert, D. M. D’Alessandro, Chem. Sci. 2020, 11, 5213.
| Crossref | GoogleScholarGoogle Scholar |
[74] U. Schroder, F. Scholz, J. Solid State Electrochem. 1997, 1, 62.
| Crossref | GoogleScholarGoogle Scholar |
[75] U. Schroder, B. Meyer, F. Scholz, Fresenius J. Anal. Chem. 1996, 356, 295.
[76] R. Murase, C. J. Commons, T. A. Hudson, G. N. L. Jameson, C. D. Ling, K. S. Murray, et al. Inorg. Chem. 2020, 59, 3619.
| Crossref | GoogleScholarGoogle Scholar | 32124614PubMed |
[77] B. W. Ding, C. Hua, C. J. Kepert, D. M. D’Alessandro, Chem. Sci. 2019, 10, 1392.
| Crossref | GoogleScholarGoogle Scholar |
[78] P. M. Usov, C. F. Leong, B. Chan, M. Hayashi, H. Kitagawa, J. J. Sutton, et al. Phys. Chem. Chem. Phys. 2018, 20, 25772.
| Crossref | GoogleScholarGoogle Scholar | 30283919PubMed |
[79] C. Hua, P. W. Doheny, B. W. Ding, B. Chan, M. Yu, C. J. Kepert, et al. J. Am. Chem. Soc. 2018, 140, 6622.
| Crossref | GoogleScholarGoogle Scholar | 29727176PubMed |
[80] C. Hua, J. Y. Ge, F. Tuna, D. Collison, J. L. Zuo, D. M. D’Alessandro, Dalton Trans. 2017, 2998.
| Crossref | GoogleScholarGoogle Scholar | 28198492PubMed |
[81] C. Hua, A. Baldansuren, F. Tuna, D. Collison, D. M. D’Alessandro, Inorg. Chem. 2016, 55, 7270.
| Crossref | GoogleScholarGoogle Scholar | 27419690PubMed |
[82] C. Hua, P. Turner, D. M. D’Alessandro, Dalton Trans. 2015, 15297.
| Crossref | GoogleScholarGoogle Scholar | 26054414PubMed |
[83] C. Hua, D. M. D’Alessandro, Supramol. Chem. 2015, 27, 792.
| Crossref | GoogleScholarGoogle Scholar |
[84] D. A. Sherman, R. Murase, S. G. Duyker, Q. Y. Gu, W. Lewis, T. Lu, et al. Nat. Commun. 2020, 11, 2808.
| Crossref | GoogleScholarGoogle Scholar | 32499512PubMed |
[85] R. W. Elliott, P. M. Usov, B. F. Abrahams, B. Chan, R. Robson, D. M. D’Alessandro, Inorg. Chem. 2018, 57, 9766.
| Crossref | GoogleScholarGoogle Scholar | 29629755PubMed |
[86] B. M. Beam, N. R. Armstrong, S. B. Mendes, Analyst 2009, 134, 454.
| Crossref | GoogleScholarGoogle Scholar | 19238279PubMed |
[87] C. Ge, W. J. Doherty Iii, S. B. Mendes, N. R. Armstrong, S. S. Saavedra, Talanta 2005, 65, 1126.
| Crossref | GoogleScholarGoogle Scholar | 18969922PubMed |
[88] D. R. Dunphy, S. B. Mendes, S. S. Saavedra, N. R. Armstrong, Anal. Chem. 1997, 69, 3086.
| Crossref | GoogleScholarGoogle Scholar | 21639329PubMed |
[89] K. Imai, T. Okazaki, N. Hata, S. Taguchi, K. Sugawara, H. Kuramitz, Anal. Chem. 2015, 87, 2375.
| Crossref | GoogleScholarGoogle Scholar | 25607737PubMed |
[90] P. A. Christensen, in Spectroscopic Properties of Inorganic and Organometallic Compounds: Techniques, Materials and Applications (Eds J. Yarwood, R. Douthwaite, S. Duckett) 2010, Vol. 41, pp. 125–165 (The Royal Society of Chemistry: London).
[91] K. Ashley, Talanta 1991, 38, 1209.
| Crossref | GoogleScholarGoogle Scholar | 18965286PubMed |
[92] S. D. Glover, J. C. Goeltz, B. J. Lear, C. P. Kubiak, Coord. Chem. Rev. 2010, 254, 331.
| Crossref | GoogleScholarGoogle Scholar |
[93] M. Krejčik, M. Daněk, F. Hartl, J. Electroanal. Chem. Interfacial Electrochem. 1991, 317, 179.
| Crossref | GoogleScholarGoogle Scholar |
[94] J. P. Bullock, D. C. Boyd, K. R. Mann, Inorg. Chem. 1987, 26, 3084.
| Crossref | GoogleScholarGoogle Scholar |
[95] W. G. Mäntele, A. M. Wollenweber, E. Nabedryk, J. Breton, Proc. Natl. Acad. Sci. USA 1988, 85, 8468.
| Crossref | GoogleScholarGoogle Scholar | 16593991PubMed |
[96] P. J. Kulesza, M. A. Malik, A. Denca, J. Strojek, Anal. Chem. 1996, 68, 2442.
| Crossref | GoogleScholarGoogle Scholar |
[97] Z. Ping, H. Neugebauer, A. Neckel, Electrochim. Acta 1996, 41, 767.
| Crossref | GoogleScholarGoogle Scholar |
[98] T. Ito, T. Hamaguchi, H. Nagino, T. Yamaguchi, J. Washington, C. P. Kubiak, Science 1997, 277, 660.
| Crossref | GoogleScholarGoogle Scholar |
[99] J. C. Salsman, C. P. Kubiak, T. Ito, J. Am. Chem. Soc. 2005, 127, 2382.
| Crossref | GoogleScholarGoogle Scholar | 15724979PubMed |
[100] H. X. Zhong, M. Ghorbani-Asl, K. H. Ly, J. C. Zhang, J. Ge, M. C. Wang, et al. Nat. Commun. 2020, 11, 1409.
[101] P. Hosseini, G. Wittstock, I. Brand, J. Electroanal. Chem. 2018, 812, 199.
| Crossref | GoogleScholarGoogle Scholar |
[102] K. Zhao, Y. M. Liu, X. Quan, S. Chen, H. T. Yu, ACS Appl. Mater. Interfaces 2017, 9, 5302.
| Crossref | GoogleScholarGoogle Scholar | 28103017PubMed |
[103] See pp. 1–12 in: D. A. Long, Raman Spectroscopy 1977 (McGraw Hill: New York, NY).
[104] M. J. L. Santos, A. G. Brolo, E. M. Girotto, Electrochim. Acta 2007, 52, 6141.
| Crossref | GoogleScholarGoogle Scholar |
[105] M. Kalbáč, L. Kavan, M. Zukalová, L. Dunsch, Carbon 2007, 45, 1463.
| Crossref | GoogleScholarGoogle Scholar |
[106] R. S. Czernuszewicz, K. A. Macor, J. Raman Spectrosc. 1988, 19, 553.
| Crossref | GoogleScholarGoogle Scholar |
[107] D. L. Jeanmaire, M. R. Suchanski, R. P. Van Duyne, J. Am. Chem. Soc. 1975, 97, 1699.
| Crossref | GoogleScholarGoogle Scholar |
[108] I. Rey, J. C. Lassègues, P. Baudry, H. Majastre, Electrochim. Acta 1998, 43, 1539.
| Crossref | GoogleScholarGoogle Scholar |
[109] R. P. Rava, T. G. Spiro, J. Phys. Chem. 1985, 89, 1856.
| Crossref | GoogleScholarGoogle Scholar |
[110] J. Ferraris, D. O. Cowan, V. Walatka, J. H. Perlstein, J. Am. Chem. Soc. 1973, 95, 948.
| Crossref | GoogleScholarGoogle Scholar |
[111] W. R. Zheng, M. J. Liu, L. Y. S. Lee, ACS Catal. 2020, 10, 81.
| Crossref | GoogleScholarGoogle Scholar |
[112] P. M. Usov, C. McDonnell-Worth, F. Zhou, D. R. MacFarlane, D. M. D’Alessandro, Electrochim. Acta 2015, 153, 433.
| Crossref | GoogleScholarGoogle Scholar |
[113] M. Fleischmann, P. J. Hendra, A. J. McQuillan, Chem. Phys. Lett. 1974, 26, 163.
| Crossref | GoogleScholarGoogle Scholar |
[114] S. Schlucker, Angew. Chem. Int. Ed. 2014, 53, 4756.
| Crossref | GoogleScholarGoogle Scholar |
[115] S. Nie, S. R. Emory, Science 1997, 275, 1102.
| Crossref | GoogleScholarGoogle Scholar | 9027306PubMed |
[116] M. Drescher, G. Jeschke, EPR Spectroscopy: Applications in Chemistry and Biology 2012 (Springer Science & Business Media: Berlin).
[117] J. Zeitouny, V. Jouikov, Phys. Chem. Chem. Phys. 2009, 11, 7161.
| Crossref | GoogleScholarGoogle Scholar | 19672525PubMed |
[118] Y.-M. Frapart, A. Boussac, R. Albach, E. Anxolabéhère-Mallart, M. Delroisse, J.-B. Verlhac, et al. J. Am. Chem. Soc. 1996, 118, 2669.
| Crossref | GoogleScholarGoogle Scholar |
[119] P. Rapta, A. Neudeck, A. Petr, L. Dunsch, J. Chem. Soc., Faraday Trans. 1998, 94, 3625.
| Crossref | GoogleScholarGoogle Scholar |
[120] A. Neudeck, A. Petr, L. Dunsch, Synth. Met. 1999, 107, 143.
| Crossref | GoogleScholarGoogle Scholar |
[121] P. Rapta, R. Fáber, L. Dunsch, A. Neudeck, O. Nuyken, Spectrochim. Acta A Mol. Biomol. Spectrosc. 2000, 56, 357.
| Crossref | GoogleScholarGoogle Scholar |
[122] U. Evans, O. Soyemi, M. S. Doescher, U. H. F. Bunz, L. Kloppenburg, M. L. Myrick, Analyst 2001, 126, 508.
| Crossref | GoogleScholarGoogle Scholar | 11340989PubMed |
[123] A. Osterholm, A. Petr, C. Kvarnstrom, A. Ivaska, L. Dunsch, J. Phys. Chem. B 2008, 112, 14149.
| Crossref | GoogleScholarGoogle Scholar | 18928313PubMed |
[124] P. Rapta, J. Lukkari, J. Tarabek, M. Salomaeki, M. Jussila, G. Yohannes, et al. Phys. Chem. Chem. Phys. 2004, 6, 434.
| Crossref | GoogleScholarGoogle Scholar |
[125] A. Staško, K. Lušpai, Z. Barbieriková, J. Rimarčík, A. Vagánek, V. Lukeš, et al. J. Phys. Chem. A 2012, 116, 9919.
| Crossref | GoogleScholarGoogle Scholar | 22974362PubMed |
[126] P. Rapta, A. Bartl, A. Gromov, A. Staško, L. Dunsch, ChemPhysChem 2002, 3, 351.
| Crossref | GoogleScholarGoogle Scholar | 12465514PubMed |
[127] L. Dunsch, P. Rapta, A. Gromov, A. Staško, J. Electroanal. Chem. 2003, 547, 35.
| Crossref | GoogleScholarGoogle Scholar |
[128] P. R. Murray, D. Collison, S. Daff, N. Austin, R. Edge, B. W. Flynn, et al. J. Magn. Reson. 2011, 213, 206.
| Crossref | GoogleScholarGoogle Scholar | 22000629PubMed |
[129] B. Valeur, in Digital Encyclopedia of Applied Physics 2009, pp. 477–531 (Wiley-VCH Verlag GmbH & Co.: Weinheim).
[130] C. W. Machan, Curr. Opin. Electrochem. 2019, 15, 42.
| Crossref | GoogleScholarGoogle Scholar |
[131] J. K. Li, S. Ghoshal, W. T. Liang, M. T. Sougrati, F. Jaouen, B. Halevi, et al. Energy Environ. Sci. 2016, 9, 2418.
| Crossref | GoogleScholarGoogle Scholar |
[132] T. M. Arruda, B. Shyam, J. S. Lawton, N. Ramaswamy, D. E. Budil, D. E. Ramaker, et al. J. Phys. Chem. C 2010, 114, 1028.
| Crossref | GoogleScholarGoogle Scholar |
[133] Q. Y. Jia, E. S. Liu, L. Jiao, S. Pann, S. Mukerjee, Adv. Mater. 2019, 31, 1805157.
| Crossref | GoogleScholarGoogle Scholar |
[134] L. Kavan, Curr. Opin. Electrochem. 2018, 11, 122.
| Crossref | GoogleScholarGoogle Scholar |
[135] S. Y. Lin, P. M. Usov, A. J. Morris, Chem. Commun. 2018, 6965.
| Crossref | GoogleScholarGoogle Scholar |
[136] J. B. Richter, C. Eßbach, I. Senkovska, S. Kaskel, E. Brunner, Chem. Commun. 2019, 6042.
| Crossref | GoogleScholarGoogle Scholar |