
Genomic Encyclopedia of Bacteria and Archaea (GEBA) VI: learning from type strains
William B Whitman A , Hans-Peter Klenk B , David R Arahal C , Rosa Aznar C , George Garrity D , Michael Pester E and Philip Hugenholtz FA Department of Microbiology, University of Georgia, Athens, GA, USA. Email: whitman@uga.edu
B School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK
C Colección Española de Cultivos Tipo (CECT), Universidad de Valencia, 46100 Burjassot (Valencia), Spain
D Department of Microbiology and Molecular Genetics, Michigan State University and NamesforLife, LLC, East Lansing, MI, USA
E DSMZ - German Collection of Microorganisms and Cell Cultures, Inhoffenstr. 7B, 38124 Braunschweig, Germany
F Australian Centre for Ecogenomics, School of Chemistry and Molecular Biosciences, The University of Queensland, Brisbane, Qld 4072, Australia
Microbiology Australia 40(3) 125-129 https://doi.org/10.1071/MA19034
Published: 12 September 2019
Type strains of species are one of the most valuable resources in microbiology. During the last decade, the Genomic Encyclopedia of Bacteria and Archaea (GEBA) projects at the US Department of Energy Joint Genome Institute (JGI) and their collaborators have worked towards sequencing the genome of all the type strains of prokaryotic species. A new project GEBA VI extends these efforts to functional genomics, including pangenome and transcriptome sequencing and exometabolite analyses. As part of this project, investigators with interests in specific groups of prokaryotes are invited to submit samples for analysis at JGI.
What are type strains? By definition, type strains are descendants of the original isolates that were the basis for species descriptions, as defined by the International Code of Nomenclature of Prokaryotes1. They exhibit all of the relevant phenotypic and genotypic properties cited in the original published taxonomic circumscriptions. Type strains are also deposited in public culture collections and are likely to remain available for the foreseeable future. Thus, the importance of type strains in nomenclature is only secondary to their value as a biological resource. By the rules of nomenclature, a type strain cannot be identical with any other type strain. Since 1987, the difference between type strains has generally been defined genetically2. In terms of genome sequences, this level of diversity is equivalent to about 70% DNA : DNA hybridisation and 95% average nucleotide identity (ANI) among the conserved DNA3,4. In terms of phenotypic similarity, species generally possess S values as defined by numerical taxonomy of >70%, which is close to the limit of significance5. Thus, any two properly described type strains must be appreciably different. If these same criteria were applied to mammals most of the primates would be members of the same species6.
During the last decade, the Genomic Encyclopedia of Bacteria and Archaea (GEBA) projects at the US Department of Energy Joint Genome Institute (JGI) and their collaborators have undertaken a program to sequence the genomes of all the type strains of prokaryotic species. As of February 2019, 16 232 validly named prokaryotic species have been formally described, most of which are represented by type strains. The genomes of 7647 of these have been sequenced, 3145 by the GEBA projects. Another 2300 genome sequencing projects are currently underway at JGI. In addition, the World Data Centre for Microorganisms (WDCM) of the World Federation of Culture Collections (WFCC) began an additional project called GCM2.0 to sequence the genomes of the remaining prokaryote type strains7. This project parallels the efforts of GEBA, and the projects are closely coordinated to ensure that there is no overlap in sequencing efforts. Currently, the WFCC has completed or has in progress more than 2600 genome sequences of type strains (www.gcm.wdcm.org).
Beginning in 2013, the GEBA project began sequencing prospective type strains during their formal description8. The WDCM also provides a similar service to make genome sequencing readily available to laboratories worldwide9. Moreover, all three of the major microbial systematics journals, International Journal of Systematic and Evolutionary Microbiology, Systematics and Applied Microbiology and Antonie van Leeuwenhoek, now either require or strongly recommend including genome sequences in the descriptions of novel species. Minimum standards for the use of genome data for taxonomy of prokaryotes have also been proposed10. For these reasons, we expect that genome sequences will be included in the description of most new species in the future. This change in policy insures that the number of type strains without sequences will no longer increase. The major challenge then is sequencing the genomes of the type strains that have been previously described without a sequence. However, given the abovementioned initiatives, it is now possible to expect that the remaining type strains will be sequenced in the near future. When this goal is realised, this biological resource can be fully utilised.
Why sequence type strains? In the larger context, only a small portion of prokaryotic diversity has ever been cultured. The genomes of all known type strains are estimated to represent no more than 15% of the total diversity11. However, given the enormity of prokaryotic diversity, this is not an insignificant fraction. Our knowledge of remaining organisms comes largely from metagenome sequencing of environmental DNA. Because the type strains are well characterised, their genome sequences provide the framework for inferring the biological properties of uncultured prokaryotes from their genome sequences. Specifically, it will address a number of complementary questions. How is gene content related to function? Is the presence of certain genes and combinations of genes a strong predictor of phenotype? What properties of prokaryotes are predicted from their phylogeny? In the absence of clear understanding of the functional annotations of genes, what properties can be predicted from those of their relatives? Can probabilistic models be developed to express the likelihood of organismal properties from gene content and phylogeny? Currently, the data to address these issues do not exist at a scale that will generalise to all prokaryotic life.
There is also a number of other, very different but equally valid reasons to sequence the genomes of microbial type strains. As more genomes become available for specific groups, the applications of genome-based systematics are revolutionising the classification of prokaryotes. Genomes provide more reliable and complete data, and allow formation of more meaningful groupings of higher taxa. For instance, genome sequences suggest that the NCBI taxonomy, which is based largely on 16S rRNA sequences, contains a large number of misclassifications within the Clostridia and Bacteroidetes (Figure 1). Already genome sequences collected in large part by the GEBA projects have led to major taxonomic revisions of the Actinobacteria, Bacteroidetes, Epsilonproteobacteria, Geodermatophilaceae and the Rhodobacteraceae13–17.

|
Because they are phylogenetically diverse, the GEBA genomes are also very useful for identifying metagenome sequences from environmental DNA18. For instance, the first thousand GEBA genomes enabled classification of more than 25 million proteins from metagenomes. The same genomes led to a >10% increase in the known protein sequence diversity in prokaryotes18,19. Most type strains were described because of their environmental, medical or commercial importance. Their genomic sequences will contribute greatly to our knowledge of the processes in which these prokaryotes play fundamental roles. Identification of prokaryotes is still a major challenge that hinders many practical applications. Genomic sequencing of type strains provides tools that greatly facilitate identification and classification.
While continuing the efforts to sequence the genomes of type strains, the newest GEBA project, GEBA VI, will go beyond genome sequencing to initiate the next stage of utilisation of this important biological resource. Because their phenotypic and physiological properties are typically well characterised, type strains are well suited for studies of functional genomics, which unite genome content with the biological properties of bacteria and archaea. The proposed studies will systematically investigate genome function of type strains by a combination of (1) genome sequencing to determine gene content, (2) transcriptomics to elucidate gene expression, (3) secreted metabolites (or exometabolomics) to examine function, and (4) pangenomics to identify the core genome and evolutionary processes.
The rationale for these projects is as follows. While genome sequences provide valuable insights into phylogeny and systematics, they are only the first layer of information available from type strains. Organism function depends critically upon gene expression. Transcriptome studies will identify the highly expressed genes central to an organism’s growth and metabolism. Many of these genes are expected to be ‘character’ genes, which play critical roles in an organism’s specific adaptation to the environment20. Exometabolomics provides a different view of function, and spent culture media will be screened for nonpolar as well as polar metabolites. Nonpolar metabolites are expected to include many secondary metabolites, such as antibiotics, polyketides and phenolics. These compounds play fundamental roles in cell-cell signaling, competition with other microorganisms, metal uptake and other important biological functions. Polar metabolites are expected to include amino acids, sugars, small organic acids and other hydrophilic compounds. These analyses will identify the components of complex culture media that are consumed, providing direct evidence for the culture’s metabolism. They will also identify compounds produced during growth by incomplete metabolism or fermentation.
Lastly, the genome content of any specific strain does not fully represent the gene content of the species21,22. Sequencing of closely related strains will identify the core and pangenome as well as the nature of evolutionary processes occurring within the taxon. The core genome is of special importance in the description of a species because it encodes those properties that are conserved across all members20. Thus, it captures the phenotypic basis for the species, including those factors which are responsible for speciation and adaptation to its environment. The dispensable genome is also interesting and provides insight into strain specific differences. For instance, in many pathogenic species, the dispensable genome encodes processes associated with host interactions. Finally, the pangenome provides important insights into the frequency of horizontal gene transfer and sources of diversity within a species23. For instance, a pangenome can be either ‘closed’ or ‘open’. The number of genes in a closed pangenome increases to a maximum value as the number of strains increases, suggesting that there is a finite limit to the genetic diversity within a species. The number of genes in an open pangenome is unbounded, and the genetic diversity is, in theory, unlimited.
To realise these goals, GEBA VI welcomes contributions of samples from individual investigators (Figure 2). To participate, submit a description of your project at our website (https://gold.jgi.doe.gov/gebaVI) or send an email to whitman@uga.edu (include ‘GEBA VI’ in the subject line). The types of projects being considered are genome sequencing of type strains, pangenome sequencing of species poorly represented in the databases, and functional genomics of type strains. The GEBA VI project will support two types of experimental designs, although other good ideas are encouraged. The first will be in-depth studies of single species, including characterisation of the pangenome or transcriptomes and exometabolomes under different growth conditions. In studies of this type, the genome of the type strain may have been sequenced in previous studies. We expect that individual investigators will be the primary contributors for these studies, and the goal is to provide sufficient data for a substantive publication. In this design, the project will support transcriptome studies of 3–4 replicates of up to four conditions. Exometabolome analyses will be dependent upon the suitability of the culture medium for analysis by mass spectroscopy. It may include analysis of the spent medium of up to four conditions for nonpolar metabolites and two conditions for polar metabolites. The pangenome is expected to comprise 5–10 reference strains. The second type of experimental design will comprise comparative studies of groups of related type strains and surveys of genome sequences, transcriptomes and exometabolomes. These designs will try to encompass collections of related species or genera. Type strains will be cultured under identical conditions for preparation of samples for the transcriptomes and exometabolomes.
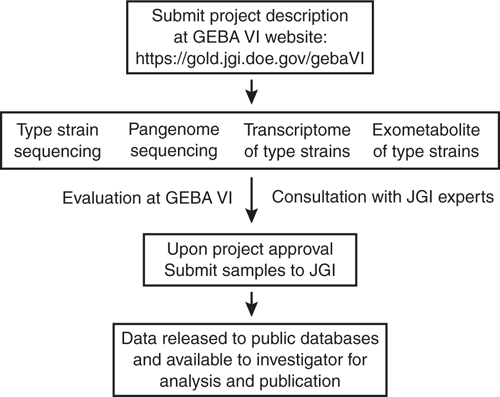
|
Our primary intention is to survey a large amount of phylogenetic diversity, and, for that reason, these studies will not be comprehensive. For instance, complete sampling of a pangenome may require hundreds of sequences. The transcriptome can only be partially elucidated in the limited studies proposed here. Similarly, production of various classes of exometabolites often depends critically on the cultivation conditions (e.g. media, time, temperature, limiting nutrients, oxygenation, pH/buffering). Historically, screening programs use batteries of complex media designed to trigger secondary metabolite production based on nutrient limitations, and spent media is extracted using a battery of solvents ranging in polarity and protonation. The cell pellet is also often extracted to recover bound metabolites. Thus, the studies proposed here are unlikely to uncover the full range of exometabolites. Nevertheless, these studies will identify candidates for subsequent in-depth studies and provide valuable comparative information.
Conflicts of interest
The authors declare no conflicts of interest.
Acknowledgement
The authors are grateful to Nikos Kyrpides, Tanja Woyke and Trent Northen for helpful discussions. This research did not receive any specific funding.
References
[1] Parker, C.T. et al. (2019) International Code of Nomenclature of Prokaryotes. Prokaryotic Code (2008 Revision). Int. J. Syst. Evol. Microbiol. 69, S7–S111.[2] Wayne, L.G. et al. (1987) Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics. Int. J. Syst. Bacteriol. 37, 463–464.
| Report of the Ad Hoc Committee on Reconciliation of Approaches to Bacterial Systematics.Crossref | GoogleScholarGoogle Scholar |
[3] Goris, J. et al. (2007) DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91.
| DNA-DNA hybridization values and their relationship to whole-genome sequence similarities.Crossref | GoogleScholarGoogle Scholar | 17220447PubMed |
[4] Konstantinidis, K.T. and Tiedje, J.M. (2005) Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 102, 2567–2572.
| Genomic insights that advance the species definition for prokaryotes.Crossref | GoogleScholarGoogle Scholar | 15701695PubMed |
[5] Jones, D. and Sneath, P.H.A. (1970) Genetic transfer and bacterial taxonomy. Bacteriol. Rev. 34, 40–81.
| 4909647PubMed |
[6] Whitman, W.B. et al. (1998) Prokaryotes: the unseen majority. Proc. Natl. Acad. Sci. USA 95, 6578–6583.
| Prokaryotes: the unseen majority.Crossref | GoogleScholarGoogle Scholar | 9618454PubMed |
[7] Wu, L. et al. (2018) The global catalogue of microorganisms 10K type strain sequencing project: closing the genomic gaps for the validly published prokaryotic and fungi species. Gigascience 7, giy026.
| The global catalogue of microorganisms 10K type strain sequencing project: closing the genomic gaps for the validly published prokaryotic and fungi species.Crossref | GoogleScholarGoogle Scholar | 30239706PubMed |
[8] Whitman, W.B. et al. (2015) Genomic Encyclopedia of Bacterial and Archaeal Type Strains, Phase III: the genomes of soil and plant-associated and newly described type strains. Stand. Genomic Sci. 10, 26.
| Genomic Encyclopedia of Bacterial and Archaeal Type Strains, Phase III: the genomes of soil and plant-associated and newly described type strains.Crossref | GoogleScholarGoogle Scholar | 26203337PubMed |
[9] Wu, L. and Ma, J. (2019) The Global Catalogue of Microorganisms (GCM) 10K type strain sequencing project: providing services to taxonomists for standard genome sequencing and annotation. Int. J. Syst. Evol. Microbiol. 69, 895–898.
| The Global Catalogue of Microorganisms (GCM) 10K type strain sequencing project: providing services to taxonomists for standard genome sequencing and annotation.Crossref | GoogleScholarGoogle Scholar | 30832757PubMed |
[10] Chun, J. et al. (2018) Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466.
| Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes.Crossref | GoogleScholarGoogle Scholar | 29292687PubMed |
[11] Wu, D. et al. (2009) A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea. Nature 462, 1056–1060.
| A phylogeny-driven genomic encyclopaedia of Bacteria and Archaea.Crossref | GoogleScholarGoogle Scholar | 20033048PubMed |
[12] Parks, D.H. et al. (2018) A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996.
| A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life.Crossref | GoogleScholarGoogle Scholar | 30148503PubMed |
[13] Montero-Calasanz, M.D.C. et al. (2017) Genome-scale data call for a taxonomic rearrangement of Geodermatophilaceae. Front. Microbiol. 8, 2501.
| Genome-scale data call for a taxonomic rearrangement of Geodermatophilaceae.Crossref | GoogleScholarGoogle Scholar |
[14] Hahnke, R.L. et al. (2016) Genome-based taxonomic classification of Bacteroidetes. Front. Microbiol. 7, 2003.
| Genome-based taxonomic classification of Bacteroidetes.Crossref | GoogleScholarGoogle Scholar | 28066339PubMed |
[15] Waite, D.W. et al. (2017) Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. nov.). Front. Microbiol. 8, 682.
| Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. nov.).Crossref | GoogleScholarGoogle Scholar | 28484436PubMed |
[16] Simon, M. et al. (2017) Phylogenomics of Rhodobacteraceae reveals evolutionary adaptation to marine and non-marine habitats. ISME J. 11, 1483–1499.
| Phylogenomics of Rhodobacteraceae reveals evolutionary adaptation to marine and non-marine habitats.Crossref | GoogleScholarGoogle Scholar | 28106881PubMed |
[17] Nouioui, I. et al. (2018) Genome-based taxonomic classification of the phylum Actinobacteria. Front. Microbiol. 9, 2007.
| Genome-based taxonomic classification of the phylum Actinobacteria.Crossref | GoogleScholarGoogle Scholar | 30186281PubMed |
[18] Mukherjee, S. et al. (2017) 1,003 reference genomes of bacterial and archaeal isolates expand coverage of the tree of life. Nat. Biotechnol. 35, 676–683.
| 1,003 reference genomes of bacterial and archaeal isolates expand coverage of the tree of life.Crossref | GoogleScholarGoogle Scholar | 28604660PubMed |
[19] Kyrpides, N.C. et al. (2014) Genomic Encyclopedia of Type Strains, Phase I: The one thousand microbial genomes (KMG-I) project. Stand. Genomic Sci. 9, 1278–1284.
| Genomic Encyclopedia of Type Strains, Phase I: The one thousand microbial genomes (KMG-I) project.Crossref | GoogleScholarGoogle Scholar | 25197443PubMed |
[20] Lapierre, P. and Gogarten, J.P. (2009) Estimating the size of the bacterial pan-genome. Trends Genet. 25, 107–110.
| Estimating the size of the bacterial pan-genome.Crossref | GoogleScholarGoogle Scholar | 19168257PubMed |
[21] Medini, D. et al. (2005) The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594.
| The microbial pan-genome.Crossref | GoogleScholarGoogle Scholar | 16185861PubMed |
[22] Vernikos, G. et al. (2015) Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154.
| Ten years of pan-genome analyses.Crossref | GoogleScholarGoogle Scholar | 25483351PubMed |
[23] Sun, Y. and Luo, H. (2018) Homologous recombination in core genomes facilitates marine bacterial adaptation. Appl. Environ. Microbiol. 84, e02545-17.
| Homologous recombination in core genomes facilitates marine bacterial adaptation.Crossref | GoogleScholarGoogle Scholar | 29934335PubMed |
Biographies
William (Barny) Whitman is an Emeritus Professor of Microbiology at the University of Georgia, USA, and trustee of Bergey’s Manual. His research interests are prokaryotic systematics and the physiology of the methanogenic Archaea.
Hans-Peter Klenk is an Emeritus Professor of Microbial Genomics and Diversity at the University of Newcastle, Newcastle upon Tyne, UK, and former Head of School of Biology at that very place. His research interests are Actinobacteria, phylogenomics and prokaryotic systematics in general.
David R Arahal is an Associate Professor of Microbiology at the University of Valencia, Spain, and senior researcher at the Spanish Type Culture Collection (CECT). His research interests are taxonomy and genomics of prokaryotes, and very especially those from aquatic environments.
Rosa Aznar is a Full Professor of Microbiology at the University of Valencia, Spain, and Director of the Spanish Type Culture Collection (CECT). Her research interest and background focus on identification and characterisation of bacteria related to food quality, food safety and functional food.
George Garrity is a Professor in the Department of Microbiology and Molecular Genetics at Michigan State University and Co-founder of NamesforLife, LLC a bioinformatics and publishing services company that was founded to commercialise a novel semantic technology developed at Michigan State University. Prior to joining Michigan State, he held a number of positions of increasing responsibility in the natural products screening program at Merck & Co.
Michael Pester is a Professor at the Technical University of Braunschweig and heads the Department of Microbiology at the Leibniz Institute DSMZ - German Collection of Microorganisms and Cell Cultures. His research interests are environmental microbiology and geomicrobiology of sulfur and nitrogen cycling.
Philip Hugenholtz is a microbiologist who has made contributions in the field of culture-independent analysis of microorganisms. He has contributed to the development and application of metagenomics, the genome-based characterisation of microbiomes, including their taxonomic classification.