

Naked eye evaluation and quantitative detection of the sugarcane leaf scald pathogen, Xanthomonas albilineans, in sugarcane xylem sap
Muhammad Umer A , Nahian Binte Aziz B C , Salma Al Jabri B , Shamsul A. Bhuiyan A C D and Muhammad J. A. Shiddiky A B D
A , Nahian Binte Aziz B C , Salma Al Jabri B , Shamsul A. Bhuiyan A C D and Muhammad J. A. Shiddiky A B D
A Queensland Micro- and Nanotechnology Centre (QMNC), Griffith University, Nathan Campus, Qld 4111, Australia.
B School of Environment and Science (ESC), Griffith University, Nathan Campus, Nathan, Qld 4111, Australia.
C Sugar Research Australia, Woodford, Qld 4514, Australia.
D Corresponding authors. Email: sbhuiyan@sugarresearch.com.au; m.shiddiky@griffith.edu.au
Crop and Pasture Science 72(5) 361-371 https://doi.org/10.1071/CP20416
Submitted: 22 October 2020 Accepted: 10 March 2021 Published: 26 May 2021
Journal Compilation © CSIRO 2021 Open Access CC BY-NC-ND
Abstract
Sugarcane leaf scald caused by the bacterium Xanthomonas albilineans is a major disease of sugarcane worldwide. Whereas erratic symptoms make phenotypic detection challenging, molecular methods require expensive instruments and labour, and longer sample-to-answer times. We report a novel method for detection of X. albilineans DNA in sugarcane xylem sap. The method involves (i) boiling lysis-based DNA extraction from sugarcane sap; (ii) magnetic purification of target sequences directly from the lysate through use of magnetic bead-bound capture probes; and (iii) DNA sandwich hybridisation platform for HRP/TMB/H2O2 reaction-based naked eye visualisation and electrochemical detection of the target. The method is sensitive (limit of detection 100 fM) and reproducible (relative standard deviation <7%) with linear dynamic range 100 fM–1 nM (R2 = 0.99). The method was tested on a range of sugarcane cultivars of known resistance ratings (susceptible, intermediate resistant, and resistant) for leaf scald disease from an inoculated field trial. Detection levels agreed with the resistance rating of cultivars tested. In addition, qPCR results strongly correlated with our assay (r = 0.91–0.99, P < 0.01) and cultivar resistance rating. We believe that our assay could be useful for rapid screening as well as sensitive quantification of target pathogen DNA in infected sugarcane plants.
Keywords: leaf scald disease, Xanthomonas albilineans detection, naked eye evaluation of leaf scald disease, electrochemical detection of leaf scald disease, sugarcane xylem sap.
Introduction
Plant pathogens are among the major threats to sugarcane productivity and biodiversity. Lost production and measures to control pests and diseases cost hundreds of millions of dollars every year (McLeod et al. 1999). Leaf scald is a major sugarcane disease caused by a xylem-inhabiting bacterium Xanthomonas albilineans (Ashby) Dowson (Garces et al. 2014). The disease has been reported in more than 60 countries; however, Australia, USA, Philippines, Vietnam and Thailand are among the most affected countries (Rott and Davis 2000). Leaf scald was first recorded in Australia in 1911 and its occurrence has since been reported in all sugarcane growing regions (Rott and Davis 1995; Davis et al. 1997). Leaf scald can cause severe yield loss or total destruction of crops if susceptible varieties are planted (Hoy 1994). Comstock et al. (1997) estimated a yield loss of 19.5–32% in Florida, and Rott (1995) reported a yield loss of 12–21% in Guadeloupe, France. Besides reductions in yield and harvestable stalk number, leaf scald can also reduce fibre quality and sucrose contents and affects the juice quality (Rott et al. 1997). Elimination of high yielding varieties in cultivar selection programs is an indirect but significant loss incurred by leaf scald. It has been reported that, in Australia, ~20% of potentially high yielding varieties are rejected because of leaf scald susceptibility (Birch 2001). The national plant biosecurity status report has identified leaf scald as a high priority pest threat (Plant Health Australia 2019).
Narrow chlorotic (white) stripes or patches of chlorotic tissue on sugarcane leaves are characteristic leaf scald symptoms. X. albilineans produces a highly potent pathotoxin, called albicidin, which is mainly responsible for these foliar symptoms (Birch and Patil 1985). Albicidin is a DNA gyrase inhibitor and blocks replication of chloroplast DNA, which ultimately leads to the inhibition of chloroplast differentiation and appearance of chlorotic symptoms on infected leaves (Cociancich et al. 2015). Albicidin also plays a key role in systemic invasion and possibly unpredictable transition from latency to active disease. Although albicidin-deficient mutant X. albilineans strains are able to colonise sugarcane efficiently, transgenic sugarcane varieties expressing the albicidin detoxification gene albD show resistance to leaf scald (Birch et al. 2000). Thus, albicidin is considered necessary for development of leaf scald.
Plants infected with X. albilineans may show two forms of symptoms, chronic and acute, and two distinct phases, latent and eclipse (Birch and Patil 1985; Rott and Davis 2000). Each of these pathogenic manifestations poses particular challenges in terms of disease diagnosis and control. The acute form, which often appears subsequent to a rainy period followed by a prolonged dry weather (Rott and Davis 1995), leads to abrupt wilting of plants resulting in death, leaving little room for preventive interventions (Rott and Davis 2000). During the latency phase, plants may remain asymptomatic for months or years before environmental conditions are favourable for the disease to trigger an outbreak (Ricaud and Ryan 1989). On the other hand, during the eclipse phase, a plant can appear diseased or healthy depending on when it is inspected because characteristic white lines on leaves continue to appear and disappear throughout the eclipse phase (Rott and Davis 1995). Many sugarcane cultivars can tolerate the pathogen without exhibiting symptoms, or the symptoms may escape detection because the disease expression may be too insignificant to be recognised (Ricaud and Ryan 1989; Rott and Davis 1995; Rott et al. 1997). Disease diagnosis as well as assessment of sugarcane test cultivars for leaf scald resistance is generally based on observation of the phenotypic symptoms. Thus, not only do latency and erratic nature of symptom expression make disease diagnosis challenging, but susceptible cultivars may also be assigned false ‘resistant’ ratings. Difficulty in accurately identifying infected plants has contributed to the worldwide spread of leaf scald via apparently ‘healthy’ planting materials (Rott 1995). As alternatives to symptom-based diagnosis, various other approaches such as isolation on selective media (Davis et al. 1994), ELISA (Comstock and Irey 1992), end-point PCR (Pan et al. 1999) and qPCR (Garces et al. 2014) have been employed for leaf scald diagnosis. However, these methods are not without shortcomings. Isolation on selective media, though very efficient especially for detecting X. albilineans in symptomless plants, is cumbersome and time-consuming (Wang et al. 1999). Immunological and molecular methods are robust and sensitive but require a centralised laboratory setup. High initial investment is required to set up laboratory infrastructure, and reliance on skilled labour significantly increases the costs of operation. In most of the cases, the testing site is located far from the disease-affected areas, therefore additional measures and costs for processing, storage and transport of samples from the field to laboratories are also involved. And last, there is a significant time lapse between sample collection and communication of the results back to farmers.
In recent years, electrochemical biosensors have emerged as potential contenders for the development of rapid, cost-efficient and amplification-free DNA/RNA detection platforms (Shiddiky et al. 2007, 2010; Boriachek et al. 2018). Although tremendous progress has been made towards the development of electrochemical methods for the detection of DNA sequences associated with human diseases and pathogens (Drummond et al. 2003; Ferapontova 2018), their application for plant pathogen detection has been relatively unexplored (Nezhad 2014; Fang and Ramasamy 2015; Khater et al. 2017; Ferapontova 2018). So far, only a few electrochemical DNA sensors for plant pathogen detection have been reported. However, most of these genosensors have certain limitations. For example, Wongkaew and Poosittisak (2014) reported a voltametric method for the detection of sugarcane white leaf (SCWL) disease. The method involved direct hybridisation of target organism genomic DNA with probes immobilised on chitosan-modified glassy carbon electrodes (GCEs) (Wongkaew and Poosittisak 2014). However, a high level of non-specific DNA binding to the electrode surface was observed on such platforms (Boriachek et al. 2018). Similarly, a highly sensitive method for the detection of the soilborne fungus Trichoderma involved complicated sensor fabrications (Siddiquee et al. 2014). On the other hand, a simple platform developed for the detection of Citrus tristeza virus (CTV) could achieve only a limited sensitivity (0.1 µM) (Khater et al. 2019). Signal amplification strategies based on redox reporters or chemical ligations sometimes lead to a loss of dynamic range (Qavi et al. 2010), which may be a crucial consideration in pathogen detection where a high degree of inter-sample variability in levels of target analyte/s is observed. Some of the other electrochemical methods of plant pathogen detection involve amplification of target DNA before electrochemical detection (Lau et al. 2017). Finally, almost all of the electrochemical methods reported so far rely on commercial kits or other complicated chemical DNA extraction procedures. These challenges are the major impediments to the development of platforms that can conduct on-farm pathogen testing and are simple enough to be used by semi-trained personnel such as farmers and extension workers.
In this study we sought to address several of these challenges. Specific aims of the study were to develop a leaf scald diagnostic method that: (i) is compatible with a simple one-step DNA isolation method; (ii) can provide naked eye/visual evaluation capability; and (iii) can provide amplification-free quantitative detection of X. albilineans DNA down to sensitivity levels suitable for routine diagnostics. We present a sandwich DNA hybridisation assay for colourimetric (naked eye) evaluation as well as electrochemical quantification of X. albilineans specific target DNA sequences in sugarcane sap.
Materials and methods
Reagents and materials
All reagents and chemicals used in this study were of analytical grade. Nuclease free distilled water (Invitrogen cat. no. 10977015; Thermo Fisher Scientific) was used for preparing all aqueous solutions. Screen printed gold electrodes (SPGEs), DropSens DRP-220AT, were purchased from Metrohm DropSens (Spain). Mercaptohexanol (MCH), Tris (2-carboxyethyl) phosphine hydrochloride (TCEP), Tris-HCl, EDTA, 3,3′,5,5′-tetramethylbenzidine (TMB), and phosphate buffered saline (PBS) tablets (0.01 M diphosphate buffer, 0.0027 M potassium chloride and 0.137 M sodium chloride; pH 7.4 at 25°C) were purchased from Sigma-Aldrich (Sydney). Horseradish peroxidase (HRP)-conjugated streptavidin (cat. no. SA10001) was purchased from Life Technologies Australia (Thermo Fisher Scientific). All synthetic probes and primers were purchased from Integrated DNA Technologies, Singapore. All electrochemical measurements were performed using a CH1040C potentiostat (CH Instruments, USA).
Sample collection and DNA isolation
Plants ~12 months old were inoculated with X. albilineans after 6 months of planting in the field. Sugarcane stalks were collected from the disease screening trial of SRA Woodford Research Station (26.929°E, 152.777°S), Woodford, Queensland, and immediately transported to the laboratory for extraction of xylem sap. Stalk samples were stored in clean plastic bags at 4°C until further analysis. Before extraction of DNA, stalks were cleaned thoroughly with Milli-Q ultrapure deionised water and 80% ethanol. Vascular sap was extracted from internode sections of stalks aseptically. Outer bark of stalks was shaved by using sterile scalpel blades. A new blade was used to cut the peeled internal tissue into small pieces (roughly 3–5 cm long, 1 cm thick), and the cut pieces of internal tissue were transferred to clean nuclease-free microcentrifuge tubes (1.5 mL). Disposable polypropylene pestles were used to extract sugarcane sap by applying pressure on the cut internal tissue pieces in the microcentrifuge tubes. To avoid cross contamination between samples, only one sample was processed at a time and fresh and sterile scalpel blades and pestles were used for each sample. Bacterial DNA was isolated from sugarcane xylem sap following the method reported by Garces et al. (2014). Briefly, collected sap samples (200 µL) were transferred to a clean microcentrifuge tube and centrifuged at 9000g for 5 min to precipitate bacterial cells. The pellet was resuspended in 100 µL lysis buffer (0.05 M KCl, 0.01 M Tris-HCl and 0.2% Tween-20; pH ~8.3) by vortexing vigorously followed by boiling at 95°C for 10 min. DNA from cultured X. albilineans was isolated using a DNeasy Plant Mini Kit (Qiagen). Isolated DNA samples were stored at –20°C until further use.
Capture and purification of target
Probes were designed targeting a 39-bp region (synthetic XALB; Supplementary Materials Table S1, available at the journal’s website) within the albicidin pathotoxin biosynthesis gene cluster XALB1 (GenBank accession no. AJ586576) (Royer et al. 2004). The target region was selected considering the pivotal role albicidin plays in leaf scald pathogenesis. The target region (synthetic target) corresponded to positions 1740266–1740304 in the X. albilineans GPE PC73 complete genome (GenBank accession no. FP565176.1) (Pieretti et al. 2009). Sequences of the probes and synthetic target sequence are shown in Table S1. Known concentrations of synthetic target sequence were prepared by diluting stock solution (100 µM) in nuclease-free H2O. DNA purified from cultured bacteria was diluted to a concentration of 1 ng µL–1, whereas DNA from xylem sap samples was used without any further dilution. Each concentration was run in triplicate throughout (from target capture to detection).
The target sequences were separated and magnetically purified following our previously described protocol with slight modifications (Koo et al. 2016; Islam et al. 2018). Unless otherwise stated, all incubations in this section were performed for 30 min at room temperature and with shaking at 300 rpm. Briefly, a sample (10 µL), or nuclease free H2O2 for no target control (NTC), was mixed with 15 μL 10 μM biotinylated capture probe 1 (CP1) and 10 μL 5× saline-sodium citrate (SSC) buffer (pH 4.0), heated at 95°C for 5 min, followed by cooling on ice and incubation. The required volume (10 µL sample–1) of Dynabeads MyOne Streptavidin C1 beads (Invitrogen) were washed three times with 1× binding and washing (B&W) buffer (10 mM Tris-HCL, pH 7.5; 1 mM EDTA, 2 M NaCl) and resuspended in 35 µL 2× B&W buffer. Washed beads were mixed with CP1-target mixture and incubated. The target-attached beads were magnetically separated, washed (two times with 1× B&W, once with 1× SSC buffer) and resuspended in 10 µL 5× SSC buffer. The captured target samples were stored at –20°C until further processing.
Sensor fabrication
Unless otherwise stated, electrodes were washed between each step using 10 mM PBS and dried by gentle air flow. The electrodes were protected from direct exposure to light throughout the experiments. The SPGEs were pre-treated electrochemically as described by Zhang et al. (2007). The effective area of working electrode was estimated by measuring the peak current obtained as a function of scan rate under cyclic voltammetric conditions for the one-electron reduction of [Fe(CN)6]3– (2.0 mM in PBS (0.5 M KCl)) (Shiddiky et al. 2009) and was determined by using the Randles–Sevcik equation:
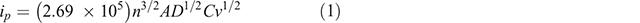
where ip is the peak current (A), n is the number of electrons transferred (Fe3+ → Fe2+, n = 1), A is the effective area of working electrode (cm2), D is the diffusion coefficient of [Fe(CN)6]3– (taken to be 7.60 × 10−5 cm2 s–1), ν is the scan rate (V s–1), and C is the concentration (mol cm–3).
Thiolated capture probes (CP2) were reduced by incubating 1 µL 1 mM TCEP with 99 µL 0.2 µM CP2. Equal volumes I-buffer (10 mM Tris-HCl + 1 mM EDTA + 0.1 M NaCl + 10 mM TCEP; pH 7.4) and reduced CP2 were mixed, and 5 µL of the mixture was placed onto the SPGE working electrode and incubated for 1 h. The working electrode was passivated with 5 µL MCH (1 mM) for 20 min.
The density of CP2 on the fabricated sensor was measured as described earlier (Wong and Gooding 2003; Shiddiky et al. 2010). Briefly, chronocoulometric (CC) measurements were taken in 50 µL E-Buffer (10 mM Tris-HCl; pH 7.0) and 50 µM ruthenium hexamine, [Ru(NH3)6]3+, (RuHex) by using following parameters: initial potential 0.2 V, final potential –0.5 V, number of steps 1, pulse width 0.25 s, and sample interval 0.002 s. Charge (Q) versus t1/2 for both CC runs was plotted and capacitive charge (QdL) and total charge (Qtotal) were obtained from the intercept at t = 0, from the E-Buffer and RuHex plots, respectively (Supplementary Fig. S1). The charge corresponding to RuHex electrostatically bound to surface-confined ssDNA (Qss) was calculated as described by Masud et al. (2017).

The probe density (Γss) was calculated by using the following equation:

where n is the number of electrons (n = 1), A is the area of the working electrode, m is the number of nucleotides in the DNA, z is the charge of the redox molecules, F is Faraday constant, and NA is Avogadro’s number.
Colourimetric and electrochemical detection
Target solutions (10 µL) were added to the sensor surface for 1 h, followed by the incubation with 10 µL biotinylated detection probe (DP) (10 µM) for 30 min and 4 µL HRP-conjugated streptavidin (0.1 ng µL–1 in PBST) for another 20 min. The electrode was then incubated with 50 µL TMB single solution (Invitrogen cat. no. 00-2023) for 10 min in the dark. Naked eye evaluation was done by observing blue colour development. The reaction was stopped by adding 4 µL stop solution (0.2 M H2SO4) to generate a yellow coloured complex. The amount of the enzymatically generated yellow coloured complex as an indicator of surface-bound target was measured by chronoamperometry at a fixed potential of +100 mV. All measurements were performed at room temperature.
Detection of leaf scald bacteria in samples collected from field trial
In order to demonstrate further the suitability of our method for field application, we analysed a series of sugarcane xylem sap samples. Samples were collected from nine cultivars of sugarcane from the SRA leaf scald screening trial. The trial was inoculated by manual decapitation of cane tops above the growing point, between the third and fourth dewlap, followed by application of infected juice within a few minutes, brushing the cut tissue with a paintbrush dipped in the inoculum. Sugarcane stalks were collected from nine cultivars (Q44, Q63, Q68, Q87, Q199, Q124, Q133, Q189, and Q208) in three categories of resistance to X. albilineans infection: susceptible, intermediate and resistant (three cultivars of each). For each cultivar, stalks from two different plants were collected initially (18 stalks in total). However, only 13 of the stalks were included in final analysis. The other five stalks were excluded mainly because of difficulties in obtaining a sufficient volume of sap for further analyses. Stalks of sugarcane cultivars were cut away from the nodes into ~six pieces, each 12 cm, in order to extract xylem saps. Xylem sap was collected from three different segments of a single stalk as described above, and sap from each segment was processed independently. In total, sap samples extracted from 13 different plants corresponding to nine different cultivars were included in final analysis.
Validation with qPCR
A qPCR assay was designed with primers targeting a region of 123 bp from positions 1740257 to 1740379 in the X. albilineans GPE PC73 complete genome. The qPCR primers encompassed the whole synthetic target region (Table S1). qPCR validation was conducted in a CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, USA) in a reaction (20 μL) using a SensiFAST Sybr No-ROX Kit (Meridian Bioscience, Cincinnati, OH, USA) and forward and reverse primers at a final concentration of 400 nM each (Table S1). The qPCR reaction conditions were as follows: initial denaturation at 95°C for 2 min; 40 cycles of 95°C for 5 s, 50°C for 30 s and 72°C for 20 s; followed by melt curve analysis (65°C to 95°C, with an increment of 0.5°C, for 5 s). Serial dilutions of known X. albilineans genomic DNA quantity (10 ng, 1 ng, 100 pg, 10 pg, 1 pg) were used to generate a standard curve for the absolute quantification of X. albilineans in sugarcane sap samples. For the validation and repeatability measurement, PROC CORR in SAS version 9.4 (SAS Institute, Cary, NC, USA) was used to calculate the correlations between electrochemical detection and qPCR measurements.
Results and discussion
Assay design
The assay is schematically outlined in Fig. 1. Most of the DNA biosensing platforms reported so far rely on costly, time-consuming and tedious DNA extraction procedures. We adopted a simple boiling lysis-based method for bacterial DNA isolation directly from sugarcane xylem sap. Target DNA sequences (region of 39 bp within the XALB1 gene cluster of X. albilineans genome) were specifically captured directly from this crude lysate by using complementary biotinylated probes (CP1) attached on the surface of streptavidin-coated magnetic beads. Subsequently, probe-bound target sequences were magnetically purified followed by heat release and magnetic purification. A self-assembled monolayer of thiolated CP2 on gold electrodes was prepared. CP2 are complementary to the target region adjacent to the CP1 complementary region. Surface density of the thiolated capture probes was in the range from 1.2 × 1012 to <~5.0 × 1012 molecules cm–2 in order to ensure efficient capture and sensitive detection of the target (see next section). The resultant target–CP2 hybrid on the gold electrode surface was reacted with biotinylated DP (CP1) to generate a sandwich CP2-DP:target assembly followed by binding of streptavidin–HRP to DNA assembly. HRP-catalysed TMB oxidation reaction yields a blue coloured product, which on the addition of concentrated acid is converted to a yellow coloured diimine compound. The intensity of the blue or yellow coloured products provides an indirect measure of target concentration and was used for naked eye visualisation (blue colour) or electrochemical quantification via fixed potential chronoamperometry of the electroactive yellow coloured diimine compound (Fig. 1).

|
Determination of probe density
Performance of DNA-probe based sensors (e.g. selectivity, sensitivity and stability) is highly dependent on the characteristics of the immobilised DNA probes, such as conformation, orientation and surface density. The assembly of DNA probes at the sensor surface is affected by various parameters such as incubation time as well as concentration and ionic strength of immobilisation buffer (I buffer) as described by Zhang et al. (2007) and Shiddiky et al. (2010). The MCH used in these experiments not only serves as a spacer by finely modulating surface density of the DNA probes but also acts as a blocker that effectively blocks non-specific gold–DNA interactions on the electrode surface and helps DNA probes to ‘stand up’ on the electrode surface, favouring the efficient hybridisation of incoming target sequences. DNA surface density was quantitatively measured by characterising the redox process of RuHex using CC readout. The electroactive label, [Ru(NH3)6]3+ (RuHex), is stoichiometrically bound to the anionic phosphate backbone of DNA strands via electrostatic interaction. RuHex complexes serve as signalling molecules whose cumulative redox charge is a direct function of the amount of DNA strand proximal to the electrode surface (Fig. S1). The probe densities obtained were in the range from 1.4 × 1012 to 5.3 × 1012 molecules cm–2, which is within the range previously recommended (1.2 × 1012 to <~5 × 1012 molecules cm–2; Zhang et al. 2007). For Fig. S1, the surface density of thiolated capture probes was calculated as 5.3 × 1012.
Assay performance
In order to demonstrate the viability of the assay, we compared the current response obtained in the presence of two designated synthetic target concentrations (1 nM and 100 fM) with that of NTC, which corresponds to a control reaction where nuclease-free water instead of synthetic target was mixed with CP1 functionalised beads, keeping all other assay components and subsequent steps consistent. As shown in Fig. 2a, an amperometric signal >10 times higher was obtained for 1 nM target DNA concentration than NTC (mean current density 2.5 vs 0.23 µA cm–2; n = 3). Similarly, low target concentration (100 fM) also showed significantly higher current response (0.71 µA cm–2) than NTC (>3 times higher). Our assay design is based on ‘sequential correlation’ whereby the target concentration in the sample is directly correlated with the number of DNA molecules hybridised to the self-assembled monolayer of CP2 on SPGEs, which in turn is proportional to the concentration of biotinylated DPs confined to the target–CP2 hybrid. This way, the streptavidin–HRP concentration immobilised onto this sandwich DNA assembly and ultimately biocatalytic oxidation of TMB are directly proportional to the target concentration in the sample. Thus, current response obtained from the electrocatalysis of the yellow coloured diimine compound provides a direct measure of target concentration. Each concentration as well as NTC were analysed in triplicate, and intra-assay variability was evaluated by determining the percentage relative standard deviation (RSD) between the current responses obtained for each replicate. As indicated by error bars in Fig. 2a, our assay showed high repeatability, with RSD values of <5% obtained. Representative chronoamperograms obtained from the analysis of known target concentrations and NTC are shown in Fig. 2b.

|
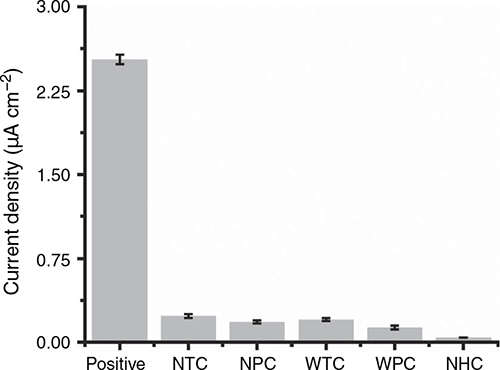
High specificity of the signal is a prerequisite for electrochemical DNA assays. Several components of the assay may be able to contribute to background signal, including: interaction of probes with non-complementary sequences (a major concern when the starting sample is not a highly purified DNA but rather a crude lysate, which is essentially a heterogenous pool of various biomolecules such as proteins and RNA as well as sugarcane genomic DNA or DNA from any other microorganisms); non-specific adsorption of target and/or non-target DNA sequences onto the bead surface; and nonspecific interaction of HRP with the gold electrodes. We systematically determined the possible effect of each assay component on the signal. As shown in Fig. 3 and Fig. S2, current response obtained for all controls was comparable with or less than NTC, thus indicating minimal interference from any of the assay components and highly specific target detection. In wrong probe control (WPC), non-complementary biotinylated and thiolated probes were used (sequences not shown), whereas in wrong target control (WTC), a synthetic DNA fragment corresponding to hsa-miR-891 was used, keeping all other components of the assay constant. Amperometric response obtained for both controls (mean current densities 0.13 µA cm–2 for WPC and 0.20 µA cm–2 for WTC; n = 3) was similar to or less than NTC, indicating that the probes used in our assay are capable of capturing X. albilineans sequences with high specificity. No probe control (NPC) comprised ‘mock purified’ high target concentration (1 nM) where magnetic beads not functionalised with biotinylated CP1 were mixed with target DNA. All subsequent assay steps remained unchanged. In no-HRP control (NHC), all assay steps were similar to the synthetic target reaction using a high concentration of target (1 nM), except at the stage of streptavidin–HRP immobilisation, where 0.1% PBST was introduced into the system rather than streptavidin–HRP. Mean current densities obtained for NPC and NHC were 0.18 and 0.042 µA cm–2, respectively. All control experiments were done in triplicate and showed high reproducibility (RSD <5%). Taken together, these results suggest high specificity of probes (WPC and WTC), low non-specific adsorption of DNA on beads or gold electrodes (NTC and NPC), and very little non-specific interaction of HRP with the sensor surface (compare NHC with other controls). Corresponding chronoamperograms for control experiments are given in Fig. S2.
|
A broad and linear dynamic range is an essential performance matrix for pathogen detection assays because high variability in target concentration is observed. Therefore, to assess the sensitivity and linearity of the assay, we analysed five different serially diluted concentrations of synthetic target DNA sequence (100 fM to 1 nM). As discussed previously, the electrochemical signal obtained in our assay is directly proportional to the analyte concentration. As shown in Fig. 4a, an increment in amperometric current response proportional to the increasing concentration of target DNA was observed. Our assay was able to detect synthetic target DNA reproducibly over a broad dynamic range between 100 fM and 1 nM. The linear regression equation was estimated to be y = 0.46 log (target concentration) + 6.64, with a correlation coefficient of R2 = 0.99, indicating good linearity of the assay (inset Fig. 4b). Limit of detection (LOD) of our assay, defined as the concentration of the analyte that can be reliably distinguished from NTC, was found to be 100 fM (S/n = >3). All of the reactions were done in triplicate and RSD was calculated. RSD values for all samples were found to be <7%, showing minimum intra-assay variability and excellent reproducibility. Our assay showed sensitivity 100 times better than one of the earlier reported sugarcane pathogen detection platforms, for which the authors reported a detection limit in the range of 10 pM (Wongkaew and Poosittisak 2014). On the other hand, a recently reported gold nanoparticle based plant virus detection platform could only achieve a lower detection limit of 100 nM; moreover, a logarithmic relationship was only observed within a very narrow range of concentrations (0.1–10 µM) (Khater et al. 2019). However, the platform reported by (Siddiquee et al. 2014) achieved attomolar-level sensitivity and detection over a very broad dynamic range (1.0 × 10−18 to 1.82 × 10−4 mol L−1). Although this platform was several orders of magnitude more sensitive than our assay, it involved complicated sensor fabrication and relied on a tedious phenol-chloroform DNA extraction process (Siddiquee et al. 2014). By comparison, our assay provided a rapid, naked eye readout streamlined with a simple field-deployable DNA isolation method and without extensive sensor fabrication steps.

|
Detection of leaf scald in samples collected from field trial
In order to further demonstrate the suitability of our method to real world applications, we analysed a series of sugarcane sap samples. Samples were collected from SRA Woodford leaf scald screening trial in an operator blind fashion. Samples were collected from varieties with various degrees of X. albilineans susceptibility (highly resistant to highly susceptible). The susceptibility/resistance status of the collected samples were known only to SRA staff. The Griffith team involved in conducting the electrochemical and molecular analyses were not aware of the sample susceptibility/resistance status during the method development stage. The target DNA was detected both colourimetrically (naked eye) and electrochemically. As shown in Fig. 5, development of blue colour was observed only in samples with known concentration of synthetic target DNA or sugarcane sap samples, whereas NTC did not show any colour development. The intensity of the colour also provided a rapid qualitative result of bacterial concentration in the sample. Bright blue colour in sample 133 (1) (cultivar Q133) indicated high bacterial concentration, whereas sample 208 (2) (cultivar Q208) showed very low intensity suggesting low bacterial concentration in the sample (Fig. 5).

|
Target DNA was quantified electrochemically by performing chronoamperometric measurements as described earlier. As shown in Fig. 6a, the sensor successfully detected varying concentrations of target sequence in the samples collected from the field trial. The target DNA detected in most of the samples was within the LOD of our assay, indicating its applicability to real-world samples. In most of the cases, detected X. albilineans DNA agreed with the resistance rating of cultivars. For example, a highly resistant variety such as Q208A (sample 208) consistently provided low response. However, some discrepancies between the expected bacterial concentrations and electrochemical response were also observed. For example, sample 44 (1), which was collected from susceptible cultivar (Q44), provided reduced current response. These discrepancies can be attributed to the age and the location of the stalks or where the samples were taken from. (Garces et al. 2014) reported variation of bacterial concentrations among types of tissues and plant ages. Nonetheless, all infected sugarcane samples collected were positive to the bacteria in our study. Figure 6b shows the amperograms for selected field trial samples.

|
Validation of assay with qPCR
A qPCR assay was designed to validate the outcomes of our biosensor platform. The qPCR primers target a segment of 114 bp within the XALB1 gene cluster (corresponding to positions 1740266–1740379 in X. albilineans GPE PC73 complete genome) that encompassed the region targeted in the electrochemical assay. The standard plot for qPCR-based absolute quantification was obtained by plotting Cq (quantification cycle) values against log of corresponding known X. albilineans starting quantity. Absolute quantification of starting DNA copy number in field trial samples was calculated by comparing with the standard plot. The qPCR results depicted an excellent concordance with the electrochemical quantification, demonstrating that the newly developed biosensing assay is suitable for detecting X. albilineans in sugarcane sap samples (Fig. 7). The qPCR values were strongly correlated (r = 0.91, P < 0.001) with the biosensor assay (Table 1). The strong correlation (r = 0.99, P < 0.0001) of Cq value of biosensor and qPCR samples showed repeatability of the test.

|

|
In this proof-of-concept study, we have demonstrated sensitive (up to femtomolar level) detection of the sugarcane pathogen X. albilineans in xylem sap samples. The assay offers some significant advantages. First, we have demonstrated that our assay is compatible with the boiling lysis-based DNA extraction method. When compared with kit-based or organic extraction-based DNA isolation methods, the boiling lysis method is not only low-cost, but also potentially field deployable because it requires relatively simple instruments (centrifuge, heat block). Also, crude lysate is a more stable sample source and more resistant to degradation and contamination than cut stalks. Thus, instead of shipping sugarcane stalks, the bacterial DNA can be extracted by using a simple on-farm setup and sent to remote laboratories for testing. In the case of colourimetric detection, the process takes <1 h and gives a first pass yes/no answer. Electrochemical assay utilises disposable screen-printed electrodes, which cost as little as ∼US$3 each, making overall per sample cost of the assay <$10. Finally, unlike most of the electrochemical assays reported previously, our method achieves sensitive target detection without complicated sensor fabrication steps.
This generic technology is flexible in the sense that it is potentially applicable to almost all types of pathogens by using specific probes. Our assay could be applicable for the diagnosis and management of diseases in other agricultural and horticultural crops, and potentially in other environments (e.g. in soil). Moreover, using multi-well screen-printed electrodes, the platform can be further extended for high throughput and/or multiplex detection of various pathogens simultaneously. The assay could also form the basis of a fully integrated, next-generation handheld device, associated with a purpose-built smartphone application. Such a device can be particularly useful in developing geographic information systems (GIS) for surveillance as well as prevalence and risk mapping of various diseases. The proposed integrated device can also be used for developing rapid screening and warning systems for exotic diseases to be implemented at entry points like airports and seaports.
Conclusion
We have developed a sensitive method for the detection of leaf scald disease of sugarcane. This method offers a dual detection where naked eye evaluation of colour change provides qualitative detection of X. albilineans, and quantification up to femtomolar levels of target DNA was achieved by electrochemistry. Our assay showed high specificity, good linear response (R2 = 0.99) for detection of target concentrations 100 fM–1 nM, and excellent repeatability (RSD <7%, n = 3). The proposed assay can potentially be used for detection of a range of sugarcane and other agricultural pathogens and can also form the basis of next generation portable integrated devices for on-farm applications.
Conflicts of interest
The authors declare no competing financial interest.
Author contributions
MU and NBA equally contributed to this work.
Funding statement
This work was jointly supported by SRA through Innovation Catalyst Project (INNOVA 06) and Griffith University ESC Research Support Scheme 2018.
References
Birch RG (2001) Xanthomonas albilineans and the antipathogenesis approach to disease control. Molecular Plant Pathology 2, 1–11.| Xanthomonas albilineans and the antipathogenesis approach to disease control.Crossref | GoogleScholarGoogle Scholar | 20572987PubMed |
Birch RG, Patil SS (1985) Preliminary characterization of an antibiotic produced by Xanthomonas albilineans which inhibits DNA synthesis in Escherichia coli. Microbiology 131, 1069–1075.
| Preliminary characterization of an antibiotic produced by Xanthomonas albilineans which inhibits DNA synthesis in Escherichia coli.Crossref | GoogleScholarGoogle Scholar |
Birch RG, Bower R, Elliott A, Hansom S, Basnayake S, Zhang L (2000) Regulation of transgene expression: progress towards practical development in sugarcane, and implications for other plant species. In ‘Developments in plant genetics and breeding’. Vol. 5. (Ed. AD Arencibia) pp. 118–125. (Elsevier)
Boriachek K, Umer M, Islam MN, Gopalan V, Lam AK, Nguyen NT, Shiddiky MJA (2018) An amplification-free electrochemical detection of exosomal miRNA-21 in serum samples. Analyst 143, 1662–1669.
| An amplification-free electrochemical detection of exosomal miRNA-21 in serum samples.Crossref | GoogleScholarGoogle Scholar | 29512659PubMed |
Cociancich S, Pesic A, Petras D, Uhlmann S, Kretz J, Schubert V, Vieweg L, Duplan S, Marguerettaz M, Noëll J, Pieretti I, Hügelland M, Kemper S, Mainz A, Rott P, Royer M, Süssmuth RD (2015) The gyrase inhibitor albicidin consists of p-aminobenzoic acids and cyanoalanine. Nature Chemical Biology 11, 195–197.
| The gyrase inhibitor albicidin consists of p-aminobenzoic acids and cyanoalanine.Crossref | GoogleScholarGoogle Scholar | 25599532PubMed |
Comstock JC, Irey MS (1992) Detection of the sugarcane leaf scald pathogen, Xanthomonas albilineans, using tissue blot immunoassay, ELISA, and isolation techniques. Plant Disease 76, 1033–1035.
| Detection of the sugarcane leaf scald pathogen, Xanthomonas albilineans, using tissue blot immunoassay, ELISA, and isolation techniques.Crossref | GoogleScholarGoogle Scholar |
Comstock JC, Wang Z, Perdomo R (1997) The incidence of leaf scald and its effect on yield components. Sugar Cane 4, 18–22.
Davis MJ, Rott P, Baudin P, Dean J (1994) Evaluation of selective media and immunoassays for detection of Xanthomonas albilineans, causal agent of sugarcane leaf scald disease. Plant Disease 78, 78–82.
| Evaluation of selective media and immunoassays for detection of Xanthomonas albilineans, causal agent of sugarcane leaf scald disease.Crossref | GoogleScholarGoogle Scholar |
Davis MJ, Rott P, Warmuth C, Chatenet M, Baudin P (1997) Intraspecific genomic variation within Xanthomonas albilineans, the sugarcane leaf scald pathogen. Phytopathology 87, 316–324.
| Intraspecific genomic variation within Xanthomonas albilineans, the sugarcane leaf scald pathogen.Crossref | GoogleScholarGoogle Scholar | 18945175PubMed |
Drummond TG, Hill MG, Barton JK (2003) Electrochemical DNA sensors. Nature Biotechnology 21, 1192–1199.
| Electrochemical DNA sensors.Crossref | GoogleScholarGoogle Scholar | 14520405PubMed |
Fang Y, Ramasamy RP (2015) Current and prospective methods for plant disease detection. Biosensors 5, 537–561.
| Current and prospective methods for plant disease detection.Crossref | GoogleScholarGoogle Scholar | 26287253PubMed |
Ferapontova EE (2018) DNA electrochemistry and electrochemical sensors for nucleic acids. Annual Review of Analytical Chemistry 11, 197–218.
| DNA electrochemistry and electrochemical sensors for nucleic acids.Crossref | GoogleScholarGoogle Scholar | 29894229PubMed |
Garces FF, Gutierrez A, Hoy JW (2014) Detection and quantification of Xanthomonas albilineans by qPCR and potential characterization of sugarcane resistance to leaf scald. Plant Disease 98, 121–126.
| Detection and quantification of Xanthomonas albilineans by qPCR and potential characterization of sugarcane resistance to leaf scald.Crossref | GoogleScholarGoogle Scholar | 30708616PubMed |
Hoy J (1994) Sugarcane leaf scald distribution, symptomatology. Plant Disease 78, 1083–1087.
| Sugarcane leaf scald distribution, symptomatology.Crossref | GoogleScholarGoogle Scholar |
Islam MN, Masud MK, Nguyen NT, Gopalan V, Alamri HR, Alothman ZA, Hossain MSA, Yamauchi Y, Lam AK, Shiddiky MJA (2018) Gold-loaded nanoporous ferric oxide nanocubes for electrocatalytic detection of microRNA at attomolar level. Biosensors & Bioelectronics 101, 275–281.
| Gold-loaded nanoporous ferric oxide nanocubes for electrocatalytic detection of microRNA at attomolar level.Crossref | GoogleScholarGoogle Scholar |
Khater M, de la Escosura-Muñiz A, Merkoçi A (2017) Biosensors for plant pathogen detection. Biosensors & Bioelectronics 93, 72–86.
| Biosensors for plant pathogen detection.Crossref | GoogleScholarGoogle Scholar |
Khater M, de la Escosura-Muñiz A, Quesada-González D, Merkoçi A (2019) Electrochemical detection of plant virus using gold nanoparticle-modified electrodes Analytica Chimica Acta 1046, 123–131.
| Electrochemical detection of plant virus using gold nanoparticle-modified electrodesCrossref | GoogleScholarGoogle Scholar | 30482289PubMed |
Koo KM, Carrascosa LG, Shiddiky MJA, Trau M (2016) Poly(A) extensions of miRNAs for amplification-free electrochemical detection on screen-printed gold electrodes. Analytical Chemistry 88, 2000–2005.
| Poly(A) extensions of miRNAs for amplification-free electrochemical detection on screen-printed gold electrodes.Crossref | GoogleScholarGoogle Scholar | 26814930PubMed |
Lau HY, Wu H, Wee EJH, Trau T, Wang Y, Botella JR (2017) Specific and sensitive isothermal electrochemical biosensor for plant pathogen DNA detection with colloidal gold nanoparticles as probes. Scientific Reports 7, 38896
| Specific and sensitive isothermal electrochemical biosensor for plant pathogen DNA detection with colloidal gold nanoparticles as probes.Crossref | GoogleScholarGoogle Scholar | 28094255PubMed |
Masud MK, Islam MN, Haque MH, Tanaka S, Gopalan V, Alici G, Nguyen NT, Lam AK, Hossain MSA, Yamauchi Y, Shiddiky MJA (2017) Gold-loaded nanoporous superparamagnetic nanocubes for catalytic signal amplification in detecting miRNA. Chemical Communications 53, 8231–8234.
| Gold-loaded nanoporous superparamagnetic nanocubes for catalytic signal amplification in detecting miRNA.Crossref | GoogleScholarGoogle Scholar |
McLeod R, McMahon G, Allsopp P (1999) Costs of major pests and diseases to the Australian sugar industry. Plant Protection Quarterly 14, 42–46.
Nezhad AS (2014) Future of portable devices for plant pathogen diagnosis. Lab on a Chip 14, 2887–2904.
| Future of portable devices for plant pathogen diagnosis.Crossref | GoogleScholarGoogle Scholar | 24920461PubMed |
Pan YB, Grisham MP, Burner DM, Legendre BL, Wei Q (1999) Development of polymerase chain reaction primers highly specific for Xanthomonas albilineans, the causal bacterium of sugarcane leaf scald disease. Plant Disease 83, 218–222.
| Development of polymerase chain reaction primers highly specific for Xanthomonas albilineans, the causal bacterium of sugarcane leaf scald disease.Crossref | GoogleScholarGoogle Scholar | 30845497PubMed |
Pieretti I, Royer M, Barbe V, Carrere S, Koebnik R, Cociancich S, Couloux A, Darrasse A, Gouzy J, Jacques M-A, Lauber E, Manceau C, Mangenot S, Poussier S, Segurens B, Szurek B, Verdier V, Arlat M, Rott P (2009) The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae. BMC Genomics 10, 616
| The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae.Crossref | GoogleScholarGoogle Scholar | 20017926PubMed |
Plant Health Australia (2019) The National Plant Biosecurity Status Report 2018 and 2019. Plant Health Australia, Canberra, ACT.
Qavi AJ, Kindt JT, Bailey RC (2010) Sizing up the future of microRNA analysis. Analytical and Bioanalytical Chemistry 398, 2535–2549.
| Sizing up the future of microRNA analysis.Crossref | GoogleScholarGoogle Scholar | 20680616PubMed |
Ricaud C, Ryan CC (1989) Leaf scald. In ‘Diseases of sugarcane’. Chapter III. (Eds C Ricaud, BT Egan, AG Gillaspie, CG Hughes) pp. 39–58. (Elsevier: Amsterdam)
Rott P (1995) Leaf scald of sugarcane. Agriculture for Development 6, 49–56.
Rott P, Davis M (1995) Recent advances in research on variability of Xanthomonas albilineans, causal agent of sugarcane leaf scald disease. In ‘Proceedings of the International Society of Sugarcane Technologists Congress’. Vol. 22, pp. 498–503. (International Society of Sugarcane Technologists)
Rott P, Davis MJ (2000) Leaf scald. In ‘A guide to sugarcane diseases’. (Eds P Rott, R Bailey, J Comstock, B Croft, A Saumtally) pp. 38–44. (CIRAD Publication Services: Montepellier, France)
Rott P, Mohamed I, Klett P, Soupa D, de Saint-Albin A, Feldmann P, Letourmy P (1997) Resistance to leaf scald disease is associated with limited colonization of sugarcane and wild relatives by Xanthomonas albilineans. Phytopathology 87, 1202–1213.
| Resistance to leaf scald disease is associated with limited colonization of sugarcane and wild relatives by Xanthomonas albilineans.Crossref | GoogleScholarGoogle Scholar | 18945019PubMed |
Royer M, Costet L, Vivien E, Bes M, Cousin A, Damais A, Pieretti I, Savin A, Megessier S, Viard M, Frutos R, Gabriel DW, Rott PC (2004) Albicidin pathotoxin produced by Xanthomonas albilineans is encoded by three large PKS and NRPS genes present in a gene cluster also containing several putative modifying, regulatory, and resistance genes. Molecular Plant-Microbe Interactions 17, 414–427.
| Albicidin pathotoxin produced by Xanthomonas albilineans is encoded by three large PKS and NRPS genes present in a gene cluster also containing several putative modifying, regulatory, and resistance genes.Crossref | GoogleScholarGoogle Scholar | 15077674PubMed |
Shiddiky MJA, Rahman MA, Shim YB (2007) Hydrazine-catalyzed ultrasensitive detection of DNA and proteins. Analytical Chemistry 79, 6886–6890.
| Hydrazine-catalyzed ultrasensitive detection of DNA and proteins.Crossref | GoogleScholarGoogle Scholar |
Shiddiky MJA, Torriero AAJ, Zhao C, Burgar I, Kennedy G, Bond AM (2009) Nonadditivity of faradaic currents and modification of capacitance currents in the voltammetry of mixtures of ferrocene and the cobaltocenium cation in protic and aprotic ionic liquids. Journal of the American Chemical Society 131, 7976–7989.
| Nonadditivity of faradaic currents and modification of capacitance currents in the voltammetry of mixtures of ferrocene and the cobaltocenium cation in protic and aprotic ionic liquids.Crossref | GoogleScholarGoogle Scholar |
Shiddiky MJA, Torriero AAJ, Zeng Z, Spiccia L, Bond AM (2010) Highly selective and sensitive DNA assay based on electrocatalytic oxidation of ferrocene bearing zinc(ii)−cyclen complexes with diethylamine. Journal of the American Chemical Society 132, 10053–10063.
| Highly selective and sensitive DNA assay based on electrocatalytic oxidation of ferrocene bearing zinc(ii)−cyclen complexes with diethylamine.Crossref | GoogleScholarGoogle Scholar |
Siddiquee S, Rovina K, Yusof NA, Rodrigues KF, Suryani S (2014) Nanoparticle-enhanced electrochemical biosensor with DNA immobilization and hybridization of Trichoderma harzianum gene. Sensing and Bio-Sensing Research 2, 16–22.
| Nanoparticle-enhanced electrochemical biosensor with DNA immobilization and hybridization of Trichoderma harzianum gene.Crossref | GoogleScholarGoogle Scholar |
Wang ZK, Comstock JC, Hatziloukas E, Schaad NW (1999) Comparison of PCR, BIO-PCR, DIA, ELISA and isolation on semiselective medium for detection of Xanthomonas albilineans, the causal agent of leaf scald of sugarcane. Plant Pathology 48, 245–252.
| Comparison of PCR, BIO-PCR, DIA, ELISA and isolation on semiselective medium for detection of Xanthomonas albilineans, the causal agent of leaf scald of sugarcane.Crossref | GoogleScholarGoogle Scholar |
Wong ELS, Gooding JJ (2003) Electronic detection of target nucleic acids by a 2,6-disulfonic acid anthraquinone intercalator. Analytical Chemistry 75, 3845–3852.
| Electronic detection of target nucleic acids by a 2,6-disulfonic acid anthraquinone intercalator.Crossref | GoogleScholarGoogle Scholar |
Wongkaew P, Poosittisak S (2014) Diagnosis of sugarcane white leaf disease using the highly sensitive DNA based voltammetric electrochemical determination. American Journal of Plant Sciences 5, 2256–2268.
| Diagnosis of sugarcane white leaf disease using the highly sensitive DNA based voltammetric electrochemical determination.Crossref | GoogleScholarGoogle Scholar |
Zhang J, Song S, Wang L, Pan D, Fan C (2007) A gold nanoparticle-based chronocoulometric DNA sensor for amplified detection of DNA. Nature Protocols 2, 2888–2895.
| A gold nanoparticle-based chronocoulometric DNA sensor for amplified detection of DNA.Crossref | GoogleScholarGoogle Scholar | 18007624PubMed |