

Chlorine chemistry in urban atmospheres: a review
C. B. Faxon A B and D. T. Allen AA Center for Energy and Environmental Resources, University of Texas, M/C 27100, 10100 Burnet Road, Austin, TX 78758, USA.
B Corresponding author. Email: cfax@utexas.edu

Cameron Faxon is currently a doctoral candidate in Chemical Engineering at the University of Texas at Austin. His research efforts involve the investigation of atmospheric chlorine chemistry with a focus on the impacts on local and regional air quality. Current research projects include the development and implementation of chlorine chemistry mechanisms for use in regional photochemical models as well as environmental chamber studies of heterogeneous processes. |

Dr David Allen is the Gertz Regents Professor of Chemical Engineering at the University of Texas at Austin. He also serves as the Director of the Center for Energy and Environmental Resources and was recently appointed as chair of the Environmental Protection Agency's Science Advisory Board (SAB). He is the author of six books and over 170 papers in areas ranging from coal liquefaction and heavy oil chemistry to the chemistry of urban atmospheres. |
Environmental Chemistry 10(3) 221-233 https://doi.org/10.1071/EN13026
Submitted: 1 February 2013 Accepted: 18 April 2013 Published: 19 June 2013
Journal Compilation © CSIRO Publishing 2013 Open Access CC BY-NC-ND
Environmental context. Atmospheric chlorine radicals can affect the chemical composition of the atmosphere through numerous reactions with trace species. In urban atmospheres, the reactions of chlorine radicals can lead to effects such as increases in ozone production, thus degrading local and regional air quality. This review summarises the current understanding of atmospheric chlorine chemistry in urban environments and identifies key unresolved issues.
Abstract. Gas phase chlorine radicals (Cl•), when present in the atmosphere, react by mechanisms analogous to those of the hydroxyl radical (OH•). However, the rates of the Cl•-initiated reactions are often much faster than the corresponding OH• reactions. The effects of the atmospheric reactions of Cl• within urban environments include the oxidation of volatile organic compounds and increases in ozone production rates. Although concentrations of chlorine radicals are typically low compared to other atmospheric radicals, the relatively rapid rates of the reactions associated with this species lead to observable changes in air quality. This is particularly evident in the case of chlorine radical-induced localised increases in ozone concentrations. This review covers five aspects of atmospheric chlorine chemistry: (1) gas phase reactions; (2) heterogeneous and multi-phase reactions; (3) observational evidence of chlorine species in urban atmospheres; (4) regional modelling studies and (5) areas of uncertainty in the current state of knowledge.
Introduction
Most management plans for the reduction of photochemical smog focus on reducing emissions of volatile organic compounds (VOCs) and nitrogen oxides (NOx), leading to reduced formation of photochemical air pollutants, such as ozone, through various chemical pathways involving HOx radicals. A growing body of observations and modelling studies, however, indicates that halogen radicals, particularly the chlorine radical and related species, can play a significant role in urban atmospheric chemistry. This may occur through both emissions of chlorine radical precursors and through the involvement of chlorinated species in VOC–NOx–HOx pathways. This review will summarise the current understanding of the role of chlorine radicals in urban photochemistry and will highlight key issues that are unresolved. The review is organised into five sections: gas phase chlorine chemistry in urban atmospheres, heterogeneous (gas-particle) and multi-phase chlorine chemistry in urban atmospheres, observations, regional modelling studies and critical gaps in current understanding.
Gas phase chlorine chemistry in urban atmospheres
Various forms of chlorine (e.g. particulate chloride, inorganic gas species, organohalogens) are present in the atmosphere. Analyses of natural emissions indicate an atmospheric loading of 23 Tg of Cl, of which 8.4 Tg comprises reactive species. The two dominant reactive species are CH3CCl3 (29 % of reactive species) and CH3Cl (43 %), with inorganic species accounting for 7 % of reactive atmospheric chlorine.[1] Other common species include HCl, CHClF2, Cl2, HOCl, CCl2=CCl2, CH2Cl2, COCl2 and CHCl3. Photodissociation of chlorinated species, the sources of which may be natural or anthropogenic, is one of the most common routes to global tropospheric chlorine radical production. Other gas phase chemical pathways to the global formation of chlorine radicals include reactions of HCl or chlorocarbons.[2,3] Gas phase species also participate in reactions involving particulate matter, which will be described in subsequent sections of this review.[3]
Although important globally, photodissociation and oxidation of many of the most common chlorinated organic species occur at rates that are generally not fast enough to contribute significant concentrations of chlorine radicals in urban atmospheres. For example, the rate constant for chlorine radical generation by the oxidation of HCl by the hydroxyl radical at 298 K is reported to be 7.8 × 10–13 cm3 molecule–1 s–1.[4,5] This rate constant is approximately two orders of magnitude lower than the rate constant for hydroxyl radical formation from the reactions of O(1D) and H2O (1.63 × 10–10).[5] Given that H2O is also typically present in the urban atmosphere at concentrations far greater than those of HCl, the result is a smaller rate of generation for chlorine radicals compared to hydroxyl radicals. For chlorocarbon photolysis, rates of reaction are very low, approaching zero, in the urban atmosphere. For example, the photolysis of CH3Cl has only been reported to occur during exposure to radiation at wavelengths below 216 nm.[6] In the urban atmosphere, radiation of this wavelength is negligible.
Production of chlorine radicals through routes other than photolysis of chlorocarbon species and oxidation of common chlorinated species such as HCl can proceed at rates that are significant in urban atmospheres, however. The photolysis of Cl2 is expected to proceed rapidly with a quantum yield of unity.[4,7,8] One comparison of radical generation from 3 ppb HONO, a photolytic source of the hydroxyl radical, and 0.1 ppb Cl2 in a polluted region estimates corresponding radical generation rates of 5 × 106 and 4 × 105 radicals cm–3 s–1 respectively for hydroxyl radicals and chlorine radicals.[8] Thus, if concentrations of species such as HOCl, ClNO2 or Cl2 are sufficiently high, photolysis reactions can provide an important source of chlorine radicals in urban atmospheres. In some regions, direct anthropogenic emissions of these chlorine radical precursors can be significant. One estimate[7] suggests that anthropogenic HOCl and Cl2 emissions in the Houston area are ~104 kg day–1, coming from cooling towers, swimming pools and industrial point sources.[9] In the vicinity of large anthropogenic sources of Cl2 or HOCl, the reactions of Cl and related radicals can significantly affect photochemistry. Thus, although photolysis of many chlorocarbon species and oxidation of the most common gas-phase chlorine species is not a significant contributor to radical production in urban atmospheres, there are situations where anthropogenic emissions of chlorine radical precursors can produce significant radical concentrations through photolysis pathways. Table 1 below shows a comparison of photolysis rates for some chlorine species and some other common atmospheric species involved in the photochemical cycle.

|
Chlorine radicals produced photolytically or through other mechanisms may react with and oxidise VOCs. The examples in Reactions 1–3 represent typical photolysis reactions and the initiating reaction for the oxidation of a generic VOC, denoted as RH.[4,5,7,8,10,11]

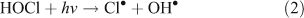
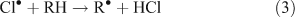
The presence of reactive chlorine species can also modify HOx levels and the HO2•/OH• ratio. This occurs from reactions resulting in the inter-conversion of OH• and HO2•. Examples of known reactions are shown in Reactions 4–8.[4,5,10,11]
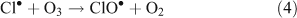
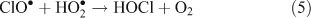
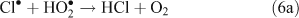

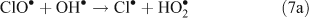
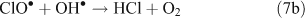
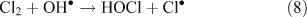
Unlike OH•, chlorine radicals are not regenerated within the oxidation cycle. However, chlorine radicals can be regenerated by heterogeneous cycling from chloride containing aerosols (discussed in the next section) or by the volatilisation and oxidation of HCl (Reaction 9).[2–5,10,11]
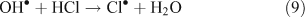
Another important difference between the hydroxyl and chlorine radicals is the rates at which they oxidise hydrocarbons. The reactivity of chlorine with most VOCs is greater than that of the hydroxyl radical, such that even concentrations of chlorine radicals an order of magnitude or more lower than hydroxyl radicals can compete with normal atmospheric concentrations of hydroxyl radicals.[12] The reaction rates of chlorine radicals with many alkanes, aromatics, alcohols and ethers typically range between one to two orders of magnitude greater than the identical reaction with the hydroxyl radical.[11,13–15] This high reactivity causes the rates of such reactions to approach the collisional limit. For alkenes and alkynes, the difference is slightly smaller (kCl/kOH ≈ 4–13). One exception to the increased reactivity of chlorine is the reaction with benzene, for which the ratio kCl/kOH is on the order of 10–4.[11,16]
Reactions of biogenic VOCs (BVOCs) with chlorine also proceed more rapidly than corresponding reactions with OH•, with rate constants typically >10–11 cm3 molecules–1 s–1.[17] The oxidation of the common biogenic, isoprene, by chlorine radicals takes place primarily through chlorine addition, with ~10 % of the reaction proceeding through the hydrogen abstraction pathway.[18] Unique reaction products such as 1-chloro-3-methyl-3-butene-2-one (CMBO), and isomers of chloromethylbutenal (CMBA) result.[18–20] Although not a BVOC, the reaction of chlorine radicals and 1,3-butadiene also produces unique tracer species, 4-chlorocrotonaldehyde (CCA) and chloromethyl vinyl ketone, with yields varying depending on the presence or absence of NO.[21] Table 2 shows a comparison of the rate parameters of OH• and Cl• for some common alkanes, alkenes and BVOCs.

|
These differences in reactivities contribute to the overall loss rates for each radical as well as the relative loss rates of different VOCs. Table 3 shows a comparison of reaction rates for a VOC mixture[22] that was formulated to represent a typical urban mixture of reactive organic gases. More details on this mixture are available in the Supplementary material (see http://www.publish.csiro.au/?act=view_file&file_id=EN13026_AC.pdf). The comparisons shown in Table 3 indicate that relative rates of reaction with chlorine and hydroxyl radicals vary among hydrocarbon species. This difference in relative rates can be used, as will be described later in this review, to infer chlorine and hydroxyl radical concentrations, based on the relative rates of reaction of selected hydrocarbons.

|
Heterogeneous and multiphase chlorine chemistry mechanisms
Chlorine in particulate matter is generally in the form of unreactive chloride; however, a variety of heterogeneous and multi-phase reaction processes can lead to the conversion of particulate chloride into gas phase, reactive chlorine species. Multiphase reactions include the reactions of dissolved gas phase species within aqueous droplets, whereas reactions taking place at the interface between two phases are considered heterogeneous.[23,24] These processes include acid displacement and reactions of N2O5, ozone and other species with chloride-containing aerosol.
Acid displacement and reactive chlorine cycling
Acid displacement involves the replacement of particulate chloride anions by anions associated with H2SO4, HNO3, methane sulfonic acid (MSA) or other acids, and has been observed in particles of marine origin.[25–28] In the case of HNO3 or H2SO4, the process may be represented as the heterogeneous reaction of a gas phase acid with NaCl aerosols.[3,27,29,30] In the examples in Reactions 10 and 11, the subscripts (g) and (cd) respectively denote gas phase and condensed phase species.
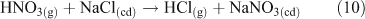
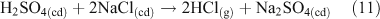
The uptake of HNO3 is more effective when the particle is aqueous or in the presence of surface adsorbed water (SAW).[29] Reduction in aerosol pH can also enhance HCl production, which may increase by up to a factor of 20 between pH 5.5 and 3.[31] The presence of MgCl2 in a particle increases SAW and thus increases the reactive uptake at lower relative humidity. It is therefore expected that this reaction will proceed at normal conditions in the marine boundary layer (MBL) and that sea salt chloride can be depleted within several hours of aerosol generation.[29,30] HCl production by acid displacement is more efficient within the smaller size fractions of typical aerosol populations.[32,33]
The efficiency of chloride loss from aerosols is often quantified in terms of a particulate chloride deficit. This is defined as the amount of particulate chloride that is necessary to balance the observed concentrations of sea salt tracer cations, such as Na+, that have no associated chloride.
Reactive uptake of gases and volatilisation of reactive chlorine
Additional multi-phase and heterogeneous chlorine reactions include the reactive uptake of other, non-acid gases, leading to the accumulation of photoreactive chlorine species. Most known mechanisms require the presence of reactive nitrogen gases.[34] Many of these reactions can take place on dry, solid salt particles as well as aqueous droplets, although the reactions are typically slower in the absence of water.[35] Some examples of these reactions, which can compete with the acid displacement reactions described above, are shown in Reactions 12–19.[11,24,30,36–40]
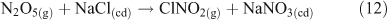
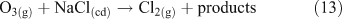
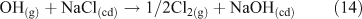
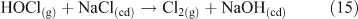
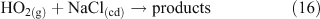
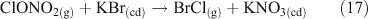
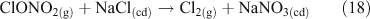
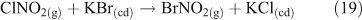
A mechanism involving the formation of a surface complex between particulate chloride and OH• (OH⋯Cl•–) (Reactions 20, 21) has been proposed to explain the processes involved in Reaction 14.[41,42]
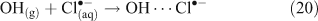
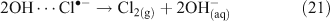
Gas phase chlorine species have also been found to react in the presence of ice surfaces or particles to produce other chlorine species (Reactions 22–24).[36,37,39,43]
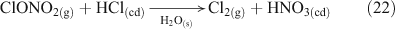
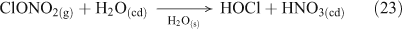
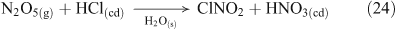
The direct uptake and scavenging of HCl by dust particles has also recently been observed, resulting in dust particles composed of up to 9 % HCl by mass.[44] In general, the cycling of chlorine can be summarised in the steps listed below.[3,45]
-
Volatilisation of Cl2, HOCl or other photolytic chlorine species from NaCl aerosols or other source.
-
Photolysis of the volatilised species to produce Cl•.
-
Hydrogen atom abstraction from non-methane hydrocarbons to produce HCl.
-
Removal of HCl by reaction with OH•, wet or dry deposition or scavenging by aerosols.
N2O5 and ClNO2 production
One heterogeneous mechanism that has been suggested and confirmed as a source of reactive chlorine (gas phase ClNO2) is the reaction of N2O5 with chloride aerosols. It is known that heterogeneous uptake of N2O5 plays a critical role in overall NOx chemistry, with up to 50 % of NOx removal being attributed to its heterogeneous mechanisms.[46] Reactions that involved chloride activation include those shown in Reactions 25–27.[38,47,48]
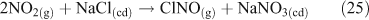
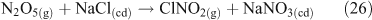
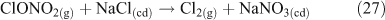
Of these three, Reaction 25 is not thought to be important.[47] However, Reaction 26 plays an important role in the activation of chloride and ClNO2 production,[49,50] although the process may involve multiple steps, as outlined below. Acid displacement by HNO3 is up to two orders of magnitude slower than the interaction with N2O5.[51] Atmospheric implications of this reaction are important, because tropospheric concentrations of N2O5 can range from 1 to 15 ppb.[36,51] The reaction results in a build up of ClNO2 in the morning, limited by N2O5 uptake. N2O5 formation, which occurs from a reversible reaction between NO3 and NO2, is favoured at night due to the photochemical instability of NO3.[51] Although ClNO2 can form by homogeneous gas phase chemistry, the reactions are extremely slow,[5] preventing significant production of ClNO2.
Hints that Reaction 26 existed arose during studies of the rates of chlorine radical production from irradiated NaCl aerosols in the presence of NOx. A smog chamber experiment using initial NO2, O3 and NaCl concentrations of 200 ppbv, 108 ppbv and 1.5 mg m–3, found a large chlorine radical source upon irradiation of the mixture after 100 min in the dark. The observed source, if occurring from the reaction between gas phase HCl and OH radical species, would have required a 1.5-ppmv HCl mixing ratio at OH concentrations of 1.7 × 107 cm–3.[52] Similar smog chamber studies following up these unexplained observations indicated that ClNO2 was produced at a high yield when the relative humidity was in the range of 71–92 %.[53]
A mechanism was proposed to explain the characteristics of the reaction, including the dependence of ClNO2 yields on particle chloride concentrations. Due to the fact that N2O5 does not react directly with chloride ions, there is no observable dependence of the uptake coefficient on chloride concentrations present in the aqueous solution. This is because aqueous chloride ions react not with aqueous N2O5 but with its dissociation products as shown below in Reactions 28–30.[47] Direct hydrolysis was found to account for approximately only 20 % of the overall reaction, and dissociation of N2O5 has since been cited as the rate limiting step.[40,47,54]
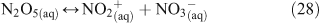
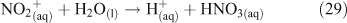
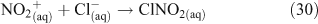
Taking this proposed mechanism a step further, Bertram and Thornton[55] proposed a reaction scheme involving the initial uptake of N2O5 into the aqueous phase and formation of a hypothetical protonated nitric acid intermediate as the initiating step in the production of ClNO2.[55] ClNO2 then partitions into the gas phase where it can undergo photolysis to produce chlorine radicals (Reactions 32–35).[46,56]
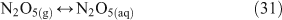
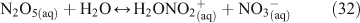
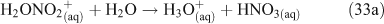
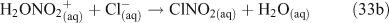
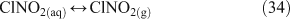
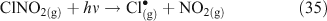
An experimental study to examine the kinetics of this mechanism concluded that either Reaction 31 or 32 is the rate limiting step at 50 % relative humidity, thus corroborating previous suggestions that mass accommodation and hydrolysis of N2O5 are the processes limiting the overall reaction rate. This same study showed that the decay of gas phase N2O5 concentrations in the presence of simulated sea salt aerosols is log-linear, implying a first order loss mechanism.[56] A coexisting reaction pathway producing Cl2, in which ClNO2 is an intermediate, is observed below pH 2.[49,54]
ClNO2 can also undergo reactions within particles instead of being released into the gas phase. The uptake and solubility of ClNO2 into aqueous particles is inhibited by the presence of chloride due to its dissociation reaction in the aqueous phase (Reaction 36).[57]
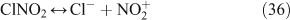
Observations of atmospheric chlorine radicals and related species
The preceding sections have summarised gas phase, heterogeneous and multi-phase reactions of chlorine radicals and related species, believed to be important in urban atmospheres. The relative importance of these pathways has been examined in a variety of observational studies. In this section, observational evidence of the presence of various atmospheric chlorine species will be explored.
Observations of chloride deficits and volatilisation
Overall, depending on region and aerosol size, particle chloride volatilisation of 70–80 % is possible.[32] A study of volatile chlorine over the North Atlantic Ocean determined that the concentrations of volatilised chlorine from aerosols, as determined from particulate chloride deficits, tended to be approximately equal to measured HCl concentrations, ranging from below detection limits up to 125 nmol m–3 (3 ppbv).[45] A separate study of chlorine chemistry in polluted air that had been advected offshore noted that the cycling of chlorine between phases can lead to a Cl2(g) increase of 90 parts per trillion by volume (pptv) in no wind scenarios and 125 pptv in high wind scenarios.[58] Shown below (Reactions 37–42) are the dominant reactions cited as being responsible for this increase. The sequence is initiated by acid displacement (Reactions 10–11).[58]
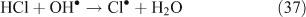
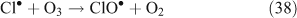
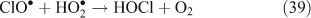
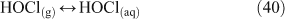
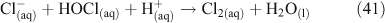

The correlation between particulate chloride deficits and HCl concentrations was observed to be much higher in air polluted with combustion products, nearing a [HCl]/[Cl– deficit] ratio of 1 in polluted air masses compared to 4.5 in cleaner air masses.[45]
Deficits vary regionally, and air in the southern hemisphere exhibits lower particulate chloride deficits whereas chloride deficits in the northern hemisphere have approached 100 %. However, once particulate chloride is removed, HCl serves as a chloride reservoir and can be scavenged by aerosols to replenish particulate chloride concentrations.[46] It is predicted that HCl has a lifetime in the MBL of 10 min before it is scavenged by sea salt aerosols.[44]
Observations of hydrogen chloride (HCl) and molecular chlorine (Cl2)
Surface concentrations of HCl in remote ocean regions range from 100 to 300 pptv, whereas peak concentrations in urban areas can be ~1500 pptv. Multiple studies note that peak HCl concentrations typically occur in the afternoon, coinciding with peak photochemical smog production and HNO3 concentrations.[59–63] This is generally attributed to the volatilisation of chloride from aerosol particles.[3,62,63] HCl generally exhibits a trend of vertical distribution, decreasing with altitude. Previous reviews of continental tropospheric HCl observations have reported peak concentrations of up to 1–3 ppbv in urban areas.[3] A marked drop off in concentration to 50–100 pptv typically occurs at altitudes above the boundary layer.[3,45]
In Virginia Key, Florida, mist chamber techniques[64] were used to measure total HCl* (HCl and possibly ClNOx species), which ranged between 40 and 268 pptv Cl. The same technique was used to measure total Cl*2 (Cl2 + some HOCl) and found concentrations ranging from below the detection limit of 26 pptv up to 254 pptv Cl. Cl*2 built up during the night and sharply decreased after sunrise corresponding with a HCl* increase. This suggests the photolysis of Cl2 or HOCl at sunrise, followed by VOC oxidation by hydrogen atom abstraction by Cl•, producing HCl. Photochemical modelling performed in this study indicated that if the observed levels of Cl*2 were completely photolysed, it would lead to Cl• concentrations of 104–105 radicals cm–3.[65] It was observed that higher levels of Cl2 and HOCl were present in winds coming from the Atlantic as opposed to those from the North, suggesting a marine origin.[65]
Other authors have also noted high concentrations of molecular chlorine in incoming marine air along the coasts of North America.[66] Using the mist chamber technique in the Hawaii boundary layer, respective HCl* and Cl2* concentrations up to 250 and 38 pptv were detected.[67] Cl2 concentrations in La Jolla, California, have been observed to average 2.3 pptv.[68] In Irvine, California, Cl2 concentrations ranging from 2.5 to 20 pptv with a 2-month mean of 3.5 pptv have been reported. Modelling the photochemistry of this observed Cl2 showed a 5–8-ppb increase in daily maximum O3 concentrations.[69] Earlier evidence of O3 production increases from the presence of chlorine was observed in industrial plumes in Telemark, Norway.[70] In Houston, Texas, the detection of the tracer species (CMBO) at peak concentrations of 12–126 pptv[71] provided evidence of chlorine radical concentrations estimated to be approximately <5 × 103 to 3.3 × 105 radicals cm–3 (2 × 10–4 to 1.3 × 10–2 pptv).[20] Associated enhanced ozone production rates of >75 ppbv h–1 were observed when small amounts of Cl2 were injected into captive ambient air during chamber experiments.[71]
Off the coast of Long Island, New York, a concentration up to 150 pptv of Cl2 was observed at night, with a rapid decline to less than 15 pptv after sunrise. Such amounts of Cl2 would lead to chlorine radical concentrations of 1.3 × 105 radicals cm–3 (5.1 × 10–3 pptv) and require a daily source of ~330 pptv Cl2. In this case, the air was again of marine origin.[66] The authors suggest that a mechanism aside from photolytically driven aqueous phase mechanisms[64,72] is needed to explain these observations. A study in the arctic coastal location of Alert, North-West Territories utilised the photoreactive halogen detector method[73] to detect levels of photolysable Cl, which ranged between <9 and 100 pptv. Concentrations were reported as Cl2, although they may have also included HOCl or other photolysable species. A nocturnal build up of photolytic chlorine species followed by a rapid decline after sunrise was once again observed. To explain the night time build up of these species, the nocturnal mechanism in Reactions 43–46 was proposed[74]:
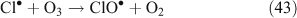
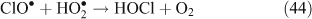
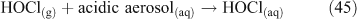
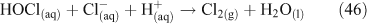
In Charlottesville, Virginia, ~250 miles (~400 km) inland, researchers using the tandem mist chamber technique[64] found HCl* concentrations ranging from <39 to 2800 pptv, with most measurements falling under 300 pptv. Most measurements of Cl2 and HOCl in the same study were under the 26 pptv Cl• detection limit, indicating that HCl (and possibly ClNOx) was the dominant form of volatile chlorine.[75] Because of results such as this, another proposed explanation of the marine origin of Cl2 has been made by suggesting a heterogeneous reaction between particulate chloride and either ozone or the hydroxyl radical.[3,34,45,74] An alternate reaction (Reaction 47) explaining the mechanism described in Reactions 20 and 21 was put forward as an explanation[45,76]:
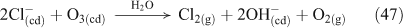
Assuming a steady-state between Cl2 volatilisation by Reaction 47 and reuptake of HCl into aerosols, this mechanism results in a Cl2 source of ~1 ppbv h–1 with corresponding net O3 production rates of 1.4 ppbv h–1 during the day.[45] Table 4 contains a summary of observations from the studies discussed in this section.

|
Reactions 43–47, or other pathways that also lead to nocturnal buildup of photolytic chlorine species, can potentially lead to increases in VOC oxidation, NO-to-NO2 conversion and O3 production early in the day. The effects of the sudden introduction of a large source of chlorine radicals would be analogous to the effect elicited by the nocturnal buildup and sunrise photolysis of a hydroxyl radical source such as HONO.
Observations of chlorine (Cl•) and chlorine monoxide (ClO•) radicals
Analysis of non-methane hydrocarbon (NMHC) decay rates has led to estimates of chlorine radical concentrations on the order of 105 molecules cm–3 (4 × 10–3 pptv) in the Pacific MBL. These concentrations were deduced from observed decay rates of VOCs such as ethane, propane and acetylene by back calculating necessary chlorine radical concentrations to compensate for a lack of sink strength from the OH• reaction alone.[77] At Appledore Island, Maine, observations of increased NMHC reactivity implied chlorine radical concentrations of 2.2 × 104 to 5.6 × 104 radicals cm–3 (8.6 × 10–4 to 2.2 × 10–3 pptv). These concentrations were deduced by measuring C2–C10 NMHC compounds, calculating the back trajectories of sampled air parcels and then calculating the chlorine radical concentration necessary to explain observed NMHC decay rates that were too fast to be fully explained by OH• concentrations.[78] In the Southern Ocean, average Cl• concentrations of 720 radicals cm–3 (2.8 × 10–5 pptv) have been reported.[12] At Great Lake, Utah, 3.8 × 108 molecules cm–3 of ClO• (15 pptv) was observed, suggesting mobilisation of chloride from salt beds in the area.[79] This observation also implied a chlorine radical concentration on the order of 105 radicals cm–3 (4 × 10–3 pptv), which would double the reactivity of the air in the area, increase OH• concentrations by up to 50 % through radical propagation and convert NOx into halogenated nitrates.[79]
Observations of nitryl chloride
Direct correlations between ClNO2, N2O5 and aerosol surface area concentrations have been noted in field observations.[80] During the Texas Air Quality Study II (TEXAQS II), concentrations of ClNO2 up to 1200 pptv were found in ship engine plumes, near NOx sources in the Houston Ship Channel and in surrounding urban areas.[80,81] Contemporaneously measured N2O5 concentrations and consumption rates suggest that anthropogenic pollutants contributed significantly to the observed ClNO2.[80]
In addition, ClNO2 concentrations up to 450 pptv have also been found as far inland as Kohler Mesa outside of Boulder, Colorado. The Kohler Mesa is 1400 km from any coastal area, but it is thought that the air masses during the measurement period were potentially influenced by long distance chlorine transport from coastal areas or inland salt beds and pollution from combustion in the Boulder, Colorado area.[46] More recent observations in Los Angeles, California, revealed concentrations of ClNO2 up to 2100 pptv.[82] In 2011, observed ClNO2 mixing ratios up to 250 pptv were observed in Calgary, Alberta, Canada.[83] Arctic observations of N2O5 and ClNO2 were made during the International Chemistry in the Arctic Lower Troposphere (ICEALOT) campaign. Respective ClNO2 and N2O5 ranges, on average, of 150–250 and 150–200 pptv were observed.[84] Table 5 summarises observations of ClNO2 in several locations.

|
Observations of chlorine tracer species
CMBO and CMBA are known tracer compounds of chlorine’s oxidation of isoprene. Observations of these compounds were made during the Texas Air Quality Study (TEXAQS) in Houston, Texas.[71] Chlorine radical concentrations were deduced from CMBO and CMBA concentrations and were determined to range between 5 × 103 and 3.3 × 105 radicals cm–3 (2 × 10–4 and 1.3 × 10–2 pptv).[20] In comparison, Maben et al.[75] approximated chlorine radical concentrations to be less than 104 radicals cm–3 (4 × 10–4 pptv) in continental air over eastern North America, at the lower end of the range of the Houston observations. During the Atlantic Stratospheric Transition Experiment/Marine Aerosol and Gas Exchange (ASTEX/MAGE), observed hydrocarbon concentrations were used to determine average chlorine radical concentrations between 0600 and 1100 hours local time, which were approximated at (3.3 ± 1.1) × 104 molecules cm3 ((1.2 ± 0.4) × 10–3 pptv). Midday concentrations were calculated to be (6.5 ± 1.4) × 104 molecules cm–3 ((2.6 ± 0.6) × 10–3 pptv), and the authors suggest a nocturnal build up of photolysable chlorine as the source.[34]
Summary of observations
Observational evidence suggests that chlorine chemistry is occurring over both oceanic and continental areas, with observed chlorine radical concentrations on the order of 103–105 molecules cm–3 (4 × 10–5 to 4 × 10–3 pptv) not being uncommon.[3,20,34,45,65,75,85] Although observations have generally supported known gas phase chemical pathways, observational evidence[46,65,66,80] hinting at a more complex role for chlorine illuminated gaps in the understanding of reactive gas phase chlorine sources in the atmosphere. Such observations have led to the proposal and investigation of heterogeneous mechanisms to explain the volatilisation of chloride from aerosol particles.[24,41,42,49,54,56,72] More recently, daytime concentrations of chlorine species such as HCl and ClNO2 on the order of 1–2 ppbv have been reported inland in regions such as Dallas–Fort Worth[59] and Boulder, Colorado.[46] These observations may suggest sources other than sea salt for atmospheric chlorine.
Modelling studies
As the importance of chlorine as an atmospheric oxidant has become increasingly apparent, atmospheric chemistry models have been supplemented with basic gas phase chlorine chemistry mechanisms. This section describes condensed chlorine mechanisms, and their use in regional photochemical modelling studies.
Condensed chlorine mechanism development
A critical component of regional models is a condensed photochemical mechanism that is computationally efficient while retaining enough accuracy be used in the study of atmospheric chemical processes. The Carbon Bond mechanism[86,87] is one of the most commonly used condensed chemical mechanisms; reactions representing chlorine chemistry in urban atmospheres were devised and added to a modified version of the Carbon Bond mechanism by Tanaka et al.[88] for use in the Comprehensive Air quality Model with extensions (CAMx). The mechanism included the reaction of chlorine with ozone and VOCs through condensed reactions for olefins and paraffins. Reactions with ethene, methane, 1,3-butadiene and isoprene were explicitly represented in order to account for the unique products of these reactions while enabling comparisons between observations and predictions of the concentrations of these molecular tracers. Photolysis reactions for Cl2 and HOCl were included, and the rates of photolysis for these species were scaled to the respective photolysis rates of NO2 and isoprene oxidation products.[88] Since the creation of this initial mechanism, chlorine reaction sets have been implemented as adjunct mechanisms in both the Carbon Bond 05 (CB05) and the SAPRC-07 mechanisms,[86,89] and these mechanisms have been used in both box[90]and regional[7,71,91–94] modelling.
Recent chlorine mechanism development has involved the addition of heterogeneous pathways, including ClNO2 production chemistry[91,92,94] and the OH•-mediated formation of gas phase Cl2.[24,41,42] Simon et al.[91] implemented a surrogate gas phase reaction for investigating ClNO2 chemistry (Reactions 28–30); Simon et al.[92] improved upon the scheme by using a heterogeneous parameterisation.[23,56,95] Parameter values cited from the literature were used and varied by region based on proximity to the Gulf Coast.[92] Further work on ClNO2 chemistry included dependencies[55] on particle liquid water, particle nitrate and particle chloride in the parameterisation of ClNO2 chemistry for modelling within the continental United States.[94] A parameterised[23,95] version of Reactions 20 and 21 was implemented to study the effect of sea salt and related chlorine emissions on coastal urban ozone in the South Coast Air Basin, California.[24]
Regional modelling of atmospheric chlorine chemistry
Photochemical models are used to probe the effects of atmospheric chlorine chemistry on a continental to global scale. The focus in this section will be on regional models because the reduced computational intensity of these models, relative to global models, has allowed more detailed treatments of chlorine chemistry.
In Houston, Texas, the inclusion of anthropogenic chlorine emissions in regional modelling revealed a maximum localised ozone increase of 16 ppb and moderate increases over a larger spatial area. The maximum modelled O3 increase coincided with a maximum CMBO mixing ratio of 59 ppt.[71] Another study found that chlorine increased 1- and 8-h ozone levels during peak ozone hours by 8 and 9 ppb. This same study also found a maximum increase during morning hours, at the times of maximum rates of Cl2 and HOCl photolysis, of 45 ppbv.[96,97] Because some of the peak enhancements of ozone concentrations predicted in these models are highly localised near chlorine radical precursor sources, the maximum predictions of the models are very sensitive to the grid cell dimensions used in the work. Wang et al.[96,97] used a nested grid with horizontal spatial resolutions as small as 4 km. In similar work, but with horizontal grid cell dimensions of 12–36 km, Sarwar and Bhave[98] modelled the effect of chlorine chemistry on ozone levels over the eastern half of the United States. The study found major effects only in the Houston, Texas, and New York–New Jersey areas, where 1-h O3 level averages increased by 12 and 6 ppbv respectively and 8-h increases were 8 and 4 ppbv. Similarly, a study of a photochemical pollution episode in September 2000 in south-east Texas, using a 4-km horizontal spatial resolution, found highly localised non-peak O3 concentration increases up to 70 ppbv, with peak hour concentration increases typically below 10 ppbv.[7] Yet another study in Texas found a 1-h average O3 level increase of 1.5 ppb from inclusion of ClNO2 production and photolysis.[91]
Modelled ClNO2 concentrations in south-east Texas have ranged from 256–1210 pptv, compared to previous field measurements in the region of 1300 pptv. A limiting factor in the modelling was the modelled availability of chloride. Scenarios assuming excess particulate chloride resulted in peak ground-level ClNO2 concentration increases of up to 3200 pptv compared to a base case scenario without excess particulate chloride.[92] More recently, modelling of ClNO2 chemistry across the continental USA revealed monthly mean 8-h O3 increases of 1–2 ppbv (3–4 %), with maximum daily 8-h maximum increases of up to 13 ppbv in the north-eastern United States. Resulting reductions in total nitrate ranged from 11 to 21 %. This study used a single domain of 12-km grid cells.[94] Regional modelling of the South Coast Air Basin (SOCAB) suggests that sea salt-derived chlorine chemistry reduces weekend ozone levels while increasing average weekday peaks, thus reducing the difference between the two.[99] A separate study in the SOCAB[24] included several chlorine reaction mechanisms (Reactions 10, 11, 20–22, 28–30) and found a maximum 1-h ozone enhancement of 12.7 ppbv with a corresponding peak in Cl2 concentrations of 12 pptv. In both of these cases, 5-km grid cells were used.[24,99] A summary of the results from the studies discussed above is shown in Table 6.

|
The concentrations predicted from these modelling studies correspond well with documented observations of reactive chlorine. However, in order to model chlorine radical concentrations at magnitudes that are consistent with observed concentrations, modellers must assume rates for the most likely heterogeneous mechanisms. Along with uncertainties in anthropogenic and natural source strength approximations, the values of these rate parameters in heterogeneous reactions remain a gap in the current understanding of urban atmospheric chlorine chemistry.
Critical gaps in current understanding
This section addresses two primary areas of uncertainty in the understanding of atmospheric chlorine chemistry. The first area of uncertainty is accurate estimation of the parameters used to model heterogeneous chlorine chemistry. The parameters involved in the heterogeneous ClNO2 mechanism that was described in previous sections is included as a case study of the range of predictions that can result from application of models, employing reaction rate parameters within accepted ranges. The second area of uncertainty is the issue of the chlorine emissions sources driving the chemistry.
Sensitivity analysis of heterogeneous ClNO2 production
The parameters that must be known to model heterogeneous reactions include[23]:
-
Particle surface area.
-
The phase(s) in or on which the reaction is taking place.
-
Chemical composition of the phases.
-
Concentrations of species involved in the reaction.
-
A reactive uptake coefficient for the gas species involved.
These physical parameters can be lumped into a single rate constant.[23,95]

Here, γobs is the observed reactive uptake coefficient, ω is the average molecular velocity of the reactant and A is the surface area density of particles (cm2 cm–3). The reactive uptake coefficient is the number of molecules lost through the surface relative to the number of molecules hitting the surface and is a unit-less parameter.[57] In addition to these parameters, the fractional yield of the product can be included on the right hand side of Eqn 48 when competitive reaction pathways are present within the condensed phase.
A wide variety of yield and reactive uptake values for reactions producing ClNO2 have been reported in the literature.[30,36,46–48,51,56,80,95,100–103] The range of heterogeneous reaction rate parameters reported in the literature was used to develop a range for the values of the reactive uptake coefficient (γ) and the ClNO2 yield (YClNO2) for the reaction of N2O5 with particulate chloride. Previous regional modelling studies of this mechanism used limited, and typically fixed, parameter values.[91,92] One recent study[94] has implemented parameterisations[46,54,55] which account for influences from particulate species such as H2O and NO3– for continental-scale modelling within the USA. These parameterisations are shown in Eqns 49–51, where f and r subscripts indicate directionality of the reversible reactions that are referenced.[54,55]
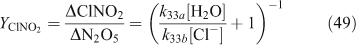
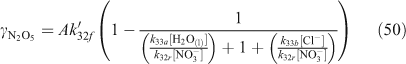
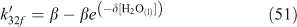
The ratio of k33b/k33a has been determined by several studies to range between 450 ± 100 and 836 ± 32.[40,50,54,55] For the other parameters involved, A is 3.2 × 10–8 s, β is (1.15 ± 0.3) × 105 s–1 and δ is (1.3 ± 0.5) × 10–1.[55] Although the introduction of parameterisations such as in Eqns 48–50 is an important advance over fixed parameter values, the parameterisation still assumes that heterogeneous rate parameters can be quantified with precision.
The objective of the sensitivity analysis described here is to quantify upper and lower limits to the effect of this mechanism with respect to O3 formation chemistry. Effects of varying VOC concentrations and composition as well as NOx availability were tested. Changes in peak O3 and ClNO2 concentrations between simulations were quantified and compared to a base case scenario lacking heterogeneous chemistry. Methods used in the sensitivity analyses are summarised in the Supplementary material.
Using well established parameter ranges, the heterogeneous reaction of N2O5 with aerosol chloride is able to reproduce ClNO2 concentrations at levels that have been observed. Depending on the combination of heterogeneous parameters, VOCs and NOx, the mechanism contributed to both peak ozone reduction as well as peak ozone increases. The range of effect was a –10.5 to 27 % change in peak O3 concentrations relative to a base case scenario with no heterogeneous reactions. The decreases in ozone typically resulted from low values of ClNO2 yield (0–15 %, depending on the amount and types of VOCs present). Relative decreases in O3 occurred in simulations including the heterogeneous chemistry due to the fact that the mechanism served as a NOx sink, particularly in cases with low ClNO2 yields. This NOx sink was not present in the simulations without heterogeneous chemistry. Combinations of yields above 25 % and reactive uptake coefficients greater than zero resulted in increases in peak ozone levels.
The effect of changing base VOC concentrations was also examined. Four scenarios with differing initial VOC concentrations were used: (1) 0 ppb C VOCs; (2) 300 ppb C VOCs; (3) 1000 ppb C VOCs and (4) 300 ppb C t-2-butene. The reason for isolating the effects of t-2-butene within a single scenario is that it rapidly reacts with NO3•, thus producing HNO3 instead of N2O5 through the reaction of NO3• with NO2. For all values of γ examined, the effect of increasing the value of the yield parameter from 0 to 100 % at a fixed γ was a linear increase in peak ozone concentrations. These results are summarised in the Supplementary material. Increasing the reactive uptake at a fixed yield value resulted in an intensification of the effect on peak ozone concentrations elicited by the value of the yield parameter. These, and additional, results are also shown in the Supplementary material.
The range of the effect that heterogeneous ClNO2 production has on peak ozone levels in the scenarios described above reflects the potential significance of this chemistry to ozone formation. However, the broad range of reported parameter values and the potential for both increases and decreases in ozone production from this mechanism highlight the importance of accurate parameter estimation. Maximum changes in peak O3 concentrations ranged from –10.5 to 27 %. ClNO2 formation by this mechanism resulted in concentrations up to 4.0 ppb at the highest combination of parameter values, with peak concentrations of 1.0–2.0 ppb being typical of more moderate parameter value combinations (e.g. 20–50 % yield and a reactive uptake of 0.03). It should be noted that this analysis excluded the consideration of effects from meteorology, mass transport and dilution. However, it serves in identifying an upper boundary of effects for the chemistry alone.
Uncertainty in emissions estimates
Traditionally, sources of molecular chlorine, and other inorganic sources of chlorine radicals, have not been included in photochemical modelling. Studies covered in the section on regional modelling[7,71,91,92,94,96–99] highlighted the effects of including chlorine emissions into the modelling of photochemical episodes. Inventories have been developed for both anthropogenic[104,105] and natural[1,32,106–108] sources. However, uncertainties exist within these inventories. Uncertainty in inventory development can stem from uncertainty in observations used to deduce source strengths and the methods or assumptions associated with extrapolating these measurements to larger scales.[1] Assumptions in emissions factors as well as approximations in their implementation into models are two additional sources of uncertainty.[106] In general, any uncertainty in the understanding of the biogeochemical processes involved in the natural chlorine cycle can lead to a corresponding uncertainty within inventories.[107]
Chang et al.[104] developed a comprehensive inventory for Houston, Texas, that evaluated industrial point sources, cooling tower use, water and wastewater treatment, swimming pool chlorination, tap water use, sea salt chloride and chlorinated organics as potential chlorine radical precursor sources in the region. The authors cite particularly high uncertainty in the emission rates of chlorinated organics and sea salt aerosols. Uncertainty in the form of volatile reactive chlorine species (HOCl and Cl2) produced by anthropogenic sources such as cooling tower operation has also been noted.[104] The contribution of inorganic chlorine from biomass burning, and particularly the anthropogenic contribution by this mechanism, is inherently uncertain due to the problem of accurately estimating the natural rate of fires in a scenario in which humans are not present. One study noted that although biomass burning could potentially contribute up to 25 % of total HCl emissions, the source strength is highly uncertain and not likely to be a significant source of particulate chloride globally.[106] Sinks and removal processes for chlorine species can also be a source of uncertainty.[107] Emissions estimates from coal combustion also suffer from uncertainty in the amount of chloride present in the coal.[105]
Summary
Most of the studies cited in this review have presumed that sea salt is the dominant source of atmospheric chlorine, however, markers of chlorine chemistry such as HCl and ClNO2, observed at inland locations such as Dallas–Fort Worth,[59] may suggest sources other than sea salt for atmospheric chlorine. An important step in improving the understanding of chlorine’s role in urban atmospheres would be to more accurately model the processes underlying emissions of the various chlorine radical precursors that have been observed. Improved emission inventories could be coupled with the increasing knowledge of heterogeneous processes in order to produce more meaningful computational predictions of the effect that chlorine species have in urban environments. As seen from the evidence related to enhanced ozone production from the presence of chlorine, this would prove extremely useful to researchers addressing issues in urban air quality.
Acknowledgement
Funding for this work was provided by the Texas Air Research Center.
References
[1] M. A. K. Khalil, R. M. Moore, D. B. Harper, J. M. Lobert, D. J. Erickson, V. Koropalov, W. T. Sturges, W. C. Keene, Natural emissions of chlorine-containing gases: reactive chlorine emissions inventory. J. Geophys. Res. 1999, 104, 8333.| Natural emissions of chlorine-containing gases: reactive chlorine emissions inventory.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjtFyltbY%3D&md5=622d92d7854f368fb912418c075faffbCAS |
[2] H. B. Singh, J. F. Kasting, Chlorine–hydrocarbon photochemistry in the marine troposphere and lower stratosphere. J. Atmos. Chem. 1988, 7, 261.
| Chlorine–hydrocarbon photochemistry in the marine troposphere and lower stratosphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXksVSqtg%3D%3D&md5=3651669869ccb231291c6560f4280c74CAS |
[3] T. E. Graedel, W. C. Keene, Tropospheric budget of reactive chlorine. Global Biogeochem. Cycles 1995, 9, 47.
| Tropospheric budget of reactive chlorine.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXktFGitLs%3D&md5=163cc328aeb9f917d79b1e22b920b659CAS |
[4] R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe, Evaluated kinetic and photochemical data for atmospheric chemistry: volume III – gas phase reactions of inorganic halogens. Atmos. Chem. Phys. 2007, 7, 981.
| Evaluated kinetic and photochemical data for atmospheric chemistry: volume III – gas phase reactions of inorganic halogens.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXjvVOhsLw%3D&md5=d6e227ebe1dfb1559d56714ba9c76c11CAS |
[5] S. P. Sander, R. R. Friedl, J. P. D. Abbatt, J. R. Barker, J. B. Burkholder, D. M. Golden, C. E. Kolb, M. J. Kurylo, G. K. Moortgat, P. H. Wine, R. E. Huie, V. L. Orkin, Chemical kinetics and photochemical data for use in atmospheric studies: evaluation number 17. JPL Publication 10-6 2011 (NASA Jet Propulsion Laboratory: Pasadena, CA) Available at http://jpldataeval.jpl.nasa.gov/pdf/JPL%2010-6%20Final%2015June2011.pdf [Verified 8 May 2013].
[6] R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe, T. J. Wallington, Evaluated kinetic and photochemical data for atmospheric chemistry: volume IV – gas phase reactions of organic halogen species. Atmos. Chem. Phys. 2008, 8, 4141.
| Evaluated kinetic and photochemical data for atmospheric chemistry: volume IV – gas phase reactions of organic halogen species.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlCnurvJ&md5=b68734802e036c33cf2306940459ee00CAS |
[7] S. Chang, D. T. Allen, Atmospheric chlorine chemistry in southeast texas: impacts on ozone formation and control. Environ. Sci. Technol. 2006, 40, 251.
| Atmospheric chlorine chemistry in southeast texas: impacts on ozone formation and control.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1GktLrI&md5=280ebbe143bf7d22a62db8c7c8a1f3ddCAS | 16433359PubMed |
[8] B. J. Finlayson-Pitts, Chlorine atoms as a potential tropospheric oxidant in the marine boundary layer. Res. Chem. Intermed. 1993, 19, 235.
| Chlorine atoms as a potential tropospheric oxidant in the marine boundary layer.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXlvVOntL4%3D&md5=52adbe7c016109977b9e1bef96c73156CAS |
[9] S. Chang, E. McDonald-Buller, Y. Kimura, G. Yarwood, J. Neece, M. Russell, P. Tanaka, D. Allen, Sensitivity of urban ozone formation to chlorine emission estimates. Atmos. Environ. 2002, 36, 4991.
| Sensitivity of urban ozone formation to chlorine emission estimates.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XnsFOrsbc%3D&md5=662c28149f34b023260ea7b91cf5766eCAS |
[10] R. T. Watson, Rate constants for reactions of ClOx of atmospheric interest. J. Phys. Chem. Ref. Data 1977, 6, 871.
| Rate constants for reactions of ClOx of atmospheric interest.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaE2sXlvVymtLk%3D&md5=69768b2bc436a6207a7a640124420310CAS |
[11] J. Stutz, B. T. Jobson, A. L. Sumner, B. K. Bailey, W. Vance, Impact of reactive halogen species on the air quality in california coastal areas, final report to the Coordinating Research Council, Inc. and the California Air Resources Board in fulfilment of contracts A-62-1 and A-62-2 and Agreement #05–307 2009 (California Air Resources Board: Sacramento, CA). Available at http://www.arb.ca.gov/research/apr/past/05-307.pdf [Verified 7 May 2013].
[12] O. W. Wingenter, D. R. Blake, N. J. Blake, B. C. Sive, F. S. Rowland, E. Atlas, F. Flocke, Tropospheric hydroxyl and atomic chlorine concentrations, and mixing timescales determined from hydrocarbon and halocarbon measurements made over the Southern Ocean. J. Geophys. Res. 1999, 104, 21 819.
| Tropospheric hydroxyl and atomic chlorine concentrations, and mixing timescales determined from hydrocarbon and halocarbon measurements made over the Southern Ocean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXmsFOnsLg%3D&md5=3ec1eaa5c84dc3f9185c25472f8347b0CAS |
[13] L. Wang, J. Arey, R. Atkinson, Reactions of chlorine atoms with a series of aromatic hydrocarbons. Environ. Sci. Technol. 2005, 39, 5302.
| Reactions of chlorine atoms with a series of aromatic hydrocarbons.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXkslGjtbo%3D&md5=bacd053a89c8a6d4d27cde71de3f5fddCAS | 16082960PubMed |
[14] L. Nelson, O. Rattigan, R. Neavyn, W. Sidebottom, J. Treacy, O. J. Nielsen, Absolute and relative rate constants for the reactions of hydroxyl radicals and chlorine atoms with a series of aliphatic alchohols and ethers at 298 K. Int. J. Chem. Kinet. 1990, 22, 1111.
| Absolute and relative rate constants for the reactions of hydroxyl radicals and chlorine atoms with a series of aliphatic alchohols and ethers at 298 K.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXotV2gtw%3D%3D&md5=3f6c39617a6701618226caaef0987720CAS |
[15] S. M. Aschmann, R. Atkinson, Rate constants for the gas-phase reactions of alkanes with Cl atoms at 296 ± 2 K. Int. J. Chem. Kinet. 1995, 27, 613.
| Rate constants for the gas-phase reactions of alkanes with Cl atoms at 296 ± 2 K.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXlslGmtbo%3D&md5=e5097de2ac1a09b36b14607de2fa5ae4CAS |
[16] O. Sokolov, M. D. Hurley, T. J. Wallington, E. W. Kaiser, J. Platz, O. J. Nielsen, F. Berho, M.-T. Rayez, R. Lesclaux, Kinetics and mechanism of the gas-phase reaction of Cl atoms with benzene. J. Phys. Chem. A 1998, 102, 10 671.
| Kinetics and mechanism of the gas-phase reaction of Cl atoms with benzene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXns1ymuro%3D&md5=19482c213d82ce3d97d685188585bb82CAS |
[17] C. E. Canosa-Mas, H. R. Hutton-Squire, M. D. King, D. J. Stewart, K. C. Thompson, R. P. Wayne, Laboratory kinetic studies of the reactions of Cl atoms with species of biogenic origin: Δ3-carene, isoprene, methacrolein and methyl vinyl ketone. J. Atmos. Chem. 1999, 34, 163.
| Laboratory kinetic studies of the reactions of Cl atoms with species of biogenic origin: Δ3-carene, isoprene, methacrolein and methyl vinyl ketone.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXlvV2kurk%3D&md5=77df57092ca4d8503bbffbcd4332224fCAS |
[18] T. Nordmeyer, W. Wang, M. L. Ragains, B. J. Finlayson-Pitts, C. W. Spicer, R. A. Plastridge, Unique products of the reaction of isoprene with atomic chlorine: potential markers of chlorine atom chemistry. Geophys. Res. Lett. 1997, 24, 1615.
| Unique products of the reaction of isoprene with atomic chlorine: potential markers of chlorine atom chemistry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXkslOltL4%3D&md5=60b56fcd0338ef920b4f051e068e04d0CAS |
[19] M. L. Ragains, B. J. Finlayson-Pitts, Kinetics and mechanism of the reaction of Cl atoms with 2-methyl-1,3-butadiene (isoprene) at 298 K. J. Phys. Chem. A 1997, 101, 1509.
| Kinetics and mechanism of the reaction of Cl atoms with 2-methyl-1,3-butadiene (isoprene) at 298 K.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXotVCluw%3D%3D&md5=74a538ef98b9f9850a0b844eae1e4413CAS |
[20] D. D. Riemer, E. C. Apel, Confirming the presence and extent of oxidation by Cl in the Houston, Texas Urban area using specific isoprene oxidation products as tracers, final report to Texas Natural Resource Conservation Commission, Contract #582034743 2002 (Texas Commission on Environmental Quality (TCEQ): Austin, TX). Available at http://tceq.info/assets/public/implementation/air/am/contracts/reports/oth/ConfirmingPresenceandExtentOfOxidationByCI.pdf [Verified 7 May 2013].
[21] W. Wang, B. J. Finlayson-Pitts, Unique markers of chlorine atom chemistry in coastal urban areas: the reaction with 1,3-butadiene in air at room temperature. J. Geophys. Res. 2001, 106, 4939.
| Unique markers of chlorine atom chemistry in coastal urban areas: the reaction with 1,3-butadiene in air at room temperature.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXltlSitb4%3D&md5=2c94b11b09506b4138bd750051cca626CAS |
[22] A. McCulloch, M. L. Aucott, C. M. Benkovits, T. E. Graedel, G. Kleiman, P. M. Midgley, Y.-F. Li, Global emissions of hydrogen chloride and chloromethane from coal combustion, inceneration and industrial activities: reactive chlorine emissions inventory. J. Geophys. Res. 1999, 104, 8391.
| Global emissions of hydrogen chloride and chloromethane from coal combustion, inceneration and industrial activities: reactive chlorine emissions inventory.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjtFylur8%3D&md5=f9a04bfaecc200630b012d40194d3704CAS |
[23] A. R. Ravishankara, Heterogeneous and multiphase chemistry in the troposphere. Science 1997, 276, 1058.
| Heterogeneous and multiphase chemistry in the troposphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXjt12ls7c%3D&md5=ed0c61ca55e2ae5650ddb05869dfd903CAS |
[24] E. M. Knipping, D. Dabdub, Impact of chlorine emissions from sea-salt aerosol on coastal urban ozone. Environ. Sci. Technol. 2003, 37, 275.
| Impact of chlorine emissions from sea-salt aerosol on coastal urban ozone.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xpt1yntL4%3D&md5=597b59aa2f47a9808c0a0bade442f818CAS | 12564898PubMed |
[25] E. Eriksson, The yearly circulation of chloride and sulfur in nature; meteorological, geochemical and pedological implications. Part I. Tellus 1959, II, 375.
[26] P. Brimblecombe, S. L. Clegg, The solubility and behaviour of acid gases in the marine aerosol. J. Atmos. Chem. 1988, 7, 1.
| The solubility and behaviour of acid gases in the marine aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXlsFGhtbg%3D&md5=70950034d0d78047b9cafb99334b59d2CAS |
[27] E. E. Gard, M. J. Kleeman, D. S. Gross, L. S. Hughes, J. O. Allen, B. D. Morrical, D. P. Fergenson, T. Dienes, M. E. Gälli, R. J. Johnson, G. R. Cass, K. A. Prather, Direct observation of heterogeneous chemistry in the atmosphere. Science 1998, 279, 1184.
| Direct observation of heterogeneous chemistry in the atmosphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXhtlSjsbs%3D&md5=d847b6f8e6ac5d1d1f6e6c777eb716b1CAS | 9469803PubMed |
[28] W. T. Sturges, L. A. Barrie, Chlorine, bromine and iodine in arctic aerosols. Atmos. Environ. 1988, 22, 1179.
| Chlorine, bromine and iodine in arctic aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXkvFyjt7s%3D&md5=5a3558fed31d7ed7c3729a5b9a8257d2CAS |
[29] T. D. Saul, M. P. Tolocka, M. V. Johnston, Reactive uptake of nitric acid onto sodium chloride aerosols across a wide range of relative humidities. J. Phys. Chem. A 2006, 110, 7614.
| Reactive uptake of nitric acid onto sodium chloride aerosols across a wide range of relative humidities.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XkvFGltro%3D&md5=bc82c2aa84afbacb15c72f7ac0628342CAS | 16774205PubMed |
[30] D. O. De Haan, B. J. Finlayson-Pitts, Knudsen cell studies of the reaction of gaseous nitric acid with synthetic sea salt at 298 K. J. Phys. Chem. A 1997, 101, 9993.
| Knudsen cell studies of the reaction of gaseous nitric acid with synthetic sea salt at 298 K.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXhs1ahsw%3D%3D&md5=24436d7a783952bc0655e9380cfe64c1CAS |
[31] W. C. Keene, R. Sander, A. A. P. Pszenny, R. Vogt, P. J. Crutzen, J. N. Galloway, Aerosol pH in the marine boundary layer: a review and model evaluation. J. Aerosol Sci. 1998, 29, 339.
| Aerosol pH in the marine boundary layer: a review and model evaluation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXis1aqsLk%3D&md5=7fa969c0d12a86bba1405f4c4d91d174CAS |
[32] D. J. Erickson, C. Seuzaret, W. C. Keene, S. L. Gong, A general circulation model based calculation of HCl and ClNO2 production from sea salt dechlorination: reactive chlorine emissions inventory. J. Geophys. Res. 1999, 104, 8347.
| A general circulation model based calculation of HCl and ClNO2 production from sea salt dechlorination: reactive chlorine emissions inventory.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjtFyltbc%3D&md5=64b16b47b2d8d926fa71f91b45c11a9aCAS |
[33] C. Volpe, M. Wahlen, A. A. P. Pszenny, A. J. Spivack, Chlorine isotopic composition of marine aerosols: implications for the release of reactive chlorine and HCl cycling rates. Geophys. Res. Lett. 1998, 25, 3831.
| Chlorine isotopic composition of marine aerosols: implications for the release of reactive chlorine and HCl cycling rates.Crossref | GoogleScholarGoogle Scholar |
[34] O. W. Wingenter, M. K. Kubo, N. J. Blake, T. W. Smith, D. R. Blake, F. S. Rowland, Hydrocarbon and halocarbon measurements as photochemical and dynamical indicators of atmospheric hydroxyl, atomic chlorine, and vertical mixing during Lagrangian flights. J. Geophys. Res. 1996, 101, 4331.
| Hydrocarbon and halocarbon measurements as photochemical and dynamical indicators of atmospheric hydroxyl, atomic chlorine, and vertical mixing during Lagrangian flights.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XhslCrtr8%3D&md5=f4ccf745aeff0cfaf2f0181f1d3f978bCAS |
[35] J. D. Raff, B. Njegic, W. L. Chang, M. S. Gordon, D. Dabdub, R. B. Gerber, B. J. Finlayson-Pitts, Chlorine activation indoors and outdoors via surface-mediated reactions of nitrogen oxides with hydrogen chloride. Proc. Natl. Acad. Sci. USA 2009, 106, 13 647.
[36] F. E. Livingston, B. J. Finlayson-Pitts, The reaction of gaseous N2O5 with solid NaCl at 298 K: estimated lower limit to the reaction probability and its potential role in tropospheric and stratospheric chemistry. Geophys. Res. Lett. 1991, 18, 17.
| The reaction of gaseous N2O5 with solid NaCl at 298 K: estimated lower limit to the reaction probability and its potential role in tropospheric and stratospheric chemistry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXhvV2nu7Y%3D&md5=1641d77d3920adbdf6dd440faff3f651CAS |
[37] B. J. Finlayson-Pitts, M. J. Ezell, J. N. Pitts, Formation of chemically active chlorine compounds by reactions of atmospheric NaCl particles with gaseous N2O5 and ClONO2. Nature 1989, 337, 241.
| Formation of chemically active chlorine compounds by reactions of atmospheric NaCl particles with gaseous N2O5 and ClONO2.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXhtl2jsLc%3D&md5=8ec9e67f4eea413754d84a5eab363728CAS |
[38] B. J. Finlayson-Pitts, The tropospheric chemistry of sea salt: a molecular-level view of the chemistry of NaCl and NaBr. Chem. Rev. 2003, 103, 4801.
| The tropospheric chemistry of sea salt: a molecular-level view of the chemistry of NaCl and NaBr.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXosVSnt78%3D&md5=0dd6e9c13b899591a1d34b120fca571dCAS | 14664634PubMed |
[39] M. J. Rossi, Atmospheric pollution: the role of heterogeneous chemical reactions. Chimia (Aarau) 1996, 50, 199.
| 1:CAS:528:DyaK28XjsVWmtLs%3D&md5=8562f08d22b58bd6d9de3504c7e6f777CAS |
[40] W. Behnke, C. George, V. Scheer, C. Zetzsch, Production and decay of ClNO2 from the reaction of gaseous N2O5 with NaCl solution: bulk and aerosol experiments. J. Geophys. Res. 1997, 102, 3795.
| Production and decay of ClNO2 from the reaction of gaseous N2O5 with NaCl solution: bulk and aerosol experiments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXhvFClsLY%3D&md5=e894f0339dbf28b93c5b3d93f7dff00fCAS |
[41] E. M. Knipping, M. J. Lakin, K. L. Foster, P. Jungwirth, D. J. Tobias, R. B. Gerber, D. Dabdub, B. J. Finlayson-Pitts, Experiments and simulations of ion-enhanced interfacial chemistry on aqueous NaCl aerosols. Science 2000, 288, 301.
| Experiments and simulations of ion-enhanced interfacial chemistry on aqueous NaCl aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXislersr4%3D&md5=2628ca6a82b90f3db9a91ac88c59a687CAS | 10764637PubMed |
[42] E. M. Knipping, D. Dabdub, Modeling Cl2 formation from aqueous NaCl particles: evidence for interfacial reactions and importance of Cl2 decomposition in alkaline solution. J. Geophys. Res. 2002, 107, 4360.
| Modeling Cl2 formation from aqueous NaCl particles: evidence for interfacial reactions and importance of Cl2 decomposition in alkaline solution.Crossref | GoogleScholarGoogle Scholar |
[43] J. N. Crowley, M. Ammann, R. A. Cox, R. G. Hynes, M. E. Jenkin, A. Mellouki, M. J. Rossi, J. Troe, T. J. Wallington, Evaluated kinetic and photochemical data for atmospheric chemistry: volume V – heterogeneous reactions on solid substrates. Atmos. Chem. Phys. 2010, 10, 9059.
| Evaluated kinetic and photochemical data for atmospheric chemistry: volume V – heterogeneous reactions on solid substrates.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXktlamsrc%3D&md5=6300f575ed1c8010f4202cd0d61140a3CAS |
[44] R. C. Sullivan, S. A. Guazzotti, D. A. Sodeman, Y. Tang, G. R. Carmichael, K. A. Prather, Mineral dust is a sink for chlorine in the marine boundary layer. Atmos. Environ. 2007, 41, 7166.
| Mineral dust is a sink for chlorine in the marine boundary layer.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtV2qsbfN&md5=75466d9348dfbbc9e40c78c7e5ddbbbaCAS |
[45] W. C. Keene, A. A. P. Pszenny, D. J. Jacob, R. A. Duce, J. N. Galloway, J. J. Schultz-Tokos, H. Sievering, J. F. Boatman, The geochemical cycling of reactive chlorine through the marine troposphere. Global Biogeochem. Cycles 1990, 4, 407.
| The geochemical cycling of reactive chlorine through the marine troposphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXksFertrc%3D&md5=6d063dfbf2a1b490c82932374099de5dCAS |
[46] J. A. Thornton, J. P. Kercher, T. P. Riedel, N. L. Wagner, J. Cozic, J. S. Holloway, W. P. Dubé, G. M. Wolfe, P. K. Quinn, A. M. Middlebrook, B. Alexander, S. S. Brown, A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry. Nature 2010, 464, 271.
| A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXjtFSmu78%3D&md5=0539b1b73ca9867628bdc24941c62cb8CAS | 20220847PubMed |
[47] F. Schweitzer, P. Mirabel, C. George, Multiphase chemistry of N2O5, ClNO2, and BrNO2. J. Phys. Chem. A 1998, 102, 3942.
| Multiphase chemistry of N2O5, ClNO2, and BrNO2.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXivVarsbo%3D&md5=c1a0696b13cb37f7c51ef237c44a790cCAS |
[48] M. Aldener, S. S. Brown, H. Stark, E. J. Williams, B. M. Lerner, W. C. Kuster, P. D. Goldan, P. K. Quinn, T. S. Bates, F. C. Fehsenfeld, A. R. Ravishankara, Reactivity and loss mechanisms of NO3 and N2O5 in a polluted marine environment: results from in situ measurements during New England Air Quality Study 2002. J. Geophys. Res. 2006, 111, D23S73.
| Reactivity and loss mechanisms of NO3 and N2O5 in a polluted marine environment: results from in situ measurements during New England Air Quality Study 2002.Crossref | GoogleScholarGoogle Scholar |
[49] J. M. Roberts, H. D. Osthoff, S. S. Brown, A. R. Ravishankara, N2O5 oxidizes chloride to Cl2 in acidic atmospheric aerosol. Science 2008, 321, 1059.
| N2O5 oxidizes chloride to Cl2 in acidic atmospheric aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtVSitrnE&md5=2d2527dc0d8c8a32f9cadd03c830480eCAS | 18599742PubMed |
[50] S. S. Brown, W. P. Dubé, H. Fuchs, T. B. Ryerson, A. G. Wollny, C. A. Brock, R. Bahreini, A. M. Middlebrook, J. A. Neuman, A. Atlas, J. M. Roberts, H. D. Osthodd, M. Trainer, F. C. Fehsenfeld, A. R. Ravishankara, Reactive uptake coefficients for N2O5 determined from aircraft measurements during the Second Texas Air Quality Study: comparison to current model parameterizations. J. Geophys. Res. 2009, 114, D00F10.
| Reactive uptake coefficients for N2O5 determined from aircraft measurements during the Second Texas Air Quality Study: comparison to current model parameterizations.Crossref | GoogleScholarGoogle Scholar |
[51] J. M. Laux, J. C. Hemminger, B. J. Finlayson-Pitts, X-ray photoelectron spectroscopic studies of the heterogeneous reaction of gaseous nitric acid with sodium chloride: kinetics and contribution to the chemistry of the marine troposphere. Geophys. Res. Lett. 1994, 21, 1623.
| X-ray photoelectron spectroscopic studies of the heterogeneous reaction of gaseous nitric acid with sodium chloride: kinetics and contribution to the chemistry of the marine troposphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXhtVymsrw%3D&md5=f3aa36bf53fef0adf8650ae4f955db4eCAS |
[52] W. Behnke, C. Zetzsch, Heterogeneous photochemical formation of Cl atoms from NaCl aerosol, NOx and ozone. J. Aerosol Sci. 1990, 21, S229.
| Heterogeneous photochemical formation of Cl atoms from NaCl aerosol, NOx and ozone.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXktFCnu74%3D&md5=c56be5ef426acdb285c67aaaa19c944fCAS |
[53] C. Zetzsch, W. Behnke, Heterogeneous photochemical sources of atomic Cl in the troposphere. Ber. Bunsenges. Phys. Chem. 1992, 96, 488.
| Heterogeneous photochemical sources of atomic Cl in the troposphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XisVGjtrY%3D&md5=015cc7166c4775900cb898d644a3f9b8CAS |
[54] J. M. Roberts, H. D. Osthoff, S. S. Brown, A. R. Ravishankara, D. Coffman, P. Quinn, T. Bates, Laboratory studies of products of N2O5 uptake on Cl– containing substrates. Geophys. Res. Lett. 2009, 36, L20808.
| Laboratory studies of products of N2O5 uptake on Cl– containing substrates.Crossref | GoogleScholarGoogle Scholar |
[55] T. H. Bertram, J. A. Thornton, Toward a general parameterization of N2O5 reactivity on aqueous particles: the competing effects of particle liquid water, nitrate and chloride. Atmos. Chem. Phys. 2009, 9, 8351.
| Toward a general parameterization of N2O5 reactivity on aqueous particles: the competing effects of particle liquid water, nitrate and chloride.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhs1Gjsbw%3D&md5=bc71da95dc773c1d4edc2c5f0ab93ac2CAS |
[56] J. A. Thornton, J. P. D. Abbatt, N2O5 reaction on submicron sea salt aerosol: kinetics, products, and the effect of surface active organics. J. Phys. Chem. A 2005, 109, 10 004.
| N2O5 reaction on submicron sea salt aerosol: kinetics, products, and the effect of surface active organics.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtV2nurvP&md5=b5f2afb7bef5c5a02914532f0e3c6196CAS |
[57] A. Frenzel, V. Scheer, R. Sikorski, Ch. George, W. Behnke, C. Zetzsch, Heterogeneous interconversion reactions of BrNO2, ClNO2, Br, and Cl2. J. Phys. Chem. A 1998, 102, 1329.
| Heterogeneous interconversion reactions of BrNO2, ClNO2, Br, and Cl2.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXntFOktA%3D%3D&md5=94046ecc310a50412df2df61d921ccdfCAS |
[58] S. Pechtl, R. von Glasow, Reactive chlorine in the marine boundary layer in the outflow of polluted continental air: a model study. Geophys. Res. Lett. 2007, 34, L11813.
| Reactive chlorine in the marine boundary layer in the outflow of polluted continental air: a model study.Crossref | GoogleScholarGoogle Scholar |
[59] R. Griffin, J. Dibb, B. Lefer, A. Steiner, Surface measurements and one-dimensional modeling related to ozone formation in the suburban Dallas–Fort Worth area, final report for AQRP Project UTA10–000875-RICE-RP24–TO2 2011 (Texas Commission on Environmental Quality (TCEQ): Austin, TX).
[60] B. R. Appel, Y. Tokiwa, V. Povard, E. L. Kothny, The measurement of atmospheric hydrochloric acid in southern California. Atmos. Environ. 1991, 25A, 525.
| 1:CAS:528:DyaK3MXhsV2it7g%3D&md5=947050052c505733de835eb84991eb7cCAS |
[61] R. M. Harrison, A. G. Allen, Measurements of atmospheric HNO3, HCl and associated species on a small network in eastern England. Atmos. Environ. 1990, 24A, 369.
| 1:CAS:528:DyaK3cXit1anu7k%3D&md5=3b961e334c5828eb98a922b568557984CAS |
[62] W. John, S. M. Wall, J. L. Ondo, A new method for nitric acid and nitrate aerosol measurement using the dichotomous sampler. Atmos. Environ. 1988, 22, 1627.
| A new method for nitric acid and nitrate aerosol measurement using the dichotomous sampler.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1cXlslGmsrw%3D&md5=a85b1ec65cc723f94dffb9f7c7fdabc7CAS |
[63] P. Matusca, B. Schwarz, K. Bächmann, Measurements of diurnal concentration variations of gaseous HCl in air in the sub-nanogram range. Atmos. Environ. 1984, 18, 1667.
| Measurements of diurnal concentration variations of gaseous HCl in air in the sub-nanogram range.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2cXlsFKltb4%3D&md5=6365cc5760b0f369d3990a9c28d98154CAS |
[64] W. C. Keene, J. R. Maben, A. A. P. Pszenny, J. N. Galloway, Measurement technique for inorganic chlorine gases in the marine boundary layer. Environ. Sci. Technol. 1993, 27, 866.
| Measurement technique for inorganic chlorine gases in the marine boundary layer.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXitFymtbY%3D&md5=914c91908624148c3c3d8a58c9472542CAS |
[65] A. A. P. Pszenny, W. C. Keene, D. J. Jacob, S. Fan, J. R. Maben, M. P. Zetwo, M. Springer-Young, J. N. Galloway, Evidence of inorganic chlorine gases other than hydrogen chloride in marine surface air. Geophys. Res. Lett. 1993, 20, 699.
| Evidence of inorganic chlorine gases other than hydrogen chloride in marine surface air.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3sXmsFCjsLg%3D&md5=d6416b09511306298125fb906db695eeCAS |
[66] C. W. Spicer, E. G. Chapman, B. J. Finlayson-Pitts, R. A. Plastridge, J. M. Hubbe, J. D. Fast, C. M. Berkowitz, Unexpectedly high concentrations of molecular chlorine in coastal air. Nature 1998, 394, 353.
| Unexpectedly high concentrations of molecular chlorine in coastal air.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXkvFKnsb4%3D&md5=2283a7013d688f66ec140f19a3995c22CAS |
[67] A. A. P. Pszenny, J. Moldanova, W. C. Keene, R. Sander, J. R. Maben, M. Martinez, P. J. Crutzen, D. Perner, R. G. Prinn, Halogen cycling and aerosol pH in the Hawaiian marine boundary layer. Atmos. Chem. Phys. 2004, 4, 147.
| Halogen cycling and aerosol pH in the Hawaiian marine boundary layer.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXjs1yit7Y%3D&md5=cb50ead68c96cc8610dc1578b69c1617CAS |
[68] B. D. Finley, E. S. Saltzman, Observations of Cl2, Br2, and I2 in coastal marine air. J. Geophys. Res. 2008, 113, D21301.
| Observations of Cl2, Br2, and I2 in coastal marine air.Crossref | GoogleScholarGoogle Scholar |
[69] B. D. Finley, E. S. Saltzman, Measurement of Cl2 in coastal urban air. Geophys. Res. Lett. 2006, 33, L11809.
| Measurement of Cl2 in coastal urban air.Crossref | GoogleScholarGoogle Scholar |
[70] Ø. Hov, The Effect of chlorine on the formation of photochemical oxidants in southern Telemark, Norway. Atmos. Environ. 1985, 19, 471.
| The Effect of chlorine on the formation of photochemical oxidants in southern Telemark, Norway.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXit1Ghtbs%3D&md5=532451be99e243590eecc401d5d3ba3fCAS |
[71] P. L. Tanaka, D. D. Riemer, S. Chang, G. Yarwood, E. C. McDonald-Buller, E. C. Apel, J. J. Orlando, P. J. Silva, J. L. Jiminez, M. R. Canagaratna, J. d. Neece, C. B. Mullins, D. T. Allen, Direct evidence for chlorine-enhanced urban ozone formation in Houston, Texas. Atmos. Environ. 2003a, 37, 1393.
| Direct evidence for chlorine-enhanced urban ozone formation in Houston, Texas.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXhslGqt7w%3D&md5=4c73484d1b9c29282f551dfb0a42e1a1CAS |
[72] K. W. Oum, M. J. Lakin, D. O. DeHaan, T. Brauers, B. J. Finlayson-Pitts, Formation of molecular chlorine from the photolysis of ozone and aqueous sea-salt particles. Science 1998, 279, 74.
| Formation of molecular chlorine from the photolysis of ozone and aqueous sea-salt particles.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXivF2nsg%3D%3D&md5=66a6e34b81b88e95b1298afbc4a49cd0CAS | 9417027PubMed |
[73] G. A. Impey, P. B. Shepson, D. R. Hastie, L. A. Barrie, Measurement technique for the determination of photolyzable chlorine and bromine in the atmosphere. J. Geophys. Res. 1997a, 102, 15 999.
| Measurement technique for the determination of photolyzable chlorine and bromine in the atmosphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXlsVKiu74%3D&md5=bb9d95bd28d9df6e257f092c61dbad7bCAS |
[74] G. A. Impey, P. B. Shepson, D. R. Hastie, L. A. Barrie, K. G. Anlauf, Measurements of photolyzable chlorine and bromine during the Polar Sunrise Experiment 1995. J. Geophys. Res. 1997, 102, 16 005.
| Measurements of photolyzable chlorine and bromine during the Polar Sunrise Experiment 1995.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXlsVKiu78%3D&md5=8fb4693d037d77757273c93457317168CAS |
[75] J. R. Maben, W. C. Keene, A. A. P. Pszenny, J. N. Galloway, Volatile inorganic Cl in surface air over eastern North America. Geophys. Res. Lett. 1995, 22, 3513.
| Volatile inorganic Cl in surface air over eastern North America.Crossref | GoogleScholarGoogle Scholar |
[76] W. Behnke, C. Zetzsch, Heterogeneous formation of chlorine atoms from various aerosols in the presence of O3 and HCl. J. Aerosol Sci. 1989, 20, 1167.
| Heterogeneous formation of chlorine atoms from various aerosols in the presence of O3 and HCl.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK3MXhsV2itb0%3D&md5=d73cf090b3f8e6fc0ce4af23c9af8961CAS |
[77] H. B. Singh, G. L. Gregory, B. Anderson, E. Browell, G. W. Sachse, D. D. Davis, J. Crawford, J. D. Bradshaw, R. Talbot, D. R. Blake, D. Thornton, R. Newell, J. Merrill, Low ozone in the marine boundary layer of the tropical Pacific Ocean: photochemical loss, chlorine atoms, and entrainment. J. Geophys. Res. 1996, 101, 1907.
| Low ozone in the marine boundary layer of the tropical Pacific Ocean: photochemical loss, chlorine atoms, and entrainment.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28Xht1Cktrk%3D&md5=f97733036a1ca987d5ee8bf9a97569dcCAS |
[78] A. A. P. Pszenny, E. V. Fischer, R. S. Russo, B. C. Sive, R. K. Varner, Estimates of Cl atom concentrations and hydrocarbon kinetic reactivity in surface air at Appledore Island, Maine (USA), during International Consortium for Atmospheric Research on Transport and Transformation/Chemistry of Halogens at the Isles of Shoals. J. Geophys. Res. 2007, 112, D10S13.
| Estimates of Cl atom concentrations and hydrocarbon kinetic reactivity in surface air at Appledore Island, Maine (USA), during International Consortium for Atmospheric Research on Transport and Transformation/Chemistry of Halogens at the Isles of Shoals.Crossref | GoogleScholarGoogle Scholar |
[79] J. Stutz, R. Ackermann, J. D. Fast, L. Barrie, Atmospheric reactive chlorine and bromine at the Great Salt Lake, Utah. Geophys. Res. Lett. 2002, 29, 1380.
| Atmospheric reactive chlorine and bromine at the Great Salt Lake, Utah.Crossref | GoogleScholarGoogle Scholar |
[80] H. D. Osthoff, J. M. Roberts, A. R. Ravishankara, E. J. Williams, B. M. Lerner, R. Sommariva, T. S. Bates, D. Coffman, P. K. Quinn, J. E. Dibb, H. Stark, J. B. Burkholder, R. K. Talukdar, J. Meagher, F. C. Fehsenfeld, S. S. Brown, High levels of nitryl chloride in the polluted subtropical marine boundary layer. Nat. Geosci. 2008, 1, 324.
| High levels of nitryl chloride in the polluted subtropical marine boundary layer.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXltlCksLo%3D&md5=68935ffb235b2829f04652ca621debe1CAS |
[81] D. D. Parrish, D. T. Allen, T. S. Bates, M. Estes, F. C. Fehsenfeld, G. Feingold, R. Ferrare, R. M. Hardesty, J. F. Meagher, J. W. Nielsen-Gammon, R. B. Pierce, T. B. Ryerson, J. H. Seinfeld, E. J. Williams, Overview of the Second Texas Air Quality Study (TexAQS II) and the Gulf of Mexico Atmospheric Composition and Climate Study (GoMACCS). J. Geophys. Res. 2009, D7, D00F13.
| Overview of the Second Texas Air Quality Study (TexAQS II) and the Gulf of Mexico Atmospheric Composition and Climate Study (GoMACCS).Crossref | GoogleScholarGoogle Scholar |
[82] T. P. Riedel, T. H. Bertram, T. A. Crisp, E. J. Williams, B. M. Lerner, A. Vlasenko, S.-M. Li, J. Gilman, J. de Gouw, D. M. Bon, N. L. Wagner, S. S. Brown, J. A. Thornton, Nitryl chloride and molecular chlorine in the coastal marine boundary layer. Environ. Sci. Technol. 2012, 46, 10 463.
| Nitryl chloride and molecular chlorine in the coastal marine boundary layer.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XksVWqu7o%3D&md5=8b3656c692b277e29fc098e858953e4fCAS |
[83] L. H. Mielke, A. Furgeson, H. D. Osthoff, Observation of ClNO2 in mid-continental urban environment. Environ. Sci. Technol. 2011, 45, 8889.
| Observation of ClNO2 in mid-continental urban environment.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXht1Wks7bJ&md5=81ea4771e855a02f941d2211355ba6f1CAS | 21877701PubMed |
[84] J. P. Kercher, T. P. Riedel, J. A. Thornton, Chlorine activation by N2O5: simultaneous, in situ detection of ClNO2 and N2O5 by chemical ionization mass spectrometry. Atmos. Meas. Tech. 2009, 2, 193.
| Chlorine activation by N2O5: simultaneous, in situ detection of ClNO2 and N2O5 by chemical ionization mass spectrometry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtFSqtL7E&md5=2ed20783f639a05c82db87e5519024f4CAS |
[85] B. T. Jobson, H. Niki, Y. Yokouchi, J. Bottenheim, F. Hopper, R. Leaitch, Measurements of C2–C6 hydrocarbons during the Polar Sunrise 1992 experiment: evidence for Cl atom and Br atom chemistry. J. Geophys. Res. 1994, 99, 25 355.
| Measurements of C2–C6 hydrocarbons during the Polar Sunrise 1992 experiment: evidence for Cl atom and Br atom chemistry.Crossref | GoogleScholarGoogle Scholar |
[86] G. Yarwood, S. Rao, M. Yocke, G. Z. Whitten, Final report: updates to the carbon bond chemical mechanism: CB05, Report Prepared for US EPA RT-04-00675 2005 (Environ: Novato, CA).
[87] G. Z. Whitten, H. Hogo, J. P. Killus, The carbon-bond mechanism: a condensed kinetic mechanism for photochemical smog. Environ. Sci. Technol. 1980, 14, 690.
| The carbon-bond mechanism: a condensed kinetic mechanism for photochemical smog.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL3cXlvFWhs7w%3D&md5=4108570e07d510ea002366162cb4773dCAS | 22296476PubMed |
[88] P. L. Tanaka, D. T. Allen, E. C. McDonald-Buller, S. Chang, Y. Kimura, C. B. Mullins, G. Yarwood, J. D. Neece, Development of a chlorine mechanism for use in the Carbon Bond IV Chemistry Model. J. Geophys. Res. 2003b, 108, 4145.
| Development of a chlorine mechanism for use in the Carbon Bond IV Chemistry Model.Crossref | GoogleScholarGoogle Scholar |
[89] W. P. L. Carter, Development of the SAPRC-07 chemical mechanism and updated ozone reactivity scales, report to the California Air Resources Board Contracts 03-318, 06-408 and 07-730 2010 (California Air Resources Board: Sacramento, CA).
[90] G. Heo, Y. Kimura, E. McDonald-Buller, W. P. L. Carter, G. Yarwood, D. T. Allen, Modeling alkene chemistry using condensed mechanisms for conditions relevant to southeast Texas, USA. Atmos. Environ. 2010, 44, 5365.
| Modeling alkene chemistry using condensed mechanisms for conditions relevant to southeast Texas, USA.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVCmtLrK&md5=7082e2d2f600d666a49253840359e7d9CAS |
[91] H. Simon, Y. Kimura, G. McGaughey, D. T. Allen, S. S. Brown, H. D. Osthoff, J. M. Roberts, D. Byun, D. Lee, Modeling the impact of ClNO2 on ozone formation in the Houston area. J. Geophys. Res. 2009, 114, D00F03.
| Modeling the impact of ClNO2 on ozone formation in the Houston area.Crossref | GoogleScholarGoogle Scholar |
[92] H. Simon, Y. Kimura, G. McGaughey, D. T. Allen, S. S. Brown, D. Coffman, J. Dibb, H. D. Osthoff, P. Quinn, J. M. Roberts, G. Yarwood, S. Kemball-Cook, D. Byun, D. Lee, Modeling heterogeneous ClNO2 formation, chloride availability, and chlorine cycling in southeast Texas. Atmos. Environ. 2010, 44, 5476.
| Modeling heterogeneous ClNO2 formation, chloride availability, and chlorine cycling in southeast Texas.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsVCmtLvE&md5=44ccd5f36133336382f90c7c17ac2ecbCAS |
[93] M. Faraji, Y. Kimura, E. McDonald-Buller, D. Allen, Comparison of the carbon bond and SAPRC photochemical mechanisms under conditions relevant to southeast Texas. Atmos. Environ. 2008, 42, 5821.
| Comparison of the carbon bond and SAPRC photochemical mechanisms under conditions relevant to southeast Texas.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXosVaisb8%3D&md5=9506e63973dace5ba7a4c6cba79901ebCAS |
[94] G. Sarwar, H. Simon, P. Bhave, G. Yarwood, Examining the impact of heterogeneous nitryl chloride production on air quality across the United States. Atmos. Chem. Phys. 2012, 12, 6455.
| Examining the impact of heterogeneous nitryl chloride production on air quality across the United States.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC38XhslSnu7jM&md5=57bd6090ff3dd74c171402ed10bb78f7CAS |
[95] D. J. Jacob, Heterogeneous chemistry and tropospheric ozone. Atmos. Environ. 2000, 34, 2131.
| Heterogeneous chemistry and tropospheric ozone.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXitlaju7s%3D&md5=bdb6a46400d00ecf9847ef2ed43c6612CAS |
[96] L. Wang, T. Thompson, E. C. McDonald-Buller, A. Webb, D. T. Allen, Photochemical modeling of emissions trading of highly reactive volatile organic compounds in Houston, Texas. 1. Reactivity based trading and potential for ozone hot spot formation. Environ. Sci. Technol. 2007a, 41, 2095.
| Photochemical modeling of emissions trading of highly reactive volatile organic compounds in Houston, Texas. 1. Reactivity based trading and potential for ozone hot spot formation.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXitFWnsrY%3D&md5=48b8cde6ee569924f919b35f4eca9ca0CAS | 17438748PubMed |
[97] L. Wang, T. Thompson, E. C. McDonald-Buller, D. T. Allen, Photochemical modeling of emissions trading of highly reactive volatile organic compounds in Houston, Texas. 2. Incorporation of chlorine emissions. Environ. Sci. Technol. 2007b, 41, 2103.
| Photochemical modeling of emissions trading of highly reactive volatile organic compounds in Houston, Texas. 2. Incorporation of chlorine emissions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXitFWnsrc%3D&md5=e194821670b7fe1814c2fd2b51afb07aCAS | 17438749PubMed |
[98] G. Sarwar, P. B. Bhave, Modeling the effect of chlorine emissions on ozone levels over the eastern United States. J. Appl. Meteorol. Climatol. 2007, 46, 1009.
| Modeling the effect of chlorine emissions on ozone levels over the eastern United States.Crossref | GoogleScholarGoogle Scholar |
[99] A. Cohan, W. Chang, M. Carreras-Sosphedra, D. Dabdub, Influence of sea-salt activated chlorine and surface-mediated renoxification on the weekend effect in the South Coast Air Basin of California. Atmos. Environ. 2008, 42, 3115.
| Influence of sea-salt activated chlorine and surface-mediated renoxification on the weekend effect in the South Coast Air Basin of California.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXkvVemsL8%3D&md5=b1c5c21e2c0d52317245f934fcf70624CAS |
[100] W. Behnke, H.-U. Krüger, V. Scheer, C. Zetzsch, Formation of atomic Cl from sea spray via photolysis of nitryl chloride: determination of the sticking coefficient of N2O5 on NaCl aerosol. J. Aerosol Sci. 1991, 22, S609.
| Formation of atomic Cl from sea spray via photolysis of nitryl chloride: determination of the sticking coefficient of N2O5 on NaCl aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38XksFCqsrY%3D&md5=bc4da99ba96f0efbb6b9e6bfc498ccf6CAS |
[101] Ch. George, J. L. Ponche, Ph. Mirabel, W. Behnke, V. Scheer, C. Zetzsch, Study of the uptake of N2O5 by water and NaCl solutions. J. Phys. Chem. 1994, 98, 8780.
| Study of the uptake of N2O5 by water and NaCl solutions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXmtFCiurk%3D&md5=e8475b3d5cccf292045d1c8684c81cdfCAS |
[102] J. H. Hu, J. P. D. Abbatt, Reaction probabilities for N2O5 hydrolysis on sulfuric acid and ammonium sulfate aerosols at room temperature. J. Phys. Chem. A 1997, 101, 871.
| Reaction probabilities for N2O5 hydrolysis on sulfuric acid and ammonium sulfate aerosols at room temperature.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2sXksVChsQ%3D%3D&md5=b9f6f6b419bdf3ffb7f6af70649ef90aCAS |
[103] M. Schütze, H. Herrmann, Determination of phase transfer parameters for the uptake of HNO3, N2O5 and O3 on single aqueous drops. Phys. Chem. Chem. Phys. 2002, 4, 60.
| Determination of phase transfer parameters for the uptake of HNO3, N2O5 and O3 on single aqueous drops.Crossref | GoogleScholarGoogle Scholar |
[104] S. Chang, P. Tanaka, E. McDonald-Buller, D. T. Allen, Emission inventory for atomic chlorine precursors in Southeast Texas, report on contract 9880077600-18 between The University of Texas and the Texas Natural Resource Conservation Commission 2001 (Texas Commission on Environmental Quality (TCEQ): Austin, TX). Available at http://www.tceq.texas.gov/assets/public/implementation/air/am/contracts/reports/oth/EmissioninventoryForAtomicChlorinePrecursors.pdf [Verified 7 May 2013].
[105] A. McCulloch, M. L. Aucott, C. M. Benkovits, T. E. Graedel, G. Kleiman, P. M. Midgley, Y.-F. Li, Global emissions of hydrogen chloride and chloromethane from coal combustion, incineration and industrial activities: reactive chlorine emissions inventory. J. Geophys. Res. 1999, 104, 8391.
| Global emissions of hydrogen chloride and chloromethane from coal combustion, incineration and industrial activities: reactive chlorine emissions inventory.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjtFylur8%3D&md5=f9a04bfaecc200630b012d40194d3704CAS |
[106] J. M. Lobert, W. C. Keene, J. A. Logan, R. Yevich, Global chlorine emissions from biomass burning: reactive chlorine emissions inventory. J. Geophys. Res. 1999, 104, 8373.
| Global chlorine emissions from biomass burning: reactive chlorine emissions inventory.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjtFylur4%3D&md5=c0072a4f80f2d54b4aac0186e7bfdfd7CAS |
[107] W. C. Keene, M. A. K. Khalil, D. J. Erickson, A. McCulloch, T. E. Graedel, J. M. Lobert, M. L. Aucott, S. L. Gong, D. B. Harper, G. Kleiman, P. Midgley, R. M. Moore, C. Seuzaret, W. T. Sturges, C. M. Benkovitz, V. Koropalov, L. A. Barrie, Y. F. Li, Composite global emissions of reactive chlorine from anthropogenic and natural sources: reactive chlorine emissions inventory. J. Geophys. Res. 1999, 104, 8429.
| Composite global emissions of reactive chlorine from anthropogenic and natural sources: reactive chlorine emissions inventory.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjtFyluro%3D&md5=98258f9c4fdf89c6e8c599d444d7fd11CAS |
[108] A. Reff, P. V. Bhave, H. Simon, T. G. Pace, G. A. Pouliot, J. D. Mobley, M. Houyoux, Emissions inventory of PM2.5 trace elements across the united states. Environ. Sci. Technol. 2009, 43, 5790.
| Emissions inventory of PM2.5 trace elements across the united states.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotFKnt74%3D&md5=5d244ef557a4c2e0bb66977ba8f37146CAS | 19731678PubMed |
[109] B. J. Finlayson-Pitts, C. J. Keoshian, B. Buehler, A. A. Ezell, Kinetics of reaction of chlorine atoms with some biogenic organics. Int. J. Chem. Kinet. 1999, 31, 491.
| Kinetics of reaction of chlorine atoms with some biogenic organics.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXktFWqtbk%3D&md5=af09709f446b6494968b8d1465ff0d30CAS |
[110] J. Stutz, M. J. Ezell, A. A. Ezell, B. J. Finlayson-Pitts, Rate constants and kinetic isotope effects in the reactions of atomic chlorine with n-butane and simple alkenes at room temperature. J. Phys. Chem. A 1998, 102, 8510.
| Rate constants and kinetic isotope effects in the reactions of atomic chlorine with n-butane and simple alkenes at room temperature.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXmvFeqtrg%3D&md5=6e22e06400926de1763ad1a554959092CAS |
[111] K. J. Gill, R. A. Hites, Rate constants for the gas-phase reactions of the hydroxyl radical with isoprene, α- and β-pinene, and limonene as a function of temperature. J. Phys. Chem. A 2002, 106, 2538.
| Rate constants for the gas-phase reactions of the hydroxyl radical with isoprene, α- and β-pinene, and limonene as a function of temperature.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xht1ert70%3D&md5=ece4ab9fb24a79612795d6bd0d5ecc91CAS |
[112] R. Atkinson, D. L. Baulch, R. A. Cox, R. F. Hampson, J. A. Kerr, J. Troe, Evaluated kinetic and photochemical data for atmospheric chemistry: supplement III. Int. J. Chem. Kinet. 1989, 21, 115.
| Evaluated kinetic and photochemical data for atmospheric chemistry: supplement III.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL1MXhvFKksrw%3D&md5=7a3d78b09b5d7e06f036cee76a0d7b98CAS |
[113] S. M. Aschmann, J. Arey, R. Atkinson, Atmospheric chemistry of three C10 alkanes. J. Phys. Chem. A 2001, 105, 7598.
| Atmospheric chemistry of three C10 alkanes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXltFKnu70%3D&md5=fc92695532e3c7369f685105e189c942CAS |
[114] W. P. L. Carter, D. Luo, I. L. Malkina, J. A. Pierce, Environmental chamber studies of atmospheric reactivities of volatile organic compounds. Effects of varying ROG surrogate and NOx, final report to California Air Resources Board, Contract A032-096 1995 (California Air Resources Board: Sacramento, CA). Available at http://www.cert.ucr.edu/~carter/pubs/explrept.pdf [Verified 7 May 2013].
[115] J. A. Manion, R. E. Huie, R. D. Levin, D. R. Burgess Jr, V. L. Orkin, W. Tsang, W. S. McGivern, J. W. Hudgens, V. D. Knyazev, D. B. Atkinson, E. Chai, A. M. Tereza, C.-Y. Lin, T. C. Allison, W. G. Mallard, F. Westley, J. T. Herron, R. F. Hampson, D. H. Frizzell, NIST Chemical Kinetics Database, NIST Standard Reference Database 17, Version 7.0 (Web Version), Release 1.4.3, Data version 2008.12 2013 (National Institute of Standards and Technology: Gaithersburg, MD). Available at http://kinetics.nist.gov/ [Verified 7 May 2013].