

Using COSMOtherm to predict physicochemical properties of poly- and perfluorinated alkyl substances (PFASs)
Zhanyun Wang A , Matthew MacLeod B , Ian T. Cousins B , Martin Scheringer A C and Konrad Hungerbühler AA Institute for Chemical and Bioengineering, Swiss Federal Institute of Technology, ETH Zurich, CH-8093 Zurich, Switzerland.
B Department of Applied Environmental Science (ITM), Stockholm University, SE-10691 Stockholm, Sweden.
C Corresponding author. Email: scheringer@chem.ethz.ch
Environmental Chemistry 8(4) 389-398 https://doi.org/10.1071/EN10143
Submitted: 24 December 2010 Accepted: 1 March 2011 Published: 19 August 2011
Journal Compilation © CSIRO Publishing 2011 Open Access CC BY-NC-ND
Environmental context. Poly- and perfluorinated alkyl substances (PFASs) include a wide range of individual compounds that are used in many consumer products, but only a few physicochemical property data are available for these chemicals. Here we provide estimates of physicochemical properties (vapour pressure, water solubility, etc.) of 130 individual PFASs derived with a quantum-chemical model. Our results provide insight into the effect of molecular structure on the properties of PFASs and a basis for estimating the environmental partitioning and fate of PFASs.
Abstract. Recently, there has been concern about the presence of poly- and perfluorinated alkyl substances (PFASs) in the environment, biota and humans. However, lack of physicochemical data has limited the application of environmental fate models to understand the environmental distribution and ultimate fate of PFASs. We employ the COSMOtherm model to estimate physicochemical properties for 130 individual PFASs, namely perfluoroalkyl acids (including branched isomers for C4–C8 perfluorocarboxylic acids), their precursors and some important intermediates. The estimated physicochemical properties are interpreted using structure-property relationships and rationalised with insight into molecular interactions. Within a homologous series of linear PFASs with the same functional group, both air–water and octanol–water partition coefficient increase with increasing perfluorinated chain length, likely due to increasing molecular volume. For PFASs with the same perfluorinated chain length but different functional groups, the ability of the functional group to form hydrogen bonds strongly influences the chemicals’ partitioning behaviour. The partitioning behaviour of all theoretically possible branched isomers can vary considerably; however, the predominant isopropyl and monomethyl branched isomers in technical mixtures have similar properties as their linear counterparts (differences below 0.5 log units). Our property estimates provide a basis for further environmental modelling, but with some caveats and limitations.
Introduction
Poly- and perfluorinated alkyl substances (PFASs) are chemicals that have partially and completely fluorinated carbon chains of various lengths. These substances have been produced in large quantities since the 1950s and are employed in a wide variety of applications, including coatings, lubricants, paints, cosmetics, surfactants and firefighting foams.[1] Recently, certain PFASs have attracted attention as environmental pollutants of high concern. They have been found in the environment and biota all over the world, including in remote regions,[2] and also in human food items.[3] Certain PFASs are suspected of having high persistence,[4,5] potential for bioaccumulation,[6–9] and toxicity,[10,11] but there is still an incomplete understanding of their sources, transport and ultimate fate in the environment.
To address this knowledge gap, environmental fate models have been applied to describe the sources and environmental distribution of some PFASs.[12–18] To date, these studies have mainly focussed on perfluorocarboxylic acids (PFCAs) with perfluorinated chain lengths of 7–12 carbons (which are often termed C8–C13 PFCAs including the carboxylic acid carbon), C8 perfluorosulfonic acid (PFOS) and a few related substances. One of the barriers to applying models to a wider range of PFASs is a lack of physicochemical property data that are required as model inputs. Important properties in this regard include: PL, liquid vapour pressure; KAW, air–water partition coefficient; KOW, octanol–water partition coefficient; KOA, octanol–air partition coefficient; SO, solubility in octanol; and SW, solubility in water.
Ideally, these physicochemical properties should be determined in laboratory experiments, and considerable efforts have been made to do so. However, experimental determination of property data for the PFASs is challenging because the property values lie near the limits of applicability of many methods, and the compounds readily adsorb to surfaces of measuring apparatus.[19] Hence, to date few experimental data are available, as summarised in the Table A1 in the Accessory publication.
Another barrier to laboratory determination of property data for PFASs is the large number of compounds of potential interest. Products with a wide variety of functional groups on fluorinated alkyl chains as short as three carbons have been produced for speciality chemical applications. In addition, during more than 50 years of global PFAS manufacturing, the two major synthetic methods (electrochemical fluorination (ECF) and telomerisation) produced PFAS mixtures with different isomeric compositions.[1] There is currently scientific interest in understanding the isomer-specific environmental and biological behaviour of PFASs, which will require isomer-specific physicochemical data. Measurement of these properties has been hindered by a lack of pure standards for branched isomers as well as challenges in developing isomer-specific analytical methods and therefore only a few attempts have been made to measure properties of individual isomers of PFCAs.[20]
As a supplement to laboratory determination, studies using computational methods have been conducted to estimate some physicochemical properties for a few PFASs.[2,21] COSMOtherm is a quantum chemistry-based method that requires no specific calibration, and thus is expected to be adaptable to estimating properties for PFASs, for which only few calibration data are available. In support of this view, properties estimated with COSMOtherm in a recent study showed good agreement with the experimental data for a limited number of PFASs.[21]
The goals of this work are to further evaluate the performance of the COSMOtherm model by comparing modelled property data with available measured data, and to extend the application of the model to a much larger set of PFASs, including branched isomers for C4–C8 PFCAs. We analyse the model results in terms of variability in calculated property data for different conformations of PFASs, the influence of functional groups and fluorinated carbon chain length on physicochemical properties, and differences of physicochemical properties for substances with different branching patterns.
The property data estimated with COSMOtherm and reported here are only for neutral PFAS monomers, and only describe absorption into a phase. They provide a basis for further environmental modelling, but only with several caveats and limitations. The data do not account for adsorption of PFASs to surfaces, or the formation of aggregates, which may occur in the environment due to the strong surfactant properties of many PFASs. Additionally, many PFASs have acidic functional groups, and therefore their overall partitioning in the environment will be determined by the fractions of the (neutral) acid and (anionic) conjugate base that are present, and the partitioning of both the neutral and anionic form. More detailed information about the limitations of these data is provided in the ‘Discussion’ section.
Methods
The COSMOtherm model and COSMO-RS theory
Detailed information about the COSMOtherm model can be found in the review by Klamt et al.[22] Briefly, the COSMOtherm model is based on the ‘Conductor-like Screening Model for Real Solvents’ (COSMO-RS) theory,[23] in which solute molecules are assumed to be embedded in a cavity within a virtual conductor environment formed by the solvent. The total energy of the molecule and the 3-D polarisation charge density (σ) on the molecular surface is calculated with a quantum chemical self-consistent field algorithm, which also calculates the strength of electrostatic, hydrogen bonding and dispersion interactions between the molecule and the virtual conductor. The strength of these interactions is then used to predict the chemical potential of molecules in the solvent using statistical thermodynamics. The physicochemical properties (PL, SW, SO, KAW, KOW and KOA) are directly estimated from the chemical potential of the solute in solutions of water and octanol, in the pure liquid phase and in the gas phase.
The COSMOtherm calculations are based on the 3-D polarisation charge density of the solute molecule. Therefore, the 3-D conformation of the molecule influences the modelled interactions with the solvent and thus affects the calculated partitioning behaviour.[24] For instance, the formation of intramolecular hydrogen bonds competes with intermolecular hydrogen bonding and thus increases the chemical potential of substances in solvents where intermolecular hydrogen bonds can be formed. We used the following steps to select molecular conformations used to calculate properties: (1) determination of starting 3-D structures by applying ‘ChemBio 3-D’ (v.12.0, Cambridge Software); (2) conformer search and categorisation of conformers using similarity-based cluster analysis (covering energy, structure, surface charge distribution of different conformers) by applying COSMOconf (v.2.1, COSMOlogic GmbH & Co. KG); (3) COSMO-RS calculation including geometry optimisation utilising ‘TURBOmole’ (v.6.0, COSMOlogic GmbH & Co. KG); (4) calculation of the properties (PL, SW, SO, KAW, KOW and KOA) with COSMOtherm (v.C2.1 release 01.10, COSMOlogic GmbH & Co. KG). For compounds having several energetically stable conformers, an overall average property value was calculated as a weighted average from the Boltzmann distribution suggested in the references.[24,25] For details see the Accessory publication (‘Equation for calculating weighting factors based on Boltzmann distribution’ section).
The computational time required for a full conformer search was prohibitive for many of the polyfluoroalkyl phosphate esters (PAPs) because of the large number of possible conformations. For instance, 39 209 possible starting conformers were found for the diPAP with the shortest carbon chain, 4 : 2 diPAP, in the first step of the conformer search. Even with the fastest semi-empirical MOPAC gas phase optimisation, each starting conformation requires ~3 min of computation time. Therefore, we have calculated physicochemical properties of only the most likely single stretched conformer of all PAPs except for 4 : 2 and 6 : 2 monoPAP.
Chemicals
We selected a set of model evaluation chemicals that included the fluorotelomer alcohols (FTOHs) and some other PFASs to evaluate the performance of COSMOtherm based on the availability of measured property data. We then extended the application of COSMOtherm to a large and diverse set of PFASs for which no or very few measured data are available. This second set of PFASs includes the most widely used perfluoroalkyl acids (PFAAs), their known precursors and some important intermediates that have been identified in degradation studies. The substances studied here are among those highlighted in a recent review of PFASs in the environment.[26] More detailed information on the chemicals, including their CAS number and structural formulae can be found in the Accessory publication (Tables A2–A27).
Evaluation of estimated property data
Two strategies were used to evaluate the calculated property data: external comparison and internal consistency checks. External comparison consists of evaluating our property estimates for the set of model evaluation compounds against measured property data. For the FTOHs, we compare our results to measured data below; for the other PFASs, we present a comparison of model results with the few available measured data in the Accessory publication (‘Comparison with available experimental data for the other PFASs’ section). Since comparison of different estimation methods is not the main focus of this work, property estimates generated with EPISuite (US EPA) and SPARC for FTOHs are not listed here, but are provided in the Accessory publication (‘Comparison of experimental data with estimates from COSMOtherm, EPISuite and SPARC’ section). Two types of internal consistency check were used. First, conformation types found by the software and their contribution to the calculated property value were evaluated by checking their 3-D geometry and corresponding weighting factors estimated for different members of a compound class. Second, the consistency of different properties of each compound was checked as suggested by Cole and Mackay[27] by evaluating the errors in the following equations (where SA is the solubility in air (mol m–3); R is the gas constant (J mol–1 K–1); and T is absolute temperature (K)):





Results
Evaluation of COSMOtherm against measured properties of FTOHs
In agreement with previous studies,[21,28,29] we found several energetically stable conformers for FTOHs. These belong to two groups: stretched conformations (SCs) and conformations with an intramolecular hydrogen bond between the hydroxylic proton and fluorine attached to the C3 carbon (electrostatically interacting conformations, EICs), Fig. 1.
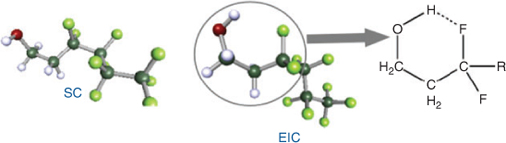
|
In general, for FTOHs with a given chain length, the EIC conformer has higher PL (~1 log unit), higher KAW (~1.5 log units), lower KOA (~1.5 log units) and lower SW (~0.5 log units) than the SC conformer. However, these two conformers have similar KOW values (for details see Table A29 in the Accessory publication). The EICs are the predominant species in the gas phase (>75% of all molecules for all FTOHs), whereas the SCs are prevalent in water and octanol (from 50 to ~80% of all molecules for different FTOHs). Detailed calculation results referring to each conformer can be found in Table A29 in the Accessory publication.
The average property values based on the weighted contribution of all conformers agree with the experimental data within one order of magnitude in all cases (Fig. 2). PL and KOA predictions are very close to the empirical data (differences are smaller than ±0.2 log units except for PL of 10 : 2 FTOH), whereas the KOW and KAW predictions are slightly lower than the experimental data (up to one log unit for KAW of 10 : 2 FTOH). Modelled SW values for 4 : 2 FTOH, 6 : 2 FTOH and 10 : 2 FTOH agree well with the experimental data, whereas the estimated value for 8 : 2 FTOH is higher than the experimental value by one log unit. Also shown in Fig. 2 are property data calculated by Arp et al.[21] with COSMOtherm. A more detailed comparison with the data from this earlier study can be found in the Accessory publication (‘Comparison of experimental data with estimates from COSMOtherm, EPISuite and SPARC’ section).
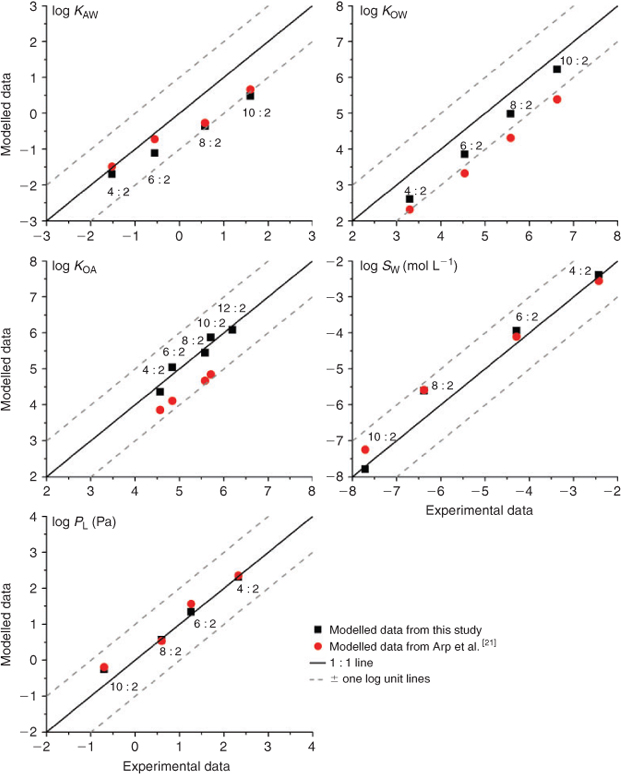
|
When inserted into Eqns 1–5, the property data estimated with COSMOtherm showed better internal consistency than the experimental KOA, KOW and KAW, which had deviations of up to 0.7 log units for 10 : 2 FTOH. In particular, KAW estimates calculated from measured PL and SW deviated by up to one order of magnitude for 10 : 2 FTOH from the experimental KAW values. The observed low internal consistency of the experimental data indicates that there is likely a measurement error in one (or all) of PL,[29] SW[30,31] or KAW.[19]
COSMO calculation results for other PFASs
A complete list of PFASs examined in this study, for which there are few or no measured physicochemical properties available, is included in the Accessory publication (see Tables A30–A33, 130 chemicals in total). For most targeted PFASs only stretched conformations (SCs) were found, since intramolecular hydrogen bonding is not possible. However, conformational analogues to the EICs of the FTOHs were found for PFCAs and perfluorinated sulfonamido ethanols (PFSEs). A detailed list is provided in Table A34 in the Accessory publication.
For PFCAs, conformations with a likely intramolecular hydrogen bond between the acidic proton and a fluorine atom attached to the β carbon (EICs) were found (Fig. 3). In contrast to the EICs of FTOHs, those of the PFCAs have lower PL than the SCs by a factor of 2–3 and constitute only a small proportion of PFCAs in both gas phase and solvents (less than 2%). A possible explanation is that the energy barrier to twist the acidic proton into a position to form the intramolecular hydrogen bond is prohibitively high, as suggested by Arp et al.[21]
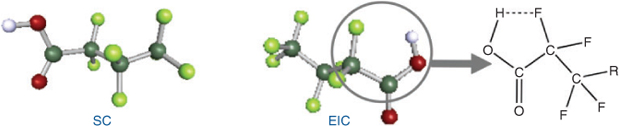
|
For some of the PFSEs, conformations with an intramolecular hydrogen bond between the hydroxyl proton and the oxygen of the sulfonic acid (OxyEICs, Fig. 4) were found. This type of hydrogen bond is likely very strong, as calculations show that the OxyEICs have little potential to act as an H donor in intermolecular interactions (blue area in Fig. 4) in comparison with the SCs. This reduces the potential for intermolecular hydrogen bonding of this conformation, and probably accounts for the finding that the OxyEIC has a higher PL than the corresponding SC, as it was observed for FTOHs. However, in spite of its higher PL, the OxyEICs are not predominant in the gas phase (in the case of EtFOSE, only 32% of molecules in the gas phase are OxyEICs). The abundance of the OxyEICs conformer is probably limited by the energetic stability of the seven-element ring.

|
The calculated KAW and KOW of the PFASs are illustrated in structure–property relationships in Fig. 5. Plots of PL, SW and SO are shown in Fig. A1 in the Accessory publication. Physicochemical properties of PFASs with the same fluorinated chain length but different functional groups, and with the same functional group but different perfluorinated chain length can be compared in Fig. 5. Note, the horizontal axis indicates the number of perfluorinated carbons, and not the total length of the carbon chain of polyfluorinated alkyl substances that include CH2 linkers.

|
In general, KAW is much more variable (~12 orders of magnitude, Fig. 5 top left) than KOW (~2 orders of magnitude, Fig. 5 bottom left) for all selected PFASs with the same perfluorinated chain length. Within a series of substances with the same functional group, both KAW and KOW increase with increasing perfluorinated chain length. In a group of substances with the same perfluorinated chain length the nature of the functional group significantly influences the partitioning behaviour of the compounds. For example, the KAW values of different PFASs are split into three groups (Fig. 5, top left), which correspond to substances (i) with an apolar functional group (no H acceptor or H donor, highest KAW values), (ii) with one monopolar or bipolar functional group (having either or both H acceptor and H donor properties, intermediate KAW values), and (iii) with two bipolar functional groups (lowest KAW values). Another notable trend in the data is that introduction of a CH2 group between a bipolar functional group and the perfluorinated chain decreases the KAW values (Fig. 5, top right).
We additionally estimated property data for the branched isomers of C4–C8 PFCAs. For C4–C7 PFCAs, we considered all possible structural isomers, and for C8 PFCA (PFOA) we estimated the four most abundant isomers found in one major commercial product, namely three monomethyl and one isopropyl isomer, which together account for 98% of the branched isomers of PFOA.[20] The properties for the branched isomers are compared with those of the corresponding linear isomer in Fig. 6.

|
The branched isomers have generally lower KOW and KOA and higher PL and SW than the linear counterparts, but can have either higher or lower KAW than the linear isomers (see Table A35 in the Accessory publication). The variability in the predicted properties of branched isomers increases with increasing chain length (C4–C7); the largest deviations from the properties of the linear isomers are, in most cases, observed for isomers with either dimethyl or monoethyl-monomethyl branching patterns. However, in technical mixtures derived from ECF, PFCA isomers with monomethyl and isopropyl branching patterns are much more abundant than others.[32] Therefore, the variability in property data shown in Fig. 6 is likely an upper limit, since in all cases the predominant isopropyl and monomethyl isomers have similar properties to their linear counterparts with differences smaller than 0.5 log units. In other words, it is likely that the branched isomers that are most predominant in PFCAs in the environment are not very different in behaviour from their corresponding linear isomers.
Discussion
Limitations of property estimations
Previous work with COSMOtherm[21–25] suggested that it was likely to be a suitable method for estimating partitioning properties of PFASs for use in environmental fate modelling. However, in this application, COSMOtherm also has limitations.
For PFASs with acidic functional groups, COSMOtherm can only estimate property data for the neutral form of the substance. For some PFAAs, especially the strongly acidic PFSAs, the neutral form represents only a small fraction of all molecules at pH values typical in the environment, and likely does not play a major role in controlling environmental behaviour. In contrast, for PFCAs it has been suggested that the neutral form may be relevant in environmental transport and partitioning processes[18] as well as in bioaccumulation.[33]
In addition, COSMOtherm estimates made here represent only the properties of PFAS monomers. However, in reality, most PFASs consist of a hydrophobic (perfluoroalkyl chain of varying length) and a hydrophilic (carboxyl, sulfate, phosphate, etc.) part. Therefore, they can adsorb on air–water,[34–37] sediment–water[38,39] and air–particle[40] interfaces as monomers, a mixture of molecular aggregates or both (depending on PFAS concentrations and prevailing pH values) even at very low concentration.[41–43] This surfactant-like nature complicates the understanding of the environmental behaviour of PFASs, as there is still no clear and coherent interpretation of how the accumulation of aggregates at interfaces influences the environmental fate of PFASs.[44–46]
Performance evaluation of COSMOtherm for FTOHs and some other PFASs
As shown in Fig. 2, most estimated property data for the FTOHs are close to the experimental data. The largest differences are up to one order of magnitude. Except for KOW, only one (SW) or two (KAW) compounds are outliers within the homologous series. Examination of possible explanations for these outliers is useful to evaluate the overall performance of COSMOtherm.
In the case of KOW, the discrepancy may reflect some systematic bias in the COSMOtherm predictions, since all estimated values are lower than the experimental data to the same extent (~0.6 log units); both the measured and estimated data series show similar changes in the partition coefficient with increasing carbon chain length.
The water solubility (SW) of 8 : 2 and 10 : 2 FTOH estimated with COSMOtherm represents substances in the subcooled-liquid phase, whereas the measurements were made on solid phase substances.[30] Therefore, to compare the data they need to be harmonised by correction with the Gibbs energies of fusion, ΔfusG, obtained from Liu and Lee[30] as suggested in a previous study.[47] After the correction, the solid phase SW (mol L–1) calculated with COSMOtherm for 10 : 2 FTOH is log SW = –7.79 (before it was log SW = –7.12) and thus close to the experimental data (log SW = –7.71). For 8 : 2 FTOH, the adjustment also reduced the discrepancy, but the solid phase SW of 8 : 2 FTOH is still ~1 log unit higher than the experimental value.
For KAW, the estimated values of 8 : 2 FTOH and 10 : 2 FTOH are ~1 log unit lower than the empirical data. However, the empirical data may have large uncertainty. In the original paper,[19] the authors only measured 4 : 2 and 6 : 2 FTOH. The reported KAW for 8 : 2 FTOH was estimated from measured SW and PL values. Since the authors have not specified which measured values of SW and PL they used, the result cannot be exactly reproduced. However, in the case of 8 : 2 FTOH, if we convert the reported subcooled-liquid PL (Pa) (log PL = 0.60[29]) into solubility in air (SA, mol L–1) (log SA = –5.79) and combine it with the reported solid phase SW (mol L–1) (log SW = –6.38[30,31]), we are able to estimate the KAW (log KAW = 0.59) and find that is very close to the reported KAW (log KAW =0.58[19]). Therefore, it is likely that the KAW of 8 : 2 FTOH was overestimated, because PL for the subcooled liquid was combined with SW for the solid state. Since the KAW value of 10 : 2 FTOH was estimated through extrapolation based on the 4 : 2–8 : 2 FTOH values, it is likely to be overestimated as well.
Additionally, we compared the available experimental data and our estimates for several other PFASs. In contrast to those for FTOHs, the experimental data for these other classes of PFASs include only some properties and only parts of each homologous series. However, the agreement is fairly consistent in that all comparisons between measurements and estimates are usually within one order of magnitude. More detailed information can be found in the Accessory publication (‘Comparison with available experimental data for the other PFASs’ section).
Structure-related intermolecular interaction affects the partitioning behaviour of PFASs
In this work, we have investigated the partitioning of PFASs between the gas phase (air) and two liquid phases (water and octanol). In the gas phase, there are no intermolecular interactions, whereas van der Waals and in many cases hydrogen-bond interactions (H bonds) of PFASs with solvent molecules occur in the liquid phases. In addition, when PFAS molecules transfer into a liquid phase, there is a free energy cost to form a cavity in the new phase (i.e. to break the interactions between solvent molecules). Therefore, the partitioning behaviour of PFASs depends on the overall effect of the intermolecular interaction strength between PFASs and solvent molecules and the free energy cost of cavity formation, which depends on the molecular volume of the PFASs for a given solvent.[48,49]
In the air–water system, the partitioning is controlled by the PFAS intermolecular interactions with water and the energy cost for forming a cavity in water to accommodate the PFAS molecule. The apolar PFASs such as perfluorinated alkanes are only capable of van der Waals interactions with water. These interactions are much weaker than H-bond interactions between water molecules that must be broken to form a cavity, and thus apolar PFASs have high air–water partition coefficients. In contrast, PFASs with monopolar (H donor or H acceptor) or bipolar (H donor and H acceptor) functional groups form H bonds with water molecules, which partially compensate for the energy cost associated with breaking H bonds between water molecules to form a cavity. These compounds therefore have air–water partition coefficients up to 12 orders of magnitude lower than the apolar PFASs. Those compounds with two bipolar functional groups have the lowest KAW values, presumably because they can form more H bonds.
The variability of KOW values among all PFASs of the same perfluorinated chain length is small (less than two log units). In the water–octanol system, the partitioning behaviour is controlled by intermolecular interactions between the PFAS molecules and the water and octanol molecules, and the energy of cavity formation in the two solvents. Water forms stronger H bonds than octanol; however, differences in KOW for PFASs with a given perfluorinated chain length are small, so it is difficult to identify clear and consistent trends attributable to the functional group.
Comparisons among the PFASs within one homologous series with the same functional group show that both KAW and KOW increase with increasing perfluorinated chain length. Compounds that have the same functional group undergo similar interactions with neighbouring molecules, and thus the difference in their partitioning is controlled by differences in the energy cost for cavity formation, which is proportional to chain length (or molecular size). Although the addition of a CF2 group increases the van der Waals interactions, it more markedly increases the free-energy costs for creating a cavity in the solvent. Water forms stronger H bonds than octanol and has a smaller molar volume, thus more H bonds must be broken to form a cavity to accommodate a PFAS. Therefore, partitioning into the phase with lower free energy costs for cavity formation (air in the air–water system and octanol in the octanol–water system) is favoured with increasing chain length, which leads to increasing KAW and KOW values.[19]
Another interesting observation is that the introduction of CH2 between the perfluorinated carbon chain and the bipolar functional group increases the polarity of the functional group leading to a lower KAW despite the higher free energy cost of cavity formation relative to a compound with a perfluorinated chain. The more CH2 groups are present, the more polar the functional group is (we compared PFSAs and FTSAs with a group of hypothetical fluorotelomer sulfonic acids (X1FTSAs) with only one CH2 between the fluorinated chain and the functional group). It is likely that the introduction of CH2 groups leads to higher negative polarity of the functional group, because the functional groups are shielded by the CH2 groups from the electron withdrawing effect of the perfluorinated chain; this effect increases with increasing number of CH2 groups between the fluorinated chain and the functional group.[50]
As shown in the ‘Results’ section, different conformations of a PFAS can have markedly different partitioning behaviour. Depending on the fraction of each conformation that is assumed to be present, the results for the partition coefficients vary considerably. To reduce this uncertainty, we conducted an extensive conformer search that covered thousands of possible candidates and took the energetically relevant fractions into account. However, for the PFSEs in particular, it is difficult to distinguish the contribution of each type of conformer to the partitioning behaviour. The EICs were found in some compounds within a homologous series (e.g. EtFOSE), but not for the others. Causes for this finding could not be identified.
Recently, Rayne and Forest used SPARC, EPISuite and the semi-empirical PM6 method to estimate some properties for branched and linear isomers, and emphasised the importance of implementing isomer-specific partitioning into environmental fate models.[2,51–53] However, they estimated properties for all theoretically possible isomers, whereas in reality only some of these occur.[32]
In our estimations, the branched isomers of PFCAs exhibit a partitioning behaviour that may differ considerably from that of the linear counterparts, if all theoretically possible branched isomers are considered. This finding is consistent with Rayne and Forest.[2] These differences can be explained by the different interaction of branched and linear isomers with the surrounding solvent. The existence of branched CF2 groups may increase the probability of EIC formation, leading to higher PL. In addition, most branched isomers have smaller molecular volume (see data in Table A36 in the Accessory publication) than the linear ones and thus need less free energy to form a cavity in the solvents, leading to higher SW. However, our results in Fig. 6 show that both isopropyl and monomethyl isomers, which are the most abundant branched isomers found in the technical products,[32] are only slightly different from the linear counterparts (within 0.5 log units).
Our results thus indicate that the consideration of branched isomers may only slightly influence the overall partitioning of a mixture of branched and linear isomers produced by the ECF process. Because large efforts (computational or laboratory) are required to determine the partitioning properties of all isomers, a simplifying approach may be taken, using only partitioning properties of linear isomers when only the overall distribution of PFASs but not the behaviour of a particular branched isomer is of interest. However, caution is warranted in using this approach since it has been found that three branched PFOS isomers degrade more rapidly than their linear analogues when subjected to UV light in the presence of water or alkaline 2-propanol.[54] Also, it has been found that the linear isomers of PFOA and PFNA are preferably accumulated in rainbow trout.[55] For such environmental processes that include isomer-specific mechanisms, it is recommended to investigate the properties of different isomers individually.
Limitations and outlook
The modelled property data reported here refer only to the neutral species of PFASs, but they can be used to estimate distribution ratios that describe partitioning of both the neutral and ionised species in environmental models.[48] However, this approach requires knowledge of the pKA of the PFAAs, which are highly uncertain for some species, most notably the PFCAs.[2,13,44,56] During our COSMOtherm calculations, we also estimated pKA for PFCAs, PFSAs, PFSiAs and PFPAs, including some branched isomers for C4–C8 PFCAs. However, we noted that the pKA estimates are highly dependent on the chosen conformations of the neutral species and the anionic species. For instance, with two different conformers of the PFOA anions (PFO), the predicted pKA values were 2.897 and 0.897. Unfortunately, with the software used, we were only able to conduct reasonable conformer searches for the neutral form of PFASs. For the anions, the conformer search remains uncertain, which leads to high uncertainty of the pKA estimates. We have listed our pKA estimates in the Accessory publication (Table A36) for those who may be interested, but these values have high and unquantifiable uncertainties. In addition, if distribution ratios are used to model partitioning of PFAS in the environment, they should consider specific sorption of anionic PFAS on organic matter (e.g. soil,[38] biota[9]).
Another limitation is that we have only estimated absorptive partitioning of PFASs. However, recent studies have shown that PFASs can also adsorb to interfaces as previously discussed and that these adsorption properties could have a strong influence on the environmental distribution of PFASs.[18] Goss has tested the possibility using COSMOtherm to predict adsorption constants for the air–water interface for a set of more than 200 organic compounds[57] that included some FTOHs and fluorotelomer olefins. Moreover, Arp and Goss have developed a COSMOtherm-based model to predict the partitioning behaviour at the gas–particle interface and evaluated it with some measurement for C6–C8 PFCAs.[40]
In summary, the property data estimates made in the current study alone are not sufficient to describe the behaviour of PFASs in environmental fate models. However, they provide a basis for further environmental modelling in some cases and insight into the relationship between chemical structure and physicochemical properties of PFASs. In particular, the quantitative structure–property relationships displayed in Fig. 5 demonstrate the correlations between the chemical structure and physicochemical properties of PFASs, and this information can be extrapolated to the likely environmental behaviour of PFASs. Information of this type may be useful to support the design of less hazardous (greener) chemicals that have the same value in use as the current generation of PFAS products.
Acknowledgement
The Swiss Federal Office for the Environment (FOEN) provided funding for this research at ETH Zurich. We thank Andreas Buser and Cecilia Pereira for helpful discussions and support in the methodological part of this work, and Erol Dedeoglu and Sebastian Strempel for IT support. We also thank the anonymous reviewers for their useful suggestions to improve the manuscript.
References
[1] K. Prevedouros, I. T. Cousins, R. C. Buck, S. H. Korzeniowski, Sources, fate and transport of perfluorocarboxylates. Environ. Sci. Technol. 2006, 40, 32.| Sources, fate and transport of perfluorocarboxylates.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXht1Gru7zK&md5=14e91a9069cf17a00b47907f105e54b6CAS |
[2] S. Rayne, K. Forest, Perfluoroalkyl sulfonic and carboxylic acids: a critical review of physicochemical properties, levels and patterns in waters and wastewaters, and treatment methods. J. Environ. Sci. Health A 2009, 44, 1145.
| Perfluoroalkyl sulfonic and carboxylic acids: a critical review of physicochemical properties, levels and patterns in waters and wastewaters, and treatment methods.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1SqurjJ&md5=99083028fb53a20b36a0de50dd4961b4CAS |
[3] B. O. Clarke, S. R. Smith, Review of ‘emerging’ organic contaminants in biosolids and assessment of international research priorities for the agricultural use of biosolids. Environ. Int. 2011, 37, 226.
| Review of ‘emerging’ organic contaminants in biosolids and assessment of international research priorities for the agricultural use of biosolids.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhsValsL3P&md5=ab1503dde70042776026237211ae1b97CAS |
[4] J. R. Parsons, M. Sáez, J. Dolfing, P. de Voogt, Biodegradation of perfluorinated compounds. Rev. Environ. Contam. Toxicol. 2008, 196, 53.
| 1:CAS:528:DC%2BD1MXmtlynsQ%3D%3D&md5=0ccfa50317e6d7891b5f4c4363367b3bCAS |
[5] T. Frömel, T. P. Knepper, Biodegradation of fluorinated alkyl substances. Rev. Environ. Contam. Toxicol. 2010, 208, 161.
| Biodegradation of fluorinated alkyl substances.Crossref | GoogleScholarGoogle Scholar |
[6] C. M. Butt, D. C. G. Muir, I. Stirling, M. Kwan, S. A. Mabury, Rapid response of Arctic ringed seals to changes in perfluoroalkyl production. Environ. Sci. Technol. 2007, 41, 42.
| Rapid response of Arctic ringed seals to changes in perfluoroalkyl production.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xht1ClurvE&md5=3df66707f18c60b34d89911214ec2fd1CAS |
[7] J. W. Martin, M. M. Smithwick, B. M. Braune, P. F. Hoekstra, D. C. G. Muir, S. A. Mabury, Identification of long-chain perfluorinated acids in biota from the Canadian Arctic. Environ. Sci. Technol. 2004, 38, 373.
| Identification of long-chain perfluorinated acids in biota from the Canadian Arctic.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXptlOgsr8%3D&md5=31028bc5f8598e0cc849bbfcf6060ae4CAS |
[8] M. Smithwick, R. J. Norstrom, S. A. Mabury, K. Solomon, T. J. Evans, I. Stirling, M. K. Taylor, D. C. G. Muir, Temporal trends of perfluoroalkyl contaminants in polar bears (Ursus maritimus) from two locations in the North American Arctic, 1972–2002. Environ. Sci. Technol. 2006, 40, 1139.
| Temporal trends of perfluoroalkyl contaminants in polar bears (Ursus maritimus) from two locations in the North American Arctic, 1972–2002.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XitVGhsg%3D%3D&md5=35a1a7a96254a9b512edec6926649e7eCAS |
[9] J. M. Conder, R. A. Hoke, W. De Wolf, M. H. Russell, R. C. Buck, Are PFCAs bioaccumulative? A critical review and comparison with regulatory criteria and persistent lipophilic compounds. Environ. Sci. Technol. 2008, 42, 995.
| Are PFCAs bioaccumulative? A critical review and comparison with regulatory criteria and persistent lipophilic compounds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXltFWmsw%3D%3D&md5=a5859b58e71eafefd64bade38f26a3c1CAS |
[10] J. P. Giesy, J. E. Naile, J. S. Khim, P. D. Jones, J. L. Newsted, Aquatic toxicology of perfluorinated chemicals. Rev. Environ. Contam. Toxicol. 2010, 202, 1.
| Aquatic toxicology of perfluorinated chemicals.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXkvF2rtw%3D%3D&md5=e6d14943f52145859cc1ff2da3441f62CAS |
[11] A. A. Jensen, H. Leffers, Emerging endocrine disrupters: perfluoroalkylated substances. Int. J. Androl. 2008, 31, 161.
| Emerging endocrine disrupters: perfluoroalkylated substances.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXltFCrsLw%3D&md5=ec262e4a5720ccd7475d50571647a8b4CAS |
[12] J. Armitage, I. T. Cousins, R. C. Buck, K. Prevedouros, M. H. Russell, M. MacLeod, S. H. Korzeniowski, Modeling global-scale fate and transport of perfluorooctanoate emitted from direct sources. Environ. Sci. Technol. 2006, 40, 6969.
| Modeling global-scale fate and transport of perfluorooctanoate emitted from direct sources.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtV2qs7%2FK&md5=69fbd98978a84fd224bc61a80d2900d9CAS |
[13] J. M. Armitage, M. MacLeod, I. T. Cousins, Comparative assessment of the global fate and transport pathways of long-chain perfluorocarboxylic acids (PFCAs) and perfluorocarboxylates (PFCs) emitted from direct sources. Environ. Sci. Technol. 2009, 43, 5830.
| Comparative assessment of the global fate and transport pathways of long-chain perfluorocarboxylic acids (PFCAs) and perfluorocarboxylates (PFCs) emitted from direct sources.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXnsVWhtL0%3D&md5=342e6bd7e649d0dae53831fb3de37fdbCAS |
[14] J. M. Armitage, M. MacLeod, I. T. Cousins, Modeling the global fate and transport of perfluorooctanoic acid (PFOA) and perfluorooctanoate (PFO) emitted from direct sources using a multispecies mass balance model. Environ. Sci. Technol. 2009, 43, 1134.
| Modeling the global fate and transport of perfluorooctanoic acid (PFOA) and perfluorooctanoate (PFO) emitted from direct sources using a multispecies mass balance model.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotlKmtw%3D%3D&md5=4c35bed062ff033d576453394497278dCAS |
[15] J. M. Armitage, U. Schenker, M. Scheringer, J. W. Martin, M. MacLeod, I. T. Cousins, Modeling the global fate and transport of perfluorooctane sulfonate (PFOS) and precursor compounds in relation to temporal trends in wildlife exposure. Environ. Sci. Technol. 2009, 43, 9274.
| Modeling the global fate and transport of perfluorooctane sulfonate (PFOS) and precursor compounds in relation to temporal trends in wildlife exposure.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVSgtbrF&md5=9fcd8f6e250be4175e9bd06e19c8ade0CAS |
[16] U. Schenker, M. Scheringer, M. MacLeod, J. W. Martin, I. T. Cousins, K. Hungerbühler, Contribution of volatile precursor substances to the flux of perfluorooctanoate to the Arctic. Environ. Sci. Technol. 2008, 42, 3710.
| Contribution of volatile precursor substances to the flux of perfluorooctanoate to the Arctic.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXks1Ont7k%3D&md5=0c810537d37242152a99af267ee97c07CAS |
[17] F. Wania, A global mass balance analysis of the source of perfluorocarboxylic acids in the Arctic Ocean. Environ. Sci. Technol. 2007, 41, 4529.
| A global mass balance analysis of the source of perfluorocarboxylic acids in the Arctic Ocean.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXlsFenu7k%3D&md5=46fdcb34f607d1fd07086ea7a0a12667CAS |
[18] E. Webster, D. A. Ellis, L. K. Reid, Modeling the environmental fate of perfluorooctanoic acid and perfluorooctanoate: an investigation of the role of individual species partitioning. Environ. Toxicol. Chem. 2010, 29, 1466.
| Modeling the environmental fate of perfluorooctanoic acid and perfluorooctanoate: an investigation of the role of individual species partitioning.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXpsFGjs7c%3D&md5=66ac16b8ee223905a4e4d7a4153ef9daCAS |
[19] K.-U. Goss, G. Bronner, T. Harner, M. Hertel, T. C. Schmidt, The partition behavior of fluorotelomer alcohols and olefins. Environ. Sci. Technol. 2006, 40, 3572.
| The partition behavior of fluorotelomer alcohols and olefins.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28Xjt1yhs70%3D&md5=1399fffab977792685257b3393cdc98cCAS |
[20] J. P. Benskin, A. O. De Silva, J. W. Martin, Isomer profiling of perfluorinated substances as a tool for source tracking: a review of early findings and future applications. Rev. Environ. Contam. Toxicol. 2010, 208, 111.
| Isomer profiling of perfluorinated substances as a tool for source tracking: a review of early findings and future applications.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlWjt77L&md5=da2f7e058f4bdd66257f7cada8195a8eCAS |
[21] H. P. H. Arp, C. Niederer, K.-U. Goss, Predicting the partitioning behavior of various highly fluorinated compounds. Environ. Sci. Technol. 2006, 40, 7298.
| Predicting the partitioning behavior of various highly fluorinated compounds.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtVWnsrnO&md5=24a9d62c4f2371b10f93cc553ba25daaCAS |
[22] A. Klamt, F. Eckert, W. Arlt, COSMO-RS: an alternative to simulation for calculating thermodynamic properties of liquid mixtures. Annu Rev Chem Biomol 2010, 1, 101.
| COSMO-RS: an alternative to simulation for calculating thermodynamic properties of liquid mixtures.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtFKrtLzJ&md5=d7a702506855169b30bab1cebd67187cCAS |
[23] A. Klamt, Conductor-like screening model for real solvents: a new approach to the quantitative calculation of solvation phenomena. J. Phys. Chem. 1995, 99, 2224.
| Conductor-like screening model for real solvents: a new approach to the quantitative calculation of solvation phenomena.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DyaK2MXjsFaisb0%3D&md5=309b3130429d90291dc5045ecdd84d4eCAS |
[24] A. Klamt, COSMO-RS: from quantum chemistry to fluid phase thermodynamics and drug design 2005 (Elsevier: Amsterdam, the Netherlands).
[25] M. Buggert, C. Cadena, L. Mokrushina, I. Smirnova, E. J. Maginn, W. Arlt, COSMO-RS calculations of partition coefficients: different tools for conformational search. Chem. Eng. Technol. 2009, 32, 977.
| COSMO-RS calculations of partition coefficients: different tools for conformational search.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotFKjur8%3D&md5=70c9bd066c9cd30d127533886518a308CAS |
[26] C. J. Young, S. A. Mabury, Atmospheric perfluorinated acid precursors: chemistry, occurrence, and impacts. Rev. Environ. Contam. Toxicol. 2010, 208, 1.
| Atmospheric perfluorinated acid precursors: chemistry, occurrence, and impacts.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXhtlWjt77K&md5=ebd2280f50b24264cf9d81990aff89e7CAS |
[27] J. G. Cole, D. Mackay, Correlating environmental partitioning properties of organic compounds: The three solubility approach. Environ. Toxicol. Chem. 2000, 19, 265.
| 1:CAS:528:DC%2BD3cXhsF2hsrw%3D&md5=db7e0f00b5eaa3b4fe5a0a78ff89efe5CAS |
[28] D. A. Ellis, S. A. Mabury, Chemical ionization pathways of polyfluorinated chemicals – a connection to environmental atmospheric processes. J. Am. Soc. Mass Spectrom. 2003, 14, 1177.
| Chemical ionization pathways of polyfluorinated chemicals – a connection to environmental atmospheric processes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXnvFahurc%3D&md5=d7f1aba62d3a0f545612996f0b3ab13cCAS |
[29] P. J. Krusic, A. A. Marchione, F. Davidson, M. A. Kaiser, C.-P. C. Kao, R. E. Richardson, M. Botelho, R. L. Waterland, R. C. Buck, Vapor pressure and intramolecular hydrogen bonding in fluorotelomer alcohols. J. Phys. Chem. A 2005, 109, 6232.
| Vapor pressure and intramolecular hydrogen bonding in fluorotelomer alcohols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXlsFWmsrg%3D&md5=a8665bac2a18fbeb9b61e3f77050db71CAS |
[30] J. Liu, L. S. Lee, Effect of fluorotelomer alcohol chain length on aqueous solubility and sorption by soils. Environ. Sci. Technol. 2007, 41, 5357.
| Effect of fluorotelomer alcohol chain length on aqueous solubility and sorption by soils.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXntVyjtLc%3D&md5=02021ea96273d6b5977e63bc593f03ffCAS |
[31] J. Liu, L. S. Lee, Solubility and sorption by soils of 8:2 fluorotelomer alcohol in water and cosolvent systems. Environ. Sci. Technol. 2005, 39, 7535.
| Solubility and sorption by soils of 8:2 fluorotelomer alcohol in water and cosolvent systems.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2MXptVCnsbc%3D&md5=9f618b61b299579eb6b05f1b9236879aCAS |
[32] S. Chu, R. J. Letcher, Linear and branched perfluorooctane sulfonate isomers in technical product and environmental samples by in-port derivatization-gas chromatography-mass spectrometry. Anal. Chem. 2009, 81, 4256.
| Linear and branched perfluorooctane sulfonate isomers in technical product and environmental samples by in-port derivatization-gas chromatography-mass spectrometry.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXlt1SgsLg%3D&md5=8daef32530d74be83b60760e13a3a182CAS |
[33] M. W. Woodcroft, D. A. Ellis, S. P. Rafferty, D. C. Burns, R. E. March, N. L. Stock, K. S. Trumpour, J. Yee, K. Munro, Experimental characterization of the mechanism of perfluorocarboxylic acids’ liver protein bioaccumulation: the key role of the neutral species. Environ. Toxicol. Chem. 2010, 29, 1669.
| 1:CAS:528:DC%2BC3cXhtlSju73E&md5=2377e93c126b9bcd25334657f6e49425CAS |
[34] C. J. McMurdo, D. A. Ellis, E. Webster, J. Butler, R. D. Christensen, L. K. Reid, Aerosol enrichment of the surfactant PFO and mediation of the water–air transport of gaseous PFOA. Environ. Sci. Technol. 2008, 42, 3969.
| Aerosol enrichment of the surfactant PFO and mediation of the water–air transport of gaseous PFOA.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXkvFKhsrg%3D&md5=5662bb6cfadb5d342ee5401a03e8351dCAS |
[35] E. Psillakis, J. Cheng, M. R. Hoffmann, A. J. Colussi, Enrichment factors of perfluoroalkyl oxoanions at the air/water interface. J. Phys. Chem. A 2009, 113, 8826.
| Enrichment factors of perfluoroalkyl oxoanions at the air/water interface.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmsFGmu7c%3D&md5=fe08472c40af099b10839f53b11c1ebdCAS |
[36] N. Rontu, V. Vaida, Surface partitioning and stability of pure and mixed films of 8–2 fluorotelomer alcohol at the air–water interface. J. Phys. Chem. C 2007, 111, 11 612.
| Surface partitioning and stability of pure and mixed films of 8–2 fluorotelomer alcohol at the air–water interface.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnvVSht70%3D&md5=131cf03c174518b9932f2dcd289d2c87CAS |
[37] N. Rontu, V. Vaida, Miscibility of perfluorododecanoic acid with organic acids at the air–water interface. J. Phys. Chem. C 2007, 111, 9975.
| Miscibility of perfluorododecanoic acid with organic acids at the air–water interface.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXmsV2ltr4%3D&md5=44823d4de7e064e411710e4d32eb1fd0CAS |
[38] C. P. Higgins, R. G. Luthy, Sorption of perfluorinated surfactants on sediments. Environ. Sci. Technol. 2006, 40, 7251.
| Sorption of perfluorinated surfactants on sediments.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD28XhtVChtrjJ&md5=1436f5eabf068622e6df6b5b5f74d81aCAS |
[39] C. You, C. Jia, G. Pan, Effect of salinity and sediment characteristics on the sorption and desorption of perfluorooctane sulfonate at sediment–water interface. Environ. Pollut. 2010, 158, 1343.
| Effect of salinity and sediment characteristics on the sorption and desorption of perfluorooctane sulfonate at sediment–water interface.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BC3cXmt1Knur4%3D&md5=0b823f6eb9ac005214ce35a26c4e57e4CAS |
[40] H. P. H. Arp, K.-U. Goss, Gas/particle partitioning behavior of perfluorocarboxylic acids with terrestrial aerosols. Environ. Sci. Technol. 2009, 43, 8542.
| Gas/particle partitioning behavior of perfluorocarboxylic acids with terrestrial aerosols.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1KrtrrN&md5=ba82e522a51ca2c71ebe020dc4b7c021CAS |
[41] J. Cheng, E. Psillakis, M. R. Hoffmann, A. J. Colussi, Acid dissociation versus molecular association of perfluoroalkyl oxoacids: environmental implications. J. Phys. Chem. A 2009, 113, 8152.
| Acid dissociation versus molecular association of perfluoroalkyl oxoacids: environmental implications.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXotVGktbs%3D&md5=aa226a3ae0344f1e3438e451bca8b521CAS |
[42] J. López-Fontán, F. Sarmiento, P. Schulz, The aggregation of sodium perfluorooctanoate in water. Colloid Polym. Sci. 2005, 283, 862.
| The aggregation of sodium perfluorooctanoate in water.Crossref | GoogleScholarGoogle Scholar |
[43] J. L. López-Fontán, A. González-Pérez, J. Costa, J. M. Ruso, G. Prieto, P. C. Schulz, F. Sarmiento, The critical micelle concentration of tetraethylammonium perfluorooctylsulfonate in water. J. Colloid Interface Sci. 2006, 294, 458.
| The critical micelle concentration of tetraethylammonium perfluorooctylsulfonate in water.Crossref | GoogleScholarGoogle Scholar |
[44] D. C. Burns, D. A. Ellis, H. Li, C. J. McMurdo, E. Webster, Experimental pKA determination for perfluorooctanoic acid (PFOA) and the potential impact of pKA concentration dependence on laboratory-measured partitioning phenomena and environmental modeling. Environ. Sci. Technol. 2008, 42, 9283.
| Experimental pKA determination for perfluorooctanoic acid (PFOA) and the potential impact of pKA concentration dependence on laboratory-measured partitioning phenomena and environmental modeling.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXhtlCgtbrP&md5=85727a753ca9f4e71649720238600e37CAS |
[45] D. C. Burns, D. A. Ellis, E. Webster, C. J. McMurdo, Response to Comment on ‘Experimental pKA determination for perfluorooctanoic acid (PFOA) and the potential impact of pKA concentration dependence on laboratory-measured partitioning phenomena and environmental modeling’. Environ. Sci. Technol. 2009, 43, 5152.
| Response to Comment on ‘Experimental pKA determination for perfluorooctanoic acid (PFOA) and the potential impact of pKA concentration dependence on laboratory-measured partitioning phenomena and environmental modeling’.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmsFGltr4%3D&md5=084734b1a4e2a00fd497010d9bffcc2aCAS |
[46] K. U. Goss, H. P. Arp, Comment on ‘Experimental pKA determination for perfluorooctanoic acid (PFOA) and the potential impact of pKA concentration dependence on laboratory-measured partitioning phenomena and environmental modeling’. Environ. Sci. Technol. 2009, 43, 5150.
| Comment on ‘Experimental pKA determination for perfluorooctanoic acid (PFOA) and the potential impact of pKA concentration dependence on laboratory-measured partitioning phenomena and environmental modeling’.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXmsFGmtb0%3D&md5=1bee01ef3e9b6551ab81fbb4197dbceeCAS |
[47] B. Schröder, L. M. N. B. F. Santos, M. A. A. Rocha, M. B. Oliveira, I. M. Marrucho, J. A. P. Coutinho, Prediction of environmental parameters of polycyclic aromatic hydrocarbons with COSMO-RS. Chemosphere 2010, 79, 821.
| Prediction of environmental parameters of polycyclic aromatic hydrocarbons with COSMO-RS.Crossref | GoogleScholarGoogle Scholar |
[48] R. P. Schwarzenbach, P. M. Gschwend, D. M. Imboden, Environmental Organic Chemistry, 2nd edn 2002 (Wiley-Interscience: Hoboken, NJ).
[49] K. Goss, R. Schwarzenbach, Rules of thumb for assessing equilibrium partitioning of organic compounds: Successes and pitfalls. J. Chem. Educ. 2003, 80, 450.
| Rules of thumb for assessing equilibrium partitioning of organic compounds: Successes and pitfalls.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD3sXit1Cjurg%3D&md5=b49a0f39230d376cb086d14d93b02f4eCAS |
[50] P. Jing, P. J. Rodgers, S. Amemiya, High lipophilicity of perfluoroalkyl carboxylate and sulfonate: implications for their membrane permeability. J. Am. Chem. Soc. 2009, 131, 2290.
| High lipophilicity of perfluoroalkyl carboxylate and sulfonate: implications for their membrane permeability.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1Ohu7k%3D&md5=44df05e9997c943b4c6282737bd2ad5eCAS |
[51] S. Rayne, K. Forest, Congener-specific organic carbon-normalized soil and sediment–water partitioning coefficients for the C1 through C8 perfluoroalkyl carboxylic and sulfonic acids. J. Environ. Sci. Health A 2009, 44, 1374.
| Congener-specific organic carbon-normalized soil and sediment–water partitioning coefficients for the C1 through C8 perfluoroalkyl carboxylic and sulfonic acids.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhsVamtL%2FF&md5=00d83d26445ea91d62d0ebec516da8f0CAS |
[52] S. Rayne, K. Forest, K. J. Friesen, Estimated congener specific gas-phase atmospheric behavior and fractionation of perfluoroalkyl compounds: rates of reaction with atmospheric oxidants, air–water partitioning, and wet/dry deposition lifetimes. J. Environ. Sci. Health A 2009, 44, 936.
| Estimated congener specific gas-phase atmospheric behavior and fractionation of perfluoroalkyl compounds: rates of reaction with atmospheric oxidants, air–water partitioning, and wet/dry deposition lifetimes.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtVChtLzK&md5=43608773d8e5c152e1aa56a8fca71113CAS |
[53] S. Rayne, K. Forest, K. J. Friesen, Estimated bioconcentration factors (BCFs) for the C4 through C8 perfluorinated alkylsulfonic acid (PFSA) and alkylcarboxylic acid (PFCA) congeners. J. Environ. Sci. Health A 2009, 44, 598.
| Estimated bioconcentration factors (BCFs) for the C4 through C8 perfluorinated alkylsulfonic acid (PFSA) and alkylcarboxylic acid (PFCA) congeners.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXjvVynsbc%3D&md5=44c3b4fe38573a8e6298783294b3a2f8CAS |
[54] T. Yamamoto, Y. Noma, S.-i. Sakai, Y. Shibata, Photodegradation of perfluorooctane sulfonate by UV irradiation in water and alkaline 2-propanol. Environ. Sci. Technol. 2007, 41, 5660.
| Photodegradation of perfluorooctane sulfonate by UV irradiation in water and alkaline 2-propanol.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXnsVCqtr8%3D&md5=ea5e9ef8460f5e0150cefa3236fde559CAS |
[55] A. O. De Silva, P. J. Tseng, S. A. Mabury, Toxicokinetics of perfluorocarboxylate isomers in rainbow trout. Environ. Toxicol. Chem. 2009, 28, 330.
| Toxicokinetics of perfluorocarboxylate isomers in rainbow trout.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXhtlCqtr4%3D&md5=2259b141a257834c30ea9ba90a127790CAS |
[56] K.-U. Goss, The pKA values of PFOA and other highly fluorinated carboxylic acids. Environ. Sci. Technol. 2008, 42, 456.
| The pKA values of PFOA and other highly fluorinated carboxylic acids.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD2sXhsVWqsLrO&md5=1335d2bcbb72f91cd933f4050d2fe345CAS |
[57] K.-U. Goss, Predicting adsorption of organic chemicals at the air–water interface. J. Phys. Chem. A 2009, 113, 12 256.
| Predicting adsorption of organic chemicals at the air–water interface.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1MXht1Wktb%2FO&md5=57e21f6d314b257a2a62b89c23fa3efbCAS |
[58] N. Carmosini, L. S. Lee, Partitioning of fluorotelomer alcohols to octanol and different sources of dissolved organic carbon. Environ. Sci. Technol. 2008, 42, 6559.
| Partitioning of fluorotelomer alcohols to octanol and different sources of dissolved organic carbon.Crossref | GoogleScholarGoogle Scholar | 1:CAS:528:DC%2BD1cXovFSktLw%3D&md5=91da447febbfc7fd02752e3d59c3ebcdCAS |