

Element 92 – Uranium
Jack Harrowfield A C and Pierre Thuéry BA ISIS, Université de Strasbourg, 8 allée Gaspard Monge, 67083 Strasbourg, France.
B NIMBE, CEA, CNRS, Université Paris-Saclay, CEA Saclay, 91191 Gif-sur-Yvette, France.
C Corresponding author. Email: harrowfield@unistra.fr
Australian Journal of Chemistry 72(5) 329-333 https://doi.org/10.1071/CH19094
Submitted: 25 February 2019 Accepted: 27 February 2019 Published: 14 March 2019
If any element of the Periodic Table could be said to be the centre of controversies and unrest over its use and over the consequences of its use, uranium would be the obvious choice.[1] While uranium minerals were used as pigments by the Romans[2] – and such use continues today in the preparation of uranium glasses, often referred to as ‘vaseline glasses’ and admired for their yellow colour and green fluorescence (Fig. 1)[3] – Becquerel’s discovery[4] of radioactivity due to uranium dramatically changed the significance attached to the nature of the element. Research[5] in the first few decades of the twentieth century largely conducted in France (Marie Curie), England (Ernest Rutherford), Germany (Otto Hahn, Lise Meitner, and Fritz Strassman) and Italy (Enrico Fermi) established that natural radioactivity was associated with transmutation of one element into another involving emission of combinations of α (helium nuclei), β (electrons) and γ (high energy radiation) particles, that transmutation could be induced by bombarding one nucleus with another, and that uranium, in particular, was just the starting point of a long decay chain ultimately leading to lead as a stable species. It was Chadwick’s 1932 discovery of the neutron, however, which led Fermi to experiment with neutron-induced transmutation and Hahn and Meitner to extend his experiments in a way which led to their 1938 discovery of nuclear fission by irradiating uranium with neutrons. The interpretation by Meitner and her nephew Otto Frisch of the processes involved and particularly of the origin of the enormous energy being released awoke the international community to the prospects, perhaps both good and bad, of this reaction and led, essentially within the context of World War II, to a race between scientists in the United States (Manhattan Project[6]) and Germany[7] to establish real applications. One of these, to use a controlled fission reaction as an energy source, was achieved by Fermi in Chicago in 1942; another, achieved within the Manhattan Project under Robert Oppenheimer’s direction, was the creation of the atomic bomb, exploited in Japan in 1945. While advocacy of peaceful uses of ‘atomic energy’ was popular immediately after World War II and led eventually to many countries (notably, France) becoming dependent upon electricity generation in nuclear power stations, it was accompanied by a nuclear weapons arms race, now between the Soviet Union and the United States (and allies), constituting the ‘Cold War’ which made, for some 50 years, the study of uranium chemistry a rather restricted subject, very much focussed on methods of treating nuclear waste. In recent times, with a wider appreciation of its prospects,[8–10] uranium chemistry has undergone remarkable developments, briefly considered below.

|
Uranium occurs naturally on Earth as three isotopes 234U, 235U, and 238U, while the isotope 233U can be synthesised by neutron irradiation of thorium. As each isotope is radioactive and has a different half-life, the natural isotopic distribution is time dependent and the current estimates are 99.3 % 238U, 0.7 % 235U, and 0.005 % 234U, although since 235U is the isotope of interest for the nuclear industry, commercially available uranium compounds contain ‘depleted’ uranium and can be considered essentially as containing just 238U. 238U is described as being weakly radioactive[9] and is an α emitter, so that its compounds can be used in the laboratory without too rigorous protection procedures provided inhalation or ingestion are avoided. 235U is the only naturally occurring ‘fissile’ nucleus, meaning that slow neutron irradiation induces a fragmentation to give a variety of lighter elements, including barium, which was the basis of Hahn’s meticulous proof of the occurrence of such fission. Since the reaction produces extra neutrons, a ‘chain reaction’ can be induced provided the amount of 235U in the uranium sample is ~3 % or higher and this of course is the basis of the use, in different ways, of uranium in nuclear reactors and atomic bombs. Anomalies in terrestrial uranium isotope distributions, first detected in samples from the Oklo mine in Gabon,[11] have been interpreted as evidence that at a time following nucleosynthesis[12] of the isotopes when the 235U level was ~3 %, some natural nuclear reactors were in operation.
The name uranium (first ‘uranit’) was given to the element by Martin Heinrich Klaproth in 1789, partly in support of a Royal Academy colleague who favoured the name Uranus for the planet, at the time a matter of dispute. Thus, the element name derives from that of the ancient Greek god of the sky. Klaproth thought that he had isolated the element from an ore sample (obtained from a mine close to the present day border between Germany and the Czech Republic) but he probably had just an oxide, and elemental uranium metal was not isolated until 1841 by Eugène Péligot using reduction of the tetrachloride with potassium.[13] The same ore, obtained from a nearby mine in what is now the Czech town of Jáchymov, was the ‘pitchblende’ or uraninite (Fig. 2) used by Marie Curie in her celebrated work which included the isolation of polonium[14] and radium. The name ‘pitchblende’ derives from the German ‘pech blende’, meaning ‘bad luck rock’ since it was intrusions of this ore which signalled the end of silver ore deposits that were the basis of the wealth of the town then known as Joachimsthal. Silver coins minted there were known as Joachimsthaler, abbreviated to simply ‘thaler’, a word which is the etymological origin of ‘dollar’.

|
Pitchblende, with the approximate formula UO2, and carnotite, K2(UO2)2(VO4)2·3H2O, are the best known uranium minerals, but there are many others,[15] widely distributed, and the crustal abundance of uranium (2.3 ppm) is not particularly low, being greater than that of tin, for example, despite the fact that it has the greatest atomic number and heaviest nucleus of any element found naturally in significant concentrations on Earth. Processing of uranium ores[16] can involve either acid (H2SO4) or base (Na2CO3) leaching, with the finally isolated product usually being ‘yellowcake’, U3O8, a mixed UV-UVI oxide. Australia, with approximately one-third of the world’s known uranium reserves, currently exports close to 6000 tonnes of yellowcake, with a value near A$1 billion, each year.[17] Treatment[18] of returned nuclear waste has been proposed as one form of uranium chemistry for Australia but has not proven to be a popular idea. Part of the research on waste treatment has involved efforts to find selective ‘uranophile’ ligands (Fig. 3), first considered[19] for their possible use in extraction of the enormous total quantities, in very dilute solution, of the uranium dissolved in the world’s oceans.
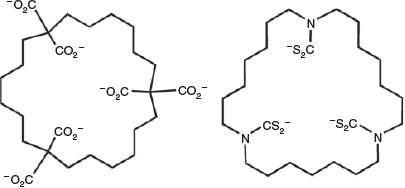
|
Putting the chemistry of nuclear applications aside, uranium chemistry still has both richness and diversity which exceed that of many familiar transition metals.[8–10] In its compounds, oxidation states from +I to +VI are well characterised, with relativistic effects in its bonding leading to significant involvement of 5f orbitals in its valence shell.[10] Compounds in the oxidation states IV and VI are most abundant, but even oxidation state V, once thought to be inherently unstable, has been shown to have many stable forms.[20] While the catalytic activity of uranium (in ammonia synthesis) was recognised by Haber as long ago as 1909,[9] the burgeoning of uranium organometallic and multidentate ligand chemistry which began in the 1960s, notably involving the synthesis of ‘uranocene’ (bis(cyclo-octatetraenide)uranium(IV)),[21] has led to the development of complexes (Fig. 4) able to activate important small molecules such as CO, CO2, and N2[10] and to catalyse a variety of reactions including olefin polymerisation, Diels-Alder additions, hydroamination, and hydrosilylation.[8] A UVI complex with a terminal nitride ligand, although not organometallic, is very active in CH bond activation and was described in 2013 as an ‘actinide milestone’.[22]

|
The combination of the valence shell, size and shape of uranium in its various oxidation states and complexes is what determines their properties and the best-known examples of stereochemistry rarely found outside the actinide series are those in the myriad complexes of UVI incorporating the linear uranyl cation UO22+, where the uranium is, with rather few exceptions,[23–25] in either pentagonal- or hexagonal-bipyramidal coordination (Fig. 5). Early crystal structure determinations established the coordination geometries of simple species such as Na[UO2(O2CCH3)3] (hexagonal-bipyramidal)[26] and [UO2(OH2)5](ClO4)2 (pentagonal-bipyramidal),[27] but recent interest in exploiting the ‘unusual’ coordination geometry of the uranyl ion for the synthesis of novel coordination polymers/metal-organic frameworks[23,28–31] has provided innumerable examples of both forms involving more complicated ligands. Both forms are also found in the large family of remarkable cluster complexes derived by peroxide complexation of the uranyl ion.[32]
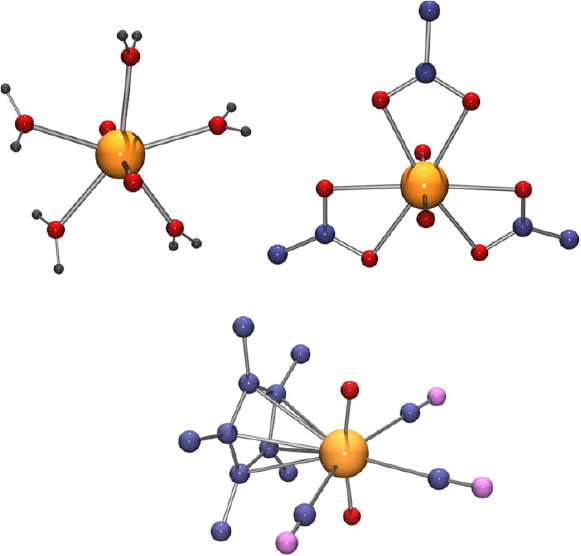
|
A property of most uranyl compounds is a green luminescence, long known in the case of uranium (‘vaseline’) glasses (Fig. 1). Typically, this luminescence displays a striking vibronic coupling (Fig. 6) due to the symmetric stretching mode of the UO22+ unit.[33] The excited uranyl ion is well known as a photo-oxidant[34] and one of the reasons[31] for investigating the synthesis of framework solids incorporating uranyl centres has been the hope of finding solid state cavities suitable for selectively binding substrates in the vicinity of the photoactive centre. Success[35] in this area has been limited to date, although simple uranyl salts deposited on supports such as titania or mesoporous silica[36] have been shown to be active photocatalysts for oxidation of a variety of substrates and quite diverse solid coordination complex systems where moderately large molecules are included within cavities (Fig. 7) are now known.[37,38] Optimism remains that real applications of uranium outside the nuclear and military domains may continue to be found, with many areas yet to be fully investigated.[39]
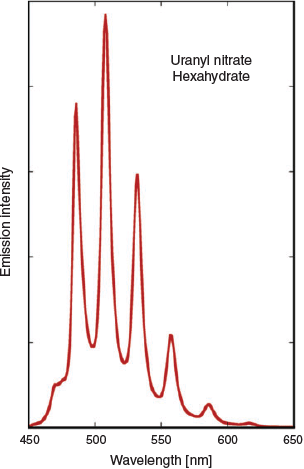
|
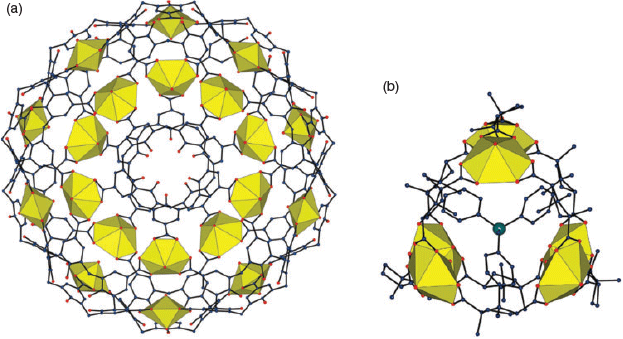
|
Conflicts of Interest
The authors declare no conflicts of interest.
References
[1] D. Muller, Uranium. Twisting the Dragon’s Tail, a television documentary produced by Gene Pool Productions for SBS Australia and PBS USA, 2015.[2] E. R. Caley, Isis 1948, 38, 190.
| Crossref | GoogleScholarGoogle Scholar | 18903949PubMed |
[3] D. Strahan, Stud. Conserv. 2001, 46, 181.
[4] H. Becquerel, Comptes Rendus Acad. Sci. 1896, 122, 420.
[5] See Ch. 1 in C. Hardy, Atomic Rise and Fall: The Australian Atomic Energy Commission 1953–1987 1999 (Glen Haven Publishing: Peakhurst) and https://en.wikipedia.org/wiki/Nuclear_fission.
[6] https://www.history.com/topics/world-war-ii/the-manhattan-project
[7] K. Mayer, M. Vallenius, K. Lützenkirchen, J. Horta, A. Nicholl, G. Rasmussen, P. van Belle, Z. Varga, R. Buda, N. Erdmann, J.-V. Kratz, N. Trautmann, L. K. Fifield, S. G. Tims, M. B. Fröhlich, P. Steier, Angew. Chem. Int. Ed. 2015, 54, 1.
[8] M. Ephritikhine, Dalton Trans. 2006, 2501.
| Crossref | GoogleScholarGoogle Scholar | 16718334PubMed |
[9] M. J. Monreal, P. L. Diaconescu, Nat. Chem. 2010, 2, 424.
| Crossref | GoogleScholarGoogle Scholar | 20414247PubMed |
[10] S. T. Liddle, Angew. Chem. Int. Ed. 2015, 54, 8604.
| Crossref | GoogleScholarGoogle Scholar |
[11] F. Gauthier-Lafaye, Comptes Rendus Physique 2002, 3, 839. https://inis.iaea.org/collection/NCLCollectionStore/_Public/07/233/7233255.pdf
[12] S. Goriely, G. Martinez-Pindo, Nucl. Phys. A 2015, 944, 158.
[13] A. J. Rossi, J. Am. Chem. Soc. 1890, 12, 128.
| Crossref | GoogleScholarGoogle Scholar |
[14] N. B. Mikeev, Chem. Zeit. 1978, 102, 277.
[15] P. C. Burns, Can. Mineral. 2005, 43, 1839.. See also
| Crossref | GoogleScholarGoogle Scholar |
[16] https://www.911metallurgist.com/blog/uranium-ore-processing-methods
[17] http://www.world-nuclear.org/information-library/country-profiles/countries- a-f/australia.aspx (a 2018 update).
[18] See F. Arnaud-Neu, M.-J. Schwing-Weil, J.-F. Dozol, in Calixarenes 2001 (Eds Z. Asfari, V. Böhmer, J. Harrowfield, J. Vicens) 2001, Ch. 35, pp. 642–662 (Kluwer Academic Publishers: Dordrecht) and references therein.
[19] (a) I. Tabushi, Y. Kobuke, K. Ando, M. Kishimoto, E. Ohara, J. Am. Chem. Soc. 1980, 102, 5947.
| Crossref | GoogleScholarGoogle Scholar |
(b) Y. Kobuke, T. Aoki, H. Tanaka, I. Tabushi, T. Kamaishi, I. Hagiwara, Ind. Eng. Chem. Res. 1990, 29, 1662.
| Crossref | GoogleScholarGoogle Scholar |
[20] C. R. Graves, J. L. Kiplinger, Chem. Commun. 2009, 3831.
| Crossref | GoogleScholarGoogle Scholar |
[21] (a) A. Streitwieser, U. Müller-Westerhoff, J. Am. Chem. Soc. 1968, 90, 7364.
| Crossref | GoogleScholarGoogle Scholar |
(b) (Crystal structure) A. Avdeef, K. N. Raymond, K. O. Hodgson, A. Zalkin, Inorg. Chem. 1972, 11, 1083.
| Crossref | GoogleScholarGoogle Scholar |
[22] T. W. Hayton, Nat. Chem. 2013, 5, 451.
| Crossref | GoogleScholarGoogle Scholar | 23695625PubMed |
[23] T. Loiseau, I. Mihalcea, N. Henry, C. Volkringer, Coord. Chem. Rev. 2014, 266–267, 69.
| Crossref | GoogleScholarGoogle Scholar |
[24] K. Cottet, P. M. Marcos, P. J. Cragg, Beilstein J. Org. Chem. 2012, 8, 201.
| Crossref | GoogleScholarGoogle Scholar | 22423288PubMed |
[25] J. Maynadié, J.-C. Berthet, P. Thuéry, M. Ephritkhine, Chem. Commun. 2006, 486.
[26] (a) I. Fankuchen, Z. Kristallogr. 1935, 90, 473.
(b) (precise determination) W. H. Zachariasen, H. A. Plettinger, Acta Crystallogr. 1959, 12, 526.
| Crossref | GoogleScholarGoogle Scholar |
[27] (a) N. W. Alcock, J. Esperas, J. Chem. Soc., Dalton Trans. 1977, 893.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. Fischer, Z. Anorg. Allg. Chem. 2003, 629, 1012.
| Crossref | GoogleScholarGoogle Scholar |
[28] P. Thuéry, J. Harrowfield, Dalton Trans. 2017, 46, 13660.
| Crossref | GoogleScholarGoogle Scholar | 28932836PubMed |
[29] J. Su, J. S. Chen, in Lanthanide Metal-Organic Frameworks (Ed. P. Cheng) 2014, Structure and Bonding, Vol. 163, pp. 265–295 (Springer: Berlin).
[30] M. B. Andrews, C. L. Cahill, Chem. Rev. 2013, 113, 1121.
| Crossref | GoogleScholarGoogle Scholar | 22881287PubMed |
[31] K. X. Wang, J. S. Chen, Acc. Chem. Res. 2011, 44, 531.
| Crossref | GoogleScholarGoogle Scholar | 21612214PubMed |
[32] J. Qiu, P. C. Burns, Chem. Rev. 2013, 113, 1097.
| Crossref | GoogleScholarGoogle Scholar | 23094705PubMed |
[33] H. D. Burrows, M. da Graça Miguel, Adv. Colloid Interface Sci. 2001, 89–90, 485.
| Crossref | GoogleScholarGoogle Scholar | 11215812PubMed |
[34] M. Sarakha, M. Bolte, H. D. Burrows, J. Phys. Chem. A 2000, 104, 3142. and references therein.
| Crossref | GoogleScholarGoogle Scholar |
[35] Y. N. Hou, X. T. Xu, N. Xing, F. Y. Bai, S. B. Duan, Q. Sun, S. Y. Wei, Z. Shi, H. Z. Zhang, Y. H. Xing, ChemPlusChem 2014, 79, 1304.
| Crossref | GoogleScholarGoogle Scholar |
[36] (a) P. O. Kolinko, T. N. Fillipov, D. N. Kozlov, V. N. Parmon, J. Photochem. Photobiol. Chem. 2012, 250, 72.
| Crossref | GoogleScholarGoogle Scholar |
(b) V. Krishna, V. S. Gamble, N. S. Gupta, P. Selvam, J. Phys. Chem. C 2008, 112, 15832.
| Crossref | GoogleScholarGoogle Scholar |
[37] S. Pasquale, S. Sattin, E. C. Escudero-Adán, M. Martinez-Belmonte, J. de Mendoza, Nat. Commun. 2012, 3, 785.
| Crossref | GoogleScholarGoogle Scholar | 22510690PubMed |
[38] P. Thuéry, Y. Atoini, J. Harrowfield, Inorg. Chem. 2019, 58, 870.
| Crossref | GoogleScholarGoogle Scholar |
[39] J. Xie, Y. Wang, W. Liu, X. Yin, L. Chen, Y. Zou, J. Diwu, Z. Chai, T. E. Albrecht-Schmitt, G. Liu, S. Wang, Angew. Chem. Int. Ed. 2017, 56, 7500.
| Crossref | GoogleScholarGoogle Scholar |