

Iron-catalysed oxidation and halogenation of organic matter in nature
Peter Comba A B D , Marion Kerscher A , Torsten Krause C and Heinz Friedrich Schöler C DA Anorganisch-Chemisches Institut, Universität Heidelberg, Im Neuenheimer Feld 270, D-69120 Heidelberg, Germany.
B Interdisciplinary Center for Scientific Computing (IWR), Universität Heidelberg, Im Neuenheimer Feld 368, D-69120 Heidelberg, Germany.
C Institut für Geowissenschaften, Universität Heidelberg, Im Neuenheimer Feld 234-236, D-69120 Heidelberg, Germany.
D Corresponding authors. Email: peter.comba@aci.uni-heidelberg.de; heinfried.schoeler@geow.uni-heidelberg.de

Peter Comba obtained a diploma in chemistry and chemical education from ETH Zürich and Ph.D. from the Université de Neuchâtel. He had research positions at the Australian National University, Canberra, the Université de Lausanne and the Universität Basel, before taking up his present position as Professor of Chemistry at the Universität Heidelberg. He is interested in fundamental transition metal coordination chemistry, involving ligand design and synthesis, preparative coordination chemistry, spectroscopy as well as theoretical and computational inorganic chemistry, with projects in bioinorganic and medicinal inorganic chemistry, molecular magnetism and molecular catalysis. |

Marion Kerscher was awarded a Ph.D. degree from the University of Heidelberg in 2003. Subsequently, she got a permanent position as a scientist in the Inorganic Chemistry Department of the University of Heidelberg, where she is working in the field of coordination chemistry and focuses on synthesis and spectroscopy. |

Torsten Krause obtained a diploma in chemistry with a thesis on analytical and pharmaceutical applications of quantum dots from the University of Marburg. From the University of Heidelberg he obtained his Ph.D. degree for research on salt lake chemistry and volatile organic compounds emitting therefrom. Today, he works at the Max Rubner-Institute, the German Federal Research Institute of Nutrition and Food within the department for safety and quality of milk and fish in Kiel. |

Heinfried Schöler studied in Bonn and obtained the diploma in chemistry and Ph.D. from the University of Bonn. He had a research position at the Medical Department of the University of Bonn. After habilitation he became Professor of Environmental Hygiene at the University of Bonn and in 1992 Professor for Environmental Organic Geochemistry at the University of Heidelberg. He is especially interested in naturally produced organohalogens (trichloroacetic acid, chlorobenzoic acids, methyl halides, chlorethene, chloroethyne, trihalomethanes) and volatile organic carbon (carbon suboxide, furan, and furan derivatives) in soils, sediments, and fluid inclusions. |
Environmental Chemistry 12(4) 381-395 https://doi.org/10.1071/EN14240
Submitted: 8 November 2014 Accepted: 2 March 2015 Published: 22 June 2015
Environmental context. Natural organohalogens produced in and released from soils are of utmost importance for ozone depletion in the stratosphere. Formation mechanisms of natural organohalogens are reviewed with particular attention to recent advances in biomimetic chemistry as well as in radical-based Fenton chemistry. Iron-catalysed oxidation in biotic and abiotic systems converts organic matter in nature to organohalogens.
Abstract. Natural and anthropogenic organic matter is continuously transformed by abiotic and biotic processes in the biosphere. These reactions include partial and complete oxidation (mineralisation) or reduction of organic matter, depending on the redox milieu. Products of these transformations are, among others, volatile substances with atmospheric relevance, e.g. CO2, alkanes and organohalogens. Natural organohalogens, produced in and released from soils and salt surfaces, are of utmost importance for stratospheric (e.g. CH3Cl, CH3Br for ozone depletion) and tropospheric (e.g. Br2, BrCl, Cl2, HOCl, HOBr, ClNO2, BrNO2 and BrONO2 for the bromine explosion in polar, marine and continental boundary layers, and I2, CH3I, CH2I2 for reactive iodine chemistry, leading to new particle formation) chemistry, and pose a hazard to terrestrial ecosystems (e.g. halogenated carbonic acids such as trichloroacetic acid). Mechanisms for the formation of volatile hydrocarbons and oxygenated as well as halogenated derivatives are reviewed with particular attention paid to recent advances in the field of mechanistic studies of relevant enzymes and biomimetic chemistry as well as radical-based processes.
Introduction
The oxidation and halogenation of organic substrates, induced or catalysed by iron complexes and oxygen-based oxidants (e.g. O2, peroxides) is of importance in biology, chemical research and industrial processes as well as in the environment. Extensive knowledge has been assembled in recent years in the area of detailed reaction mechanisms, substrate and product selectivity and the optimisation of active iron species and reaction conditions. It is of importance to note that although many of the reactions and mechanisms in the field of biological processes, chemical reactions and environmentally relevant transformations are similar in terms of reaction conditions, structures of the active iron complexes, product distribution and yields, there are also large and important differences: in biological processes, substrate and product selectivity generally are close to 100 % and turnover frequencies need to be enormous, i.e. close to absolute selectivity, close to quantitative yield, and very fast reactions are the rule. In industrial processes, high yields and product selectivities are of importance, but a broad scope with respect to substrates is also desirable, and high catalyst stability, i.e. large turnover numbers are of importance for commercial reasons. In environmental transformations, very low yields of environmentally relevant products need to be considered. Owing to the large surface of the earth, yields in the parts per billion range, which in biological and industrial processes are completely insignificant, may lead to enormous amounts of products, accumulated in the atmosphere where they influence the terrestrial ecosystem. Although there are many differences in the various fields in terms of relevance and reaction conditions and, therefore, also in terms of reaction mechanisms, it appears that, to a large extent, general concepts that have emerged in recent years, in particular from biomimetic coordination chemistry, are generally relevant. However, it seems that the different communities are not sufficiently aware of important results assembled in the different research fields. Therefore, we review important recent results of iron-induced oxidation processes of organic substrates, obtained specifically in the fields of bioinorganic, biomimetic and mechanistic inorganic chemistry, as well as in the area of environmental organic chemistry. Thorough mechanistic work has been the focus of biomimetic coordination chemistry and trace analysis is of specific importance in environmental chemistry. It appears that mechanistic ideas emerging from biomimetic studies may be adapted with much profit to novel products and reactions detected in environmental sciences, and this could lead to a shift of some of the paradigms but care needs to be taken in terms of generalisation and overinterpretation. Also, there is some hope that various of the novel environmentally relevant reactions found in recent years may lead to interesting new discoveries in the area of coordination and biochemistry. Therefore, although the present review is primarily addressed at environmental scientists, bio- and inorganic chemists may also find it useful.
Abiotic processes in natural environments
Natural and anthropogenic organic matter is continuously transformed by abiotic and biotic processes in the biosphere. These reactions include oxidation, i.e. complete mineralisation, or reduction of organic matter, depending on the redox milieu, which is subdivided into oxic, suboxic and anoxic. Products of these transformation processes are, among others, volatile substances with atmospheric relevance, e.g. CO2, alkanes and halogenated alkanes. Also, organic acids play a key role in the mobilisation and bioavailability of minerals. Key reactions are metal ion-induced radical and non-heme iron-based processes.
Iron in nature
With a mean abundance of 4.2 %, iron is, after the redox-inactive aluminium, the second most abundant metal in the continental crust.[1] In primary minerals such as biotite, olivine and magnetite, iron is mostly present in its +2 oxidation state. Through weathering processes, it is liberated in the presence of water, oxidised by oxygen and immobilised after hydrolysis as FeIII oxide-hydroxides (ferrihydrite, goethite and hematite), which are responsible for the brown or red colour of soils. The iron content of soils is in the range 0.5–5 %.[2] FeIII oxides are very stable weathering products and reside in soil under aerobic conditions. When microbial oxidation of organic material takes place, FeIII oxide serves as an electron acceptor and is reductively dissolved.[3] Diffusion into aerobic environments again leads to precipitation of FeIII oxide-hydroxides. Reductive dissolution and oxidative precipitation are parts of the Fe redox cycling under changing redox conditions.[4] FeIII is stable in aqueous soil solutions in oxic environments at pH < 3.5, whereas at pH > 4, FeIII concentrations are extremely low and limited to soluble FeIII complexes of organic ligands. In conclusion, iron availability in oxic soils is determined by the interaction of poorly crystalline iron minerals and soluble organic ligands such as humic acids,[5] microbially produced siderophores[6] and root exudates.[7]
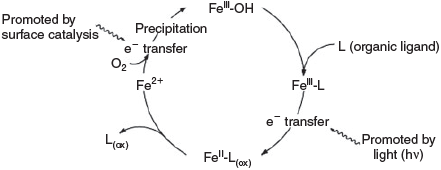
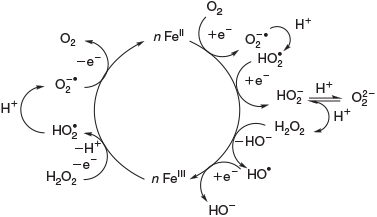
Iron coordination compounds play an important role in proteins and enzymes.[8–10] These are involved in electron transfer, oxidation and oxygenation. Other redox-active metal ions such as copper, vanadium, nickel and manganese play similar roles in biotic systems but iron and copper are the most abundant.[11,12] Abiotic processes are believed to follow similar pathways and are responsible for the oxidation and oxygenation of organic matter. Copper- and iron-based oxygen activation and oxidation chemistry[8–10] are ubiquitous, high-valent manganese model chemistry is gaining more attention recently[13–18] and various studies on high-valent nickel chemistry also have recently been published.[19–21] Here, we concentrate on iron chemistry. Fig. 1 shows a general scheme for iron cycling in natural environments.[22]
H2O2 in nature
H2O2 has multifarious sources in the biosphere. It is abundant in the atmospheric gas phase, clouds and water droplets[23] and is transported to soils and surface waters by rain.[24] Elevated H2O2 concentrations are found during thunderstorms, implying formation through electrostatic discharge,[25–27] and near fire plumes.[28] Moreover, the reaction of hydroperoxyl radicals with each other (Eqn 1) is an important atmospheric radical termination, producing H2O2.[29]
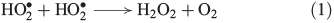
Another source for atmospheric H2O2 is the reaction of ozone with isoprene, monoterpenes and alkenes by a Criegée biradical, expressed in Eqns 2 and 3 (R represents H, alkyl or acyl).[30]
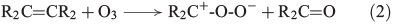
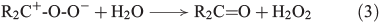
Photochemical reactions are also a common source of H2O2 in terrestrial[31] and marine[32] aqueous environments.[33] The underlying mechanism is the photo-ionisation or photo-excitation of dissolved organic matter by sunlight, producing free electrons or excited states that can reduce dissolved oxygen to superoxide (Eqn 4, OM, organic matter).[34] As shown in Eqn 5, superoxide may also be formed by metal-based reduction, involving iron[35–37] or manganese,[38] but this reaction is inhibited under acidic conditions and in the presence of chloride.[39–42]
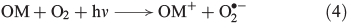
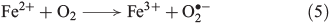
Superoxide can disproportionate, yielding H2O2 (Eqn 6)[43] or it may be reduced further to H2O2 (Eqn 7).[44]
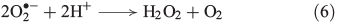
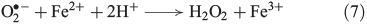
Other known biogenic H2O2 sources in soil and aquatic systems are the release of H2O2 by fungi[45] and brown algae.[46] H2O2 also plays a significant role in bacterial metabolism.[47]
Halides in nature
Weathering processes liberate large amounts of halides from primary rocks and minerals. These are dissolved in water and transported via rivers to the oceans as the final sink. One litre of ocean water contains 0.5 mol chloride, 1 mmol bromide and 1 μmol iodide (the molar ratio of Cl– : Br– : I– is 500 000 : 1000 : 1).[48] Part of the halides is mobilised by sea-spray or as organohalogens and enters the terrestrial environment. Therefore, the atmospheric deposition of halides is dependent on the distance from the ocean and on the amount of precipitation.
In addition, primary rocks and evapotranspiration contribute to the halide content of soils. The mean chloride content of soils in humid climates is in the range 100–300 mg kg–1; the corresponding bromide and iodide contents are 5–50 and 3–30 mg kg–1 respectively.[2] The molar ratio of Cl– : Br– : I– in soil is ~100 : 5 : 1.[48] In conclusion, the molar ratio in soil is distinctly shifted relative to ocean water, especially for iodide, which is enriched by a factor of ~5000.
Relevant oxidation reactions
The reaction of halides with OH radicals and H2O2
In the presence of FeII, aqueous H2O2 may produce OH radicals (see below). Halides are known to react with OH radicals, yielding reactive halogen species. A general reaction mechanism is shown in Eqns 8 and 9 with X = Br,[49] Cl[50,51] and I.[52]
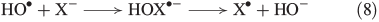
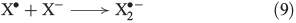
Owing to increasing electronegativity, the reactivity of OH radicals with halides decreases from iodide to chloride.[53] The recombination of halogen radicals is described in Eqn 10.[54–57]

Recently, the mechanism for Cl2 evolution was summarised,[58] involving the reactions shown in Eqns 11 and 12.
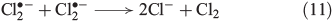
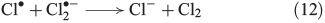
The oxidative power of FeIII is high enough to oxidise iodide through a clock reaction directly to I2.[59]
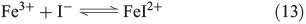
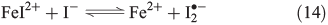
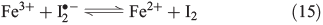
Counteracting the formation of reactive halogen species is their reductive depletion by FeII[51] as well as by H2O2[58] to halides.
The reaction of H2O2 with halides is not restricted to an iron-catalysed Fenton-type reaction but halides are also directly oxidised by H2O2. H2O2 reacts in the presence of iodide under acidic conditions to form hypoiodous acid, described by the modified ‘Iodine Clock’ reaction shown in Eqn 16,[60] which is based on the Landolt reaction.[61,62] This process is probably the main source of iodocarbons in the metabolism of brown algae.[63]
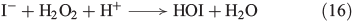
A similar mechanism was postulated for the reaction of bromide and chloride with H2O2, as illustrated in Eqns 17[64] and 18[65] respectively.
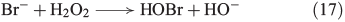
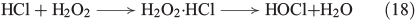
The reactivity between halides and H2O2, described in Eqns 17 and 18, decreases from I– to Cl–.[53] Furthermore, H2O2 is able to reduce the hypohalous acids to hydrogen halides under evolution of oxygen, reducing the yield of HOCl[66] and HOBr.[67] Hypoiodous acid or iodine even catalyse the decomposition of H2O2, as illustrated in Fig. 2.[68,69]
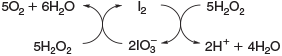
This illustrates the complicated mechanisms of Fenton-type processes and the corresponding side reactions in the presence of halides, yielding reactive halogen species like X, X2–, X2, XOH and XO–. In addition, X2, XOH and XO– are in pH-dependent equilibria with one another (Eqn 19).[70]

Oxidants for the natural oxidation of organic matter
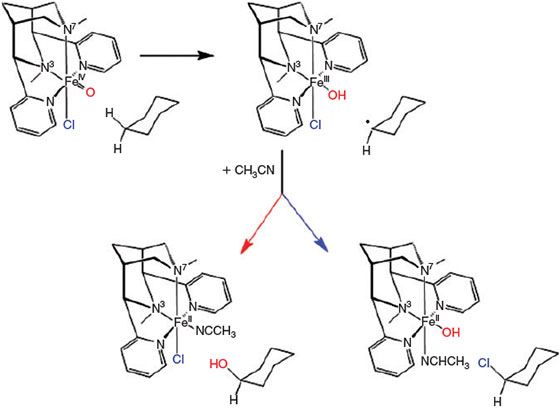
The oxidation chemistry of FeII in water has been studied extensively since Fenton’s discovery that FeII salts in the presence of H2O2 efficiently oxidise organic matter in acidic aqueous solution.[71–73] The involvement of OH radicals has been suggested and the OH radicals are often believed to be the relevant oxidant.[74] However, on the basis of experimental observations in the 1930s, high-valent iron-oxo species have also been suggested to be involved in Fenton reactions,[75] and this was confirmed by density functional theory (DFT) calculations a decade ago.[76] At approximately the same time, the first reports of low-molecular-weight ferryl complexes and non-heme iron enzymes appeared.[54–57] Important milestones in this area are the first report of the spectroscopic[77] and structural[78] characterisation of intermediate-spin non-heme ferryl model complexes as well as a detailed mechanistic study of the α-keto-acid-dependent enzyme TauD with a high-spin ferryl active site.[79–82] Especially interesting, therefore, are the observations of high-spin low-molecular-weight ferryl species[83–87] and the proposal of catalytically active intermediate-spin dihydroxo-iron(IV) complexes.[84] An important observation is that ferryl complexes can be trapped in pure water, prepared from the FeII precursor and H2O2 under typical Fenton conditions. Therefore, in the first phase of the stoichiometric reaction, this Fenton-type process does not involve OH radicals (see Fig. 3).[88] Also of importance are reports on the halogenation of unactivated substrates with ferryl model systems.[89,90] It needs to be pointed out that non-heme iron enzymes generally activate dioxygen and that in most of the model reactions discussed here, the ferryl species are obtained from the FeII precursors by oxygen atom transfer, electron transfer or oxidation with H2O2. There are only few examples of direct oxidation by dioxygen, and recent results indicate that in at least one example, this is not based on oxygen activation but rather on radical-based autoxidation.[91]
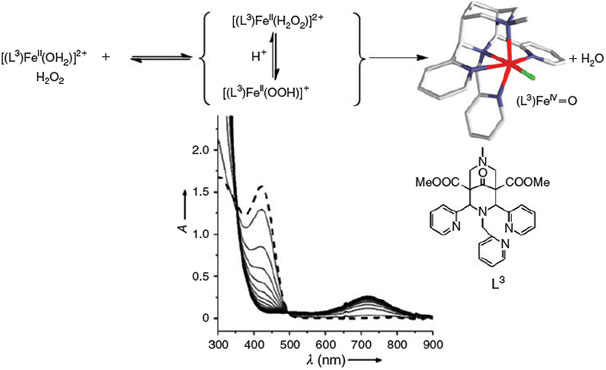
|
There is no doubt that ferryl complexes such as that shown in Fig. 3 and, more generally, other high-valent metal complexes, are strong and very efficient oxidants and that they are available in natural abiotic aquatic systems – i.e. under Fenton-type conditions usually assumed in soils, water and aerosols – and therefore, they obviously play an important role in various environmental compartments. However, OH radicals are also produced in FeII/H2O2 systems and there is no doubt either that these are also of importance in the oxidation of organic matter. Although the product selectivity of in vitro oxidation reactions may indicate whether there is a preferred ferryl or OH radical-based process,[92] such differentiation seems to be difficult in environmental transformations.
The ‘classical’ mechanism of OH radical-based Fenton processes (aqueous Fe2+/H2O2 mixtures) was proposed by Haber and Weiss (Eqn 20).[74]
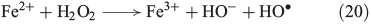
Another reaction of importance is the reduction of FeIII by H2O2, and this completes the catalytic iron cycle of the Fenton reaction[93] (see Fig. 4).
Alternatively to the FeII catalysed decomposition of H2O2, photolysis by UV radiation (λ < 400 nm) can generate OH radicals. Radiation at 290–400 nm enhances Fenton-like reactions by reduction of FeIII(OH)2+ to FeII and the concomitant generation of free OH radicals in this light-induced Fenton or photo-Fenton reaction (Eqn 21).[94]
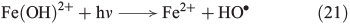
Importantly, metal complex-based Fenton chemistry is not limited to iron as redox catalyst. The amount of oxidised species formed in Fenton-like reactions at circumneutral pH increases according to the order NiII < MnII < FeIII < CoII < CrIII < CuII.[95] In reactions of CuII with H2O2, OH radicals are believed to only play a minor role, and CuII is proposed to be of major importance.[96,97]
In summary, it appears that, depending on the conditions, the active species may involve OH radicals, but in the presence of suitable ligands, high-valent metal complexes may be the more relevant oxidant. Specifically, an FeII complex may be oxidised to FeIII and then coordinate H2O2. The resulting FeIII hydroperoxido complexes themselves are known to be strong oxidants[98] but may also produce ferryl species and OH radicals (homolytic cleavage of the O–O bond).[99,100] Moreover, coordination of H2O2 to the FeII precursor and subsequent homolytic cleavage of the O–O bond may lead to an FeIV(OH)2 complex, a tautomer of the reactive ferryl complex.[84,101] Which of these pathways and which of the resulting oxidants leads to the observed oxidation of organic matter in soils is not clear in most of the reactions studied so far (see sections below), but it obviously depends on the reaction conditions, most importantly on the ligands available.[75,102,103]
Abiotic oxidation of organic matter
A key initial reaction in the context of abiotic natural environments is the degradation of humic acid, and it is believed that the iron-catalysed oxygenation of catechol- and hydroquinone-derived subunits is of importance. The biochemistry of the relevant iron-based catechol dioxygenases (mononuclear non-heme iron enzymes) has been studied extensively.[104–107] Three different reactions are possible when dioxygen reacts with a catecholate derivative bound to a non-heme iron centre, (1) the oxidation to the ortho-quinone (catecholase activity, i.e. a simple two-electron oxidation), and dioxygenation, (2) leading to intradiol cleavage, or (3) leading to extradiol cleavage. Various biomimetic systems with dioxygenase reactivity have been investigated, and these have helped to elucidate the biologically relevant catalytic mechanisms.[108–113]
A wide variety of published work on the stoichiometric and catalytic oxidation of organic molecules with low-molecular-weight non-heme iron model complexes is available,[57,114,115] and this includes oxygen-transfer reactions, e.g. sulfoxidation,[91,116] oxidative demethylation,[117] and methane formation[118] as well as the oxidation of alkanes and alkenes,[91] which are of specific interest in the degradation of organic matter in nature. An interesting recently described reaction, related to published work on demethylation and well-studied sulfoxidation reactions, is methane formation from methionine under oxidative conditions.[118]
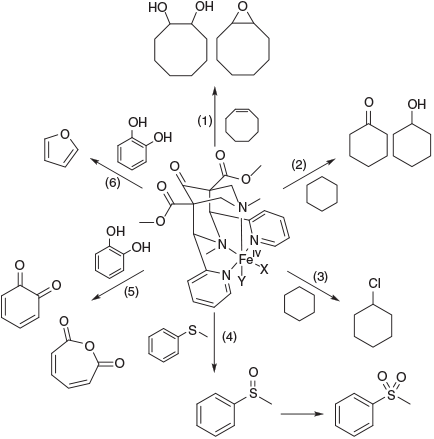
As an example of these biomimetic studies with well-defined systems and their complexes, we describe a few processes of bispidine-type ligands relevant to environmental transformations, also because some of the environmentally relevant mechanistic work has been done with these ligands. Note that in soils, the relevant organic matter is humic substances, and these are believed to coordinate to FeII and FeIII to enable chemical reactions similar to the structurally well-defined model systems described here. With bispidine-type ligands, the formation of ferryl complexes has been confirmed in some detail (see also Fig. 3),[57,88,101,119] and the mechanism of olefin epoxidation and dihydroxylation has been studied, both experimentally and by quantum-chemical methods.[120,121] Careful product analyses, including 18O labelling studies (separately with H218O, 18O2 and H218O2) led to proposing the mechanisms shown in Fig. 5, and these were confirmed by DFT calculations. The most interesting features are that (i) the attack by the ferryl oxygen at the double bond is asymmetrical, leading to a carbon-based radical intermediate, which may be quenched by O2 in ambient atmosphere to selectively yield an epoxide with cyclooctene as substrate.[122] (ii) With the tetradentate ligand system, the initial FeIV species is the novel intermediate-spin FeIV(OH)2 complex (Fig. 5),[84,101] and this may tautomerise to the high-spin ferryl complex. The ferryl complex oxidises the double bond to the epoxide with the same mechanism described above for the pentadentate bispidine ferryl complexes, and the dihydroxo complex yields the cis-diol products (see Fig. 5).[84,101]

|
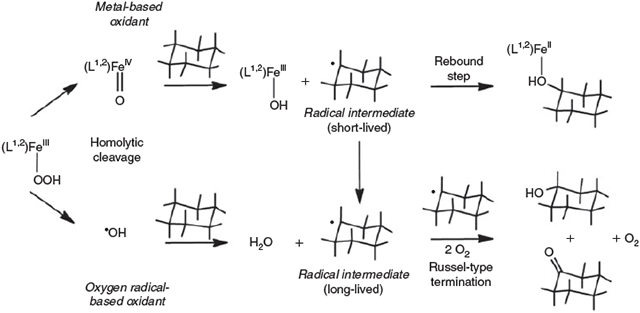
The very high FeIV/III redox potentials[123,124] suggest that the bispidine-based ferryl complexes should even be able to oxidise the strong C–H bonds of alkanes.[89,124] With bispidines and several other ligand systems,[125–127] various possible high-valent iron species were proposed to be responsible for substrate activation, i.e. FeV=O (heterolytic O–O cleavage in the FeIII-OOH intermediate), FeIV=O together with OH radicals (homolytic O–O cleavage in the FeIII precursor) or the products of direct oxidation of the FeII precursor to the catalytically active FeIV oxidant (see above).[89,92,128,129] In the catalytic oxidation of alkanes, it is believed that metal-based oxidants (either FeIV=O or FeV=O) predominantly yield the alcohol products, i.e. hydrogen abstraction by the ferryl oxidant leads to carbon-based radicals that are rebound to the FeIII-OH intermediate to produce the alcohol (see Fig. 6 and Fig. 7 below).[130] In contrast, OH radical-based processes lead to carbon-based substrate radicals that are trapped by O2 to generate equimolar amounts of alcohol and ketone (Fig. 6).[131,132] Product analyses of oxidation reactions initiated by the bispidine complexes, including 18O labelling studies, the determination of kinetic isotope effects and experiments with adamantane as substrate to determine the preference for tertiary over secondary alcohol formation (stabilisation of the tertiary radical intermediate), supported by DFT calculations, lead to the proposed mechanism shown in Fig. 6.[92]
|
|
The more classical analysis of Fenton reactions describes the oxidation of organic matter by an initial abstraction of a H atom by an OH radical (Eqn 22),[103] and radicals are also known to react with unsaturated C=C bonds (Eqn 23).[133]
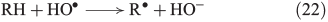
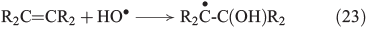
The propagation of radical chain reactions is summarised in Eqns 24–28.[133]
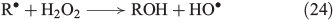
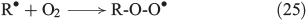
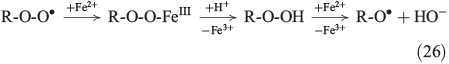
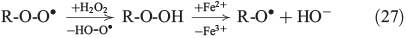
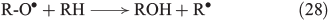
The chain reactions are terminated by iron oxidation or reduction as well as possible dimerisation of carbon radicals (Eqns 29–31).[103,133]
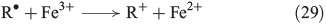
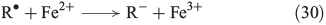
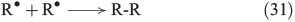
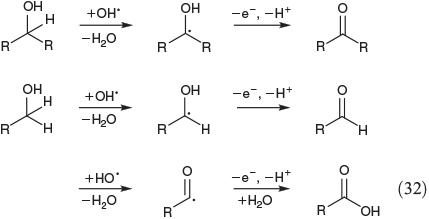
Eqn 32 shows the FeIII-mediated pathway for the oxidation of primary and secondary alcohols. Secondary alcohols are oxidised to ketones[134] whereas the less-reactive primary alcohols are converted to aldehydes and furtheron to carboxylic acids (Eqn 32).[135]
There are two pathways for biological iron-catalysed halogenation processes,[10,55,57,89,136–138] involving heme-based chloroperoxidases or non-heme halogenases (as with other oxidation processes discussed here, nature also uses other redox-active metal centres, e.g. in vanadium-based haloperoxidases). Chloroperoxidases generate a metal-bound hypochlorite ion and hypochlorous acid that are capable of halogenating electron-rich organic substrates. Owing to their high-spin ferryl centres, spectroscopically characterised as reactive intermediates, the αKG-dependent non-heme halogenases can also halogenate less reactive electron-poor alkanes.[136] Similarly to the hydroxylation reactions discussed above, the ferryl intermediates abstract a hydrogen atom from the substrate and the rebound step then transfers a halogen atom from the FeIII-Cl intermediate to the substrate radical (see Fig. 7).
Owing to the similarity of the enzyme active sites and relevant intermediates, it appears that the radical intermediate in the halogenases may compete in the rebound step with the FeIIIOH and FeIIICl sites. However, attempts to detect alcohol products in halogenases failed, and the selectivity required in biological reactions seems to be accomplished by the enzyme-enforced substrate positioning.[137,138] Low-molecular-weight non-heme iron model systems have been shown to be able to halogenate alkanes and alkenes in stoichiometric[89,139] and catalytic processes.[89,140] With the tetradentate bispidine L1 (Fig. 7), the mechanism was studied in some detail, and the kinetic isotope effect as well as the product distribution with adamantane as substrate (relative stability of secondary and tertiary carbon-based radicals) indicated that the rate-determining step indeed is hydrogen atom abstraction, a scenario that is also supported by DFT calculations.[89] It appears that the high halogenation selectivity (~90 %) is due to a lower activation barrier for the FeIII-Cl v. FeIII-OH rebound, and it is proposed that this is related to the relative redox potentials of OH– v. Cl–, i.e. to electronic effects.[89,139] These mechanistic interpretations were challenged in a recent study of another low-molecular-weight non-heme iron halogenase model system, where the first step of the halogenase process, i.e. hydrogen atom abstraction, was characterised unambiguously, but did not produce any halogenated alkanes.[90] Preliminary DFT calculations indicate that this may be related to the thermodynamics of the rebound step, i.e. that halogenation is thermodynamically an uphill process whereas hydroxylation is a downhill process. Because the preliminary DFT calculations indicate that halogenation as well as hydroxylation are thermodynamically possible with the bispidine catalyst, it appears that the first step in the proposed mechanism in Fig. 7 (formation of a carbon-based radical and an FeIII-chlorido-hydroxy intermediate, ready for the rebound step) is unambiguous. Whether or not the product-forming second step is a rebound or a radical-based reaction is not easy to prove experimentally. Alternatives may involve halide oxidation, as recently studied with two non-heme iron model complexes,[141] or typical Gif chemistry as discussed below.[142] Importantly, there are recently published observations that the so-far generally assumed rebound mechanism for non-heme iron model systems (see Fig. 7) is at least in one case not relevant.[143]
In the presence of halides, several halogenation mechanisms are conceivable. Eqn 33 shows the iron-mediated ligand transfer of a halide to an organic radical,[144] whereas the reaction in Eqn 34 is based on halogen radicals.[145]
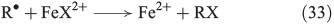
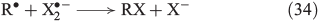
Further important reactions are the electrophilic addition of halogens to alkenes (Eqn 35)[146–148] and, in analogy to OH radicals, the addition of free halogen radicals to unsaturated carbon bonds (Eqn 36).
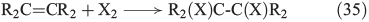
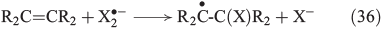
Apart from oxidation by radicals, there is another FeIV-based process. Postulated mechanisms for the hydroxylation and halogenation of hydrocarbons by Gif chemistry are shown in Eqns 37 and 38.[142,149]
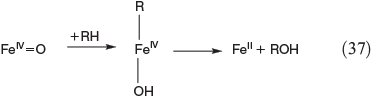
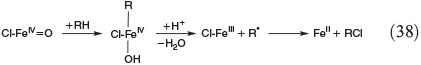
Model reactions
Chemical reactions in soil and natural aqueous systems are complicated by heterogeneity and poorly defined environmental parameters (temperature, pH, concentrations, as well as type and concentrations of relevant species present). To decipher natural abiotic processes, it is inevitable that the reactions must be mimicked under constrained laboratory conditions with a reasonable amount of reactants and parameters. In the last two decades, degradation mechanisms of organic model compounds under Fenton-like conditions with a variety of iron species have been elucidated. Some of the results, which partially redefine the production and flow of small organic compounds in environmental compartments, are discussed in the final sections of the present account.
Organic model compounds
Organic matter in the environment is an umbrella designation for a multitude of organic molecules, resulting from the metabolism of residues of plants and animals, as well as cells and tissues of various organisms. These compounds are found at different stages of degradation. The major fraction of natural organic matter is classified as humic acid and comprises compounds of high molecular weight with a complex structure with numerous carboxylic and phenolic groups. Humic acids are closely related to the lower-molecular-weight fulvic acids. A proposed model of humic substances is shown in Fig. 8, and this is composed of lignin, peptides, cellulose and chitin groups with various metal coordination sites.[150]

Because organic matter in soil has a highly complex and ill-defined structure, chemical reactions are generally studied with well-defined small molecular model compounds with the relevant structural elements and redox properties. Widely accepted models are, for example, catechol, hydroquinone, guaiacol, resorcinol and 2,3-dihydroxybenzoic acid.
Modelling of the catalytically active iron species
The most relevant form of iron species in soil model systems are soluble FeII and FeIII salts, e.g. sulfates, chlorides or carbonates. Several experiments have also been done with iron minerals.[151,152] In a recent series of studies, biomimetic non-heme iron complexes were used to model the coordination of iron cations to natural organic matter in soils. Iron bispidine complexes were chosen for these experiments because they provide a well-defined coordination sphere, enforced by the rigid adamantane-derived backbone (see Figs 3, 5 and 7 above),[123,153,154] and the FeII–bispidine–H2O2 system has been studied extensively and thoroughly with trapped and well-characterised metastable intermediates.[57,155,156]
Environmentally relevant organic reaction products
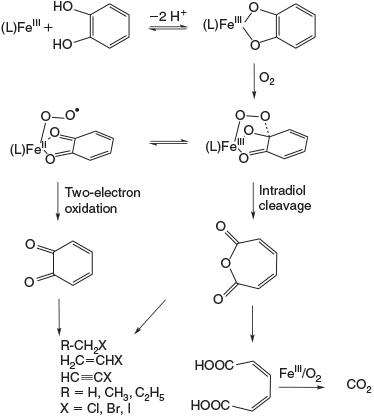
Although oxidation processes under Fenton- or Fenton-like conditions were intensively studied for hazardous waste treatment,[157,158] their relevance for natural abiotic chemistry was not appreciated until the abiotic formation of halomethanes was studied in detail (Fig. 9).[151] A key feature was the observation that guaiacol is oxidised by FeIII to chloromethane among other products.
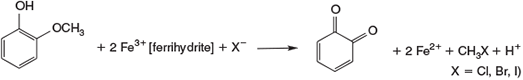
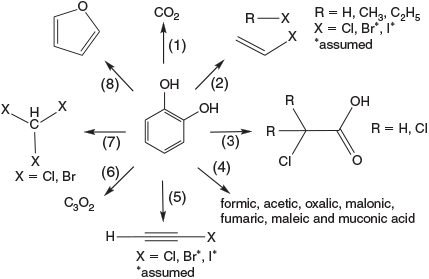
The detailed investigation of the degradation of catechol derivatives under Fenton-like conditions led to the identification of the environmentally relevant series of abiotically formed products shown in Fig. 10.
|
A closer look at the oxidative capabilities of FeIII demonstrated the mineralisation of catechol to CO2,[159] and the formation of chloroethene and chloroalkanes from catechol under Fenton-like conditions in the presence of chloride.[160] The degradation of catechol to mono- and dicarboxylic acids was observed,[162,163,164] and chloroethyne,[165] carbon suboxide[166] and trihalomethanes[167] were also detected. All reaction channels in Fig. 10 may have similar probabilities, and the product yields in the terrestrial environment are in the parts per million or up to percentage range, depending on the substrate concentration and environmental parameters such as pH, Eh and humidity.
For the liberation of chloromethane from guaiacol,[151] a mechanism for the formation of halogenated alkanes was proposed,[170] based on nucleophilic substitution promoted by FeIII and formation of 1,2-benzoquinone. The latter is assumed to be an intermediate in the mineralisation process[159] and the formation of saturated and unsaturated halocarbons,[160,165] see Fig. 11 and Abiotic oxidation of organic matter section.
|
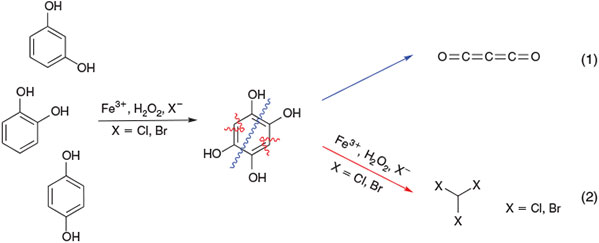
Based on studies with several phenolic derivatives (catechol, hydroquinone, and recorcinol),[166,167] the formation of 1,2,4,5-tetrahydroxybenzene and, depending on where the C–C bonds in this intermediate is cleaved, the release of carbon suboxide and trihalomethanes were postulated (Fig. 12).
|
The well-known haloform reaction[171] of methyl ketones in alkaline conditions yields trihalomethanes after ring cleavage. Alternatively, the degradation of 1,3-dihydroxybenzene (resorcinol) by direct chlorination of aromatic residues is proposed to produce trichloromethane and trichloroacetic acid (Fig. 13).[172]
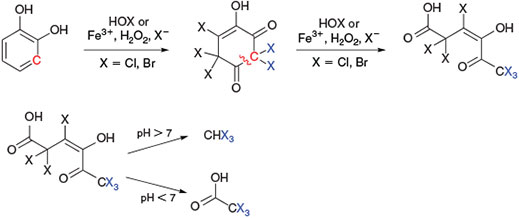
This mechanism can explain the formation of haloacetic acid[161] but, in contrast to the experimental conditions, it assumes basic conditions for the release of trihalomethanes.[167] A review of the literature on natural trichloromethane formulation in peatlands, tundra and forest soils found strong evidence that also in acidic environments the liberation of trichloromethane is conclusive.[173]
A non-halogenated volatile compound emerging from catechol is furan. By isotopic labelling,[168] it was demonstrated that the oxygen atom in furan arises from H2O2, leading to the degradation pathway proposed in Fig. 14.
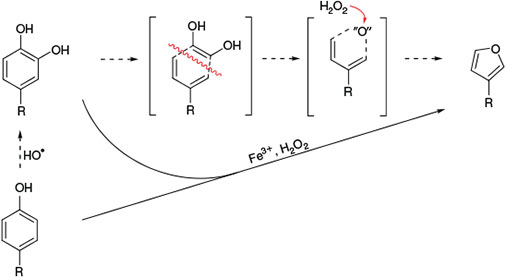
The degradation of catechol to furan was also studied with bispidine model systems. Several the thoroughly studied reactions with bispidine–ferryl complexes are shown in Fig. 15 (epoxidation and cis-dihydroxylation of alkenes,[84,121] oxidation[92,120] and halogenation[89] of cyclohexane, sulfoxidation[116] and catechol oxidation[113]). Although these earlier mechanistic studies were generally performed in organic solvents (usually acetonitrile), more recent work has also involved aqueous chemistry and in particular also the environmentally relevant conditions described above, and this confirmed the formation of furan by a ferryl oxidant.[169]
|
Conclusions
Iron-induced and iron-catalysed oxidation of organic substrates in nature, in biotic and abiotic systems, leads to alkanes and alkenes as well as oxygenated and halogenated hydrocarbons. From abiotic systems, these volatile products are liberated into the atmosphere, and a large number of related interesting and novel products have been found in recent years. Field and laboratory studies have helped to trace them back to degradation processes of humic substances as one of the main contributors of organic matter in the soil. Inorganic model chemistry of biological processes (biomimetic chemistry) has helped to solve important mechanistic problems in the area of high-valent metal-oxo, in particular iron-oxo, chemistry in the last decade. It is therefore attractive to transfer some of these important and novel results from bioinorganic to environmental chemistry. Although one needs to be very cautious in reinterpreting earlier mechanistic interpretations that are primarily based on OH radical and ‘swimming pool’ chemistry, it appears that ferryl-based as opposed to OH radical-driven processes are in general more important than so far appreciated in environmental science.
Acknowledgements
We are grateful for the financial support of the German Science Foundation (DFG Forschergruppe 763, HaloProc), to our coworkers for their excellent contributions and to our HaloProc colleagues for inspiring discussions.
References
[1] S. R. Taylor, Abundance of chemical elements in the continental crust: a new table. Geochim. Cosmochim. Acta 1964, 28, 1273.| Abundance of chemical elements in the continental crust: a new table.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF2cXkslGntrk%3D&md5=3e25224851e8257822b113989df26ac0CAS |
[2] F. Scheffer, H. P. Blume, G. W. Brümmer, P. Schachtschabel, R. Horn, E. Kandeler, I. Kögel-Knabner, R. Krezschmar, K. Stahr, B. M. Wilke, C. Welp, S. Thiele-Bruhn, Lehrbuch der Bodenkunde 2010 (Spektrum Verlag: Heidelberg).
[3] D. R. Lovley, Dissimilatory FeIII and MnIV reduction. Microbiol. Rev. 1991, 55, 259.
| 1:CAS:528:DyaK3MXltFSjtLY%3D&md5=43dc1620953c864b549fbf2d6ad75097CAS | 1886521PubMed |
[4] W. Stumm, J. J. Morgan, Aquatic Chemistry 1981 (Wiley: New York).
[5] T. R. Khan, C. H. Langford, G. B. Skippen, Complexation and reduction as factors in the link between metal ion concentrations and organic matter in the Indian River. Org. Geochem. 1984, 7, 261.
| Complexation and reduction as factors in the link between metal ion concentrations and organic matter in the Indian River.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXhslSiu7g%3D&md5=58ccee0f83e89bc9e13a5bacea28a8a2CAS |
[6] B. Goodell, J. Jellison, L. Liu, G. Daniel, A. Paszczynski, F. Fekete, S. Krishnamurthy, L. Jun, G. Xu, Low-molecular-weight chelators and phenolic compounds isolated from wood decay fungi and their role in the fungal biodegradation of wood. J. Biotechnol. 1997, 53, 133.
| Low-molecular-weight chelators and phenolic compounds isolated from wood decay fungi and their role in the fungal biodegradation of wood.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXjvVWntrw%3D&md5=71a22e5e001ae23631929cff7193950eCAS |
[7] V. Römheld, H. Marschner, Mechanisms of iron uptake by peanut plants: reduction, chelate splitting, and release of phenolics. Plant Physiol. 1983, 71, 949.
| Mechanisms of iron uptake by peanut plants: reduction, chelate splitting, and release of phenolics.Crossref | GoogleScholarGoogle Scholar | 16662934PubMed |
[8] B. Meunier (Ed.), Metal-Oxo and Metal-Peroxo Species in Catalytic Oxidations 2000, Vol. 97 (Springer Verlag: Berlin).
[9] B. Meunier (Ed.), Biomimetic Oxidations Catalyzed by Transition Metal Complexes 2000 (Imperial College Press: London).
[10] S. P. de Visser, D. Kumar (Eds), Iron-Containing Enzymes: Versatile Catalysts of Hydroxylation Reactions in Nature 2011 (RSC Publishing: Cambridge, UK).
[11] R. J. P. Williams, J. J. R. Fraústo da Silva, The Natural Selection of the Chemical Elements 1997 (Oxford University Press: Oxford, UK).
[12] K. D. Karlin, S. Itoh (Eds), Copper–Oxygen Chemistry 2011 (Wiley: New York).
[13] S. C. Sawant, X. Wu, J. Cho, K.-B. Cho, S. H. Kim, M. S. Seo, Y. M. Lee, M. Kubo, T. Ogura, S. Shaik, W. Nam, Water as an oxygen source: synthesis, characterization, and reactivity studies of a mononuclear non-heme manganese(IV) oxo complex. Angew. Chem. Int. Ed. 2010, 49, 8190.
| Water as an oxygen source: synthesis, characterization, and reactivity studies of a mononuclear non-heme manganese(IV) oxo complex.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlalu7zN&md5=ae6f436cd6b82fe5fd99e0258c03db96CAS |
[14] W. Liu, X. Huang, M. J. Cheng, R. J. Nielsen, W. A. Goddard, J. T. Groves, Oxidative aliphatic C–H fluorination with fluoride ion catalyzed by a manganese porphyrin. Science 2012, 337, 1322.
| Oxidative aliphatic C–H fluorination with fluoride ion catalyzed by a manganese porphyrin.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtlaitL%2FM&md5=0df375bf3402ae353660c06d71023f84CAS | 22984066PubMed |
[15] T. Taguchi, R. Gupta, B. Lassalle-Kaiser, D. W. Boyce, V. K. Yachandra, W. B. Tolman, J. Yano, M. P. Hendrich, A. S. Borovich, Preparation and properties of a monomeric high-spin MnV–oxo complex. J. Am. Chem. Soc. 2012, 134, 1996.
| Preparation and properties of a monomeric high-spin MnV–oxo complex.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38Xks1ahtw%3D%3D&md5=ca06425122fa1ba49aea8491d6cf2217CAS | 22233169PubMed |
[16] D. Leto, R. Ingram, V. W. Daya, T. A. Jackson, Spectroscopic properties and reactivity of a mononuclear oxomanganese(IV) complex. Chem. Commun. 2013, 5378.
| Spectroscopic properties and reactivity of a mononuclear oxomanganese(IV) complex.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXnvVWrs7w%3D&md5=206f0de46779c90b1ab8005dcf5a129dCAS |
[17] H. M. Neu, T. Yang, R. A. Baglia, T. H. Yosca, M. T. Green, M. G. Quesne, S. P. de Visser, D. P. Goldberg, Oxygen-atom transfer reactivity of axially ligated MnV-oxo complexes: evidence for enhanced electrophilic and nucleophilic pathways. J. Am. Chem. Soc. 2014, 136, 13 845.
| Oxygen-atom transfer reactivity of axially ligated MnV-oxo complexes: evidence for enhanced electrophilic and nucleophilic pathways.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhsFyjtb3P&md5=9b2a28d85b9d5ba69c04e5082a513928CAS |
[18] P. Barman, A. K. Vardhaman, B. Martin, S. J. Wörner, C. V. Sastri, P. Comba, Infuence of ligand architecture on oxidation reactions by high-valent non-heme manganese–oxo complexes using water as a source of oxygen. Angew. Chem. Int. Ed. 2015, 54, 2095.
| Infuence of ligand architecture on oxidation reactions by high-valent non-heme manganese–oxo complexes using water as a source of oxygen.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXmsVw%3D&md5=aa737c922570a91b1e6da684f3caf0b4CAS |
[19] T. Nagataki, Y. Tachi, S. Itoh, NiII(TPA) as an efficient catalyst for alkane hydroxylation with m-CPBA. Chem. Commun. 2006, 4016.
| NiII(TPA) as an efficient catalyst for alkane hydroxylation with m-CPBA.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XpvF2mtr4%3D&md5=f57a4f65a4ff097c8872b61801fa77edCAS |
[20] T. Nagataki, K. Ishii, Y. Tachi, S. Itoh, Ligand effects on NiII-catalysed alkane hydroxylation with m-CPBA. Dalton Trans. 2007, 1120.
| Ligand effects on NiII-catalysed alkane hydroxylation with m-CPBA.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXisVKksL4%3D&md5=b0cf364286445810ef8e693fc5a891e3CAS | 17339995PubMed |
[21] M. Sankaralingam, M. Balamurugan, M. Palaniandavar, P. Vadivelu, C. H. Suresh, Nickel(II) complexes of pentadentate N5 ligands as catalysts for alkane hydroxylation by using m-CPBA as oxidant: a combined experimental and computational study. Chemistry 2014, 20, 11 346.
| Nickel(II) complexes of pentadentate N5 ligands as catalysts for alkane hydroxylation by using m-CPBA as oxidant: a combined experimental and computational study.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhtlWmtLrN&md5=5ca6899d34f038436f3ee76fe6920bccCAS |
[22] W. Stumm, B. Sulzberger, The cycling of iron in natural environments: considerations based on laboratory studies of heterogeneous redox processes. Geochim. Cosmochim. Acta 1992, 56, 3233.
| The cycling of iron in natural environments: considerations based on laboratory studies of heterogeneous redox processes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XmtF2gtrk%3D&md5=ba59f863196550869ef3d5afd56e6497CAS |
[23] K. J. Olszyna, J. F. Meagher, E. M. Bailey, Gas-phase, cloud and rain-water measurements of hydrogen peroxide at a high-elevation site. Atmos. Environ. 1988, 22, 1699.
| Gas-phase, cloud and rain-water measurements of hydrogen peroxide at a high-elevation site.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXlvFKhu7g%3D&md5=eef4a89aeea45e51262e4303dc2129beCAS |
[24] K. Yoshizumi, K. Aoki, I. Nouchi, T. Okita, T. Kobayashi, S. K. Amakura, M. Tajima, Measurements of the concentration in rainwater and of the Henry’s Law constant of hydrogen peroxide. Atmos. Environ. 1984, 18, 395.
| Measurements of the concentration in rainwater and of the Henry’s Law constant of hydrogen peroxide.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXitFGiur0%3D&md5=f1da7ef6223472fce06265e0c7992424CAS |
[25] G. L. Kok, Measurements of hydrogen peroxide in rainwater. Atmos. Environ. 1980, 14, 653.
| Measurements of hydrogen peroxide in rainwater.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3cXlslWmt7w%3D&md5=0aaa685d452465d8bf0af6bfee2ff6cfCAS |
[26] Y. Zuo, Y. Deng, Evidence for the production of hydrogen peroxide in rainwater by lightning during thunderstorms. Geochim. Cosmochim. Acta 1999, 63, 3451.
| Evidence for the production of hydrogen peroxide in rainwater by lightning during thunderstorms.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXotVCktr0%3D&md5=5a6cb92faa501faf6cc0b34633825e1eCAS |
[27] Y. Deng, Y. Zuo, Factors affecting the levels of hydrogen peroxide in rainwater. Atmos. Environ. 1999, 33, 1469.
| Factors affecting the levels of hydrogen peroxide in rainwater.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXhsVykt74%3D&md5=8463880e95e5fdd93ad5b220e4754d59CAS |
[28] G. L. Kok, K. R. Darnall, A. M. Winer, J. N. Pitts, B. W. Gay, Ambient air measurements of hydrogen peroxide in the California south coast air basin. Environ. Sci. Technol. 1978, 12, 1077.
| Ambient air measurements of hydrogen peroxide in the California south coast air basin.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE1cXmtF2qur4%3D&md5=ea25ea7acf57decc95d3d24214faba29CAS |
[29] H. Levy, Normal atmosphere: large radical and formaldehyde concentrations predicted. Science 1971, 173, 141.
| Normal atmosphere: large radical and formaldehyde concentrations predicted.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3MXks1Kgu7o%3D&md5=c39fa65b42bf1b39114025de749b3d1aCAS | 17739642PubMed |
[30] R. I. Martinez, J. T. Herron, R. E. Huie, The mechanism of ozone–alkene reactions in the gas phase. A mass spectrometric study of the reactions of eight linear and branched-chain alkenes. J. Am. Chem. Soc. 1981, 103, 3807.
| The mechanism of ozone–alkene reactions in the gas phase. A mass spectrometric study of the reactions of eight linear and branched-chain alkenes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3MXksVWrtbY%3D&md5=e684abc89b2039fa6f2e41d0448cf69bCAS |
[31] W. M. Draper, D. G. Crosby, The photochemical generation of hydrogen peroxide in natural waters. Arch. Environ. Contam. Toxicol. 1983, 12, 121.
| The photochemical generation of hydrogen peroxide in natural waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3sXhs1Kmuro%3D&md5=1a84381acc5ea2dc5f9bacd04ba0daa7CAS |
[32] B. H. Yocis, D. J. Kieber, K. Mopper, Photochemical production of hydrogen peroxide in Antarctic waters. Deep Sea Res. Part I Oceanogr. Res. Pap. 2000, 47, 1077.
| Photochemical production of hydrogen peroxide in Antarctic waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXis1Witbo%3D&md5=270ecbe9c1294832e844fb3c96b8a335CAS |
[33] B. C. Faust, C. Anastasio, J. M. Allen, T. Arakaki, Aqueous-phase photochemical formation of peroxides in authentic cloud and fog waters. J. Geophys. Res., D, Atmospheres 1993, 260, 73.
| 1:CAS:528:DyaK3sXhvFOmtr0%3D&md5=fb4bfba4a81194d5f2b0f3f14bf454fbCAS |
[34] W. J. Cooper, R. G. Zika, R. G. Petasne, J. M. C. Plane, Photochemical formation of hydrogen peroxide in natural waters exposed to sunlight. Environ. Sci. Technol. 1988, 22, 1156.
| Photochemical formation of hydrogen peroxide in natural waters exposed to sunlight.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXltlCmsb0%3D&md5=d607cc53fd87bd907776259808b668f5CAS | 22148607PubMed |
[35] J. Weiss, Elektronenübergangsprozesse im Mechanismus von Oxydations- und Reduktionsreaktionen in Lösungen. Naturwissenschaften 1935, 23, 64.
| Elektronenübergangsprozesse im Mechanismus von Oxydations- und Reduktionsreaktionen in Lösungen.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA2MXjtVaitQ%3D%3D&md5=80f3120671ec41d0bd5b3fc7f2325148CAS |
[36] S. D. Aust, L. A. Morehouse, C. E. Thomas, Role of metals in oxygen radical reactions. J. Free Radic. Biol. Med. 1985, 1, 3.
| Role of metals in oxygen radical reactions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXksFSjurc%3D&md5=7dbe92b05e21d6e48486cff40802c97dCAS | 3013969PubMed |
[37] B. M. Voelker, F. M. M. Morel, B. Sulzberger, Iron redox cycling in surface waters: effects of humic substances and light. Environ. Sci. Technol. 1997, 31, 1004.
| Iron redox cycling in surface waters: effects of humic substances and light.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXhvFGgsbs%3D&md5=4b2b2c15b33a46660a891ef38f1140f9CAS |
[38] M. A. Kessick, J. J. Morgan, Mechanism of autoxidation of manganese in aqueous solution. Environ. Sci. Technol. 1975, 9, 157.
| Mechanism of autoxidation of manganese in aqueous solution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2MXptVGqug%3D%3D&md5=91c03f29f433e384c8ce87e4b6bedcd6CAS |
[39] W. Stumm, G. F. Lee, Oxygenation of ferrous iron. Ind. Eng. Chem. 1961, 53, 143.
| Oxygenation of ferrous iron.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF3MXnslymsQ%3D%3D&md5=67307e4e191305687da8e26aaf953717CAS |
[40] F. J. Millero, The effect of ionic interactions on the oxidation of metals in natural waters. Geochim. Cosmochim. Acta 1985, 49, 547.
| The effect of ionic interactions on the oxidation of metals in natural waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXhtFOjtrs%3D&md5=844811656ee4bc12757621fd0363f42fCAS |
[41] F. J. Millero, Effect of ionic interactions on the oxidation of FeII and CuI in natural waters. Mar. Chem. 1989, 28, 1.
| Effect of ionic interactions on the oxidation of FeII and CuI in natural waters.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXht1Clsb8%3D&md5=a52decd2853c023cfd3b5050d9df91e3CAS |
[42] J. M. Trapp, F. J. Millero, The oxidation of iron(II) with oxygen in NaCl brines. J. Solution Chem. 2007, 36, 1479.
| The oxidation of iron(II) with oxygen in NaCl brines.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtlKmtrrK&md5=6d42539245faeb475957f6105370d298CAS |
[43] R. G. Petasne, R. G. Zika, Fate of superoxide in coastal sea water. Nature 1987, 325, 516.
| Fate of superoxide in coastal sea water.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2sXht1Gms70%3D&md5=3f303e2564bb613fc6557e3ed3646116CAS |
[44] J. D. Rush, B. H. J. Bielski, Pulse radiolytic studies of the reaction of perhydroxyl/superoxide O2– with iron(II)/iron(III) ions. The reactivity of HO2/O2– with ferric ions and its implication on the occurrence of the Haber–Weiss reaction. J. Phys. Chem. 1985, 89, 5062.
| Pulse radiolytic studies of the reaction of perhydroxyl/superoxide O2– with iron(II)/iron(III) ions. The reactivity of HO2/O2– with ferric ions and its implication on the occurrence of the Haber–Weiss reaction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXls1Krur4%3D&md5=18359111429b73774965af17294705e1CAS |
[45] J. W. Koenigs, Production of hydrogen peroxide by wood-rotting fungi in wood and its correlation with weight loss, depolymerization, and pH changes. Arch. Microbiol. 1974, 99, 129.
| Production of hydrogen peroxide by wood-rotting fungi in wood and its correlation with weight loss, depolymerization, and pH changes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2MXivVegtQ%3D%3D&md5=3db68fb75a8e070f74f6806baf30747dCAS |
[46] F. C. Küpper, D. G. Müller, A. F. Peters, B. Kloareg, P. Potin, Oligoalginate recognition and oxidative burst play a key role in natural and induced resistance of sporophytes of laminariales. J. Chem. Ecol. 2002, 28, 2057.
| Oligoalginate recognition and oxidative burst play a key role in natural and induced resistance of sporophytes of laminariales.Crossref | GoogleScholarGoogle Scholar | 12474900PubMed |
[47] D. L. Pardieck, E. J. Bouwer, A. T. Stone, Hydrogen peroxide use to increase oxidant capacity for in situ bioremediation of contaminated soils and aquifers: a review. J. Contam. Hydrol. 1992, 9, 221.
| Hydrogen peroxide use to increase oxidant capacity for in situ bioremediation of contaminated soils and aquifers: a review.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XhvVSgsrw%3D&md5=a839d5f96bd00dd6b543f48b5bb290ddCAS |
[48] W. J. M. Bowen, Environmental Chemistry of the Elements 1979 (Academic Press: London).
[49] M. S. Matheson, W. A. Mulac, J. L. Weeks, J. Rabani, The pulse radiolysis of deaerated aqueous bromide solutions. J. Phys. Chem. 1966, 70, 2092.
| The pulse radiolysis of deaerated aqueous bromide solutions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF28XktlGns7Y%3D&md5=0d7e51ea393a764e7ef76f6139985a57CAS |
[50] M. Anbar, J. K. Thomas, Pulse radiolysis studies of aqueous sodium chloride solutions. J. Phys. Chem. 1964, 68, 3829.
| Pulse radiolysis studies of aqueous sodium chloride solutions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF2MXhtlWgug%3D%3D&md5=53f5f4222c1e6b36d03989a2cbf9505eCAS |
[51] G. G. Jayson, B. J. Parsons, A. J. Swallow, Some simple, highly reactive, inorganic chlorine derivatives in aqueous solution. Their formation using pulses of radiation and their role in the mechanism of the Fricke dosimeter. J. Chem. Soc., Faraday Trans. I 1973, 69, 1597.
| Some simple, highly reactive, inorganic chlorine derivatives in aqueous solution. Their formation using pulses of radiation and their role in the mechanism of the Fricke dosimeter.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE3sXlsV2qsLg%3D&md5=9a1479392664a2baa7f1b09420495eb9CAS |
[52] J. Weiss, Reaction mechanism of the enzymes catalase and peroxidase in the light of the theory of chain reactions. J. Phys. Chem. 1937, 41, 1107.
| Reaction mechanism of the enzymes catalase and peroxidase in the light of the theory of chain reactions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA1cXjslWj&md5=f2204f6a722fb17c820ac22297ee8819CAS |
[53] M. G. Evans, N. S. Hush, N. Uri, The energetics of reactions involving hydrogen peroxide, its radicals, and its ions. Q. Rev. Chem. Soc. 1952, 6, 186.
| The energetics of reactions involving hydrogen peroxide, its radicals, and its ions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG38XltFWruw%3D%3D&md5=2f6ae251618855701e5e79adb4c229a4CAS |
[54] M. M. Abu-Omar, A. Loaiza, N. Hontzeas, Reaction mechanisms of mononuclear non-heme iron oxygenases. Chem. Rev. 2005, 105, 2227.
| Reaction mechanisms of mononuclear non-heme iron oxygenases.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXivV2lu7s%3D&md5=0d296d8256108f6d02fc5ec6551aad75CAS | 15941213PubMed |
[55] C. Krebs, D. Galonić Fujimori, C. T. Walsh, J. M. Bollinger, Non-heme FeIV–oxo intermediates. Acc. Chem. Res. 2007, 40, 484.
| Non-heme FeIV–oxo intermediates.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmtVKktr8%3D&md5=35754357f3193f874bc088652a9ecba2CAS | 17542550PubMed |
[56] A. R. McDonald, L. Que, High-valent non-heme iron-oxo complexes: synthesis, structure, and spectroscopy. Coord. Chem. Rev. 2013, 257, 414.
| High-valent non-heme iron-oxo complexes: synthesis, structure, and spectroscopy.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhtlOls77E&md5=b2bc3237d9a5398c2b5eb27a97473a92CAS |
[57] P. Comba, in Molecular Catalysis (Eds. L. H. Gade, P. Hofmann) 2014, pp. 123–145 (Wiley-VCH: Weinheim).
[58] X. Y. Yu, J. R. Barker, Hydrogen peroxide photolysis in acidic aqueous solutions containing chloride ions. I. Chemical mechanism. J. Phys. Chem. A 2003, 107, 1313.
| Hydrogen peroxide photolysis in acidic aqueous solutions containing chloride ions. I. Chemical mechanism.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXpvVaitQ%3D%3D&md5=4351ab862bb7a84646f77f33db06db91CAS |
[59] A. J. Fudge, K. W. Sykes, 25. The reaction between ferric and iodide ions. Part I. Kinetics and mechanism. J. Chem. Soc. 1952, 1952, 119.
[60] C. L. Copper, E. Koubek, A kinetics experiment to demonstrate the role of a catalyst in a chemical reaction: a versatile exercise for general or physical chemistry students. J. Chem. Educ. 1998, 75, 87.
| A kinetics experiment to demonstrate the role of a catalyst in a chemical reaction: a versatile exercise for general or physical chemistry students.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXhtFyitw%3D%3D&md5=c2e3af811368e4573c7231471c2c203fCAS |
[61] H. Landolt, Ueber die Zeitdauer der Reaction zwischen Jodsäure und schwefliger Säure. Ber. Dtsch. Chem. Ges. 1886, 19, 1317.
| Ueber die Zeitdauer der Reaction zwischen Jodsäure und schwefliger Säure.Crossref | GoogleScholarGoogle Scholar |
[62] H. Landolt, Ueber die Zeitdauer der Reaction zwischen Jodsäure und schwefliger Säure. Ber. Dtsch. Chem Ges. 1887, 20, 745.
| Ueber die Zeitdauer der Reaction zwischen Jodsäure und schwefliger Säure.Crossref | GoogleScholarGoogle Scholar |
[63] F. C. Küpper, L. J. Carpenter, G. B. McFiggans, C. J. Palmer, T. J. Waite, E.-M. Boneberg, S. Woitsch, M. Weiller, R. R. Abela, D. Grolimund, P. Potin, A. Butler, G. W. Luther, P. M. Kroneck, W. Meyer-Laucke, M. C. Feiters, Iodide accumulation provides kelp with an inorganic antioxidant impacting atmospheric chemistry. Proc. Natl. Acad. Sci. USA 2008, 105, 6954.
| Iodide accumulation provides kelp with an inorganic antioxidant impacting atmospheric chemistry.Crossref | GoogleScholarGoogle Scholar | 18458346PubMed |
[64] V. M. Solyanikov, E. T. Denisov, Formation of radicals in the reaction of hydrogen peroxide with the bromide anion. Bull. Academy Sci. USSR. Chem Sci. 1968, 17, 1415.
[65] V. I. Skudaev, A. B. Solomonov, A. I. Morozovskii, N. A. Isakov, Oxidation of hydrogen chloride with hydrogen peroxide in aqueous solution. Russ. J. Appl. Chem. 2008, 81, 14.
| Oxidation of hydrogen chloride with hydrogen peroxide in aqueous solution.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXislWgsL8%3D&md5=66a08f93bf598565f3530c683f6e83bfCAS |
[66] R. E. Connick, The interaction of hydrogen peroxide and hypochlorous acid in acidic solutions containing chloride ion. J. Am. Chem. Soc. 1947, 69, 1509.
| The interaction of hydrogen peroxide and hypochlorous acid in acidic solutions containing chloride ion.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaH2sXis12ltA%3D%3D&md5=7aedb95b79c3824af0c18f1db8207b6cCAS |
[67] U. Von Gunten, Y. Oliveras, Kinetics of the reaction between hydrogen peroxide and hypobromous acid: implication on water treatment and natural systems. Water Res. 1997, 31, 900.
| Kinetics of the reaction between hydrogen peroxide and hypobromous acid: implication on water treatment and natural systems.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXitVykurc%3D&md5=04e984ba33966c3a1d7aa4c2a4d71aeaCAS |
[68] W. C. Bray, H. A. Liebhafsky, Reactions involving hydrogen peroxide, iodine and iodate ion. I. Introduction. J. Am. Chem. Soc. 1931, 53, 38.
| Reactions involving hydrogen peroxide, iodine and iodate ion. I. Introduction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA3MXhtlaqtQ%3D%3D&md5=0e7a9730d1ba725c906908ef8af0831eCAS |
[69] M. G. Peard, C. F. Cullis, A periodic chemical reaction. The reaction between hydrogen peroxide and iodic acid. Trans. Faraday Soc. 1951, 47, 616.
| A periodic chemical reaction. The reaction between hydrogen peroxide and iodic acid.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG3MXlvV2jsA%3D%3D&md5=2110efc63e381971d035c2d6ac71e1e9CAS |
[70] M. Barcellos da Rosa, Untersuchungen heterogener troposphärenrelevanter Reaktionen von Schwefel- und Halogenverbindungen 2003, Ph.D. thesis, University Bayreuth.
[71] H. J. H. Fenton, LXXIII. Oxidation of tartaric acid in presence of iron. J. Chem. Soc. Trans. 1894, 65, 899.
| LXXIII. Oxidation of tartaric acid in presence of iron.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaD28XmtlCnsQ%3D%3D&md5=df6c09b18b8ca208e8aafc79b1a660efCAS |
[72] C. Walling, Fenton’s reagent revisited. Acc. Chem. Res. 1975, 8, 125.
| Fenton’s reagent revisited.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2MXhs1Sksro%3D&md5=7de1be590ea780d04e97a0f4f03cb8c3CAS |
[73] C. Walling, Intermediates in the reactions of Fenton-type reagents. Acc. Chem. Res. 1998, 31, 155.
| Intermediates in the reactions of Fenton-type reagents.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXitVejtLo%3D&md5=1ccd7b7e59d5637fb75100c8dd2a2eb3CAS |
[74] F. Haber, J. Weiss, Über die Katalyse des Hydroperoxydes. Naturwissenschaften 1932, 20, 948.
| Über die Katalyse des Hydroperoxydes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA3sXhslWktg%3D%3D&md5=b6f2b23820e30169f201002f916825cfCAS |
[75] W. C. Bray, M. H. Gorin, Ferryl ion, a compound of tetravalent iron. J. Am. Chem. Soc. 1932, 54, 2124.
| Ferryl ion, a compound of tetravalent iron.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA38Xjtlaquw%3D%3D&md5=5bcd1ea463dae0b13d274585bc2024e1CAS |
[76] F. Buda, B. Ensing, M. C. M. Gribnau, E. J. Baerends, DFT study of the active intermediate in the Fenton reaction. Chem. Eur. J. 2001, 7, 2775.
| DFT study of the active intermediate in the Fenton reaction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXltFantrg%3D&md5=bb52a2f650b3b4c8d734f6739ef228e8CAS | 11486953PubMed |
[77] C. A. Grapperhaus, B. Miener, E. Bill, T. Weyhermüller, K. Wieghardt, Mononuclear (nitrido)iron(V) and (oxo)iron(IV) complexes via photolysis of [(cyclam-acetato)FeIII(N3)]+ and ozonolysis of [(cyclam-acetato)FeIII(O3SCF3)]+ in water/acetone mixtures. Inorg. Chem. 2000, 39, 5306.
| Mononuclear (nitrido)iron(V) and (oxo)iron(IV) complexes via photolysis of [(cyclam-acetato)FeIII(N3)]+ and ozonolysis of [(cyclam-acetato)FeIII(O3SCF3)]+ in water/acetone mixtures.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXnsFGltLc%3D&md5=5603cb8cf2e40fd92af9a34a59627607CAS | 11187471PubMed |
[78] J. U. Rohde, J. H. In, M. H. Lim, W. W. Brennesse, M. R. Bukowski, A. Stubna, E. Münck, W. Nam, L. Que, Crystallographic and spectroscopic characterization of a non-heme Fe(IV)=O complex. Science 2003, 299, 1037.
| Crystallographic and spectroscopic characterization of a non-heme Fe(IV)=O complex.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXhtFarsLs%3D&md5=0f0a7b632e06e08c7359d5b49753e730CAS | 12586936PubMed |
[79] D. A. Proshlyakov, T. F. Henshaw, G. R. Monterosso, M. J. Ryle, R. P. Hausinger, Direct detection of oxygen intermediates in the non-heme Fe enzyme taurine/alpha-ketoglutarate dioxygenase. J. Am. Chem. Soc. 2004, 126, 1022.
| Direct detection of oxygen intermediates in the non-heme Fe enzyme taurine/alpha-ketoglutarate dioxygenase.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXksFOmug%3D%3D&md5=3a2819b30122340a4c353ad83e105238CAS | 14746461PubMed |
[80] P. J. Riggs-Gelasco, J. C. Price, R. B. Guyer, J. H. Brehm, E. W. Barr, J.-M. Bollinger, C. Krebs, EXAFS spectroscopic evidence for an FeO unit in the FeIV intermediate observed during oxygen activation by taurine:α-ketoglutarate dioxygenase. J. Am. Chem. Soc. 2004, 126, 8108.
| EXAFS spectroscopic evidence for an FeO unit in the FeIV intermediate observed during oxygen activation by taurine:α-ketoglutarate dioxygenase.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXkslWhsb0%3D&md5=185bf5414111b64f5a3889d496151b3dCAS | 15225039PubMed |
[81] J. C. Price, E. W. Barr, B. Tirupati, J. M. Bollinger, C. Krebs, The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin FeIV complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli. Biochemistry 2003, 42, 7497.
| The first direct characterization of a high-valent iron intermediate in the reaction of an α-ketoglutarate-dependent dioxygenase: a high-spin FeIV complex in taurine/α-ketoglutarate dioxygenase (TauD) from Escherichia coli.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXktVCjsL0%3D&md5=66578c1fbd006e5afb26ba7b238b026aCAS | 12809506PubMed |
[82] J. C. Price, E. W. Barr, T. E. Glass, C. Krebs, J. M. Bollinger, Evidence for hydrogen abstraction from C1 of taurine by the high-spin FeIV intermediate detected during oxygen activation by taurine:α-ketoglutarate dioxygenase (TauD). J. Am. Chem. Soc. 2003, 125, 13 008.
| Evidence for hydrogen abstraction from C1 of taurine by the high-spin FeIV intermediate detected during oxygen activation by taurine:α-ketoglutarate dioxygenase (TauD).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXnslagt7w%3D&md5=8c72d8a8e153cbe35391ce29cdafa843CAS |
[83] O. Pestovsky, S. Stoian, E. L. Bominaar, X. Shan, E. L. Q. Münck, E. Münck, L. Que, Aqueous FeIV=O: spectroscopic identification and oxo-group exchange. Angew. Chem. Int. Ed. 2005, 44, 6871.
| Aqueous FeIV=O: spectroscopic identification and oxo-group exchange.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1amu7fL&md5=a44afacf5587e1c3991be5ff5dee1341CAS |
[84] J. Bautz, P. Comba, C. Lopez de Laorden, M. Menzel, G. Rajaraman, Biomimetic high-valent non-heme iron oxidants for the cis-dihydroxylation and epoxidation of olefins. Angew. Chem. Int. Ed. 2007, 46, 8067.
| Biomimetic high-valent non-heme iron oxidants for the cis-dihydroxylation and epoxidation of olefins.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1KnsrvM&md5=1bb5ac1a19464992a42fd5676b01a891CAS |
[85] J. England, M. Martinho, E. R. Farquhar, J. R. Frisch, E. L. Bominaar, E. Münck, L. Que, A synthetic high-spin oxoiron(IV) complex: generation, spectroscopic characterization, and reactivity. Angew. Chem. Int. Ed. 2009, 48, 3622.
| A synthetic high-spin oxoiron(IV) complex: generation, spectroscopic characterization, and reactivity.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtVKgsbw%3D&md5=b163dcd676363ab13dda2a01679f5791CAS |
[86] D. C. Lacy, R. Gupta, K. L. Stone, J. Greaves, J. W. Ziller, M. P. Hendrich, A. S. Borovik, Formation, structure, and EPR detection of a high-spin FeIV–oxo species derived from either an FeIII–oxo or FeIII–OH complex. J. Am. Chem. Soc. 2010, 132, 12 188.
| Formation, structure, and EPR detection of a high-spin FeIV–oxo species derived from either an FeIII–oxo or FeIII–OH complex.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtVSmsLfI&md5=56b201668b85a8012b56586c1db127c9CAS |
[87] J. P. Bigi, W. H. Harman, B. Lassalle-Kaiser, D. M. Robles, T. A. Stich, J. B. Yano, D. Britt, C. J. Chang, A high-spin iron(IV)-oxo complex supported by a trigonal non-heme pyrrolide platform. J. Am. Chem. Soc. 2012, 134, 1536.
| A high-spin iron(IV)-oxo complex supported by a trigonal non-heme pyrrolide platform.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XivVaqsg%3D%3D&md5=a287b86e7172ce8bbab39b7bf128513bCAS | 22214221PubMed |
[88] J. Bautz, M. R. Bukowski, M. Kerscher, A. Stubna, P. Comba, A. Lienke, E. Münck, L. Que, Formation of an aqueous oxoiron(IV) complex at pH 2–6 from a non-heme iron(II) complex and H2O2. Angew. Chem. Int. Ed. 2006, 45, 5681.
| Formation of an aqueous oxoiron(IV) complex at pH 2–6 from a non-heme iron(II) complex and H2O2.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XpsVWnurg%3D&md5=57d483ac565d2247d530a3f07a04598aCAS |
[89] P. Comba, S. Wunderlich, Iron-catalyzed halogenation of alkanes: modeling of non-heme halogenases by experiment and DFT calculations. Chemistry 2010, 16, 7293.
| Iron-catalyzed halogenation of alkanes: modeling of non-heme halogenases by experiment and DFT calculations.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXotVert70%3D&md5=8354c37cca40fef591eeb0db6bb4c4a8CAS | 20458709PubMed |
[90] O. Planas, M. Clémancey, J.-M. Latour, A. Company, M. Costas, Structural modeling of iron halogenases: synthesis and reactivity of halide–iron(IV)–oxo compounds. Chem. Commun. 2014, 10 887.
| Structural modeling of iron halogenases: synthesis and reactivity of halide–iron(IV)–oxo compounds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhtF2ktrzJ&md5=0d263728e79de441ce310298cfaafc50CAS |
[91] P. Comba, Y. M. Lee, W. Nam, A. Waleska, Catalytic oxidation of alkanes by iron bispidine complexes and dioxygen: oxygen activation versus autoxidation. Chem. Commun. 2014, 50, 412.. [Special Web Themed Issue ‘Biological Oxidation Reactions’]
| 1:CAS:528:DC%2BC3sXhvVyqs73J&md5=4acbfb67297d23400a9d2fc1e97d8638CAS |
[92] P. Comba, M. Maurer, P. Vadivelu, Oxidation of cyclohexane by high-valent iron bispidine complexes: tetradentate versus pentadentate ligands. Inorg. Chem. 2009, 48, 10 389.
| Oxidation of cyclohexane by high-valent iron bispidine complexes: tetradentate versus pentadentate ligands.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1CgtrzF&md5=1647bf27a9a99bbd2196bfe1b0dd92b3CAS |
[93] W. G. Barb, J. H. Baxendale, P. George, K. R. Hargrave, Reactions of ferrous and ferric ions with hydrogen peroxide. Part II. The ferric ion reaction. Trans. Faraday Soc. 1951, 47, 591.
| Reactions of ferrous and ferric ions with hydrogen peroxide. Part II. The ferric ion reaction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaG3MXlvV2jsw%3D%3D&md5=20eb0500a7908184f05a63111c810268CAS |
[94] S. M. Kim, A. Vogelpohl, Degradation of organic pollutants by the photo-Fenton-process. Chem. Eng. Technol. 1998, 21, 187.
| Degradation of organic pollutants by the photo-Fenton-process.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXhs1Citbo%3D&md5=0f592a4fc96c2cdbef271ec3fece90a3CAS |
[95] M. Strlič, J. Kolar, V.-S. Šelih, D. Kočar, B. Pihlar, A comparative study of several transition metals in Fenton-like reaction systems at circumneutral pH. Acta Chim. Slov. 2003, 50, 619.
[96] G. R. A. Johnson, N. B. Nazhat, R. A. Saadalla-Nazhat, Reaction of the aquocopper(I) ion with hydrogen peroxide: evidence against hydroxyl free radical formation. J. Chem. Soc. Chem. Commun. 1985, 407.
| Reaction of the aquocopper(I) ion with hydrogen peroxide: evidence against hydroxyl free radical formation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXksVensLw%3D&md5=345a28f3054620241f2e2ea38d0ff73cCAS |
[97] A. N. Pham, G. Xing, C. J. Miller, T. D. Waite, Fenton-like copper redox chemistry revisited: hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production. J. Catal. 2013, 301, 54.
| Fenton-like copper redox chemistry revisited: hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXlslWms7k%3D&md5=19440afa6f2d20a7abea7f2e1aba2465CAS |
[98] A. Ansari, A. Kaushik, G. Rajaraman, Mechanistic insights on the ortho-hydroxylation of aromatic compounds by non-heme iron complex: a computational case study on the comparative oxidative ability of ferric-hydroperoxo and high-valent FeIV=O and FeV=O intermediates. J. Am. Chem. Soc. 2013, 135, 4235.
| Mechanistic insights on the ortho-hydroxylation of aromatic compounds by non-heme iron complex: a computational case study on the comparative oxidative ability of ferric-hydroperoxo and high-valent FeIV=O and FeV=O intermediates.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXhvVyms7g%3D&md5=c129252a4c7b0887ea948b3c6df3493eCAS | 23373840PubMed |
[99] S. Goldstein, D. Meyerstein, G. Czapski, The Fenton reagents. Free Radic. Biol. Med. 1993, 15, 435.
| The Fenton reagents.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXhvFWkt7w%3D&md5=1e93a21d9d6c8283bdeb4afb7a53b000CAS | 8225025PubMed |
[100] S. Goldstein, D. Meyerstein, Comments on the mechanism of the ‘Fenton-like’ reaction. Acc. Chem. Res. 1999, 32, 547.
| Comments on the mechanism of the ‘Fenton-like’ reaction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXhvVeisrY%3D&md5=cb25558b4358d1d2d58d6b59f8480b08CAS |
[101] P. Comba, G. Rajaraman, H. Rohwer, A density functional theory study of the reaction of the biomimetic iron(II) complex of a tetradentate bispidine ligand with H2O2. Inorg. Chem. 2007, 46, 3826.
| A density functional theory study of the reaction of the biomimetic iron(II) complex of a tetradentate bispidine ligand with H2O2.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXjvFyju78%3D&md5=1994b512575ac7684067c7d59c3bb2a4CAS | 17425302PubMed |
[102] I. Yamazaki, L. H. Piette, EPR spin-trapping study on the oxidizing species formed in the reaction of the ferrous ion with hydrogen peroxide. J. Am. Chem. Soc. 1991, 113, 7588.
| EPR spin-trapping study on the oxidizing species formed in the reaction of the ferrous ion with hydrogen peroxide.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXmsl2iur0%3D&md5=52d2d0524ade06e10f55c21b0a22aed8CAS |
[103] F. Gozzo, Radical and non-radical chemistry of the Fenton-like systems in the presence of organic substrates. J. Mol. Catal. Chem. 2001, 171, 1.
| Radical and non-radical chemistry of the Fenton-like systems in the presence of organic substrates.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXktF2nt7w%3D&md5=1551a132ecea461bbe5675ddfc1eca8cCAS |
[104] J. D. Lipscomb, A. M. Orville (Eds) Mechanistic Aspects of Dihydroxybenzoate Dioxygenases 1992, Vol. 28 (Marcel Dekker: New York).
[105] J. Que Jr, Iron carriers and iron proteins, in Iron Carriers and Iron Proteins (Ed. T. M. Loehr) 1989, pp. 243–524 (VCH: New York).
[106] H. J. Krüger, Iron-containing models of catechol dioxygenases, in Biomimetic Oxidations Catalyzed by Transition Metal Complexes (Ed. B. Meunier) 2000, pp. 363–413 (Imperial College Press: Oxford).
[107] E. I. Solomon, M. Y. M. Pau, R. K. Hocking, Nature of the catecholate–Fe(III) bond: high affinity binding and substrate activation in bioinorganic chemistry, in Computational Inorganic and Bioinorganic Chemistry (Eds E. I. Solomon, R. A. Scott, R. B. King) 2009, pp. 241–253 (Wiley: New York).
[108] M. G. Weller, U. Weser, Fe(NTA)-catalyzed dioxygenation of 4-t-butyl catechol, the mechanism of non-heme ferric dioxygenases. Inorg. Chim. Acta 1985, 107, 243.
| Fe(NTA)-catalyzed dioxygenation of 4-t-butyl catechol, the mechanism of non-heme ferric dioxygenases.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL28XislCh&md5=986c9e7edf17b6cf9d070ea7a5d20e16CAS |
[109] W. O. Koch, H. J. Krüger, A highly reactive and catalytically active model system for intradiol-cleaving catechol dioxygenases: structure and reactivity of iron(III) catecholate complexes of N,N′-dimethyl-2,11-diaza[3.3](2,6)pyridinophane. Angew. Chem. Int. Ed. Engl. 1996, 34, 2671.
| A highly reactive and catalytically active model system for intradiol-cleaving catechol dioxygenases: structure and reactivity of iron(III) catecholate complexes of N,N′-dimethyl-2,11-diaza[3.3](2,6)pyridinophane.Crossref | GoogleScholarGoogle Scholar |
[110] M. Duda, M. Pascal, B. Krebs, A highly reactive functional model for catechol 1,2-dioxygenase:reactivity studies of iron(III) catecholate complexes of bis[(2-pyridyl)methyl][(1-methylimidazol-2-yl)methyl]amine. Chem. Commun. 1997, 835.
| A highly reactive functional model for catechol 1,2-dioxygenase:reactivity studies of iron(III) catecholate complexes of bis[(2-pyridyl)methyl][(1-methylimidazol-2-yl)methyl]amine.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXjtlGks78%3D&md5=9d87b07690559b67e24003f7b1f1358fCAS |
[111] P. Mialane, L. Tchertanov, F. Banse, J. Sainton, J. J. Girerd, Aminopyridine iron catecholate complexes as models for intradiol catechol dioxygenases. Synthesis, structure, reactivity, and spectroscopic studies. Inorg. Chem. 2000, 39, 2440.
| Aminopyridine iron catecholate complexes as models for intradiol catechol dioxygenases. Synthesis, structure, reactivity, and spectroscopic studies.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXjt1WktLk%3D&md5=e1e53e2c00d1a96a6532dc6a2becc554CAS | 11196993PubMed |
[112] R. Mayilmurugan, K. Visvaganesan, E. Suresh, M. Palaniandavar, Complexes of tripodal monophenolate ligands as models for non-heme catechol dioxygenase enzymes: correlation of dioxygenase activity with ligand stereoelectronic properties. Inorg. Chem. 2009, 48, 8771.
| Complexes of tripodal monophenolate ligands as models for non-heme catechol dioxygenase enzymes: correlation of dioxygenase activity with ligand stereoelectronic properties.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVaitr%2FO&md5=6e876ca12096541269e038e4ee148963CAS | 19694480PubMed |
[113] P. Comba, H. Wadepohl, S. Wunderlich, Oxidation versus dioxygenation of catechol: the iron–bispidine system. Eur. J. Inorg. Chem. 2011, 5242.
| Oxidation versus dioxygenation of catechol: the iron–bispidine system.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlyrsbbK&md5=77f65cde687f991a8d43df541c5ca7f0CAS |
[114] W. Nam, High-valent iron(IV)–oxo complexes of heme and non-heme ligands in oxygenation reactions. Acc. Chem. Res. 2007, 40, 522.
| High-valent iron(IV)–oxo complexes of heme and non-heme ligands in oxygenation reactions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXkslOlsbc%3D&md5=77027c3f561ed6dd57179a378574d021CAS | 17469792PubMed |
[115] Z. Codola, J. Lloret-Fillol, M. Costas, Aminopyridine iron and manganese complexes as molecular catalysts for challenging oxidative transformations. Prog. Inorg. Chem. 2014, 59, 447.
| Aminopyridine iron and manganese complexes as molecular catalysts for challenging oxidative transformations.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2MXosFGnurw%3D&md5=4d0c06a24255b3519279314943f027aeCAS |
[116] M. Jaccob, P. Comba, M. Maurer, P. Vadivelu, P. Venuvanalingam, A combined experimental and computational study on the sulfoxidation by high-valent iron bispidine complexes. Dalton Trans. 2011, 11 276.
| A combined experimental and computational study on the sulfoxidation by high-valent iron bispidine complexes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXhtlejt7jP&md5=9b4213d047f9f3fca3a47b4707f60a50CAS |
[117] P. Comba, S. Kuwata, G. Linti, H. Pritzkow, M. Tarnai, H. Wadepohl, CH activation with cobalt complexes of tetradentate bispidine ligands. Chem. Commun. 2006, 2074.
| CH activation with cobalt complexes of tetradentate bispidine ligands.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XktlWltLc%3D&md5=899010948197c67b637814eebe2e3c95CAS |
[118] F. Althoff, K. Benzing, P. Comba, C. McRoberts, D. R. Boyd, S. Greiner, F. Keppler, Abiotic methanogenesis from organosulphur compounds under ambient conditions. Nat. Commun. 2014, 5, 4205.
| Abiotic methanogenesis from organosulphur compounds under ambient conditions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXitVWgtrzI&md5=61ad5b45fd8ab1c4c15b034ca8554f6bCAS | 24957135PubMed |
[119] M. Bukowski, P. Comba, C. Limberg, M. Merz, L. Que, T. Wistuba, Bispidine ligand effects on iron/hydrogen peroxide chemistry. Angew. Chem. Int. Ed. 2004, 43, 1283.
| Bispidine ligand effects on iron/hydrogen peroxide chemistry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXitlKgtLs%3D&md5=a65b5ca30eb95a131962ace4c1abf962CAS |
[120] P. Comba, M. Maurer, P. Vadivelu, Oxidation of cyclohexane by a high-valent iron bispidine complex: a combined experimental and computational mechanistic study. J. Phys. Chem. A 2008, 112, 13 028.
| Oxidation of cyclohexane by a high-valent iron bispidine complex: a combined experimental and computational mechanistic study.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtFChsbfM&md5=75dc1c5e9141f61560ab0ce55f5be32cCAS |
[121] M. R. Bukowski, P. Comba, A. Lienke, C. Limberg, C. L. de Laorden, R. Mas-Balleste, M. Merz, L. Que, Catalytic epoxidation and 1,2-dihydroxylation of olefins with bispidine-iron(II)/H2O2 systems. Angew. Chem. Int. Ed. 2006, 45, 3446.
| Catalytic epoxidation and 1,2-dihydroxylation of olefins with bispidine-iron(II)/H2O2 systems.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XlsVKjsLw%3D&md5=9a8a68eccbca74835b5555dc7a9648f1CAS |
[122] D. E. Van Sickle, F. R. Mayo, R. M. Arluck, Liquid-phase oxidations of cyclic alkenes. J. Am. Chem. Soc. 1965, 87, 4824.
| Liquid-phase oxidations of cyclic alkenes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaF2MXkvFOrsLs%3D&md5=e4b1bab9675970f72a485c6a3f918cc3CAS |
[123] P. Comba, S. Fukuzumi, H. Kotani, S. Wunderlich, Electron-transfer properties of an efficient non-heme iron oxidation catalyst with a tetradentate bispidine ligand. Angew. Chem. Int. Ed. 2010, 49, 2622.
| Electron-transfer properties of an efficient non-heme iron oxidation catalyst with a tetradentate bispidine ligand.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXktVajsbs%3D&md5=0ac93e493af4869d0522172a4213bf8dCAS |
[124] D. Wang, K. Ray, M. J. Collins, E. R. Farquhar, J. R. Frisch, L. Gómez, T. A. Jackson, M. Kerscher, A. Waleska, P. Comba, M. Costas, L. Que, Non-heme oxoiron(IV) complexes of pentadentate N5 ligands: spectroscopy, electrochemistry, and oxidative reactivity. Chem. Sci. 2013, 4, 282.
| Non-heme oxoiron(IV) complexes of pentadentate N5 ligands: spectroscopy, electrochemistry, and oxidative reactivity.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhslKkurzL&md5=82140f87b466035973b355837d754107CAS | 23227304PubMed |
[125] G. Roelfes, M. Lubben, R. Hage, L. Que, B. L. Feringa, Catalytic oxidation with a non-heme iron complex that generates a low-spin FeIIIOOH intermediate. Chemistry 2000, 6, 2152.
| Catalytic oxidation with a non-heme iron complex that generates a low-spin FeIIIOOH intermediate.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXkvVWgs7c%3D&md5=4a4a27caac600671d97e8df3e2c8e069CAS | 10926220PubMed |
[126] K. Kim, K. Chen, J. Kim, J. Que, Stereospecific alkane hydroxylation with H2O2 catalyzed by an iron(II)–tris(2-pyridylmethyl)amine complex. J. Am. Chem. Soc. 1997, 119, 5964.
| Stereospecific alkane hydroxylation with H2O2 catalyzed by an iron(II)–tris(2-pyridylmethyl)amine complex.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXktVejsLs%3D&md5=08ef661d75ab0744ff5ab3317c840432CAS |
[127] K. Chen, L. Que, Evidence for the participation of a high-valent iron–oxo species in stereospecific alkane hydroxylation by a non-heme iron catalyst. Chem. Commun. 1999, 1375.
| Evidence for the participation of a high-valent iron–oxo species in stereospecific alkane hydroxylation by a non-heme iron catalyst.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXksVOis7k%3D&md5=837063811476f5d9a097d022c81f416aCAS |
[128] T. A. Jackson, L. Que, Jr, Structural and functional models for oxygen-activating nonheme iron enzymes, in Concepts and Models in Bioinorganic Chemistry (Eds H.-B. Kraatz, N. Metzler-Nolte) 2006, pp. 260–286 (Wiley-VCH: Weinheim).
[129] K. Chen, M. Costas, L. Que, Spin-state tuning of non-heme iron-catalyzed hydrocarbon oxidations: participation of FeIII-OOH and FeV=O intermediates. J. Chem. Soc., Dalton Trans. 2002, 672.
| Spin-state tuning of non-heme iron-catalyzed hydrocarbon oxidations: participation of FeIII-OOH and FeV=O intermediates.Crossref | GoogleScholarGoogle Scholar |
[130] J. T. Groves, G. A. McClusky, Aliphatic hydroxylation via oxygen rebound. Oxygen transfer catalyzed by iron. J. Am. Chem. Soc. 1976, 98, 859.
| Aliphatic hydroxylation via oxygen rebound. Oxygen transfer catalyzed by iron.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE28XovFaluw%3D%3D&md5=5bd4b7304e5cd5bb27021c04f1949168CAS |
[131] R. M. Burger, Cleavage of nucleic acids by bleomycin. Chem. Rev. 1998, 98, 1153.
| Cleavage of nucleic acids by bleomycin.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXis12htrc%3D&md5=e8f283bbae99e43555d70183227abbdfCAS | 11848928PubMed |
[132] J. Stubbe, J. W. Kozarich, W. Wu, D. E. Vanderwall, Bleomycins: a structural model for specifity, binding, and dubble strand cleavade. Acc. Chem. Res. 1996, 29, 322.
| Bleomycins: a structural model for specifity, binding, and dubble strand cleavade.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28Xjs1GktLY%3D&md5=90445fe5323c718fe959024f5e9976aeCAS |
[133] E. Neyens, J. Baeyens, A review of classic Fenton’s peroxidation as an advanced oxidation technique. J. Hazard. Mater. 2003, 98, 33.
| A review of classic Fenton’s peroxidation as an advanced oxidation technique.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXhslKjsLw%3D&md5=b480c84d4024991649021e73adff4f3fCAS | 12628776PubMed |
[134] J. H. Merz, W. A. Waters, Electron-transfer reactions. The mechanism of oxidation of alcohols with Fenton’s reagent. Discuss. Faraday Soc. 1947, 2, 179.
| Electron-transfer reactions. The mechanism of oxidation of alcohols with Fenton’s reagent.Crossref | GoogleScholarGoogle Scholar |
[135] K. Sato, M. Aoki, J. Takagi, R. Noyori, Organic solvent- and halide-free oxidation of alcohols with aqueous hydrogen peroxide. J. Am. Chem. Soc. 1997, 119, 12 386.
| Organic solvent- and halide-free oxidation of alcohols with aqueous hydrogen peroxide.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXmsVKj&md5=9c0ac7fbf4f70a17455ff7ef6f4e12ceCAS |
[136] D. G. Fujimori, E. W. Barr, M. L. Matthews, G. M. Koch, J. R. Yonce, C. T. Walsh, J. M. Bollinger, C. Krebs, P. J. Riggs-Gelasco, Spectroscopic evidence for a high-spin Br–FeIV–Oxo intermediate in the α-ketoglutarate-dependent halogenase CytC3 from Streptomyces. J. Am. Chem. Soc. 2007, 129, 13 408.
| Spectroscopic evidence for a high-spin Br–FeIV–Oxo intermediate in the α-ketoglutarate-dependent halogenase CytC3 from Streptomyces.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtFKks73N&md5=51ed8f827654b058a19c413175312e4dCAS |
[137] L. C. Blasiak, F. H. Vaillancourt, C. T. Walsh, C. L. Drennan, Crystal structure of the non-haem iron halogenase SyrB2 in syringomycin biosynthesis. Nature 2006, 440, 368.
| Crystal structure of the non-haem iron halogenase SyrB2 in syringomycin biosynthesis.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XitlKgurc%3D&md5=67120e7f524e42f8a24cf18e377552a5CAS | 16541079PubMed |
[138] M. L. Matthews, C. S. Neumann, L. A. Milesc, T. L. Grovea, S. J. Bookera, C. Krebs, C. T. Walsh, J. M. Bollinger, Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2. Proc. Natl. Acad. Sci. USA 2009, 106, 17 723.
| Substrate positioning controls the partition between halogenation and hydroxylation in the aliphatic halogenase, SyrB2.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVSjsLvK&md5=a1bffeacf7eefb46dffcf41fa559aad5CAS |
[139] T. Kojima, R. A. Leising, S. Yan, L. Que, Alkane functionalization at non-heme iron centers. Stoichiometric transfer of metal-bound ligands to alkane. J. Am. Chem. Soc. 1993, 115, 11 328.
| Alkane functionalization at non-heme iron centers. Stoichiometric transfer of metal-bound ligands to alkane.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXhs1eis7Y%3D&md5=76b6e9da113b2bb2d49e46d0b20a2a12CAS |
[140] S. Rana, S. Bag, P. Patra, D. Maiti, Catalytic electrophilic halogenations and haloalkoxylations by non-heme iron halides. Adv. Synth. Catal. 2014, 356, 2453.
| Catalytic electrophilic halogenations and haloalkoxylations by non-heme iron halides.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXht1GqsLjF&md5=c414b00b46aabcc4d286ec1fe65a4bfeCAS |
[141] A. K. Vardhaman, P. Barman, S. Kumar, C. V. Sastri, D. Kumar, S. P. de Visser, Mechanistic insight into halide oxidation by non-heme iron complexes. Haloperoxidase versus halogenase activity. Chem. Commun. 2013, 10 928.
| Mechanistic insight into halide oxidation by non-heme iron complexes. Haloperoxidase versus halogenase activity.Crossref | GoogleScholarGoogle Scholar |
[142] D. H. R. Barton, B. Hu, D. K. Taylor, R. U. R. Wahl, The selective functionalization of saturated hydrocarbons. Part 32. Distinction between the FeII–FeIV and FeIII–FeV manifolds in Gif chemistry. The importance of carboxylic acids for alkane activation. Evidence for a dimeric iron species involved in Gif-type chemistry. J. Chem. Soc., Perkin Trans. 2 1996, 1031.
| The selective functionalization of saturated hydrocarbons. Part 32. Distinction between the FeII–FeIV and FeIII–FeV manifolds in Gif chemistry. The importance of carboxylic acids for alkane activation. Evidence for a dimeric iron species involved in Gif-type chemistry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XjsFCiu74%3D&md5=76e7ee03031f38420496827ddb28183aCAS |
[143] K.-B. Cho, X. Wu, Y.-M. Lee, Y. H. Kwan, S. Shaik, W. Nam., Evidence for an alternative to the oxygen rebound mechanism in C–H activation by non-heme FeIVO complexes. J. Am. Chem. Soc. 2012, 134, 20 222.
| Evidence for an alternative to the oxygen rebound mechanism in C–H activation by non-heme FeIVO complexes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhslOmu7bJ&md5=595cf9208805d2a05ab8ea3023416c27CAS |
[144] F. Minisci, F. Fontana, Mechanism of the Gif–Barton-type alkane functionalization by halide and pseudohalide ions. Tetrahedron Lett. 1994, 35, 1427.
| Mechanism of the Gif–Barton-type alkane functionalization by halide and pseudohalide ions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXisVekt78%3D&md5=f942beb6aa526daf5e023c3d155ec6b7CAS |
[145] J. Kiwi, A. Lopez, V. Nadtochenko, Mechanism and kinetics of the OH-radical intervention during Fenton oxidation in the presence of a significant amount of radical scavenger (Cl–). Environ. Sci. Technol. 2000, 34, 2162.
| Mechanism and kinetics of the OH-radical intervention during Fenton oxidation in the presence of a significant amount of radical scavenger (Cl–).Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXisVOqu7s%3D&md5=f353e5408c531354abb5dfd6d28b35baCAS |
[146] D. S. Tarbell, P. D. Bartlett, The mechanism of addition reactions. Chloro- and bromo-beta-lactones from dimethylmaleic and dimethylfumaric acids. J. Am. Chem. Soc. 1937, 59, 407.
| The mechanism of addition reactions. Chloro- and bromo-beta-lactones from dimethylmaleic and dimethylfumaric acids.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA2sXhtlSitQ%3D%3D&md5=a98cb35b982ad6418799ec2e140a91fdCAS |
[147] I. Roberts, G. E. Kimball, The halogenation of ethylenes. J. Am. Chem. Soc. 1937, 59, 947.
| The halogenation of ethylenes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA2sXjtVCrtQ%3D%3D&md5=12cb4df81587297225c4d12f1cc6244aCAS |
[148] M. F. Ruasse, Bromonium ions or beta-bromocarbocations in olefin bromination. A kinetic approach to product selectivities. Acc. Chem. Res. 1990, 23, 87.
| Bromonium ions or beta-bromocarbocations in olefin bromination. A kinetic approach to product selectivities.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3cXhsFOgsb0%3D&md5=57bf980cfe7d2bade3721824c91ce5bdCAS |
[149] H. C. Tung, C. Kang, D. T. Sawyer, Nature of the reactive intermediates from the iron-induced activation of hydrogen peroxide: agents for the ketonization of methylenic carbons, the monooxygenation of hydrocarbons, and the dioxygenation of aryl olefins. J. Am. Chem. Soc. 1992, 114, 3445.
| Nature of the reactive intermediates from the iron-induced activation of hydrogen peroxide: agents for the ketonization of methylenic carbons, the monooxygenation of hydrocarbons, and the dioxygenation of aryl olefins.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XhvFentbs%3D&md5=c106299f6f26eebf7fe26530185c8e1dCAS |
[150] H. F. Schöler, F. Keppler, Abiotic formation of organohalogens during early diagenetic processes, in The Handbook of Environmental Chemistry. The Natural Production of Organohalogen Compounds (Eds O. Hutzinger, G.W. Gribble). 2003, Vol. 3, pp. 63–84 (Springer: Heidelberg).
[151] F. Keppler, R. Eiden, V. Niedan, J. Pracht, H. F. Schöler, Halocarbons produced by natural oxidation processes during degradation of organic matter. Nature 2000, 403, 298.
| Halocarbons produced by natural oxidation processes during degradation of organic matter.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXns1ChsA%3D%3D&md5=c99967dc51e17d29c8d517dfdab9200dCAS | 10659846PubMed |
[152] F. Althoff, A. Jugold, F. Keppler, Methane formation by oxidation of ascorbic acid using iron minerals and hydrogen peroxide. Chemosphere 2010, 80, 286.
| Methane formation by oxidation of ascorbic acid using iron minerals and hydrogen peroxide.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmslWhurY%3D&md5=3e227c978529caf476b8a9b07296118fCAS | 20444486PubMed |
[153] P. Comba, B. Nuber, A. Ramlow, The design of a new type of very rigid tetradentate ligand. J. Chem. Soc., Dalton Trans. 1997, 347.
| The design of a new type of very rigid tetradentate ligand.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXhtlGlurk%3D&md5=052a9187f013ffc74012c734c1f48db7CAS |
[154] H. Börzel, P. Comba, K. S. Hagen, Y. D. Lampeka, A. Lienke, G. Linti, H. Pritzkow, L. V. Tsymbal, Iron coordination chemistry with tetra-, penta- and hexadentate bispidine-type ligands. Inorg. Chim. Acta 2002, 337, 407.
| Iron coordination chemistry with tetra-, penta- and hexadentate bispidine-type ligands.Crossref | GoogleScholarGoogle Scholar |
[155] P. Comba, M. Kerscher, W. Schiek, Bispidine coordination chemistry. Prog. Inorg. Chem. 2007, 55, 613.
| Bispidine coordination chemistry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXns1OrtLg%3D&md5=85e1f26efbeef2a2dcd1d47b53a972deCAS |
[156] P. Comba, M. Kerscher, Computation of structures and properties of transition metal compounds. Coord. Chem. Rev. 2009, 253, 564.
| Computation of structures and properties of transition metal compounds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXisFSks78%3D&md5=fd2f49215d0219a231243d15de000fb4CAS |
[157] C. P. Huang, C. Dong, Z. Tang, Advanced chemical oxidation: its present role and potential future in hazardous waste treatment. Waste Manag. 1993, 13, 361.
| Advanced chemical oxidation: its present role and potential future in hazardous waste treatment.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXit1Glt7g%3D&md5=f667ec0374978c9e92dd43d2c7c78332CAS |
[158] F. J. Potter, J. A. Roth, Oxidation of chlorinated phenols using Fenton’s reagent. Hazard. Waste Hazard. Mater. 1993, 10, 151.
| Oxidation of chlorinated phenols using Fenton’s reagent.Crossref | GoogleScholarGoogle Scholar |
[159] J. Pracht, J. Boenigk, M. Isenbeck-Schröter, F. Keppler, H. F. Schöler, Abiotic FeIII-induced mineralization of phenolic substances. Chemosphere 2001, 44, 613.
| Abiotic FeIII-induced mineralization of phenolic substances.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXkvVehtro%3D&md5=7bd42157ddb2a72ed13d180c26690cffCAS | 11482648PubMed |
[160] F. Keppler, R. Borchers, J. Pracht, S. Rheinberger, H. F. Schöler, Natural formation of vinyl chloride in the terrestrial environment. Environ. Sci. Technol. 2002, 36, 2479.
| Natural formation of vinyl chloride in the terrestrial environment.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XivVGgtLo%3D&md5=810e268850daecade50562ef616034a3CAS | 12075808PubMed |
[161] I. J. Fahimi, F. Keppler, H. F. Schöler, Formation of chloroacetic acids from soil, humic acid and phenolic moieties. Chemosphere 2003, 52, 513.
| Formation of chloroacetic acids from soil, humic acid and phenolic moieties.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjsVGntrk%3D&md5=ca850a8b72d3eb0a3fc45390cf1e8d8dCAS | 12738276PubMed |
[162] J. A. Zazo, J. A. Casas, A. F. Mohedano, M. A. Gilarranz, J. J. Rodríguez, Chemical pathway and kinetics of phenol oxidation by Fenton’s reagent. Environ. Sci. Technol. 2005, 39, 9295.
| Chemical pathway and kinetics of phenol oxidation by Fenton’s reagent.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtFCitLfP&md5=e678b1b22e9affc801dac942a908597dCAS | 16382955PubMed |
[163] A. M’Hemdi, B. Dbira, R. Abdelhedi, E. Brillas, S. Ammar, Mineralization of catechol by Fenton and photo-Fenton processes. CLEAN – Soil, Air, Water 2012, 40, 878.
| 1:CAS:528:DC%2BC38XhtVGgsbjF&md5=2397ce7a165719bef0766cd8b39e2d05CAS |
[164] S. Studenroth, S. G. Huber, K. Kotte, H. F. Schöler, Natural abiotic formation of oxalic acid in soils: results from aromatic model compounds and soil samples. Environ. Sci. Technol. 2013, 47, 1323.
| 1:CAS:528:DC%2BC3sXntVWltg%3D%3D&md5=c6bd4a26721f59d3e3666dd35ecf77f6CAS | 23311299PubMed |
[165] F. Keppler, R. Borchers, J. T. G. Hamilton, G. Kilian, J. Pracht, H. F. Schöler, De novo formation of chloroethyne in soil. Environ. Sci. Technol. 2006, 40, 130.
| De novo formation of chloroethyne in soil.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1emsbfN&md5=df461d1e6e3a92b10726b1aa3307e38bCAS | 16433342PubMed |
[166] S. G. Huber, G. Kilian, H. F. Schöler, Carbon suboxide, a highly reactive intermediate from the abiotic degradation of aromatic compounds in soil. Environ. Sci. Technol. 2007, 41, 7802.
| Carbon suboxide, a highly reactive intermediate from the abiotic degradation of aromatic compounds in soil.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtFGisrzF&md5=8f221e0e60cd005662a97978752bfdf3CAS | 18075091PubMed |
[167] S. G. Huber, K. Kotte, H. F. Schöler, J. Williams, Natural abiotic formation of trihalomethanes in soil: results from laboratory studies and field samples. Environ. Sci. Technol. 2009, 43, 4934.
| Natural abiotic formation of trihalomethanes in soil: results from laboratory studies and field samples.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmtVGnsbY%3D&md5=878e371b694f0cd74e15b81156df73f5CAS | 19673288PubMed |
[168] S. G. Huber, S. Wunderlich, H. F. Schöler, J. Williams, Natural abiotic formation of furans in soil. Environ. Sci. Technol. 2010, 44, 5799.
| Natural abiotic formation of furans in soil.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXos1Wgurw%3D&md5=24e5250700bdaac70f32fa667427b83cCAS | 20614942PubMed |
[169] T. Krause, C. Tubbesing, K. Benzing, H. F. Schöler, Model reactions and natural occurrence of furans from hypersaline environments. Biogeosciences 2014, 11, 2871.
| Model reactions and natural occurrence of furans from hypersaline environments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC2cXhs1Sju73J&md5=7cb50be77d560436d32122398576f2cbCAS |
[170] F. Keppler, R. Borchers, P. Elsner, I. J. Fahimi, J. Pracht, H. F. Schöler, Formation of volatile iodinated alkanes in soil: results from laboratory studies. Chemosphere 2003, 52, 477.
| Formation of volatile iodinated alkanes in soil: results from laboratory studies.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXjsVGntro%3D&md5=4a8e296dbc8d396521a3bcdc2400460cCAS | 12738273PubMed |
[171] R. C. Fuson, B. A. Bull, The haloform reaction. Chem. Rev. 1934, 15, 275.
| The haloform reaction.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaA2MXit1OisQ%3D%3D&md5=096a6a75bad4543608dde17551d82ac9CAS |
[172] E. J. Hoekstra, E. W. B. de Leer, U. A. T. Brinkman, Findings supporting the natural formation of trichloroacetic acid in soil. Chemosphere 1999, 38, 2875.
| Findings supporting the natural formation of trichloroacetic acid in soil.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXitlOlur4%3D&md5=aef80a7b7725e477987c3dd59ae670c1CAS |
[173] W. Z. Tang, C. P. Huang, The effect of chlorine position of chlorinated phenols on their dechlorination kinetics by Fenton’s reagent. Waste Manag. 1995, 15, 615.
| The effect of chlorine position of chlorinated phenols on their dechlorination kinetics by Fenton’s reagent.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XntlShurg%3D&md5=d57127a1dbf885789f927d73210de32eCAS |