

Role of sea salt aerosols in the formation of aromatic secondary organic aerosol: yields and hygroscopic properties
Ross Beardsley A , Myoseon Jang A C , Baber Ori A , Yunseok Im A , Carrie A. Delcomyn B and Ned Witherspoon BA Department of Environmental Engineering Sciences, University of Florida, PO Box 116450, Gainesville, FL 32611, USA.
B Science, Technology, Analysis and Simulation Department, Naval Surface Warfare Center, Panama City, FL 32407, USA.
C Corresponding author. Email: mjang@ufl.edu
Environmental Chemistry 10(3) 167-177 https://doi.org/10.1071/EN13016
Submitted: 24 January 2013 Accepted: 27 April 2013 Published: 28 June 2013
Environmental context. In the coastal and ocean environment, oil spills and ship movement can produce hazardous, organic aerosols. In this study, the role of sea salt in the formation processes of crude-oil-derived organic aerosols derived was explored, and it was found that sea salt can greatly increase the formation and growth of these toxic aerosols. Understanding of this process is crucial for evaluating the effect of oil spills and ship movements on air quality and human health.
Abstract. Dual, large (52 m3), outdoor chambers were used to investigate the effect of aerosol aqueous phase chemistry on the secondary organic aerosol (SOA) yields of the photooxidation products of aromatic hydrocarbons in the coastal environment. Toluene and 1,3,5-trimethylbenzene were photochemically oxidised in the presence and absence of inorganic seeds (sea salt aerosol (SSA) or NaCl) at low NOx conditions. Overall, the presence of SSA, which was shown to contain water even at low relative humidities (RHs), led to higher SOA yields than the presence of NaCl seeds and the seedless condition. The results suggest that SOA yields in the coastal environment will be higher than those produced in terrestrial environment. To study the effect of SOA formation on the chemical composition of SSA, inorganic species were measured using a particle-into-liquid-sampler coupled to an ion chromatograph. The hygroscopic properties of the SSA internally mixed with SOA were analysed using a Fourier-transform infrared spectrometer. The fresh SSA shows a weak phase transition whereas no clear phase transition appeared in the aged SSA. The depletion of Cl– due to the accommodation of nitric acid and carboxylic acids on the surface of SSA coincides with changes in aerosol hygroscopic properties.
Additional keywords: aerosol water content, toluene, 1,3,5-trimethylbenzene.
Introduction
Secondary organic aerosol (SOA), formed by the semi-volatile products of the atmospheric oxidation of volatile organic compounds (VOC), make up a significant fraction of the particulate matter in the atmosphere. Enormous effort has been devoted to the characterisation of SOA, the determination of the kinetic mechanisms of SOA formation and the development of a SOA model, but current SOA models, which are primarily built around partitioning theory, underestimate organic aerosol budgets suggesting that there are missing mechanisms of SOA formation.[1,2] These underestimations of SOA mass are likely caused by the failure of SOA models to account for aerosol phase chemistry involving atmospheric organic compounds.
Several recent studies have highlighted the importance of aerosol phase chemistry in SOA formation and growth. Jang and Kamens[3] discovered high molecular weight (MW) oligomer formation in the particle phase due to heterogeneous acid-catalysed reactions that lead to increased SOA formation. Since then, several other SOA formation mechanisms that help compensate for the underestimation of volatility models have been found by researchers. Blando and Turpin[4] proposed that aqueous chemistry within clouds plays an important role in SOA formation, hypothesising that organic gas can react within water droplets to form lower-volatility compounds. After cloud evaporation these compounds remain in hygroscopic aerosols to form SOA. Ervens et al.[5] and Lim et al.[6] found that cloud processing of soluble organic isoprene products by the OH radical leads to several carboxylic acids that form a considerable amount of SOA. In addition, Altieri et al.[7] reported that the aqueous reaction of pyruvic acid leads to the formation of oligomers.
More recently, the role of the aqueous phase of aerosols in SOA formation has been investigated. Volkamer et al.[8] noted hygroscopic aerosols as a sink for volatile organic gases after finding that measured glyoxal concentrations over Mexico City were lower than predictions of conservative models. Furthermore, Ervens et al.[9] found that glyoxal can form low volatility products through oligomerisation in the aqueous phase of aerosols. Glyoxal is much more volatile than compounds traditionally considered in partitioning models suggesting that aqueous phase chemistry can help account for higher SOA concentrations than model predictions.[8,10] Although it has been found that the aqueous phase reactions of organic vapours can lead to higher molecular weight products than the equivalent reactions in the gas phase, the role of the liquid phase of aerosols in SOA formation is still largely unknown.
Aromatic volatile organic compounds (AVOCs) make up ~15 % of the anthropogenic non-methane hydrocarbon budget[11] and produce a wide range of highly reactive, low volatility products that are more reactive than the parent compounds.[12] Although AVOCS, which are released during the combustion and evaporation of fuels, are primarily a concern in urban areas, accidental oil spills and ship movement emit these compounds into the coastal and ocean environment. In the case of an oil spill, a large amount of AVOCs quickly evaporate and form condensable organic matter (OM). Within 24 h, 25–30 % of the oil in a spill at sea can evaporate[13] creating high potential for SOA formation and growth.[14]
Once released into the coastal and marine environment, AVOCs are exposed to the gases and aerosols of the marine boundary layer (MBL). Sea salt aerosols (SSA) are the most abundant aerosols in the coastal atmosphere and thus act as pre-existing particulate matter for SOA formation. Unlike NaCl aerosols that have a clear phase transition (efflorescence relative humidity (ERH) and deliquescence relative humidity (DRH)), SSA have a very low ERH and have been shown to hold water at low RH.[15] Consequently, SSAs act as a medium for aqueous-phase reactions in marine environments. In order to accurately predict SOA yields in the marine and coastal environment, the role of SSA in SOA formation must be investigated.
In the present study, toluene and 1,3,5-trimethylbenzene (TMB) were photochemically oxidised in the presence of NOx with and without seed aerosols using dual outdoor chambers. Toluene and TMB were chosen as representative aromatic VOCs of oil because of their respective high SOA yield and high concentration in crude oil.[16] The SOA yields of each of the representative AVOCS were determined in the absence of seed, in the presence of SSA and in the presence of NaCl. NOx was injected into the chamber at a low concentration to more closely simulate the marine condition. The objectives of this study were to: (1) investigate the effect of SSA on aromatic SOA formation in the marine environment and (2) illustrate the chemical composition change of SSA due to the formation of SOA from the photochemical reaction of AVOC in the presence of NOx.
Experimental
Outdoor chamber experiments
Outdoor chamber experiments were conducted to study the SOA produced under ambient sunlight, and diurnal temperature and RH patterns. These experiments were conducted in the University of Florida Atmospheric Photochemical Outdoor Reactor (UF-APHOR) dual chamber facility (52 m3 each) located on the roof of the Black Hall of the Engineering School for Sustainable Infrastructure and Environment at the University of Florida. The aerosol and gaseous compounds produced in the chamber were directly carried through the sampling conduits from the chamber to the various analytical instruments in the Atmospheric Chemistry Laboratory allowing for characterisation of the photochemical oxidation products (Fig. 1). The dual chambers allow for the direct comparison of aromatic SOA yields in the presence and absence of varying inorganic seed aerosol under similar meteorological conditions. Before each experiment, the chambers were flushed with clean air for 24 h.
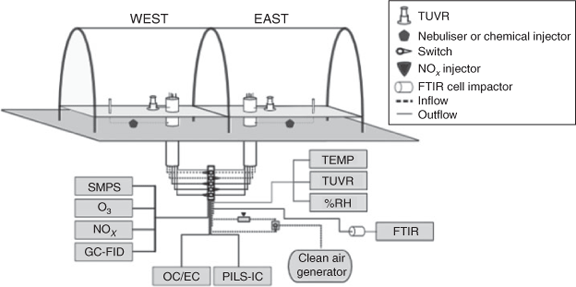
|
Either toluene (Reagent ACS, Acros, Thermo Fisher Scientific, New Jersey, USA) or TMB (99 % Extra Pure, Acros) was introduced to the chamber using a U-shaped glass chemical injector under a clean air stream. Approximately 300 ppb of carbon tetrachloride (Spectrophotometric grade 99+ %, Acros) was also added to the chamber as a non-reactive tracer to account for dilution of gaseous compounds. The saline water used in this project was collected from Jensen Beach, FL, at a water depth of at least 20 feet (~6 m) and then kept in a refrigerator at 4 °C. Prior to each chamber experiment, the sea salt water was diluted 20 times with deionised water and atomised into the chamber using a nebuliser as a wet seed (LC STAR, Pari Respiratory Equipment Inc., Midlothian, VA, USA). NOx (1 % nitrogen oxide, AirGas, Gainesville, FL) was introduced directly before sunrise. Experimental conditions for the outdoor APHOR runs are displayed in Table 1.

|
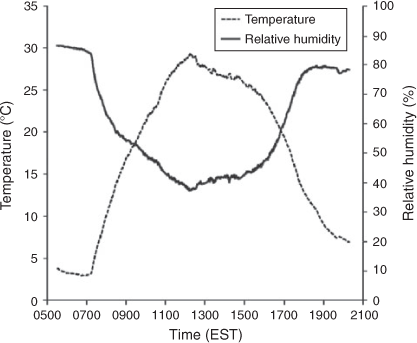
Temperature and humidity were monitored using a hygrometer integrated with a data acquisition system (CR1000 Measurement and Control System, Campbell Scientific, Logan, UT, USA). Solar UV-radiation ranging from 0.295 to 0.385 μm was measured using a total ultraviolet radiometer (TUVR, EPPLEY Laboratory Inc., Newport, RI, USA). In the outdoor APHOR runs, humidity and temperature follow natural, diurnal patterns. Initially RH is high but falls with increasing temperatures caused by the rising sun. As the sun sets in the evening the RH once again rises with decreasing temperature (see Fig. 2).
|
Indoor chamber experiments
In order to verify the results of the APHOR chamber runs under controlled temperature and RH, indoor chamber runs were performed in a 2-m3 Teflon film chamber equipped with UV-visible lamps (Solarc Systems Inc., FS40T12/UVB, Minesing, ON, Canada). The chamber was flushed with clean air before each experiment using clean air generators (Aadco Model 737, Cleves, OH, USA; Whatman Model 75-52, Florham Park, NJ). Humidity was controlled by passing clean air through a water bubbler. Temperature and humidity were measured using an electronic thermo-hygrometer (Hanna Instruments, Milan, Italy). The UV lamps were not turned on until the injected chemical concentrations stabilised. For the wet seeded experiments, the RH of the chamber was controlled to be above the DRH of NaCl, and for the dry seeded runs the RH was kept well below the ERH of the respective seeds (Table 2).

|
Instrumentation
Both aromatic hydrocarbon and CCl4 concentrations were monitored using an HP-5890 GC-FID with a DB5 fused silica capillary column (15 m, 0.53-mm internal diameter, 1.5-μm thickness, J & W Scientific Inc., Cat# 1255012, Folsom, CA, USA). The gas chromatography (GC) oven temperature was fixed at 80 °C. The carrier gas (nitrogen) flow rate was 10.0 mL min–1. The particle concentration was monitored with a scanning mobility particle sizer (SMPS) (TSI, Model 3080, Shoreview, MN, USA) integrated with a condensation nuclei counter (TSI, Model 3025A and Model 3022). NOx and ozone concentrations were monitored with a Chemiluminescence NO/NO2 analyser (Teledyne, model 200E, Thousand Oaks, CA) and photometric ozone analyser (Teledyne, model 400E). In measuring organic carbon (OC) data, aerosols were analysed throughout each experiment with a semi-continuous OC aerosol analyser (Sunset Laboratory Model 4, Hillsborough, NC, USA) utilising the NIOSH 5040 method. The detection limit of OC analysis is 0.3 μg m–3 (for 120-min sampling) and the associated error is ±10 %.
The inorganic ion content of the aerosols was measured using a particle-into-liquid-sampler coupled to both an anion and cation chromatograph (PILS-IC) (Metrohm, Riverview, FL). The detection limit of PILS-IC is 0.2 μg m–3 and the associated error is ±6 %. The cation eluent was 2.0 mM HNO3 and the anion eluent was 9.57 mM NaCO3 and 2.4 mM NaHCO3. The ion chromatography (IC) column used for cations was a Metrosep C 4 100 and for anions was an IonPac AS9-HC. Both a basic denuder coated with 1 % glycerol and 2 % Na2CO3 in ethanol/water (1 : 1) (to remove gaseous inorganic acids) and an acidic denuder coated with 1 % glycerol and 1 % citric acid in ethanol/water (1 : 1) (to remove gas phase ammonia) were placed upstream of the PILS. LiBr was used as the internal standard.
All aerosol data such as OC, concentrations of inorganic chemical species, and SMPS data were corrected for wall loss and dilution. For correcting aerosol loss in the presence of a seed, the decay data of sodium ions in the aerosol, as measured by the PILS-IC, were used because the sodium ion is chemically stable. The seedless aerosol data were corrected for dilution and wall loss using the first order decay method.[17] The gaseous data were corrected for dilution using CCl4 decay data.
Aerosol water content
Aerosol water content was measured for both fresh and aged SSA and NaCl seeds during experiment Tol 4. Each chamber was sampled twice (before the injection of AVOC/NOx and after SOA formation) by impacting aerosols onto a silicon Fourier transform infrared (FTIR) window (13 × 2 mm, Sigma–Aldrich, St Louis, MO, USA) to allow for comparison of the water content of the seeds. After the impacted mass of each sample was measured using an analytical balance, the FTIR window was placed in a custom made flow chamber within the FTIR (Nicolet Magma 560, Madison, WI, USA). The flow chamber allows for humidity control through the manipulation of air flow (held between 0.5 and 1.5 L min–1) between a water bubbler and clean air from a dry air tank (Breathing Quality Air, Airgas). FTIR spectra were measured for an incrementally decreasing percentage of RH ranging from >90 % to <15 % in absorption mode. A 0.02 M aqueous solution of pure NaCl was also impacted onto a silicon FTIR cell and its spectra were measured over the same range of percentage of RH. The peak heights at 1650 cm–1 were then determined. Correlation between the FTIR measured peak height of the pure NaCl at 1650 cm–1 and the water content of the pure NaCl aerosols at 85 % RH, predicted by the inorganic thermodynamic model E-AIM,[18–20] allowed for water mass calculations of all measured samples.
Results and discussion
SOA yields
The SOA yield (Y) of each experiment is determined as[21]:

where Δ[OM] is the measured change of the concentration of OM (μg m–3) within the aerosols, which was determined from OC data using a conversion factor of 1.8[22] and Δ[HC] is the consumption of AVOC (change in concentration of the hydrocarbon (HC) of interest). The aromatic SOA yield of each chamber was determined as a function of time.
SSA effect on SOA yields using outdoor chamber
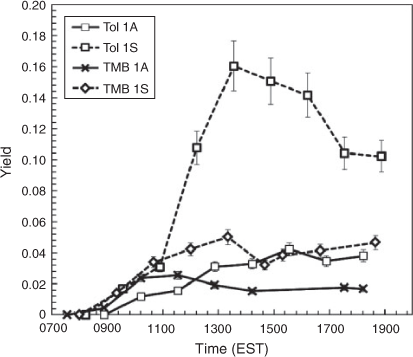
The effect of SSA on the SOA yields of AVOC was determined by comparing the calculated SOA yields of toluene (YTOL) and 1,3,5-TMB (YTMB) in the absence and presence of SSA (see Tol 1 and TMB 1 in Table 1). As shown in Fig. 3, the presence of SSA significantly increased YTOL and YTMB relative to the seedless condition. Considering that all other conditions between each experimental set were equivalent, the differences in SOA yield must stem from the existence of SSA. Reactive oxygenated compounds such as aldehydes and bicarbonyls can be dissolved in the aqueous phase served by SSA aerosol, hydrated and react further by oligomerisation to increase SOA yields.
|
Overall, YTOL for each experimental set was found to be higher than YTMB under the same conditions, as has been shown by several studies,[12,23–25] contradicting partitioning theory in which TMB should produce higher yields due to a lower product volatility (additional methyl groups). This effect can be explained by the reactivity of the SOA products. TMB oxidation products are primarily ketones, which are less reactive than the aldehydes derived from toluene photooxidation.[26] For example, toluene produces large amounts of glyoxal, which has been shown in the aqueous phase to form gem-diols and hydrates that form oligomers.[9] Conversely, TMB produces high concentrations of methyl-glyoxal, which is less reactive than glyoxal.
SOA yields: SSA v. NaCl seed using outdoor chamber
To further investigate the potential of SSA to increase aromatic SOA yields through aerosol aqueous phase reactions, SSA was compared to NaCl as an inorganic seed. The similar composition of the seeds has led to the use of NaCl as a representative of SSA in studies focused on SOA formation in the ocean environment, but in reality the hygroscopic properties of the two seeds differ (see SSA chemical composition by aerosol aging below). NaCl is a typical hygroscopic salt that loses water with decreasing percentage RH and becomes completely dry at its ERH (~55 % for NaCl[15,27–29]). SSAs have been shown to hold water at a much lower percentage RH than NaCl.[15,30] The observed hygroscopic differences are likely due to the minor constituents in sea salt, primarily the chlorides and sulfates of magnesium and calcium, which are known to form hydrates. Lee and Hsu[30] found that SSA retains water at all RHs including zero and attribute this to the presence of MgCl2·6H2O. In addition, Zhao et al.[31] proposed that the presence of MgSO4 in fresh SSAs promotes the uptake of water at low RH.
As is shown in the maximum [OM] column of Table 1, YTol (Tol 2 and Tol 3) and YTMB (TMB 2) were higher for the SSA condition than the NaCl condition in all cases. The overall productivity of aerosol aqueous phase reactions depends directly on the amount of aerosol phase water. The overall amount of water within SSA is greater than NaCl and is available over a broader range of RHs (see Water content by aerosol aging below). Hence, the results provided here support the theory that the accommodation of the reactive oxygenated products is higher onto SSA than NaCl due to increased water availability. The differences of YTol between experimental sets Tol 2 and Tol 3 are due to temperature differences of the experimental runs. TMB once again produced a much lower Y, as was expected.
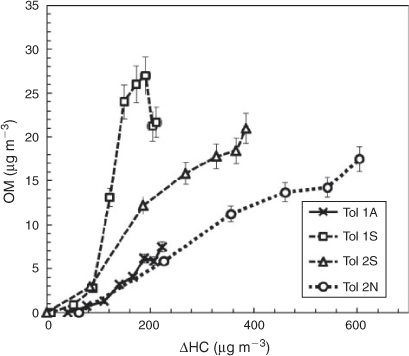
Although outdoor chambers allow for more realistic simulation of the ambient atmosphere, the dynamic change of conditions during an experiment complicates the comparison of Y between different experimental sets. To allow for a more direct comparison of YTol for all seed conditions, Fig. 4 displays the [OM] as a function of Δ[HC] for the Tol 1 and Tol 2 experimental sets, which were performed under similar temperature ranges. As can be seen in Fig. 4, the NaCl seed aerosol produced similar OM growth to the seedless condition whereas the presence of SSA produced the highest [OM] of the toluene runs. In Tol 1S, a decrease in [OM] is seen in the late afternoon. Net SOA production is from both formation and decomposition. It is likely that the decrease in [OM] seen is due to fragmentation of SOA products during the later stages of oxidation. The late decrease in [OM] is more typically seen for the TMB–NOx system due to the high reactivity of TMB with OH radicals, but is also observed in this case for Tol 1S.
|
SOA yields: SSA v. NaCl seed using indoor chamber
To confirm the results of the APHOR chamber runs, an indoor chamber was utilised. Tol–NOx SOA experiments were performed both without seeds and in the presence of wet or dry SSA or NaCl. As seen in Table 2, the Y of dry SSA seeds (24.16 %) exceeded the Y of the seedless (17.30 %) and dry NaCl seed (17.45 %) runs. The Y of the wet seeded cases were similar for NaCl (27.74 %) and SSA (28.36 %) as is expected as the seeds will both have a very high fraction of water at that RH. The results confirm the findings that the presence of water in SSA below the ERH of NaCl will increase the SOA yields of aromatics and other hydrocarbons with soluble secondary products.
SOA yields in the presence of inorganic aerosol: outdoor chamber v. indoor chamber
Indoor chambers are widely used for studies of SOA formation because they allow for controlled experiments, but they fail to replicate the dynamic nature of the actual atmosphere. Under ambient atmospheric conditions, RH and temperature often follow inverse, diurnal cycles (Fig. 2). Therefore, atmospheric aerosols will experience significant changes in water content and SOA volatility that are difficult to replicate within an indoor smog chamber. In the case of the seeded experiments, the inorganic seed (SSA or NaCl) was nebulised from solution into chambers as wet seeds at high RH before sunrise. With sunrise, surging temperatures both increase the volatility of the SOA and lower the RH to below the ERH of NaCl. Also, the spectrum of the natural UV light facilitated in the outdoor chamber is quite different from that of the artificial lamps used in indoor smog chambers. Such differences will likely influence photochemical reactions rates and aging making the comparison of indoor and outdoor data difficult.
The indoor smog chamber datasets were added to this study in order to confirm our conclusions under controlled temperature and RH, but the outdoor datasets are likely to be more representative of actual atmospheric processes. When comparing the yields of the present study to the SOA yields of datasets of other studies on the AVOC–NOx system, differences in temperature, RH, oxidant concentration, and UV light must be considered. For example, Ng et al.[32] found higher yields for the toluene–NOx system than those observed here, but there were many significant differences. Their study was at very low RH and at lower temperatures. Also, very high oxidant concentrations due to the use of HONO in their study increased the oxidation rate of SOA products effectively reducing wall loss of the slow reacting system. In this study, low concentrations of NO were injected in order to approach atmospherically relevant conditions and temperatures were significantly higher for many of the runs. The Y of the indoor runs of this study are more comparable to the Y found in Ng et al., but are lower due to the increased wall loss of a slower reacting system (NOx system) in a much smaller chamber and shorter experimental times.
SSA chemical composition by aerosol aging
Over time, atmospheric aerosols undergo aging through interactions with reactive gases, such as nitric and sulfuric acid, influencing hygroscopic properties and SOA formation. The photochemical NOx cycle in the presence of VOCs leads to the production of HNO3, which is known to condense onto aerosols, form NaNO3 and expel HCl(g).[33,34] Unlike NaCl, which crystalises upon reaching its ERH, NaNO3 and NaNO3–NaCl mixtures have been shown to remain as amorphous solids at low percentage RH.[35] Numerous studies have shown that the presence of surface water increases the reactivity and condensation rates of HNO3[33,35,36] suggesting that the aging of SSAs in the presence of NOx will be greater than that of NaCl.
Fig. 5 presents the inorganic composition of SSA and NaCl as a function of time for low seed (Tol 2) and high seed runs (Tol 3). In both cases the expected change in composition can be seen, with the quick depletion of Cl– and subsequent increase in NO3– following the NOx cycle. In the high seed case (Tol 3) the concentration of NO3– continuously increases, whereas in the low seed case (Tol 2) NO3– quickly peaks and then declines. However, in both cases, the summed molar concentration by both Cl– and NO3– declines with time. It is thought that depletion of the measured anions is due to the formation of organic acid salts, such as sodium carboxylates, and the subsequent volitisation of HNO3. Under the high seed case, HNO3 remains in the aerosol because enough Cl– is present. In Tol 2, after the initial depletion of Cl–, HCl(g) does not continue to volitise whereas HNO3(g) does. A possible explanation for this behaviour is that the aerosols become more viscous over time due to the formation of high molecular weight OC and thus bulk diffusion rates are reduced.[37] In this case, anion substitution by organic acids could favour surface available NO3– over the Cl– locked within the aerosol core. In Tol 3, this behaviour is not seen because of cooler temperatures, which favour the condensation of semivolatile HNO3, and a much higher seed concentration, which limits the thickness of the organic coating on individual aerosols. The formation of organic salts in SSA was also found by Laskin et al. in aged SSA from polluted areas and was shown to significantly affect particle hygroscopic properties.[38]

|
The hygroscopic properties of organic salts associated with SSA-based SOA have been shown to vary. Some organic salts, such as sodium acetate, showed both ERH and DRH, whereas others, such as sodium malonate, show neither an ERH or DRH.[39] Therefore, the hygroscopic properties of SSA may be significantly affected by organic aerosol growth, but the effect is complex depending upon the concentration of each organic species within the aerosol.
Water content by aerosol aging
Fig. 6 displays the water mass fraction (WMF) of all aerosol samples as a function of RH. The WMF is the ratio of water mass to the total inorganic aerosol mass (inorganic mass + water mass) of the sampled aerosols. The particle water content was measured using a FTIR spectrometer (see the Experimental section). For the fresh NaCl aerosol and SSA, the total aerosol mass impacted on the FTIR window comprises water and inorganic salt. Hence, the inorganic mass was calculated as the difference of the total impacted mass and the water mass. For aged NaCl and SSA, the impacted aerosol mass comprised SOA, inorganic salt and water. In order to estimate inorganic salt mass, the total impacted aerosol mass was first subtracted by water mass at a given humidity and then multiplied by the ratio of inorganic salt mass (PILS-IC) to the total dry mass (inorganic salt mass + OM).
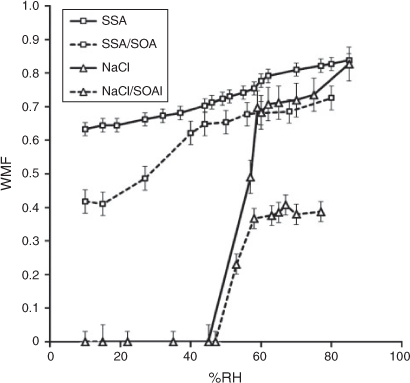
|
At greater than 80 % RH, the fresh SSA and NaCl (SSA and NaCl in Fig. 6) are both composed of greater than 80 % water by mass with indistinguishable WMF (considering error). With deceasing RH, the NaCl particle sample clearly shows an ERH between 55 and 58 %. The fresh SSA seems to show a slight ERH between 55 and 65 %, but continues to contain a large fraction of water (>0.5 WMF). After SOA loading by the photooxidation of toluene–NOx, both SSA–SOA and NaCl–SOA show a significant decrease in WMF. At a RH greater than 80 %, the WMF of SSA and NaCl drop by 0.2 and 0.5. The ERH of NaCl–SOA occurs within the same range as NaCl, but no clear ERH appeared in SSA–SOA showing considerable reduction of WMF between 20 and 40 % RH. Both SSA and SSA–SOA contain a substantial amount of water at all measured values. Such a high water content in SSA would increase SOA yields through aerosol phase reactions of oxidation products over the course of the photooxidation of toluene.
Fig. 7 presents selected FTIR spectra of SSA with and without SOA loading at two different RH levels. In spectra A and B SSA–Tol SOA from the afternoon samples are shown at high (84 %) and low (10 %) RH. There is a clear reduction in the water peaks at 3416 and 1640 cm–1, but the presence of organics complicates water mass calculations. The FTIR spectrum of toluene SOA (spectrum D at 10 % RH) in SSA–SOA was obtained by subtracting spectrum C (SSA at 10 % RH) from spectrum B (SSA–SOA at 10 % RH) to eliminate the remaining water, which can be attributed to hydrates present in SSA, leaving the organic peaks. Much insight can be gained from comparing the remaining organics present in the internally mixed SSA–Tol SOA to a Tol SOA spectrum. Toluene–NOx SOA characteristically shows a broad peak between 3300 and 2500 cm–1 due to O–H stretching of the carboxylic acids present, as well as a peak between 3500 and 3100 cm–1 representative of the O–H stretching of hydroxy groups.[40] The FTIR spectrum of SSA–Tol SOA has a small peak between 3300 and 2500 cm–1 that lacks the representative tailing caused by the O–H stretching of carboxylic acids providing evidence for the presence of the sodium carboxylates suggested in the previous section. This is further supported by the C=O stretching at 1650 cm–1, which represents the C=O stretching of a carboxylate, and by the strong peak present at 1365 cm–1, which is caused by the C–O stretching within sodium carboxylates. FTIR data in Fig. 7 confirm the depletion of Cl ions due to the formation of sodium carboxylates. The displacement of Cl ions with carboxylic acids leads to significant changes in the hygroscopic properties as shown in Fig. 6.
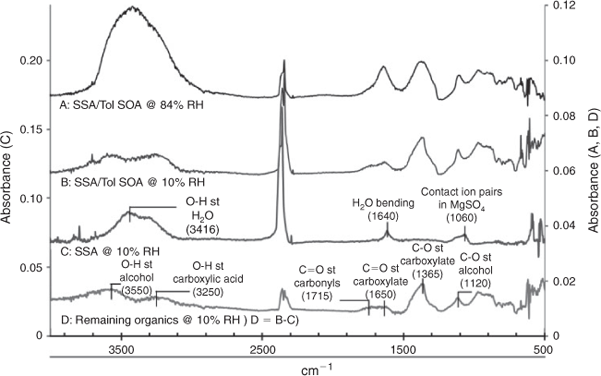
|
Interpretation of OM by traditional partitioning theory
In order to better quantify the effect of the aerosol water content on SOA yields, the measured [OM] ([OM]exp) was compared to the [OM] predicted for the toluene–NOx system by the semi-empirical, two-product model ([OM]Odum), described by Odum et al.[21] This model applies gas–particle partitioning theory to predict SOA yields using the organic aerosol mass concentration (Mo), OM partitioning coefficients (KOM,i),[41] and gas phase stoichiometric product coefficients (αi) of two surrogate products. The equilibrium partitioning coefficients of the two products of the model, KOM,1 and KOM,2, were fit to the seedless toluene experiments (Tol 1A, 5A and 6A) along with the corresponding stoichiometric mass coefficients, α1 and α2. The ratio of α1 to α2 was fixed and determined through lumping of toluene products by vapor pressure using the master chemical mechanism (MCM 3.1).[11,42]
The temperature dependence of KOM,i, was determined using the method described by Takekawa et al.[25]:
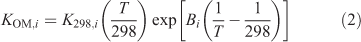
where K298,i is the KOM,i at 298 K, T is the temperature (K) and Bi is the ratio of the enthalpy of vaporisation to the gas constant. Yields were calculated over the period of the experiment using Odum’s two product model as is shown in Eqn 3 using the fitted parameters and the measured values of Mo.
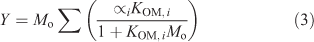
The parameters were fit to the toluene only condition (Tol 1A, 5A and 6A) so that the deviation from the traditional gas–particle partitioning model in the presence of aqueous aerosols could be determined (Δ[OM]). [OM]Odum was calculated using Eqn 4.
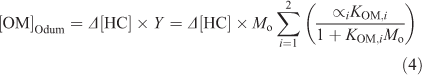
Although the two-product model is dated and the chemistry of SOA formation between the seeded and seedless experiments is different, use of the model allows for a crude estimation of the positive increase of SOA formation due to the presence of the hygroscopic aerosols under changing experimental conditions. Fig. 8 presents [OM]exp and [OM]Odum from experiments Tol 1 and Tol 3. Tol 1A was used in the parameterisation of the model and thus the OM formation is well predicted by the model. In Tol 1S, [OM]exp quickly outpaces [OM]Odum due to the presence of SSA and thus Δ[OM] is largely significant. By comparing Δ[OM] between the NaCl–SOA and SSA–SOA in Tol 3, it can be seen that the aqueous phase reactions in the SSA are more substantial, but the presence of either seed leads to higher OM formation than what is predicted by the traditional model.

|
Atmospheric implications
As of 1995, 37 % of the world’s population lived within 100 km of a coast and that fraction has continued to increase.[43] Growing coastal populations and increasing numbers of oil spills reassert the need for a comprehensive understanding of SOA formation in the coastal environment including the role of SSA, the most prevalent aerosol. This study begins to address the aforementioned need by expanding aerosol phase chemistry to the formation of organic aerosol that is internally mixed with SSA in the coastal environment under conditions close to the ambient coastal atmosphere. NOx levels used in this experiment were held at levels relevant to the coastal environment (between 20 and 80 ppb),[44,45] but the measured SOA concentrations were much higher than those seen in the ambient atmosphere due to the high seed concentrations used. Also, the mean particle diameter of the SSA in these experiments ranged from 75 to 150 nm, whereas the majority of SSA in the MBL are larger than 1 μm.[46] Although there are some deviations from the coastal atmosphere within this study, the finding that the presence of SSA leads to increased yields is widely applicable to the coastal environment. SSA will contain water under all conditions in the coastal and ocean environments and will thus act as a constant source for aqueous phase reactions that lead to increased SOA yields.
This study also exposed important differences between the measured SOA yields of outdoor and indoor chambers. SOA yields derived from outdoor chamber runs are considerably lower than those from indoor chamber studies meaning that indoor chamber derived SOA yields utilised in regional SOA models are likely overestimated, but SOA formation in the presence of hygroscopic salts is underestimated. Future development of a comprehensive SOA model that accounts for aerosol aqueous phase reactions and SOA formation under dynamic atmospheric conditions is required for accurate prediction of global organic aerosol budgets.
Conclusions
Several conclusions have been drawn based on the datasets of this study and are summarised as follows: (1) Under atmospheric conditions, the presence of SSA was shown to significantly increase the SOA yields of aromatic hydrocarbons, compared to the seedless condition or the presence of NaCl seeds. These differences are likely due to dissimilarities in the amount of aerosol phase water, which influences the potential for aqueous phase reactions. (2) SOA coated on inorganic aerosol will influence hygroscopic properties of inorganic seed and thus SOA yields. (3) The linked diurnal patterns of RH and temperature dynamically affect the water content of atmospheric aerosols and volatility of SOA, and are thus important in predicting SOA formation in the presence of hygroscopic seeds.
Acknowledgments
This work was supported by grants from the Navy (N61331-11-1-G001) and National Science Foundation (ATM-0852747).
References
[1] R. Volkamer, J. Jimenez, F. San Martini, K. Dzepina, Q. Zhang, D. Salcedo, L. Molina, D. Worsnop, M. Molina, Secondary organic aerosol formation from anthropogenic air pollution: rapid and higher than expected. Geophys. Res. Lett. 2006, 33, L17811.| Secondary organic aerosol formation from anthropogenic air pollution: rapid and higher than expected.Crossref | GoogleScholarGoogle Scholar |
[2] J. de Gouw, A. Middlebrook, C. Warneke, P. Goldan, W. Kuster, J. Roberts, F. Fehsenfeld, D. Worsnop, M. Canagaratna, A. Pszenny, W. Keene, M. Marchewka, S. Bertman, T. Bates, Budget of organic carbon in a polluted atmosphere: results from the New England Air Quality Study in 2002. J. Geophys. Res. – Atmos. 2005, 110, D16305.
| Budget of organic carbon in a polluted atmosphere: results from the New England Air Quality Study in 2002.Crossref | GoogleScholarGoogle Scholar |
[3] M. Jang, N. Czoschke, S. Lee, R. Kamens, Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions. Science 2002, 298, 814.
| Heterogeneous atmospheric aerosol production by acid-catalyzed particle-phase reactions.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38XotFSntLk%3D&md5=f347a4de506d13b874097c68a4f53a39CAS | 12399587PubMed |
[4] J. Blando, B. Turpin, Secondary organic aerosol formation in cloud and fog droplets: a literature evaluation of plausibility. Atmos. Environ. 2000, 34, 1623.
| Secondary organic aerosol formation in cloud and fog droplets: a literature evaluation of plausibility.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXitFOjt7c%3D&md5=0d4ea5e801d14c0a230234974956c55dCAS |
[5] B. Ervens, G. Feingold, G. Frost, S. Kreidenweis, A modeling study of aqueous production of dicarboxylic acids: 1. Chemical pathways and speciated organic mass production. J. Geophys. Res. – Atmos. 2004, 109, D15205.
| A modeling study of aqueous production of dicarboxylic acids: 1. Chemical pathways and speciated organic mass production.Crossref | GoogleScholarGoogle Scholar |
[6] H. Lim, A. Carlton, B. Turpin, Isoprene forms secondary organic aerosol through cloud processing: model simulations. Environ. Sci. Technol. 2005, 39, 4441.
| Isoprene forms secondary organic aerosol through cloud processing: model simulations.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXktVWrsrg%3D&md5=555691f199f9159e51b9ddbac358d181CAS | 16047779PubMed |
[7] K. Altieri, A. Carlton, H. Lim, B. Turpin, S. Seitzinger, Evidence for oligomer formation in clouds: reactions of isoprene oxidation products. Environ. Sci. Technol. 2006, 40, 4956.
| Evidence for oligomer formation in clouds: reactions of isoprene oxidation products.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xmslaqsrg%3D&md5=223633d12584346acb444ec9baab548fCAS | 16955892PubMed |
[8] R. Volkamer, F. Martini, L. Molina, D. Salcedo, J. Jimenez, M. Molina, A missing sink for gas-phase glyoxal in Mexico City: formation of secondary organic aerosol. Geophys. Res. Lett. 2007, 34, L19807.
| A missing sink for gas-phase glyoxal in Mexico City: formation of secondary organic aerosol.Crossref | GoogleScholarGoogle Scholar |
[9] B. Ervens, R. Volkamer, Glyoxal processing by aerosol multiphase chemistry: towards a kinetic modeling framework of secondary organic aerosol formation in aqueous particles. Atmos. Chem. Phys. 2010, 10, 8219.
| Glyoxal processing by aerosol multiphase chemistry: towards a kinetic modeling framework of secondary organic aerosol formation in aqueous particles.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXktlams7k%3D&md5=1afbc328cc960690297abd736a8f9463CAS |
[10] R. Volkamer, P. Ziemann, M. Molina, Secondary organic aerosol formation from acetylene (C2H2): seed effect on SOA yields due to organic photochemistry in the aerosol aqueous phase. Atmos. Chem. Phys. 2009, 9, 1907.
| Secondary organic aerosol formation from acetylene (C2H2): seed effect on SOA yields due to organic photochemistry in the aerosol aqueous phase.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXlt1yisr4%3D&md5=259699adfbd26129d7ae39b83b9a4f4dCAS |
[11] C. Bloss, V. Wagner, M. Jenkin, R. Volkamer, W. Bloss, J. Lee, D. Heard, K. Wirtz, M. Martin-Reviejo, G. Rea, J. Wenger, M. Pilling, Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons. Atmos. Chem. Phys. 2005, 5, 641.
| Development of a detailed chemical mechanism (MCMv3.1) for the atmospheric oxidation of aromatic hydrocarbons.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXktlyrtrs%3D&md5=cfca9a376e970cd5a081af5c9e6eb92aCAS |
[12] D. Johnson, M. Jenkin, K. Wirtz, M. Martin-Reviejo, Simulating the formation of secondary organic aerosol from the photooxidation of aromatic hydrocarbons. Environ. Chem. 2005, 2, 35.
| Simulating the formation of secondary organic aerosol from the photooxidation of aromatic hydrocarbons.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXisV2it7k%3D&md5=42bcdafc9f08812a691c3f65ac65c302CAS |
[13] P. Sebastião, C. Soares, Modeling the fate of oil spills at sea. Spill Sci. Technol. Bull. 1995, 2, 121.
| Modeling the fate of oil spills at sea.Crossref | GoogleScholarGoogle Scholar |
[14] J. de Gouw, A. Middlebrook, C. Warneke, R. Ahmadov, E. Atlas, R. Bahreini, D. Blake, C. Brock, J. Brioude, D. Fahey, F. Fehsenfeld, J. Holloway, M. Le Henaff, R. Lueb, S. McKeen, J. Meagher, D. Murphy, C. Paris, D. Parrish, A. Perring, I. Pollack, A. Ravishankara, A. Robinson, T. Ryerson, J. Schwarz, J. Spackman, A. Srinivasan, L. Watts, Organic aerosol formation downwind from the deepwater horizon oil spill. Science 2011, 331, 1295.
| Organic aerosol formation downwind from the deepwater horizon oil spill.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3MXivVWks7Y%3D&md5=a9fe3cc2647a24e0ef0f7f485aa9bd48CAS | 21393539PubMed |
[15] J. Jang, M. Jang, W. Mui, C. Delcomyn, M. Henley, J. Hearn, Formation of active chlorine oxidants in saline–oxone aerosol. Aerosol Sci. Technol. 2010, 44, 1018.
| Formation of active chlorine oxidants in saline–oxone aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFKht77N&md5=10a449a816a7668c779393652fa74c81CAS |
[16] Z. Wang, B. P. Hollebone, M. Fingas, B. Fieldhouse, L. Sigouin, M. Landriault, P. Smith, J. Noonan, G. Thouin, Characteristics of spilled oils, fuels, and petroleum products: 1. Composition and properties of selected oils, US EPA report 2003 (US Environmental Protection Agency: Research Triangle Park, NC).
[17] P. McMurry, D. Grosjean, Gas and aerosol wall losses in teflon film smog chambers. Environ. Sci. Technol. 1985, 19, 1176.
| Gas and aerosol wall losses in teflon film smog chambers.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaL2MXmtVSiur8%3D&md5=be9edb2f0700a220977835f36411c92aCAS | 22280133PubMed |
[18] A. Wexler, S. Clegg, Atmospheric aerosol models for systems including the ions H+, NH4+, Na+, SO42–, NO3–,Cl–, Br–, and H2O. J. Geophys. Res. – Atmos. 2002, 107, 4207.
| Atmospheric aerosol models for systems including the ions H+, NH4+, Na+, SO42–, NO3–,Cl–, Br–, and H2O.Crossref | GoogleScholarGoogle Scholar |
[19] S. Clegg, K. Pitzer, P. Brimblecombe, Thermodynamics of multicomponent, miscible, ionic solutions. Mixtures including unsymmetrical electrolytes. J. Phys. Chem. 1992, 96, 9470.
| Thermodynamics of multicomponent, miscible, ionic solutions. Mixtures including unsymmetrical electrolytes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK38Xmt1Wju7c%3D&md5=1dcaeb3666d20e3c44184de21cf54d4dCAS |
[20] S. Clegg, P. Brimblecombe, A. Wexler, Thermodynamic model of the system H+–NH4+–Na+–SO42––NO3––Cl––H2O at 298.15 K. J. Phys. Chem. A 1998, 102, 2155.
| Thermodynamic model of the system H+–NH4+–Na+–SO42––NO3––Cl––H2O at 298.15 K.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXhtlekt7c%3D&md5=333a2382b2a342998df311cb4b47e7edCAS |
[21] J. Odum, T. Hoffmann, F. Bowman, D. Collins, R. Flagan, J. Seinfeld, Gas/particle partitioning and secondary organic aerosol yields. Environ. Sci. Technol. 1996, 30, 2580.
| Gas/particle partitioning and secondary organic aerosol yields.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XjslOiurc%3D&md5=13f353ecb6ce96f5a2ec49a13a669298CAS |
[22] B. Turpin, H. Lim, Species contributions to PM2.5 mass concentrations: revisiting common assumptions for estimating organic mass. Aerosol Sci. Technol. 2001, 35, 602.
| 1:CAS:528:DC%2BD3MXkvVarsb8%3D&md5=734178c87fb0ae8d587bbf9009e2f6d0CAS |
[23] J. Odum, T. Jungkamp, R. Griffin, R. Flagan, J. Seinfeld, Aromatics, reformulated gasoline, and atmospheric aerosol formation. Abstr. Pap. Am. Chem. S. 1997, 214, 107..
[24] G. Cao, M. Jang, Effects of particle acidity and UV light on secondary organic aerosol formation from oxidation of aromatics in the absence of NOx. Atmos. Environ. 2007, 41, 7603.
| Effects of particle acidity and UV light on secondary organic aerosol formation from oxidation of aromatics in the absence of NOx.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXht1yis7vK&md5=3f7a0ff6170b04ee7d2e283c09263a05CAS |
[25] H. Takekawa, H. Minoura, S. Yamazaki, Temperature dependence of secondary organic aerosol formation by photo-oxidation of hydrocarbons. Atmos. Environ. 2003, 37, 3413.
| Temperature dependence of secondary organic aerosol formation by photo-oxidation of hydrocarbons.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXkvFems7o%3D&md5=c27f99ae278efd105eeed894695a3415CAS |
[26] M. Jang, N. Czoschke, A. Northcross, Semiempirical model for organic aerosol growth by acid-catalyzed heterogeneous reactions of organic carbonyls. Environ. Sci. Technol. 2005, 39, 164.
| Semiempirical model for organic aerosol growth by acid-catalyzed heterogeneous reactions of organic carbonyls.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXhtVCmsLvL&md5=99f4af4d06c4eeb3f17dbde518c9b79eCAS | 15667091PubMed |
[27] G. Biskos, A. Malinowski, L. Russell, P. Buseck, S. Martin, Nanosize effect on the deliquescence and the efflorescence of sodium chloride particles. Aerosol Sci. Technol. 2006, 40, 97.
| Nanosize effect on the deliquescence and the efflorescence of sodium chloride particles.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtFKjsr8%3D&md5=bafe26016f8b141f344251cff4e59924CAS |
[28] I. Tang, H. Munkelwitz, Composition and temperature-dependence of the deliquesence properties of hygroscopic aerosols. Atmos. Environ., A Gen. Topics 1993, 27, 467.
| Composition and temperature-dependence of the deliquesence properties of hygroscopic aerosols.Crossref | GoogleScholarGoogle Scholar |
[29] M. Wise, G. Biskos, S. Martin, L. Russell, P. Buseck, Phase transitions of single salt particles studied using a transmission electron microscope with an environmental cell. Aerosol Sci. Technol. 2005, 39, 849.
| Phase transitions of single salt particles studied using a transmission electron microscope with an environmental cell.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXhtF2ktLfE&md5=22ebb36733d91d0611a0e152b9714178CAS |
[30] C. Lee, W. Hsu, The measurement of liquid water mass associated with collected hygroscopic particles. J. Aerosol Sci. 2000, 31, 189.
| The measurement of liquid water mass associated with collected hygroscopic particles.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3cXivFSjtg%3D%3D&md5=af26f4c7984086f80a3dffef0bcb3b65CAS |
[31] L. Zhao, Y. Zhang, Z. Wei, H. Cheng, X. Li, Magnesium sulfate aerosols studied by FTIR spectroscopy: hygroscopic properties, supersaturated structures, and implications for seawater aerosols. J. Phys. Chem. A 2006, 110, 951.
| Magnesium sulfate aerosols studied by FTIR spectroscopy: hygroscopic properties, supersaturated structures, and implications for seawater aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XlsFKj&md5=3c9361b234a70baf6d6546157d1ca2f6CAS | 16419995PubMed |
[32] N. Ng, J. Kroll, A. Chan, P. Chhabra, R. Flagan, J. Seinfeld, Secondary organic aerosol formation from m-xylene, toluene, and benzene. Atmos. Chem. Phys. 2007, 7, 3909.
| Secondary organic aerosol formation from m-xylene, toluene, and benzene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhtV2gtb7E&md5=905e2695355c481874dfc444c2fa4ee7CAS |
[33] H. ten Brink, Reactive uptake of HNO3 and H2SO4 in sea-salt (NaCl) particles. J. Aerosol Sci. 1998, 29, 57.
| Reactive uptake of HNO3 and H2SO4 in sea-salt (NaCl) particles.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1cXmtFCgsw%3D%3D&md5=a29fcfdc34ebca23c88aedaac39b40e7CAS |
[34] D. Weis, G. Ewing, The reaction of nitrogen dioxide with sea salt aerosol. J. Phys. Chem. A 1999, 103, 4865.
| The reaction of nitrogen dioxide with sea salt aerosol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK1MXjsFCqtb8%3D&md5=8e57755b1f8899b26d461ecbffd103acCAS |
[35] R. Hoffman, A. Laskin, B. Finlayson-Pitts, Sodium nitrate particles: physical and chemical properties during hydration and dehydration, and implications for aged sea salt aerosols. J. Aerosol Sci. 2004, 35, 869.
| Sodium nitrate particles: physical and chemical properties during hydration and dehydration, and implications for aged sea salt aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2cXlsVShtb0%3D&md5=9803f57b42d4f59acf08d59def7e955cCAS |
[36] P. Beichert, B. Finlayson-Pitts, Knudsen cell studies of the uptake of gaseous HNO3 and other oxides of nitrogen on solid NaCl: the role of surface-adsorbed water. J. Phys. Chem. 1996, 100, 15218.
| Knudsen cell studies of the uptake of gaseous HNO3 and other oxides of nitrogen on solid NaCl: the role of surface-adsorbed water.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK28XltV2ksL8%3D&md5=42bfc75071c5f46b3517111602d06b74CAS |
[37] M. Shiraiwa, J. H. Seinfeld, Equilibration timescale of atmospheric secondary organic aerosol partitioning. Geophys. Res. Lett. 2012, 39, L24801.
| Equilibration timescale of atmospheric secondary organic aerosol partitioning.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3sXotVOgsg%3D%3D&md5=6ca82ab7aa9a089b9dd5bdb127016112CAS |
[38] A. Laskin, R. Moffet, M. Gilles, J. Fast, R. Zaveri, B. Wang, P. Nigge, J. Shutthanandan, Tropospheric chemistry of internally mixed sea salt and organic particles: surprising reactivity of NaCl with weak organic acids. J. Geophys. Res. – Atmos. 2012, 117, D15302.
| Tropospheric chemistry of internally mixed sea salt and organic particles: surprising reactivity of NaCl with weak organic acids.Crossref | GoogleScholarGoogle Scholar |
[39] C. Peng, C. Chan, The water cycles of water-soluble organic salts of atmospheric importance. Atmos. Environ. 2001, 35, 1183.
| The water cycles of water-soluble organic salts of atmospheric importance.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXntVOktw%3D%3D&md5=109e84d10fcb0a03ba7a96f12f271310CAS |
[40] M. Jang, R. Kamens, Characterization of secondary aerosol from the photooxidation of toluene in the presence of NOx and 1-propene. Environ. Sci. Technol. 2001, 35, 3626.
| Characterization of secondary aerosol from the photooxidation of toluene in the presence of NOx and 1-propene.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3MXlvFynurw%3D&md5=b4dc3f829d8470f78be9fb3bcb131989CAS | 11783638PubMed |
[41] J. Pankow, An absorption model of gas-particle partitioning of organic compounds in the atmosphere. Atmos. Environ. 1994, 28, 185.
| An absorption model of gas-particle partitioning of organic compounds in the atmosphere.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2cXisFajs78%3D&md5=c57a2c978502e6cc9c92a9ebca46e43aCAS |
[42] M. Jenkin, S. Saunders, V. Wagner, M. Pilling, Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds. Atmos. Chem. Phys. 2003, 3, 181.
| Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part B): tropospheric degradation of aromatic volatile organic compounds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXns1Sntrw%3D&md5=0145ea2e3046ed18a736df85992d5c73CAS |
[43] J. Cohen, C. Small, A. Mellinger, J. Gallup, J. Sachs, Estimates of coastal populations. Science 1997, 278, 1209.
| Estimates of coastal populations.Crossref | GoogleScholarGoogle Scholar |
[44] S. Saito, I. Nagao, H. Tanaka, Relationship of NOx and NMHC to photochemical O3 production in a coastal and metropolitan areas of Japan. Atmos. Environ. 2002, 36, 1277.
| Relationship of NOx and NMHC to photochemical O3 production in a coastal and metropolitan areas of Japan.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD38Xhs1Ont74%3D&md5=ea6817e97d94862fa2d479882381d3aaCAS |
[45] Y. Itano, H. Bandow, N. Takenaka, A. Asayama, H. Tanaka, S. Wakamatsu, K. Murano, Daily variation and effect on inland air quality of the strong NOx emissions from ships in the Osaka Bay, Japan. Terr. Atmos. Ocean Sci. 2005, 16, 1177.
[46] E. R. Lewis, S. E. Schwartz, Sea Salt Aerosol Production: Mechanisms, Methods, Measurements, and Models 2004 (American Geophysical Union: Washington, DC).