

Improved Fenton Therapy Using Cancer Cell Hydrogen Peroxide*
Hadi Ranji-Burachaloo A B , Qiang Fu A , Paul A. Gurr A , Dave E. Dunstan B C and Greg G. Qiao A C
A B , Qiang Fu A , Paul A. Gurr A , Dave E. Dunstan B C and Greg G. Qiao A C
A Polymer Science Group, Department of Chemical Engineering, The University of Melbourne, Parkville, Vic. 3010, Australia.
B Complex Fluids Group, Department of Chemical Engineering, The University of Melbourne, Parkville, Vic. 3010, Australia.
C Corresponding authors. Email: davided@unimelb.edu.au; gregghq@unimelb.edu.au
Australian Journal of Chemistry 71(10) 826-836 https://doi.org/10.1071/CH18281
Submitted: 7 June 2018 Accepted: 18 August 2018 Published: 19 September 2018
Abstract
Fenton cancer therapy as a new methodology for the treatment of tumour cells is largely restricted owing to the low stability, high aggregation, and poor selectivity of reported nanoparticles. In this study, an improved approach for the selective destruction of cancer cells is reported. Metal–organic framework (MOF) nanoparticles were synthesized and reduced via a hydrothermal method, and then PEGylated through the surface-initiated atom transfer radical polymerization (SI-ATRP) reaction to produce a PEGylated reduced MOF (P@rMOF). The ratio of PEG to nanoparticles was used to optimize the size and aggregation of the nanoparticles, with 2P@rMOF (2 : 1 mass ratio) having the smallest hydrodynamic diameter. The nanoparticles were further conjugated with folic acid for cell targeting. In vitro cell uptake experiments demonstrated that the internalization of 2P@rMOF-FA nanoparticles into cancer cells (HeLa) was almost 3-fold that of normal cells (NIH-3T3). In the presence of 2P@rMOF-FA, the HeLa cell viability decreased dramatically to 22 %, whereas the NIH-3T3 cell viability remained higher than 80 % after 24 h incubation. The selectivity index for 2P@rMOF-FA is 4.48, which is significantly higher than those reported in the literature for similar strategies. This work thus demonstrates the most stable and selective nanoparticle system for the treatment of cancer cells using the cell’s own H2O2.
Introduction
Complete removal of tumour cells without damage to normal tissues is the ideal goal of cancer therapy. In the past decades, nanotechnology has been making significant improvements in this field by designing agents that are responsive to the tumour microenvironment.[1,2] For example, the acidic nature of cancer cells has been utilized for many drug delivery systems in which drugs are released in tumour cells at low pH.[3,4] In addition, another difference between normal and cancer cells is the high level of reactive oxygen species (ROS) within cancer cells, which has been used by different therapy methods for destroying tumour cells selectively.[5,6] ROS are radicals, ions or molecules that have a single unpaired electron in their structures, and they can be categorized into two groups of free oxygen radicals and non-radical ROS.[7] Hydrogen peroxide (H2O2) is generally regarded as the most abundant and stable non-radical ROS in cancer cells.[8] However, it can be converted to hydroxyl radicals (OH•), which are much more toxic than other ROS.[9] As OH• has a short half-life (10−9 s) and high reactivity, it does not diffuse from the generation site and instead oxidizes proteins, lipids, and DNA within the biological system.[10,11]
Recent studies have shown that Fe-based nanoparticles are able to treat cancer cells by the generation of OH• according to the Fenton reaction consuming the H2O2.[12–17] However, the reported nanoparticles were unable to treat cancer cells using endogenous H2O2 and required the addition of exogenous sources to be effective. Additives such as ascorbic acid, β-lapachone, cinnamaldehyde, and cisplatin were used to undergo redox cycles to generate high H2O2 levels inside the cancer cells. The combination of these agents with either iron oxide particles[12,15] or ferrocene[18] was used as a new source of ROS to enhance the effectiveness of anticancer drugs. It was reported that FePt nanoparticles did not require external hydrogen peroxide sources to treat cancer cells.[19] However, there are serious concerns relating to the nanoparticles’ biocompatibility, stability, and low selectivity owing to potential metal (Pt) contamination.[20,21] Further to this, our group demonstrated that reduced iron metal–organic framework nanoparticles conjugated with folic acid (rMOF-FA) have the ability to treat cancer cells by generation of OH• inside cancer cells using the cell’s own H2O2 without the need for external hydrogen peroxide sources.[22] The selectivity index for this novel nanoparticle (2.45) was higher than commercial drugs but lower than other novel strategies.[23–26] In order to increase their stability, biocompatibility, treatment effectiveness, and selectivity, a surface PEGylation of rMOF-FA was investigated.
Surface-initiated atom transfer radical polymerization (SI-ATRP) is a popular method for constructing well-defined core–shell structures.[27] By applying this method, the shell thickness, grafting density, and chemical composition of layers can be easily controlled.[28] This method has previously been utilized for drug delivery systems,[29,30] in which the coating of nanoparticles with compatible polymers increases their biocompatibility and improves treatment selectivity.[31,32] MOF/polymer core–shell composite nanoparticles (P@MOF) prepared by an SI-ATRP method were reported by our group in which the polymer shell was shown to effectively improve the dispersity and the catalytic activity of the MOF nanoparticles in water.[33]
Herein, we report a novel stable polymer-grafted MOF conjugated with folic acid (P@rMOF-FA) nanoparticles for selective cancer treatment. By applying an SI-ATRP method and varying the ratio of monomer to MOF, three different sizes of nanoparticles (1P@rMOF, 2P@rMOF, and 3P@rMOF) were prepared. Transmission electron microscopy (TEM) analysis of the nanoparticles confirmed the average hydrodynamic diameter of the particles to be 100, 40, and 65 nm for 1P@rMOF, 2P@rMOF, and 3P@rMOF respectively. After conjugation with folic acid, the three P@rMOF nanoparticles were completely stable in aqueous solution and showed significant peroxidase-like activity at low H2O2 concentration. HeLa (cervical cancer cells) and NIH-3T3 (non-cancerous fibroblasts) cell lines were used for cellular uptake, ROS, and in vitro cytotoxicity studies. The results demonstrated that decreasing the size of the nanoparticles and modification of the surface increased the cellular uptake and ROS levels in cancer cells. Finally, it was shown that 2P@rMOF-FA nanoparticles have greater treatment effectiveness and selectivity compared with 1P@rMOF and 3P@rMOF nanoparticles.
Experimental
Materials
FeCl3.6H2O (97 %, Sigma), 2-aminobenzene-1,4-dicarboxylic acid (H2N-BDC; 99 %, Sigma), hydroquinone (99 % , Sigma), Pluronic F-127 (Sigma), folic acid (97 %, Sigma), 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC; 98 %, Acros), N-hydroxysuccinimide (NHS; 98 %, Sigma), dimethyl sulfoxide (DMSO; AR, Ajax Finechem), trimethylamine (TEA; 99.5 %, Sigma), α-bromoisobutyryl bromide (BiBB; 98 %, Sigma), poly(ethylene glycol) methyl ether methacrylate (PEGMA; average Mn 475, Sigma), copper(ii) bromide (CuBr2; 99 %, Sigma), copper(i) bromide (CuBr; 99 %, Sigma), sodium ascorbate (98 %, Sigma), propargylamine (98 %, Sigma), sodium azide (NaN3; 99 %, Chem-Supply), N,N,N′,N″,N″-pentamethyldiethylenetriamine (PMDTA; Sigma, 99 %), acetic acid (AcOH; Chem-Supply), phosphate buffered saline tablets (Sigma), syringe filters (cellulose acetate, Dismic, 0.2 µm), hydrogen peroxide (H2O2; 30 wt-%, Chem-Supply), 3,3′,5,5′-tetramethylbenzidine (TMB; 99 %, Sigma), Dulbecco’s modified Eagle medium (DMEM; Gibco BRL Invitrogen), foetal bovine serum (FBS; Gibco BRL Invitrogen), GlutaMAXTM (Gibco BRL Invitrogen), penicillin (Gibco BRL Invitrogen), trypsin–EDTA (1×, Gibco BRL Invitrogen), 96-well plates (black, white), 2′,7′-dichlorofluorescein diacetate (DCFH-DA; 97 %, Sigma), Cell Counting Kit-8 solution (CCK-8; Sigma), ethanol (AR, Chem-Supply), and methanol (AR, Chem-Supply) were used as received. Tetrahydrofuran (THF) was distilled from benzophenone and sodium metal under argon. Tris[2-(dimethylamino)ethyl]amine (Me6Tren) was prepared according to a literature procedure.[34]
Instrumentation
TEM characterizations were performed on an LVEM5 transition electron low voltage microscope (Delong America) operating at a voltage of 5 kV, with the samples prepared by dropping the solution (0.1 mg mL−1 solution) onto carbon-coated copper grids for ~30 s, then drying for 30 min. X-ray diffraction (XRD) patterns of the samples were recorded on a Bruker D8 Avance instrument with CuKα radiation (40 kV, 40 mA) and a nickel filter, and the samples were exposed at a scanning rate of 2θ = 0.020° s−1 in the range of 5–45°. X-ray photoelectron spectroscopy (XPS) analysis was performed on a VG Escalab 220i-XL spectrometer under ultrahigh vacuum (6 × 10−9 mbar [6 × 10−7 Pa]). A fixed photon energy (AlKα 1486.6 eV) was used. A survey scan was performed between 0 and 1200 eV with a resolution of 1.0 eV and pass energy of 100 eV. High-resolution scans for Fe 2p (699 to 739 eV) were also conducted with a resolution of 0.1 eV and a pass energy of 20 eV. Attenuated total reflectance Fourier transform infrared (ATR FT-IR) was performed on dried samples using a Bruker Tensor 27 in the mid-infrared range (400–4000 cm−1). The instrument was equipped with OPUS 6.5 software. Measurements were made in transmittance mode. Dynamic light scattering (DLS) measurements were conducted on a Wyatt DynaPro NanoStar DLS/SLS instrument with a GaAs laser (658 nm) at an angle of 90° and a temperature of 25 ± 0.1°C. Stable spectra were determined at sample concentrations of 1 mg mL−1. Thermogravimetric analysis (TGA) was performed on a PerkinElmer Pyris-1 thermogravimetric analyser, and the samples were heated from 30 to 800°C at a heating rate of 2 K min−1 under an air flow. Zeta-potentials of the nanoparticles were analysed with a Zetasizer Nano ZS (Malvern Instruments). Particles (800 µL of 0.1 mg mL−1 solution) were placed into a disposable zeta cell, and zeta-potential measurements were conducted at room temperature. 1H NMR analysis was performed using a Varian unity Plus 400 MHz NMR spectrometer using deuterated solvent as reference. Inductively coupled plasma optical emission spectrophotometry (ICP-OES) was performed on a PerkinElmer Optima 4300 DV using calibration curves generated from standard solutions (0.01–5 ppm). Finally, UV-vis spectrometry (UV-vis) was performed on a Shimadzu UV-1800 spectrometer using quartz cuvettes with a 1-cm path length.
Preparation of NH2-MIL-88B(Fe) MOF Nanoparticles
NH2-MIL-88B(Fe) MOF nanoparticles were synthesized using the hydrothermal method previously described.[35] Briefly, a surfactant solution of Pluronic F-127 (640 mg) in Milli-Q water (60 mL) was prepared followed by addition of FeCl3.6H2O (714 mg, 2.64 mmol). The reaction mixture was stirred for 2 h, then H2N-BDC (240 mg, 1.32 mmol) was added to the solution, and the mixture stirred for a further 2 h. Finally, the reaction mixture was transferred into an autoclave and heated at 110°C for 16 h. The mixture was washed 3 times with ethanol, centrifuged (5323 g, 1 h), and then dried under vacuum to afford raw MOF nanoparticles as a brown powder (570 mg, 60 % yield).
Hydrothermal Reduction of MOF to rMOF Nanoparticles
According to our previous study,[36] preformed raw MOF (100 mg) and hydroquinone (1 g) were dissolved in Milli-Q water (10 mL).[36] The mixture was transferred into an autoclave and heated at 150°C for 16 h. After the autoclave had cooled to ambient temperature, the black precipitate (rMOF) was washed with methanol, centrifuged (5323 g, 1 h), and dried under vacuum to afford rMOF as a black powder (45 mg, 45 % yield).
Preparation of Br-Functionalized rMOF Nanoparticles
According to our previous study,[33] preformed raw rMOF (50 mg) was dispersed in anhydrous THF (5 mL) by sonification. TEA (47 µL, 34 mg, 0.37 mmol) and BiBB (23 µL, 43 mg, 0.18 mmol) were dissolved in THF (1 mL) sequentially. The TEA solution was injected into the rMOF suspension under a nitrogen atmosphere. The BiBB solution was then added dropwise to the mixture at room temperature and it was subsequently stirred at 50°C for 24 h. The reaction mixture was filtered, rinsed with THF and methanol to remove the unreacted precursors and TEA.HBr salt by-product, and then dried under reduced pressure at 40°C to afford Br@MOF (44 mg, 85 % yield).
PEGylation of rMOF Using the SI-ATRP Reaction
The P@rMOF samples were synthesized by SI-ATRP using Br@rMOF as the initiator.[33,37,38] Typically, Br@rMOF (40 mg) and varying amounts of PEGMA (111, 222, 333 µL) were sonicated in Milli-Q water (14 mL). A mixture of CuBr2 (5.8 mg, 0.026 mmol), Me6Tren (34.8 µL, 30 mg, 0.13 mmol), and sodium ascorbate (15.3 mg, 0.077 mmol) dissolved in Milli-Q water (2 mL) was then added and the reaction mixture stirred at 30°C for 24 h. The resultant solution was purified by dialysis (cut-off MW 3500) and dried by lyophilization to obtain the P@rMOF (~60 % yield).
Preparation of Azide End-Functionalized P@rMOF Nanoparticles
P@rMOF nanoparticles (20 mg) containing bromide end groups were dissolved in anhydrous DMF (6.7 ml). NaN3 (43.6 mg, 10 equiv.) was then added and the reaction mixture stirred at 30°C for 24 h.[39] This solution was purified by dialysis (cut-off MW 3500) and dried by lyophilization to obtain the product (40 % yield).
Synthesis of Alkyne-Functionalized Folic Acid
Folic acid (1.0 g, 2.2 mmol) was dissolved in anhydrous DMF (10 mL) and cooled in a water/ice bath. NHS (0.26 g, 2.5 mmol) and EDC (0.40 g, 2.5 mmol) were added, and the resulting mixture was stirred in an ice bath for 30 min. A solution of propargylamine (0.12 g, 2.3 mmol) in DMF (5.0 mL) was added, the mixture was allowed to warm to room temperature and then stirred for 24 h. The reaction mixture was poured into water (100 mL) and stirred for 30 min to form a precipitate. The orange-yellow precipitate was filtered, washed with acetone, and dried under vacuum for 6 h (50 % yield).[40,41]
Click Reaction of Azide-P@rMOF Nanoparticles with Alkyne–Folic Acid
P@rMOF-N3 (20 mg) and alkyne-folic acid (96.16 mg) were added to DMSO (10 mL) in a dry 25-mL round-bottom flask, and the solution was degassed by bubbling N2 through it for 30 min. CuBr/PMDTA (1 : 1 molar ratio) (14.4 and 17.42 mg respectively) catalyst solution (100 mM in degassed DMF, 1 mL) was then added, and the reaction mixture was stirred at 60°C for 24 h. The mixture was washed twice with DMSO, centrifuged (8800 g, 1 h), and then dried under vacuum to afford final P@rMOF-FA nanoparticles as a black powder (30 % yield).[40]
Peroxidase-Like Activity Assay
Measurements were carried out in AcOH buffer solution (3 mL, 0.1 M, pH 5.0) containing the different nanoparticles (1P@rMOF-FA, 2P@rMOF-FA, or 3P@rMOF-FA, 50 µg mL−1), H2O2 (500 µM), and TMB (250 µM) at 37°C. The solutions were incubated for a predefined time period, and the absorbance spectra then were observed using a UV-vis spectrometer.[42] In addition, the peroxidase-like activity of 2P@rMOF-FA (50 µg mL−1) was determined while varying the pH from 3 to 7.4 (pH 3–6 AcOH buffer and pH 6–7.4 phosphate buffer) at an absorbance of 652 nm.
Cell Culture
HeLa and NIH-3T3 cells were maintained in DMEM supplemented with 10 % FBS, GlutaMAXTM (2 mM), and penicillin (100 units mL−1). Cells were passaged every 3–4 days using 0.25 % trypsin–EDTA at subconfluence and incubated at 37°C, 5 % CO2, and 90 % humidity. Cell passages 5–15 were used for cell experiments.[25]
Cellular Uptake Test
HeLa and NIH-3T3 cells were seeded into black 12-well plates (1 × 105 cells per well) in cell culture medium (supplemented DMEM). The medium was replaced with serum-free medium (1 mL) containing the different nanoparticles (1P@rMOF-FA, 2P@rMOF-FA, 3P@rMOF-FA, 50 µg mL−1) after 12 h and incubated for a predefined time period. Finally, the medium was removed and the cells were rinsed twice with PBS. For quantitative evaluation of cellular uptake, the cells were then harvested with trypsin and centrifuged at 109 g for 5 min. The supernatant was removed, and the pellet was digested with fresh aqua regia (0.5 mL) for 10 min and then diluted to a total volume of 5 mL with Milli-Q water. The concentrations of internalized iron nanoparticles were measured by ICP-OES, and are reported as the concentration of nanoparticles per cell.[43,44]
Reactive Oxygen Species Assay
Confocal Microscopy Images
HeLa and NIH-3T3 cells were seeded into eight-well-chamber glass-bottom slides (Laboratory-Tek, Chambered no. 1.0 borosilicate coverglass) (2.5 × 104 cells per well) in cell culture medium (supplemented DMEM). The medium was replaced with serum-free medium (300 μL) containing DCFH-DA (20 μM) after 12 h and incubated for 30 min. The cells were further rinsed twice with PBS and incubated with 2P@rMOF-FA (50 µg mL−1) for 2 h. The cells were carefully rinsed again and the intracellular ROS levels were evaluated by detecting the fluorescence of DCFH-DA (λex 488 and λem 525 nm) with confocal laser scanning microscopy (Nikon A1R).[45] Images were generated by optical sectioning in the z direction and were analysed using ImageJ software.
Time-Dependent Fluorescence Intensity
HeLa and NIH-3T3 cells were seeded into black 96-well plates (1 × 104 cells per well) in cell culture medium (supplemented DMEM). The medium was replaced with serum-free medium (100 μL) containing DCFH-DA (20 μM) after 12 h and incubated for 30 min. The cells were further rinsed twice with PBS and incubated with different nanoparticles (1P@rMOF-FA, 2P@rMOF-FA, or 3P@rMOF-FA, 50 µg mL−1) for a predefined time period. Finally, the cells were carefully rinsed again and the fluorescence intensity (λex 488/λem 525 nm) measured using a fluorescence plate reader (Tecan M200 Infinite Pro).
Cell Viability Assay
The cell viability tests were analysed by the standard cell counting kit-8 (CCK-8) assay method. HeLa and NIH-3T3 cells were seeded into 96-well plates (1 × 104 cells per well) in cell culture medium. After 12 h, the medium was replaced with 100 μL of fresh medium containing various concentrations of nanoparticles (1P@rMOF-FA, 2P@rMOF-FA or 3P@rMOF-FA, 0–100 µg mL−1) and incubated for a further 24 h. The cells were then washed twice with PBS and incubated with 110 μL fresh medium containing 10 μL CCK-8 solution for a further 3 h. Finally, the medium was removed and the absorbance at 460 nm was measured using a microplate reader (Tecan M200 Infinite Pro). Note that all experiments were conducted in triplicate, and error bars shown represent the standard error of independent experiments. The cell viability (%) was calculated with the following formula, where [A] is the average absorbance:
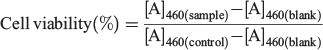
Selectivity Index
The degree of selectivity of the nanoparticles against cancer cells was calculated using the selectivity index as follows:
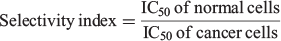
where IC50 is the calculated half-maximum inhibitory concentration value.
Statistical Analysis
Data are shown as averages and standard deviations. Student’s t-tests were used to analyse the statistical differences between samples for cytotoxicity and hydrogen peroxide concentration measurements and were considered significant at P < 0.05.
Results and Discussion
Material Synthesis and Characterization
In the present study, P@rMOF-FA nanoparticles were prepared via a four-step process, as shown in Scheme 1. Initially, NH2-MIL-88B(Fe) was synthesized using the hydrothermal procedure previously reported.[35] The raw MOF material was then reduced in the presence of hydroquinone to produce rMOF as black crystals, containing both Fe2+ and Fe3+ ions.[36] rMOF nanoparticles were then functionalized with BiBB followed by polymerization with PEGMA (Mn 475 Da) using SI-ATRP to produce P@rMOFs with varying polymer chain lengths.[33,37,38] Finally, the obtained P@rMOF was conjugated with folic acid via azide-alkyne click chemistry to afford a series of P@rMOF-FA nanoparticles.[40]

|
MOF and rMOF Preparation
TEM was used to determine the shape and size of the MOF and rMOF nanoparticles. From TEM imaging, MOF particles had needle-shaped morphologies (Fig. 1a) with an average length of 250 nm and a width of 50 nm. However, rMOF (Fig. 1b) showed an irregular shape and size. The particle size distribution based on measurements from six TEM images was in the range of 20–60 nm (Fig. S1, Supplementary Material). This may be attributed to partial structural damage during the reduction process.[36] The crystal structure of MOF and rMOF were verified using XRD analysis. As shown in Fig. 1c(i), the diffraction profile of the MOF represents the crystalline structure, with the characteristic peaks at 2θ of 9.2, 10.5, 13.5, 17.4, 18.2, and 20.2° corresponding to NH2-MIL-88B(Fe) with an octahedral structure.[35,46] However, the diffraction profile of the rMOF changed after reduction and shows a different crystalline structure, with characteristic peaks at 2θ of 8.5, 12.1, 20.9, 24.3, and 27.1°(Fig. 1c(ii)). XPS analysis was utilized to verify the oxidation states of iron in MOF and rMOF crystals. The resultant peaks of Fe 2p3/2 and Fe 2p1/2 for the MOF and rMOF are shown in Fig. 1d. The presence of FeIII in the MOF structures is verified by the peak positions of Fe 2p3/2 and Fe 2p1/2 at 712.1 and 725.3 eV respectively (Fig. 1d(i)).[47] After reduction, the Fe 2p1/2 peak shifts to 726.2 eV, and the Fe 2p3/2 satellite peak at 716.8 eV appears (Fig. 1d(ii)). These results verify that FeIII in the MOF structure was partially reduced to FeII in the reduction step. Deconvolution of the rMOF signal in the Fe 2p3/2 region was performed and showed that 31 % of FeIII in the MOF particle structure was converted to FeII (Fig. 1d(ii)).

|
SI-ATRP
SI-ATRP of the rMOF nanoparticles (P@rMOF) was carried out in two steps as shown in Scheme 2. Initially, the rMOF was conjugated with BiBB to obtain the bromo-functionalized rMOF (Br@rMOF).[33] Polymerization of the functionalized rMOFs using the macro-monomer PEGMA under SI-ATRP conditions afforded PEGylated nanoparticles P@rMOF.

|
TEM, FTIR spectroscopic, TGA, and zeta potential analyses were used to confirm the introduction of PEGMA chains on the rMOF nanoparticles. As shown in Fig. 2a, a dense and uniform polymeric shell surrounding the rMOF core was observed after SI-ATRP. The IR spectrum of P@rMOF exhibited distinctive peaks at 2870 cm−1 (C–H stretching), 1720 cm−1 (C=O stretching), and 1100 cm−1 (C–O stretching) in comparison with the spectrum of bare rMOF, indicating the successful formation of polymer on the rMOF particles (Fig. 2b).[29] TGA in an air atmosphere was used to study the decomposition pattern and thermal stability of the rMOF and three different P@rMOF nanoparticles (Fig. 2c). P@rMOF nanoparticles displayed more weight loss than rMOF particles owing to the grafted decomposable polymer chains. Measurements revealed the relative amount of Fe is 20.8 wt-% for rMOF and 10.2, 7.0, and 5.2 wt-% for the three different P@rMOF nanoparticles. Calculations verified that the mass ratio of conjugated PEG to rMOF nanoparticles was ~1, 2, and 3, and they were hence named 1P@rMOF, 2P@rMOF, and 3P@rMOF respectively. In addition, zeta potential results for MOF, rMOF, 1P@rMOF, 2P@rMOF, and 3P@rMOF, shown in Fig. 2d, revealed the negative charge on the surface of the nanoparticles increased from −24 to −34 mV when the MOF particles were reduced to rMOF, thus making them more stable in aqueous solution. However, after polymerization, the surface charge of 1P@rMOF, 2P@rMOF, and 3P@rMOF decreased to −9.5, −7.8, and −5.8 mV respectively, suggesting that the surface of the rMOF was decorated with the PEG polymer layer.[48] Finally, DLS was used to determine the hydrodynamic diameter of the synthesized particles (Fig. 2e). The average hydrodynamic diameter (DH) of rMOF is 221 nm, indicating aggregation of the particles in aqueous solution due to the interparticle interaction of rMOF particles.[49,50] After polymerization on the surface of the rMOFs using SI-ATRP, the hydrodynamic diameter of P@rMOF decreased. As presented in Fig. 2e, the largest particles were achieved for 1P@rMOF (100 nm), where interparticle interactions were still possible. As the length of the polymer chain increased, this interparticle interaction was minimized, leading to the smallest particle size (DH) 2P@rMOF (40 nm); however, with a further increase in polymer chain length in 3P@rMOF, the impact of polymer chain interactions, most likely through entanglement, led to an increase in particle size to 65 nm. rMOF modification with hydrophilic polymer hindered interparticle aggregation and led to improved solubility in aqueous solution.[33] No obvious stratification or aggregation of the optimized 2P@rMOF nanoparticles in solution was observed after 30 days (Fig. 2f).

|
Folic Acid Conjugation
In the final step, folic acid was conjugated to these three P@rMOF particles via a three-step process (Scheme 3). Initially, alkyne-terminated folic acid was synthesized by a simple amidation reaction according to a published procedure, with preferential attachment (60–90 %) at the γ-position over the α-position, as shown in Scheme 3.[41,51] The bromine group of P@rMOF was substituted with an azide functionality using sodium azide.[39] Folic acid was then conjugated on the P@rMOF nanoparticles via a Cu-catalyzed azide-alkyne cycloaddition reaction (CuAAC) to form P@rMOF-FA.[52]

|
The alkyne-functionalized folic acid was characterized using 1H-NMR (Fig. S2, Supplementary Material). Two strong signals observed at 3.80 and 2.82 ppm are consistent with the –CONH–CH2C=CH protons of the final product. The IR spectrum of 2P@rMOF-N3 exhibited a distinctive peak at 2050 cm−1 (N3 stretching), in comparison with the spectrum of P@rMOF, indicating the successful replacement of Br with azide functionality (Fig. S3, Supplementary Material).[53] Successful conjugation of the folic acid was verified with the disappearance of the azide peak after the click reaction. Finally, UV-vis spectroscopy was used to confirm the folic acid groups on the 2P@rMOF-FA nanoparticles. Normalized UV-vis spectra of FA, 2P@rMOF, and 2P@rMOF-FA are shown in Fig. 3. The FA suspension displays a strong peak at λ = 285 nm, corresponding to the π–π* transition of the pterin ring.[54] A strong increase and shift in the relative intensity of the peak at 285 nm after the modification of 2P@rMOF surface using folic acid indicates the successful formation of 2P@rMOF-FA.

|
Hydroxyl Radical Generation
In our previous study, we demonstrated that rMOF nanoparticles with a mesoporous structure can produce OH• both by reacting at the surface (heterogeneous catalysis) and by releasing iron in an acidic environment (homogeneous catalysis).[22] In the present report, TMB as a classic chromogenic reagent was used to detect the hydroxyl radical by its oxidizing TMB and producing a blue product ox-TMB with a λmax = 652 nm.[42,55] The absorbance spectra of the solution containing 3P@rMOF-FA nanoparticles, H2O2, and TMB relative to time are shown in Fig. S4 (Supplementary Material). A strong absorption peak at 652 nm appears, demonstrating the production of ox-TMB. The absorbance at 652 nm as a function of time for the 1P@rMOF-FA, 2P@rMOF-FA, and 3P@rMOF-FA nanoparticles is presented in Fig. 4a. It shows that there is a dramatic increase in absorbance for all P@rMOF-FA nanoparticles after only 10 min at a low concentration of H2O2 (500 µM), demonstrating the intrinsic peroxidase-like activity of P@rMOF-FA. Also, owing to a relative decrease in Fe content for each nanoparticle, the activity decreases with an increase in polymerization from 1P- to 3P@rMOF-FA. Finally, the relative peroxidase-like activity of 2P@rMOF-FA (50 µg mL−1) at various pH values is shown in Fig. 4b. The activity dramatically increases when the pH decreases from 7.4 to 3. Owing to the lower stability of pristine NH2-MIL-88B(Fe) under acidic conditions,[22] P@rMOF-FA nanoparticles are stable at neutral pH (physiological pH) but rapidly release iron in an acidic environment (tumour tissue). The significant activity of P@rMOF-FA at low pH and [H2O2] can facilitate selective killing of the cancer cells using the cell’s own hydrogen peroxide.

|
Cancer Studies
1P@rMOF, 2P@rMOF, and 3P@rMOF nanoparticles as a new type of therapeutic agent were utilized for cancer studies. For this aim, cellular uptake, ROS, and in-vitro cytotoxicity studies were observed and are reported for both HeLa (cervical cancer cells) and NIH-3T3 (non-cancerous fibroblasts) cell lines.
Cell Uptake Study
The cellular internalization of different P@rMOF-FA was studied by exposing the nanoparticles to HeLa and NIH-3T3 cells, followed by extensive washing to remove nanoparticles adsorbed to the cell surface. The intracellular concentration of iron after incubation with the nanoparticles 1P@rMOF-FA, 2P@rMOF-FA, and 3P@rMOF-FA was quantified for both normal and cancer cells using ICP-OES. After incubation for 5 h, the cellular uptake of different nanoparticles was higher than that for 1 and 3 h, which shows time-dependent cellular uptake (Fig. S5, Supplementary Material). In addition, the cellular uptake of nanoparticles for cancer cells was significantly higher than normal cells over the duration of the testing. The nanoparticles are able to take advantage of the overexpression of folate receptors and are transported into the tumour cells by receptor-mediated endocytosis.[56] As shown in Fig. 5, after 5 h of incubation, nanoparticle concentrations in the HeLa cells were 2.55, 10.00 and 9.75 pg cell−1 for 1P@rMOF-FA, 2P@rMOF-FA, and 3P@rMOF-FA respectively. This equates to percentage uptake of the cells of 0.51, 2.00, and 1.95 % for 1P@rMOF-FA, 2P@rMOF-FA and 3P@rMOF-FA nanoparticles respectively, based on the total amount of nanoparticles added. This is consistent with previous reports, demonstrating that small nanoparticles are able to penetrate very deeply and achieve great uptake into tumour cells.[57]

|
ROS Study
The concentration of ROS, which consist of superoxide anion radical (O2•−), singlet oxygen (1O2), hydrogen peroxide (H2O2), and hydroxyl radical (OH•), is one of the most important differences between normal and cancer cells.[58] High concentrations of ROS have the ability to damage cellular constituents effectively.[59] In the present report, the intracellular ROS levels in HeLa and NIH-3T3 cells were analysed using DCFH-DA as an oxidative probe after incubation with the different nanoparticles. Time-dependent fluorescent intensities from DCFH-DA-labelled HeLa cells in cubated with 50-µg mL−1 1P@rMOF-FA, 2P@rMOF-FA, and 3P@rMOF-FA nanoparticles are shown in Fig. 6a. Owing to the time-dependent cellular nanoparticle uptake, cells incubated for 5 h showed higher intensities than after 1 and 3 h. Importantly, the relative amount of Fe in 2P@rMOF-FA nanoparticles is higher than in 3P@rMOF-FA (Fig. 2c), when both nanoparticles showed similar uptake after 5 h (Fig. 5). In this case, 2P@rMOF-FA has a higher relative release of Fe within the HeLa cell to react with the high level of H2O2 and produce more ROS (e.g. OH•). In addition, owing to the low concentration of H2O2 in normal cells, a weak intensity and thus reduced uptake can be seen in the NIH-3T3 cells treated with 50-µg mL−1 1P@rMOF-FA, 2P@rMOF-FA, and 3P@rMOF-FA nanoparticles over various times (Fig. S6, Supplementary Material). The results were then verified using confocal microscopy images. As shown in Fig. 6b, control NIH-3T3 and HeLa cells did not show a statistically significant impact on ROS generation, and this could be due to the low hydroxyl radical concentration in untreated cells. Weak emissions can be seen in the 2P@rMOF-FA-treated NIH-3T3 cells; however, bright green fluorescence signals were observed inside 2P@rMOF-FA-treated HeLa cells, indicating higher intracellular OH• concentrations. This implies 2P@rMOF-FA can react with high concentrations of hydrogen peroxide inside the HeLa cells and increase OH• levels. Generation of OH• subsequently induced endoplasmic reticulum stress, DNA damage, and genomic instability and cell death.[60,61]

|
Cell Viability Study
The in vitro cell viability of the P@rMOF-FA was assessed using the CCK-8 approach.[62] HeLa and NIH-3T3 cells were incubated with various concentrations of nanoparticles for 24 h and then processed with fresh medium containing CCK-8 solutions for a further 3 h to determine cell viability. The final results for 2P@rMOF-FA are shown in Fig. 7a. In the nanoparticle concentration range of 0–60 µg mL−1, negligible cytotoxicity on NIH-3T3 was observed, and all viabilities were more than 90 % after 24 h incubation. In addition, minor cytotoxicity was seen at high concentrations of nanoparticles, and the viability of normal cells decreased to 87 and 82 % at concentration of 80 and 100 µg mL−1 respectively. Importantly, Fig. 7a showed a dose-dependent toxicity towards HeLa cells, with cell viability decreasing dramatically at low concentrations. The viability of cancer cells decreases to 22 % at concentration of 100-µg mL−1 2P@rMOF-FA. Consequently, nanoparticles are statistically significantly more toxic to HeLa than NIH-3T3 cells. The calculated half-maximum inhibitory concentration value (IC50) for HeLa cells was 60 µg mL−1, which was significantly lower than for NIH-3T3 at 270 µg mL−1 (Fig. 7b). At low concentrations, 2P@rMOF-FA nanoparticles were able to be internalized into HeLa cells using the overexpression of folate receptors on the surface, react with the cell’s own hydrogen peroxide, increase intracellular hydroxyl radical levels, and treat the tumour cells. The in vitro cell viability results for nanoparticles 1P@rMOF-FA and 3P@rMOF-FA are shown in Figs S7 and S8 (Supplementary Material) respectively.

|
The selectivity index (the ratio of IC50 of normal cells to cancer cells) of the P@rMOF-FA nanoparticles prepared in this study were compared with rMOF-FA and other novel strategies, summarized in Table 1.

|
Based on our previous study, the selectivity index for rMOF-FA was 2.45, which is higher than current commercial drug treatments such as quercetin, cisplatin, and doxorubicin,[63–66] as well as previously reported similar-strategy nanoparticles.[19,67] However, it was smaller than other novel strategies that used chemotherapy and gene therapy where cisplatin@polypetide and siRNA@Lipopolyplex nano particles were employed (Table 1). As shown in the present study, decreasing the size of the nanoparticles and modifying the surface to reduce aggregation and increase solubility led to greater performance in biocompatibility, treatment effectiveness, and selectivity. Importantly, the selectivity index for 2P@ rMOF-FA is in most cases greater than or comparable with the reported novel strategies for the treatment of HeLa and NIH-3T3 cells. Consequently, these results demonstrate that the improvement of Fenton therapy based on 2P@rMOF-FA nanoparticles makes them a promising candidate for further development as active agents for treating various types of cancer cells.
Conclusion
In summary, stable P@rMOF-FA nanoparticles as potential therapeutic agents have been demonstrated to effectively treat a cancer cell line (HeLa). The MOF-based nanoparticles were prepared by a hydrothermal method followed by PEGylation through SI-ATRP and conjugation with folic acid, and fully characterized using TEM, XRD, XPS, DLS, FTIR, TGA, zeta potential, and UV-vis spectrometry and analyses. P@rMOF-FAs were shown to react with H2O2 and produce OH• according to the Fenton reaction. It was found that the activity of nanoparticles dramatically increased when the pH decreased from 7.4 to 5. In addition, 2P@rMOF-FA nanoparticles had the greatest efficacy and selectivity relative to 1P@rMOF-FA and 3P@rMOF-FA nanoparticles according to in vitro results. A cellular uptake study showed that the amount of internalized 2P@rMOF-FA nanoparticle in HeLa cells (cancer cells) is 3 times higher than in NIH-3T3 cells. This cell toxicity study also demonstrated that the viability of cancer cells decreased dramatically in the range of 0–100 µg mL−1, a concentration range without obvious toxic effects on NIH-3T3. Owing to the biocompatibility, treatment effectiveness, and selectivity of 2P@rMOF-FA, it may function as a new selective agent for cancer treatment using the cell’s own H2O2.
Supplementary Material
rMOF size distribution, 1H NMR spectra of alkyne-functionalized folic acid, FTIR spectra of P@rMOF, 2P@rMOF-N3, and 2P@rMOF-FA, activity of 3P@rMOF-FA nanoparticle over time, quantitative analysis of intracellular uptake of internalized nanoparticles, time dependent intracellular ROS production, and cytotoxicity of 1P@rMOF-FA and 3P@rMOF-FA toward HeLa and NIH-3T3 cells are available on the Journal’s website.
Conflicts of Interest
The authors declare no conflicts of interest.
Acknowledgements
H.R.-B. would like to acknowledge the University of Melbourne for a Melbourne International Research Scholarship (MIRS) and the Melbourne International Fee Remission Scholarship (MIFRS). The authors thank the Particulate Fluids Processing Centre (PFPC) and Materials Characterisation and Fabrication Platform (MCFP). The authors would also like to thank Mr Ke Xie for help with TGA measurements.
References
[1] M. E. Davis, D. M. Shin, Nat. Rev. Drug Discov. 2008, 7, 771.| Crossref | GoogleScholarGoogle Scholar |
[2] I. Brigger, C. Dubernet, P. Couvreur, Adv. Drug Deliv. Rev. 2002, 54, 631.
| Crossref | GoogleScholarGoogle Scholar |
[3] F. Muhammad, M. Guo, W. Qi, F. Sun, A. Wang, Y. Guo, G. Zhu, J. Am. Chem. Soc. 2011, 133, 8778.
| Crossref | GoogleScholarGoogle Scholar |
[4] M. Zan, J. Li, S. Luo, Z. Ge, Chem. Commun. 2014, 7824.
| Crossref | GoogleScholarGoogle Scholar |
[5] S. S. Lucky, K. C. Soo, Y. Zhang, Chem. Rev. 2015, 115, 1990.
| Crossref | GoogleScholarGoogle Scholar |
[6] A. S. Thakor, S. S. Gambhir, CA Cancer J. Clin. 2013, 63, 395.
| Crossref | GoogleScholarGoogle Scholar |
[7] G.-Y. Liou, P. Storz, Free Radic. Res. 2010, 44, 479.
| Crossref | GoogleScholarGoogle Scholar |
[8] B. Kumar, S. Koul, L. Khandrika, R. B. Meacham, H. K. Koul, Cancer Res. 2008, 68, 1777.
| Crossref | GoogleScholarGoogle Scholar |
[9] D. Wang, F. Peng, J. Li, Y. Qiao, Q. Li, X. Liu, Mater. Today 2017, 20, 238.
| Crossref | GoogleScholarGoogle Scholar |
[10] J. Cadet, T. Douki, J.-L. Ravanat, Free Radic. Biol. Med. 2010, 49, 9.
| Crossref | GoogleScholarGoogle Scholar |
[11] M. L. Circu, T. Y. Aw, Free Radic. Biol. Med. 2010, 48, 749.
| Crossref | GoogleScholarGoogle Scholar |
[12] Q. An, C. Sun, D. Li, K. Xu, J. Guo, C. Wang, ACS Appl. Mater. Interfaces 2013, 5, 13248.
| Crossref | GoogleScholarGoogle Scholar |
[13] J. Fu, Y. Shao, L. Wang, Y. Zhu, Nanoscale 2015, 7, 7275.
| Crossref | GoogleScholarGoogle Scholar |
[14] G. Huang, H. Chen, Y. Dong, X. Luo, H. Yu, Z. Moore, E. A. Bey, D. A. Boothman, J. Gao, Theranostics 2013, 3, 116.
| Crossref | GoogleScholarGoogle Scholar |
[15] P. A. Ma, H. Xiao, C. Yu, J. Liu, Z. Cheng, H. Song, X. Zhang, C. Li, J. Wang, Z. Gu, Nano Lett. 2017, 17, 928.
| Crossref | GoogleScholarGoogle Scholar |
[16] Y. Peng, Z. Wang, W. Liu, H. Zhang, W. Zuo, H. Tang, F. Chen, B. Wang, Dalton Trans. 2015, 12871.
| Crossref | GoogleScholarGoogle Scholar |
[17] C. Zhang, W. Bu, D. Ni, S. Zhang, Q. Li, Z. Yao, J. Zhang, H. Yao, Z. Wang, J. Shi, Angew. Chem. Int. Ed. 2016, 55, 2101.
| Crossref | GoogleScholarGoogle Scholar |
[18] B. Kwon, E. Han, W. Yang, W. Cho, W. Yoo, J. Hwang, B.-M. Kwon, D. Lee, ACS Appl. Mater. Interfaces 2016, 8, 5887.
| Crossref | GoogleScholarGoogle Scholar |
[19] C. Xu, Z. Yuan, N. Kohler, J. Kim, M. A. Chung, S. Sun, J. Am. Chem. Soc. 2009, 131, 15346.
| Crossref | GoogleScholarGoogle Scholar |
[20] J. Wang, L. Yue, Z. Hu, Z. Dai, Y. Qi, X. Zheng, Z. Li, D. Yu, RSC Adv. 2016, 6, 107331.
| Crossref | GoogleScholarGoogle Scholar |
[21] L. Yue, J. Wang, Z. Dai, Z. Hu, X. Chen, Y. Qi, X. Zheng, D. Yu, Bioconjug. Chem. 2017, 28, 400.
| Crossref | GoogleScholarGoogle Scholar |
[22] H. Ranji-Burachaloo, F. Karimi, K. Xie, Q. Fu, P. A. Gurr, D. E. Dunstan, G. G. Qiao, ACS Appl. Mater. Interfaces 2017, 9, 33599.
| Crossref | GoogleScholarGoogle Scholar |
[23] A. C. R. Grayson, J. Ma, D. Putnam, Mol. Pharm. 2006, 3, 601.
| Crossref | GoogleScholarGoogle Scholar |
[24] M. Machacek, A. Cidlina, V. Novakova, J. Svec, E. Rudolf, M. Miletin, R. Kučera, T. Simunek, P. Zimcik, J. Med. Chem. 2015, 58, 1736.
| Crossref | GoogleScholarGoogle Scholar |
[25] S. J. Shirbin, K. Ladewig, Q. Fu, M. Klimak, X. Zhang, W. Duan, G. G. Qiao, Biomacromolecules 2015, 16, 2463.
| Crossref | GoogleScholarGoogle Scholar |
[26] J. Wang, D. Chen, B. Li, J. He, D. Duan, D. Shao, M. Nie, Sci. Rep. 2016, 6, 26126.
[27] X. Fan, L. Lin, J. L. Dalsin, P. B. Messersmith, J. Am. Chem. Soc. 2005, 127, 15843.
| Crossref | GoogleScholarGoogle Scholar |
[28] S. M. Kang, I. S. Choi, K.-B. Lee, Y. Kim, Macromol. Res. 2009, 17, 259.
| Crossref | GoogleScholarGoogle Scholar |
[29] A. P. Majewski, A. Schallon, V. Jérôme, R. Freitag, A. H. Müller, H. Schmalz, Biomacromolecules 2012, 13, 857.
| Crossref | GoogleScholarGoogle Scholar |
[30] H. Y. Cho, S. E. Averick, E. Paredes, K. Wegner, A. Averick, S. Jurga, S. R. Das, K. Matyjaszewski, Biomacromolecules 2013, 14, 1262.
| Crossref | GoogleScholarGoogle Scholar |
[31] F. Hu, K. G. Neoh, L. Cen, E.-T. Kang, Biomacromolecules 2006, 7, 809.
| Crossref | GoogleScholarGoogle Scholar |
[32] H. S. Kim, Y. J. Son, W. Mao, K. W. Leong, H. S. Yoo, Nano Lett. 2018, 18, 314.
| Crossref | GoogleScholarGoogle Scholar |
[33] K. Xie, Q. Fu, Y. He, J. Kim, S. Goh, E. Nam, G. Qiao, P. Webley, Chem. Commun. 2015, 15566.
| Crossref | GoogleScholarGoogle Scholar |
[34] E. H. H. Wong, M. P. van Koeverden, E. Nam, S. N. Guntari, S. H. Wibowo, A. Blencowe, F. Caruso, G. G. Qiao, Macromolecules 2013, 46, 7789.
| Crossref | GoogleScholarGoogle Scholar |
[35] M.-H. Pham, G.-T. Vuong, A.-T. Vu, T.-O. Do, Langmuir 2011, 27, 15261.
| Crossref | GoogleScholarGoogle Scholar |
[36] Q. Fu, K. Xie, S. Tan, J. Ren, Q. Zhao, P. Webley, G. Qiao, Chem. Commun. 2016, 12226.
| Crossref | GoogleScholarGoogle Scholar |
[37] K. Matyjaszewski, J. Xia, Chem. Rev. 2001, 101, 2921.
| Crossref | GoogleScholarGoogle Scholar |
[38] Y. Zhu, H. S. Sundaram, S. Liu, L. Zhang, X. Xu, Q. Yu, J. Xu, S. Jiang, Biomacromolecules 2014, 15, 1845.
| Crossref | GoogleScholarGoogle Scholar |
[39] J. M. Ren, K. Satoh, T. K. Goh, A. Blencowe, K. Nagai, K. Ishitake, A. J. Christofferson, G. Yiapanis, I. Yarovsky, M. Kamigaito, Angew. Chem. Int. Ed. 2014, 53, 459.
| Crossref | GoogleScholarGoogle Scholar |
[40] L. Jiang, Z.-m. Gao, L. Ye, A.-y. Zhang, Z.-g. Feng, Polymer 2013, 54, 5188.
| Crossref | GoogleScholarGoogle Scholar |
[41] X. Shen, Z. Ge, Y. Pang, J. Solid State Chem. 2015, 222, 37.
| Crossref | GoogleScholarGoogle Scholar |
[42] L. Gao, J. Zhuang, L. Nie, J. Zhang, Y. Zhang, N. Gu, T. Wang, J. Feng, D. Yang, S. Perrett, Nat. Nanotechnol. 2007, 2, 577.
| Crossref | GoogleScholarGoogle Scholar |
[43] S. Her, L. Cui, R. G. Bristow, C. Allen, Radiat. Res. 2016, 185, 549.
| Crossref | GoogleScholarGoogle Scholar |
[44] X. Xie, J. Liao, X. Shao, Q. Li, Y. Lin, Sci. Rep. 2017, 7, 3827.
[45] L. Zhang, L. Laug, W. Munchgesang, E. Pippel, U. Gösele, M. Brandsch, M. Knez, Nano Lett. 2010, 10, 219.
| Crossref | GoogleScholarGoogle Scholar |
[46] L. Shi, T. Wang, H. Zhang, K. Chang, X. Meng, H. Liu, J. Ye, Adv. Sci. 2015, 2, 1500006.
[47] P. Singh, K. Shiva, H. Celio, J. B. Goodenough, Energy Environ. Sci. 2015, 8, 3000.
| Crossref | GoogleScholarGoogle Scholar |
[48] S. Guo, Y. Huang, Q. Jiang, Y. Sun, L. Deng, Z. Liang, Q. Du, J. Xing, Y. Zhao, P. C. Wang, ACS Nano 2010, 4, 5505.
| Crossref | GoogleScholarGoogle Scholar |
[49] A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke, P. Behrens, Chem. – Eur. J. 2011, 17, 6643.
| Crossref | GoogleScholarGoogle Scholar |
[50] Q.-L. Zhu, Q. Xu, Chem. Soc. Rev. 2014, 43, 5468.
| Crossref | GoogleScholarGoogle Scholar |
[51] A. F. Trindade, R. F. Frade, E. M. Maçôas, C. Graça, C. A. Rodrigues, J. M. Martinho, C. A. Afonso, Org. Biomol. Chem. 2014, 12, 3181.
| Crossref | GoogleScholarGoogle Scholar |
[52] L. Jiang, Z.-m. Gao, L. Ye, A.-y. Zhang, Z.-g. Feng, Polymer 2013, 54, 5188.
| Crossref | GoogleScholarGoogle Scholar |
[53] S.-W. Kuo, J.-L. Hong, Y.-C. Huang, J.-K. Chen, S.-K. Fan, F.-H. Ko, C.-W. Chu, F.-C. Chang, J. Nanomater. 2012, 2012, 7.
[54] A. H. Thomas, G. Suárez, F. M. Cabrerizo, R. Martino, A. L. Capparelli, J. Photochem. Photobiol. Chem. 2000, 135, 147.
| Crossref | GoogleScholarGoogle Scholar |
[55] Z. Chen, J.-J. Yin, Y.-T. Zhou, Y. Zhang, L. Song, M. Song, S. Hu, N. Gu, ACS Nano 2012, 6, 4001.
| Crossref | GoogleScholarGoogle Scholar |
[56] W. B. Liechty, N. A. Peppas, Eur. J. Pharm. Biopharm. 2012, 80, 241.
| Crossref | GoogleScholarGoogle Scholar |
[57] S. D. Steichen, M. Caldorera-Moore, N. A. Peppas, Eur. J. Pharm. Sci. 2013, 48, 416.
| Crossref | GoogleScholarGoogle Scholar |
[58] J. A. Magee, E. Piskounova, S. J. Morrison, Cancer Cell 2012, 21, 283.
| Crossref | GoogleScholarGoogle Scholar |
[59] S. C. Gupta, D. Hevia, S. Patchva, B. Park, W. Koh, B. B. Aggarwal, Antioxid. Redox Signal. 2012, 16, 1295.
| Crossref | GoogleScholarGoogle Scholar |
[60] S. Li, K. Chang, K. Sun, Y. Tang, N. Cui, Y. Wang, W. Qin, H. Xu, C. Wu, ACS Appl. Mater. Interfaces 2016, 8, 3624.
| Crossref | GoogleScholarGoogle Scholar |
[61] P. A. Cerutti, Lancet 1994, 344, 862.
| Crossref | GoogleScholarGoogle Scholar |
[62] S. Bian, X. Sun, A. Bai, C. Zhang, L. Li, K. Enjyoji, W. G. Junger, S. C. Robson, Y. Wu, PLoS One 2013, 8, e60184.
| Crossref | GoogleScholarGoogle Scholar |
[63] M. Danihelová, M. Veverka, E. Šturdík, S. Jantová, Interdiscip. Toxicol. 2013, 6, 209.
| Crossref | GoogleScholarGoogle Scholar |
[64] W. A. G. W. N. Hafiza, S. Y. Latifah, OncoTargets Ther. 2014, 7, 1375.
[65] Y. Huang, T. Yang, W. Zhang, Y. Lu, P. Ye, G. Yang, B. Li, S. Qi, Y. Liu, X. He, Int. J. Nanomedicine 2014, 9, 4581.
[66] K. Tomankova, K. Polakova, K. Pizova, S. Binder, M. Havrdova, M. Kolarova, E. Kriegova, J. Zapletalova, L. Malina, J. Horakova, Int. J. Nanomedicine 2015, 10, 949.
| Crossref | GoogleScholarGoogle Scholar |
[67] S. K. Maji, A. K. Mandal, K. T. Nguyen, P. Borah, Y. Zhao, ACS Appl. Mater. Interfaces 2015, 7, 9807.
| Crossref | GoogleScholarGoogle Scholar |
* Greg G. Qiao is the recipient of the 2017 RACI Applied Research Award.