

Consequences of Subtle Chiral Effects: From ‘Majority-Rules’ to ‘Minority-Rules’
Patrick J. M. Stals A B C , Müge Artar A B , Pim Vendrig A , Anja R. A. Palmans A B and E. W. Meijer A B DA Laboratory of Macromolecular and Organic Chemistry, Eindhoven University of Technology, PO Box 513, NL 5600 MB Eindhoven, The Netherlands.
B Institute for Complex Molecular Systems, Eindhoven University of Technology, PO Box 513, NL 5600 MB Eindhoven, The Netherlands.
C DSM Coating Resins, Sluisweg 12, 5145 PE Waalwijk, The Netherlands.
D Corresponding author. Email: e.w.meijer@tue.nl
Australian Journal of Chemistry 68(4) 622-626 https://doi.org/10.1071/CH14596
Submitted: 29 September 2014 Accepted: 22 October 2014 Published: 22 December 2014
Abstract
Mixing experiments were conducted on dilute solutions of asymmetrically substituted benzene-1,3,5-tricarboxamides (BTAs) with stereogenic methyl groups ranging from the α- to the δ-position with respect to the amide in one of the three side groups. While normally the majority compound determines the helical sense preference of the formed supramolecular polymers, we find here that several combinations show a helical preference governed by the minority compound. BTAs with the methyl substituent at the α- and γ-position overrule the helical preference of BTAs with the methyl substituent at the β- and δ-position. This new effect is referred to as a ‘minority-rules’ system.
Introduction
The chiral methyl group using the three isotopes of hydrogen – protium, deuterium, and tritium – as introduced by John Warcup Cornforth is one of the hallmarks in stereochemistry and the stereochemical effects in enzyme catalysis.[1] This seminal work initiated many beautiful studies concerning the limits of the expression of chirality and how subtle chiral effects can influence structure and reactivity of molecules. More recently, these effects are used in supramolecular aggregates in exploring how small molecules self-assemble into helical supramolecular polymers. These studies have significantly increased our understanding of the molecular factors that determine the thermodynamics and kinetics of self-assembly processes governed by non-covalent interactions. Amplification of chirality can occur at the supramolecular level, which means that a small chiral bias on the molecular level results in the formation of one helical sense only.[2] First investigated by Green and coworkers for helical covalent polymers,[3] two types of systems that show amplification of chirality have been distinguished: the ‘majority-rules’ and the ‘sergeant-and-soldiers’ system. In a ‘sergeants-and-soldiers’ system, the helicity of large numbers of achiral units (the soldiers) is controlled by a few chiral units (the sergeants), whereas in a ‘majority-rules’ system, a slight excess of one enantiomer leads to a strong bias towards the preferred helicity of the majority enantiomer. In addition, chiral non-racemic solvents are also able to bias the helicity of helical, supramolecular polymers.[4] Benzene-1,3,5-tricarboxamides (BTAs) have been particularly versatile to study amplification of chirality both at the supramolecular as well as at the molecular level.[5] BTAs are synthetically easily accessible, and self-assemble cooperatively in apolar solvents through strong 3-fold hydrogen bonding between the amides of adjacent BTAs.[6] As a result, one-dimensional helical aggregates are formed and in the absence of chiral information, both helical senses occur in equal amounts.[6a] Introducing a stereogenic centre in one or more of the alkyl side-chains results in a bias for the formation of either a right-handed (P) or a left-handed (M) helical aggregate. Circular dichroism (CD) spectroscopy emerged from these studies as a powerful tool to study both the mechanism of the self-assembly process as well as providing detailed information on the conformation of the supramolecular polymer.[7] The shape and sign of the Cotton effect measured therein characterise the nature and helicity of the self-assembled structure, whereas the size of the Cotton effect is a quantitative measure for the excess helicity.[6]
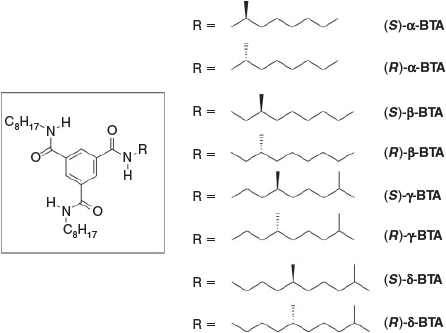
In recent years, we focussed on asymmetrically substituted BTAs in which there is only one stereocentre in one of the alkyl side-chains (Fig. 1).[8] By varying the position of this chiral centre from the α-carbon (α-BTA) next to the amide to the β-carbon (β-BTA), γ-carbon (γ-BTA), and δ-carbon (δ-BTA), an odd–even effect was observed in the self-assembly of these BTAs; keeping the absolute stereochemistry identical, but moving the stereocentre from the α- to the δ-position resulted in aggregates with alternating helical senses. Additionally, these BTAs show a pronounced ‘majority-rules’ effect when mixing two enantiomers. Because the propensity to form one type of helical stack is very strong, the minor enantiomer in scalemic mixtures adjusts its helical preference to that of the major enantiomer. Only at a critical enantiomeric excess (eecr) of around 0.2, the minor enantiomer starts forming helical stacks of the opposite helicity.
|
It is an intriguing question what will happen when mixing BTAs that have opposite helical preferences, but with the stereocentres on different carbon atoms. The directing power of the stereogenic methyl group at different positions may differ, resulting in a weaker or stronger bias for one helical sense. Here, we investigate amplification of chirality in mixtures of BTAs with the stereocentres on different carbons. We observe that BTAs with the stereocentre on an odd position (α or γ) next to an amide dominate the helical sense preference when mixed with BTAs with the stereocentre on an even position (β or δ). This is the first observation in which a minor component in a mixture can bias the helical sense preference in a supramolecular polymerisation process. We refer to this phenomenon as the ‘minority-rules’ principle.
Results and Discussion
All eight molecules were synthesised, purified, and characterised following procedures published before.[8] In the CD measurements, heptane was selected as the solvent of choice. The shapes of the CD spectra are identical for all BTAs investigated, indicating that the conformations of the helical supramolecular polymers are identical for all BTA derivatives (no differences in the dihedral angle connecting the amide with the central benzene unit).[8b] The sign of the Cotton effect depends on the configuration of the stereocentre in combination with the position of the methyl group: (S)-α-BTA, (R)-β-BTA, (S)-γ-BTA, and (R)-δ-BTA show negative Cotton effects, indicative for M-helical aggregates,[9] whereas their enantiomers show positive Cotton effects. Fig. 2a illustrates the alternating signs in the CD spectra of (R)-α-BTA, (R)-β-BTA, (R)-γ-BTA, and (R)-δ-BTA (c = 30 µM). This alternation in sign is the result of the odd–even effect, in which a stereogenic methyl group at the α-position biases a different helical sense than when it is at a β-position.[8b] In all cases, the molar circular dichroism Δϵ is +/– 40 L mol–1 cm–1 at 223 nm indicating that all BTAs are fully aggregated in structures with similar conformation under the applied conditions.

|
Methyl-substituted BTAs show a pronounced ‘majority-rules’ effect when mixing the two enantiomers. In methylcyclohexane, we have previously observed that the ee at which the CD effect is no longer constant (eecr) is 0.25, 0.19, and 0.26 for the α-, β-, and γ-BTA derivative, respectively.[5a] Though the alkane solvent participates in the formation of the helical aggregates, the effect of the alkane structure on the amplification of supramolecular chirality in BTAs has not yet been evaluated. We therefore first repeated the ‘majority-rules’ experiment for α- and β-BTAs – as representatives for BTAs with stereocentre at the odd or even position – in heptane; the results are shown in Fig. 2b. As expected, at an ee of zero (in other words when equal amounts of R- and S-enantiomers are present in the mixture), the CD effect is also zero because equal amounts of P- and M-helical aggregates are present. The values for eecr are at 0.25 and 0.18 for the α- and β-BTA, respectively. This suggests that the solvent does not have a significant effect on the magnitude of the ‘majority-rules’ effect.
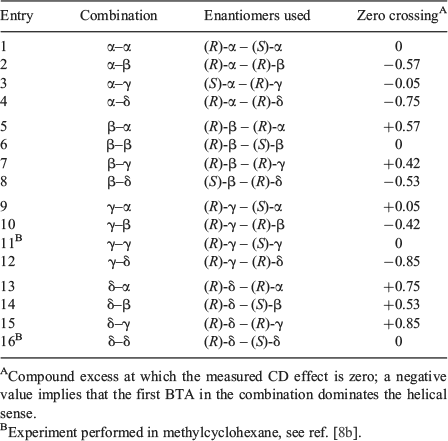
When mixing BTAs that have opposite helical preferences but with the stereocentre on different carbon atoms, the two compounds are no longer enantiomers, but different compounds. Therefore, we introduce the term compound excess (ce), defined in analogy to ee, to quantify the ratio between the two BTAs in the mixture. For example, for a mixture of (R)-α-BTA and (R)-β-BTA in which we start the titration with pure (R)-α-BTA, the ce is defined as (Rα – Rβ/Rα + Rβ) wherein Rα and Rβ represent the amount of the α- and β-derivative, respectively. Six titrations were conducted that represent all possibilities between BTAs with the stereogenic methyl groups differing in configuration and in position. In all cases, pairs were selected that adopt opposite helical senses. To account for the lower optical purities for both (S)-δ-BTA and (S)-β-BTA, we only used (R)-δ-BTA and (R)-β-BTA (ee > 97 %) in the titrations with α- and γ-BTAs. Furthermore, because both (R)-δ-BTA and (R)-β-BTA have the same helical sense preference, we used (S)-β-BTA (ee = 60 %)[8a] for mixing with (R)-δ-BTA and corrected for the lower ee in the calculation for the ce.[10] The results of these mixing experiments are shown in Fig. 3. To allow a more easy comparison, the mirror image results are plotted for two combinations in Fig. 3.

|
Surprisingly, only the mixture of BTAs containing stereocentres at the odd positions (α-BTA and γ-BTA) show a CD effect of close to zero at ce = 0. In all other combinations, the zero crossing is far from ce = 0, indicating that one BTA-derivative in the mixture has a significantly stronger helical preference than the other. This leads to the interesting situation in which it is possible for the minority in a mixture of two chiral compounds to dictate the helical preference of the majority. For example, in Fig. 3a, the CD effect only starts to decrease at ce = –0.56 for the (R)-α+(R)-δ mixture. This means that >3.5 times more (R)-δ-BTA compared with (R)-α-BTA is required for the majority compound to start forming aggregates of the opposite helicity. We refer to this novel effect in chiral amplification as a ‘minority-rules’ system, because in contrast to the ‘majority-rules’ system, the minority component now dominates the helical preference.
Closer inspection of Fig. 3a–c reveals that BTAs with the stereocentre on the odd-position (α-BTA and γ-BTA) dominate the overall helicity in the mixtures containing BTAs with the stereocentre on an even-position (β-BTA and δ-BTA). Additionally, within the odd- or even-series, BTAs with the chiral centre closer to the amide group (α-BTA and β-BTA) dominate their γ-BTA and δ-BTA counterparts, respectively. To quantify the strength of this ‘minority-rules’ effect, we determine at which point the net CD effect of each mixture is zero. The resulting data are summarised in Table 1 and visualised in Fig. 3d. There appears to be no clear trends in the magnitude of the ‘minority-rules’ effect. Though the results clearly show that δ-BTA is by far the weakest director in all mixtures, α-BTA – with the stereocentre closest to the amide group – is still a weaker director than γ-BTA in combination with δ-BTA. On the other hand, in mixtures of α-BTA or γ-BTA with β-BTA, α-BTA shows the stronger ‘minority-rules’ effect, whereas in the mixture of α-BTA and γ-BTA, the former seems to be slightly stronger although the difference is small. Because all BTAs are fully self-assembled under the conditions employed[8] and the conformation of the supramolecular BTA-based polymers is identical in heptane, as evidenced by the similarly shaped CD spectra, other effects must be responsible for this non-linear behaviour.
|
The origin of the differences in magnitude of the ‘minority rules’ effect is not what one would intuitively expect based on results previously obtained in helical polymers. In a series of helical polyisocyanides, Amabilino and coworkers observed that the helical sense preference, expressed as the magnitude of the Cotton effect, diminished rapidly when the stereocentre was positioned further away from the polymer backbone.[11] This reduced induction was attributed to the increased rotational freedom of the stereogenic centre when more intervening methylene units were present. In helical poly(N-propargylamides), in which the helical superstructure is stabilised by hydrogen bonds, Masuda and coworkers introduced methylene units between the stereogenic methyl group and the amide bond.[12] Also here, a fast decrease of the Cotton effect was found when more methylene units were present between the stereocentre and the amide. The decrease in the efficiency in which the chiral information was transmitted from the side chain to the main chain caused the polymer to adopt a less defined screw sense, resulting in a reduction of the helical preference. All these observations account for the reduction in helical preference in case of β- and δ-BTA compared with α-BTA. It is likely that the methyl at the α-position has less rotational freedom and is sterically more dominating than when the methyl is further away, rationalising why α-BTA is a strong helical director. The strong helical preference of γ-BTA, however, is remarkable. For this case, we propose that long-range stereochemical effects enhance the conformational preference of the amide bonds, and hence a more pronounced bias of the helical sense is obtained. Such remote stereochemical effects are well known to bias specific conformations over long distances in a non-intuitive manner.[13]
Conclusion
In conclusion, we here report that in a series of asymmetrically substituted BTAs with stereogenic methyl groups ranging from the α- to the δ-position with respect to the amide, the helical preference of the supramolecular polymers can be governed by the minority compound in a mixture. This is in contrast to previous studies in which in a mixture of enantiomers, the majority compound always dominates the helical sense preference. We refer to this new effect as a ‘minority-rules’ system. The effect relies on mixing compounds with opposite helical preferences, but in which the position of the stereogenic methyl group differs. We find that BTAs with the methyl substituent at the α- and γ-position overrule the helical preference of BTAs with the methyl substituent at the β- and δ-position, indicative of an odd–even effect. Several questions remain, however, the most intriguing one being why the γ-BTA shows such a strong helical preference. In the future, it would be fascinating to evaluate if this ‘minority-rules’ effect also holds in other self-assembling systems, and to unravel which molecular factors are responsible for the observed behaviour. Moreover, these results can be useful to get a better understanding of the stereochemical effects in natural structures based on isoprene.
Experimental
Instrumentation, Materials, and Methods
All spectroscopic measurements were performed with analytical reagent (AR) grade n-heptane obtained from Biosolve. The synthesis and characterisation of N-((R)-1-methylheptyl)-N′,N′-di(n-octyl)benzene-1,3,5-tricarboxamide ((R)-α-BTA),[8a] N-((S)-1-methyl-heptyl)-N′,N′-di(n-octyl)benzene-1,3,5-tricarboxamide ((S)-α-BTA),[8a] N-((R)-2-methyloctyl)-N′,N′-di(n-octyl)-benzene-1,3,5-tricarboxamide ((R)-β-BTA, ee = 97 %),[5a] N-((R)-2-methyloctyl)-N′,N′-di(n-octyl)-benzene-1,3,5-tricarboxamide ((R)-β-BTA, ee = 60 %),[8a] N-((S)-2-methyloctyl)-N′,N′-di(n-octyl)-benzene-1,3,5-tri-carboxamide ((S)-β-BTA),[8a] N-((R)-3,7-dimethyloctyl)-N′,N′-di(n-octyl)benzene-1,3,5-tricarboxamide ((R)-γ-BTA),[8a] N-((S)-3,7-dimethyloctyl)-N′,N′-di(n-octyl)benzene-1,3,5-tri-carboxamide ((S)-γ-BTA),[8a] and N-((S)-4,8-dimethylnonyl)-N′,N′-di-(n-octyl)benzene-1,3,5-tricarboxamide ((S)-δ-BTA)[8b] have been reported elsewhere. The synthesis of N-((R)-4,8-dimethylnonyl)-N′,N′-di-(n-octyl)benzene-1,3,5-tricarboxamide ((R)-δ-BTA) was performed analogously to that of its (S)-enantiomer according to ref. [8b].
Circular dichroism measurements were performed on a Jasco J-815 spectropolarimeter where the sensitivity, time constant, and scan rate were chosen appropriately. The temperature during the measurement was controlled with a PFD-425S/15 Peltier-type temperature controller with a temperature range of 263–383 K and an adjustable temperature slope. In all measurements, the temperature was kept constant at 293 K. In all experiments, the linear dichroism was also measured and in all cases, no linear dichroism was observed. Cells with an optical path length of 1 cm were used. Solutions were prepared by weighing in the necessary amount of compound for a 30 µM concentration and this amount was transferred to a 25 mL volumetric flask. Three-quarters of the flask capacity was filled with n-heptane, and the flask was placed in an oscillation bath at 40°C for 60 min, after which the flask was allowed to cool to room temperature and filled up to its meniscus.
Acknowledgements
We would like to acknowledge the seminal contributions of Sir John Warcup Cornforth, all of them being so inspirational for our generations and without his outstanding stereochemical studies, our research would have been different. This paper is dedicated to him. This work was financially supported by The Netherlands Organization for Scientific Research (NWO – TOP grant: 10007851 and NWO – ECHO Grant: 713.011.001), the Dutch Ministry of Education, Culture and Science (Gravity program 024.001.035), the European Research Council (FP7/2007–2013, ERC Grant Agreement 246829), and the NRSC-C.
References
[1] J.W. Cornforth, Asymmetry and Enzyme Action, Nobel Lecture, 12 December 1975.[2] (a) A. R. A. Palmans, E. W. Meijer, Angew. Chem., Int. Ed. 2007, 46, 8948.and references cited therein.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsVCju7rM&md5=864e0778ac76f1fb75ccdfd02b1347c2CAS |
(b) A. Lohr, F. Wuerthner, Angew. Chem., Int. Ed. 2008, 47, 1232.
| Crossref | GoogleScholarGoogle Scholar |
(c) T. E. Kaiser, V. Stepanenko, F. Wuerthner, J. Am. Chem. Soc. 2009, 131, 6719.
| Crossref | GoogleScholarGoogle Scholar |
(d) F. Helmich, C. C. Lee, A. P. H. J. Schenning, E. W. Meijer, J. Am. Chem. Soc. 2010, 132, 16753.
| Crossref | GoogleScholarGoogle Scholar |
(e) H. Cao, X. Zhu, M. Liu, Angew. Chem., Int. Ed. 2013, 52, 4122.
| Crossref | GoogleScholarGoogle Scholar |
(f) M. Peterca, M. R. Imam, C.-H. Ahn, V. S. K. Balagurusamy, D. A. Wilson, B. M. Rosen, V. Percec, J. Am. Chem. Soc. 2011, 133, 2311.
| Crossref | GoogleScholarGoogle Scholar |
(g) F. Garcia, P. M. Viruela, E. Matesanz, E. Orti, L. Sanchez, Chem. – Eur. J. 2011, 17, 7755.
| Crossref | GoogleScholarGoogle Scholar |
(h) T. Seki, A. Asano, S. Seki, Y. Kikkawa, H. Murayama, T. Karatsu, A. Kitamura, S. Yagai, Chem. – Eur. J. 2011, 17, 3598.
| Crossref | GoogleScholarGoogle Scholar |
(i) C. Kulkarni, R. Munirathinam, S. J. George, Chem. – Eur. J. 2013, 19, 11270.
| Crossref | GoogleScholarGoogle Scholar |
(j) B. Nie, T.-G. Zhan, T.-Y. Zhou, Z.-E. Xiao, G.-F. Jiang, X. Zhao, Chem. – Asian J. 2014, 9, 754.
| Crossref | GoogleScholarGoogle Scholar |
(k) J. Kang, D. Miyajima, Y. Itoh, T. Mori, H. Tanaka, M. Yamauchi, Y. Inoue, S. Harada, T. Aida, J. Am. Chem. Soc. 2014, 136, 10640.
| Crossref | GoogleScholarGoogle Scholar |
(l) R. Fasel, M. Parschau, K.-H. Ernst, Nature 2006, 439, 449.
| Crossref | GoogleScholarGoogle Scholar |
[3] (a) M. M. Green, M. P. Reidy, R. D. Johnson, G. Darling, D. J. O’Leary, G. Wilson, J. Am. Chem. Soc. 1989, 111, 6452.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. M. Green, B. A. Garetz, B. Munoz, H. Chang, S. Hoke, R. G. Cooks, J. Am. Chem. Soc. 1995, 117, 4181.
| Crossref | GoogleScholarGoogle Scholar |
(c) K. Tang, M. M. Green, K. S. Cheon, J. V. Selinger, B. A. Garetz, J. Am. Chem. Soc. 2003, 125, 7313.
| Crossref | GoogleScholarGoogle Scholar |
[4] (a) M. M. Green, C. Khatri, N. C. Peterson, J. Am. Chem. Soc. 1993, 115, 4941.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXkslentLc%3D&md5=f6c2da375282ef395ce6a6584b27f6f8CAS |
(b) A. R. A. Palmans, J. A. J. M. Vekemans, E. E. Havinga, E. W. Meijer, Angew. Chem., Int. Ed. Engl. 1997, 36, 2648.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. Nakashima, J. R. Koe, K. Torimitsu, M. Fujiki, J. Am. Chem. Soc. 2001, 123, 4847.
| Crossref | GoogleScholarGoogle Scholar |
(d) B. Isare, M. Linares, L. Zargarian, S. Fermandjian, M. Miura, S. Motohashi, N. Vanthuyne, R. Lazzaroni, L. Bouteiller, Chem. – Eur. J. 2010, 16, 173.
| Crossref | GoogleScholarGoogle Scholar |
(e) S. J. George, Z. Tomovic, A. P. H. J. Schenning, E. W. Meijer, Chem. Commun. 2011, 47, 3451.
| Crossref | GoogleScholarGoogle Scholar |
(f) I. Destoop, H. Xu, C. Oliveras-Gonzales, E. Ghijsens, D. B. Amabilino, S. De Feyter, Chem. Commun. 2013, 49, 7477.
| Crossref | GoogleScholarGoogle Scholar |
[5] (a) M. M. J. Smulders, P. J. M. Stals, T. Mes, T. F. E. Paffen, A. P. H. J. Schenning, A. R. A. Palmans, E. W. Meijer, J. Am. Chem. Soc. 2010, 132, 620.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsFKgtbfE&md5=7f7c4ab68ff2efa983af8277a05af4abCAS |
(b) M. M. J. Smulders, I. A. W. Filot, J. M. A. Leenders, P. van der Schoot, A. R. A. Palmans, A. P. H. J. Schenning, E. W. Meijer, J. Am. Chem. Soc. 2010, 132, 611.
| Crossref | GoogleScholarGoogle Scholar |
(c) S. Cantekin, H. M. M. ten Eikelder, A. J. Markvoort, M. A. J. Veld, P. A. Korevaar, M. M. Green, A. R. A. Palmans, E. W. Meijer, Angew. Chem., Int. Ed. 2012, 51, 6426.
| Crossref | GoogleScholarGoogle Scholar |
(d) P. J. M. Stals, P. A. Korevaar, M. A. J. Gillissen, T. F. A. de Greef, C. F. C. Fitié, R. P. Sijbesma, A. R. A. Palmans, E. W. Meijer, Angew. Chem., Int. Ed. 2012, 51, 11297.
| Crossref | GoogleScholarGoogle Scholar |
(e) M. A. J. Veld, D. Haveman, A. R. A. Palmans, E. W. Meijer, Soft Matter 2011, 7, 524.
| Crossref | GoogleScholarGoogle Scholar |
(f) P. J. M. Stals, J. C. Everts, R. de Bruijn, I. A. W. Filot, M. M. J. Smulders, R. Martin-Rapun, E. A. Pidko, T. F. A. de Greef, A. R. A. Palmans, E. W. Meijer, Chem. – Eur. J. 2010, 16, 810.
| Crossref | GoogleScholarGoogle Scholar |
[6] (a) M. M. J. Smulders, A. P. H. J. Schenning, E. W. Meijer, J. Am. Chem. Soc. 2008, 130, 606.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsVehur7E&md5=4af1b285f05115786a92f21d10431123CAS |
(b) S. Cantekin, T. F. A. de Greef, A. R. A. Palmans, Chem. Soc. Rev. 2012, 41, 6125.
| Crossref | GoogleScholarGoogle Scholar |
[7] G. Gottarelli, S. Lena, S. Masiero, S. Pieraccini, G. P. Spada, Chirality 2008, 20, 471.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXis12hu78%3D&md5=0c1ad6c6b62aa59967bb00f06f5ecbaeCAS | 17918751PubMed |
[8] (a) P. J. M. Stals, M. M. J. Smulders, R. Martín-Rapún, A. R. A. Palmans, E. W. Meijer, Chem. – Eur. J. 2009, 15, 2071.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXisVaiur0%3D&md5=af30ed6c12f0de14fb22c647517e4e2aCAS |
(b) Y. Nakano, T. Hirose, P. J. M. Stals, E. W. Meijer, A. R. A. Palmans, Chem. Sci. 2012, 3, 148.
| Crossref | GoogleScholarGoogle Scholar |
[9] M. M. J. Smulders, T. Buffeteau, D. Cavagnat, M. Wolffs, A. P. H. J. Schenning, E. W. Meijer, Chirality 2008, 20, 1016.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtFKgsLnL&md5=0d49746b27aaf08aa7d145954cb5c6cfCAS |
[10] For mixing (S)-β-BTA with (R)-δ-BTA, the following formula was used to calculate the ce: (Sβ – (Rβ + Rδ))/(Sβ + Rβ + Rδ), in which Sβ, Rβ, and Rδ are the respective moles of the specified BTA in the titration point.
[11] D. B. Amabilino, E. Ramos, J.-L. Serrano, T. Sierra, J. Veciana, J. Am. Chem. Soc. 1998, 120, 9126.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXls1ajs7k%3D&md5=3626dfdbd46b79ccbec4cbb2205d1642CAS |
[12] J. Tabei, M. Shiotsuki, F. Sanda, T. Masuda, Macromolecules 2005, 38, 5860.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXltFCgt78%3D&md5=c83978814fa6d3e4d49720802bb6395eCAS |
[13] (a) J. Clayden, A. Lund, L. Vallverdu, M. Helliwell, Nature 2004, 431, 966.
| Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXoslSltrs%3D&md5=c7d5414c8f14797de276ebc0c2c4b66eCAS | 15496918PubMed |
(b) J. Clayden, Chem. Soc. Rev. 2009, 38, 817.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. Clayden, N. Vassiliou, Org. Biomol. Chem. 2006, 4, 2667.
| Crossref | GoogleScholarGoogle Scholar |