

Chiral 1-D coordination polymer chains featuring 1,1′-binaphthyl
Hui Min Tay A B , Shannon Thoonen A C and Carol Hua A *
A B , Shannon Thoonen A C and Carol Hua A *
A
B
C
Abstract
Four 1-D chain coordination polymers containing bent 1,1′-binaphthyl ligands were synthesised with NiII, CuII and AgI. The use of (R)-4,4′-(2,2′-diethoxy-[1,1′-binaphthalene]-6,6′)dipyridine as a ligand yielded isostructural 1-D looping chains with NiII and CuII, whereas the use of AgI yielded both linear and helical 1-D chains. Changing the dipyridyl coordination groups to dicarboxylates in (S)-6,6′-dicarboxyl-2,2′-diethoxy-1,1′-binaphthalene yielded a 1-D looping chain with a CuII paddlewheel motif. The AgI 1-D chain features two crystallographically distinct 1-D chain morphologies with a triple helix and linear strips. The packing arrangement of the 1-D chains differs because of the intermolecular interactions present, with the steric bulk of the ethoxy substituent on the 1,1′-binaphthyl enabling the formation of large void spaces.
Keywords: 1,1′-binaphthyl, 1-D looping chains, chirality, coordination polymers, helical 1-D chains, packing, self-assembly, transition metals.
Introduction
Chirality has an integral role in nature, with many important biomolecules including enzymes and sugars consisting of chiral building blocks. The ability to detect or separate chiral molecules is particularly important in the pharmaceutical, food and agriculture industries as the presence of the incorrect enantiomer may lead to detrimental side effects. Carefully designed chiral molecular systems are therefore necessary to enable their detection and separation by the specific interactions of chiral molecules with other chiral systems.
1,1′-Binaphthyl has attracted significant attention as a source of chirality due to its modularity, robustness and versatility.1 In contrast to the point chirality featured in most chiral molecules including amino acids, 1,1′-binaphthyl possesses axial chirality which is caused by the hindered rotation of two naphthyl rings around a C–C bond, making the system particularly robust to racemisation from changes in pH or chemical environment.2 Racemisation of 1,1′-binaphthyl typically does not occur until high temperatures are reached.3
The use of 1,1′-binaphthyl in chiroptical and enantioseparation applications has been widely explored in smaller molecular systems,4 however, there exists significant advantages for the incorporation of 1,1′-binaphthyl as the source of chirality in larger supramolecular structures including coordination polymers (CPs), metal–organic frameworks (MOFs) and covalent organic frameworks (COFs).2,5–8 These advantages include increased stability, the ability to operate in the solid state and opportunities for greater selectivity. A relatively small number of studies have involved MOFs and CPs containing 1,1′-binaphthyl systems and include 1-D,9–14 2-D and 3-D systems.15–24 Despite the limited reports of MOFs and CPs containing 1,1′-binaphthyl, they have been used in myriad applications including asymmetric catalysis,15,16,23 chemical sensing,17 chiroptical fluorescence sensing18,20 and hydrogen storage.19
The formation of 1-D chains with 1,1′-binaphthyl-containing ligands most commonly occurs with 6,6′-substituted species due to the bent orientation of the ligand.9–14 The 1-D chains formed are most commonly formed as loops where a pair of 1,1′-binaphthyl ligands bind to the metal centre forming a parallelogram-shaped void. 1-D looping chains containing 1,1′-binaphthyl have been used in a number of stereoselective applications including circularly polarised luminescence and two-photon excited fluorescence in a CdII 1-D chain CP,11 spin-crossover in an interlocked 1-D chain containing poly[n]catenane with FeII 12 and as a chiral stationary phase with high-performance liquid chromatography (HPLC) as a MOF–silica composite with CuII.13 Of particular importance in all applications is the ability of the framework to interact with chiral guests, which is facilitated by diffusion of the guests into the voids. Guest interaction with the framework structures can be promoted by engineering the 1-D channels or facilitating larger accessible voids through altering the packing of supramolecular components.
In this work we report the synthesis of four 1-D chains with NiII, CuII and AgI containing two bent 1,1′-binaphthyl ligands, (R)-4,4′-(2,2′-diethoxy-[1,1′-binaphthalene]-6,6′)dipyridine ((R)-L1) and (S)-6,6′-dicarboxyl-2,2′-diethoxy-1,1′-binaphthalene ((S)-H2L2) (Fig. 1). The 1-D chains synthesised using NiII and CuII are structurally similar, featuring a 1-D looping chain with pairs of 1,1′-binaphthyl ligands coordinating to the metal centres. The 1-D chain obtained with AgI contains an open structure where the 1-D chains stack with each other to form a helix. The stacking of the 1-D chains is investigated in each case to rationalise the formation of large voids and channels.
Results and discussion
L1 was synthesised using a Suzuki–Miyaura cross coupling reaction between (R)-6,6′-dibromo-2,2′-diethoxy-1,1′-binaphthalene and 4-pyridyl boronic acid to yield the product as a white solid in 97% yield. H2L2 was synthesised according to literature methods,25,26 with additional ligand synthesis details provided in the Supplementary material. (R)-4,4′-(2,2′-Diethoxy-[1,1′-binaphthalene]-6,6′)dipyridine ((R)-L1) was dissolved in DMF and combined with either Ni(NO3)2∙6H2O or Cu(NO3)2∙3H2O in diffusion reactions at room temperature to yield the formation of [Ni((R)-L1)2(H2O)2] (1) and [Cu((R)-L1)2(H2O)2] (2) respectively. Frameworks 1 and 2 are isostructural and crystallised in the orthorhombic P21212 space group (Fig. 2 and Supplementary Fig. S1; Supplementary Table S1). The bulk purity of 1 and 2 were verified by powder X-ray diffraction (PXRD) (Supplementary Fig. S2 and S3). The coordination geometry around both the NiII and CuII centres is octahedral, featuring four N-donors from four (R)-L1 ligands and two O-donors from coordinated water molecules in the axial positions (Fig. 2a; Supplementary Table S2). Typical Ni–N (2.096(3), 2.111(3) Å) and Ni–O bond lengths (2.093(3) Å) are observed in 1 whereas Jahn–Teller distortion occurs in 2 with elongated Cu–O bond lengths of 2.437(5) Å and shorter Cu–N bond lengths of 2.015(5) and 2.026(5) Å. The effect of the Jahn–Teller distortion of CuII is reflected in the cell parameters for 1 and 2; a = 12.954(3), b = 16.633(3), c = 18.387(4) Å for 1 and a = 12.662(3), b = 16.357(3) and c = 18.472(4) Å for 2 where the shortening of the a and b axes may be attributed to the contraction in the Cu–N bonds and the lengthening of the c axis to the longer Cu–O bonds. The (R)-L1 ligands adopt a bent conformation with the two naphthalene rings lying at 73.6° for 1 and 72.1° for 2 relative to each other, clearly displaying the origin of 1,1′-binaphthyl’s axial chirality (Fig. 2b). Each (R)-L1 ligand connects two MII centres through the pyridyl groups.
Crystal structure of [Ni((R)-L1)2(H2O)2] (1) showing (a) the coordination environment around NiII, (b) the looping 1-D chain and stacking of the 1-D chain as viewed down the (c) c axis and (d) b axis.
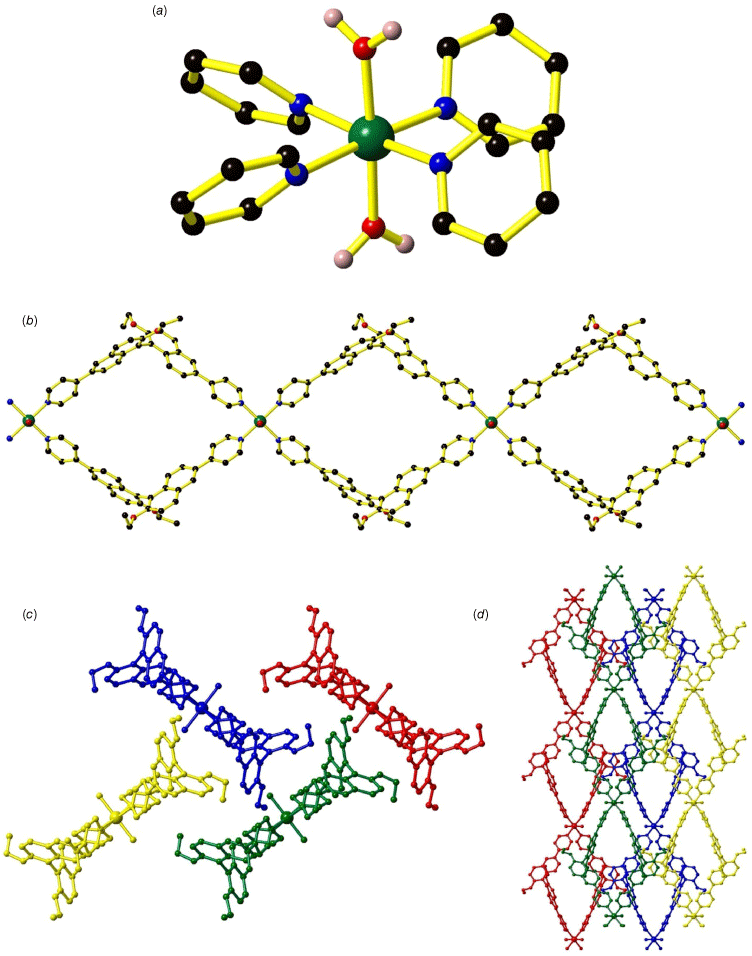
Adjacent MII centres are linked by pairs of (R)-L1 ligands to form a looping 1-D chain containing parallelogram-shaped voids (Fig. 2b) with dimensions 18.4 × 12.2 Å for 1 and 18.5 × 11.8 Å for 2. The steric bulk provided by the ethoxy substituent on the 1,1′-binaphthyl ring prevents close packing of the 1-D chains. The 1-D chains pack in an AABB manner with each ‘layer’ of 1-D chains orientated in a perpendicular arrangement relative to its neighbours above and below (Fig. 2c, d). No catenation occurs through the looping chains. Intermolecular interactions are observed between the stacked 1-D chains with CH–π interactions between the ethoxy substituent and an adjacent naphthyl ring from a neighbouring 1-D chain dominating (d = 3.854 Å for 1, d = 3.773 Å for 2). The preclusion of close packing results in relatively large accessible void spaces of 40.2% for 1 and 35.3% for 2, which is reflected in the thermal gravimetric analysis (TGA) with a mass loss below 150°C of 25% for 1 and 22.5% for 2 (Supplementary Fig. S8 and S9). These mass losses below 150°C are attributed to the liberation of the DMF and H2O molecules present in the voids of the crystal structure. A gradual mass loss above 300 and 250°C for 1 and 2 respectively represents decomposition of the framework structure. FT-IR spectra of 1 and 2 are similar, being dominated by the features of the ligand (Supplementary Fig. S5–S6).
A previous example of a CP exists containing an analogous dipyridyl 1,1′-binaphthyl ligand with substitution at the 6 and 6′ positions but which contains a OCH2OCH3 group in place of the ethoxy substituent, (R)-2,2′-bis(methoxymethoxy)6,6′-bis(4-pyridyl)-1,10-binaphthyl) ((R)-L3).11 In this report, (R)-L3 was combined with CdII to yield [Cd(NO3)2(R)-L3] (CSD refcodes: ZIJKIP and XITQUP).11 [Cd(NO3)2(R)-L3] also features a 1-D looping chain, but with a 7-c coordination geometry around the CdII centre from four N-donors on the equatorial plane and from four L1 ligands and three O-donors from two NO3− in the axial positions. One of the NO3− ligands is bound in a bidentate manner, whereas the other NO3− is bound in a monodentate manner. A torsion angle of 72.8° exists between the naphthyl rings in (R)-L3 in this structure, and it has voids with dimensions of 18.8 × 12.4 Å (for THF solvate) which is similar to 1 and 2. Modest accessible void spaces of 21.1% are present in [Cd(NO3)2(R)-L3] that are smaller than those observed for 1 and 2. The differences in the accessible void spaces can be attributed to both the stacking of the 1-D chains and the lower steric bulk from the smaller OEt group on (R)-L1 when compared to the OCH2OCH3 group in (R)-L3.
(R)-2,2′-Bis(methoxymethoxy)-6,6′-bis(4-pyridylethynyl)-1,10-binaphthyl ((R)-L4) containing an additional alkynyl group between the pyridyl and the 1,1′-binaphthyl has also been incorporated into a 1-D chain.12 [Fe(NCBH3)2((R)-L4)2]∙6CH3CN (CSD refcode: VUCXID with MeCN, VUCYAW with MeOH) was synthesised through the reaction of (R)-L4 with [Fe(NCBH3)2] to yield looping 1-D chains in an ABAC manner. The longer (R)-L4 enabled interlocking of the 1-D chains in a catenane-type motif and displayed spin crossover behaviour of the iron centre.12
Changing the dipyridyl coordinating groups in (R)-L1 to dicarboxylate groups, (S)-H2L2, resulted in the formation of [Cu2((S)-L2)2(DMF)2]‧2H2O (3) (Fig. 3). Framework 3 was synthesised using a solvothermal reaction of (S)-H2L2 with Cu2(NO3)2‧3H2O in an acidified DMF/H2O mixture (3:1) at 85°C to yield green needles. The bulk purity of 3 was confirmed using PXRD (Supplementary Fig. S4) and characterised with FT-IR (Supplementary Fig. S7). Framework 3 displays a number of similarities to 1 and 2 with the largest difference being that of the CuII coordination environment due to the carboxylate coordinating groups. The naphthyl rings of 1,1′-binaphthyl in 3 are orientated at ~105° relative to each other, reflecting the axial chirality of the ligand. The structure of 3 includes dinuclear CuII paddlewheel units, with the carboxylate groups of (S)-L2 ligands bridging two CuII centres in a bidentate manner (Fig. 3a). The apical sites of each CuII paddlewheel unit are occupied by DMF molecules with each CuII centre adopting a square pyramidal coordination geometry (Fig. 3b; Supplementary Table S3). The octahedral coordination geometry of 3 contrasts with 1 and 2, where water molecules are located in the axial positions. The CuII–O bond lengths in 3 range from 1.934(4) Å to 2.116(4) Å, typical for 5-coordinate CuII centres. The CuII paddlewheel units are linked by pairs of (S)-L2 ligands to form looping 1-D chains (Fig. 3c).
Crystal structure of [Cu2((S)-L2)2(DMF)2]‧2H2O (3) showing (a) the dinuclear CuII paddlewheel unit, (b) the (S)-L1 ligand coordinated to CuII ions, (c) the 1-D chain where the DMF coordinated in the axial positions has been omitted for clarity and the packing of the 1-D chains down the (d) a axis and (e) c axis. Atoms have been denoted as follows: carbon (black), copper (light blue), oxygen (red). Hydrogens have been omitted for clarity.
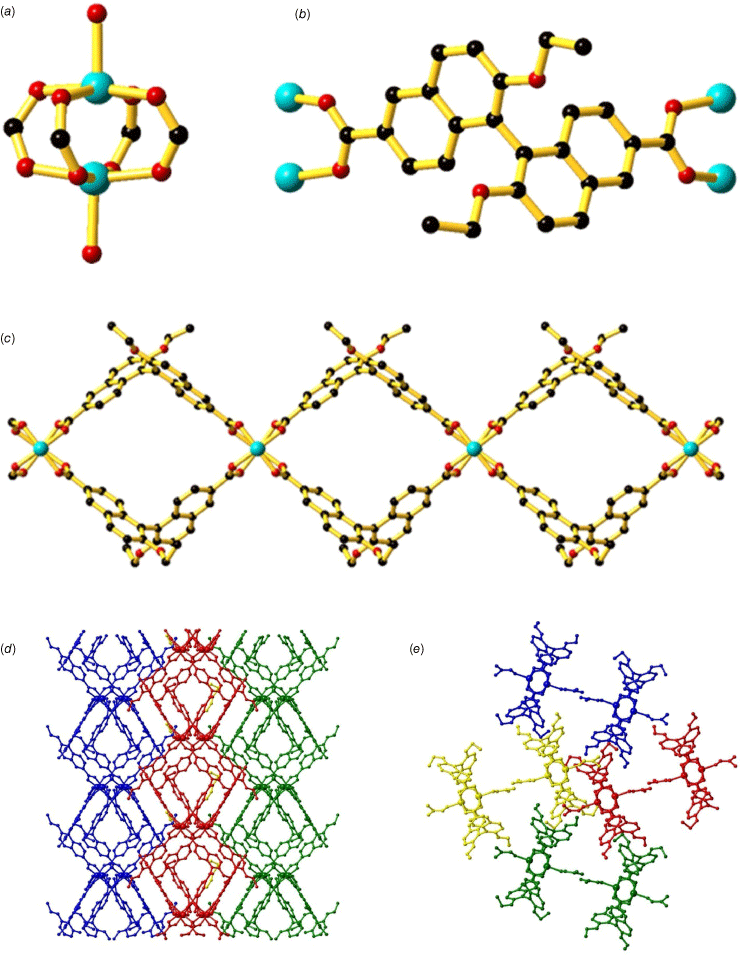
The 1-D chains pack in an ABAB fashion, with each ‘layer’ of 1-D chains orientated in the same direction. In a similar manner to 1 and 2, the adjacent layers of 1-D chains are positioned perpendicular to each other such that it appears the 1-D chains are running in the opposite direction. The steric bulk provided by the ethoxy substituents on the 1,1′-binaphthyl prevents close packing of the 1-D chains, resulting in an accessible void space of 33.8% (Fig. 3d, e). A gradual mass loss of ~16% is observed in the TGA (Supplementary Fig. S10) under 200°C which can be assigned to the loss of DMF molecules within the voids as well as the coordinated DMF molecule. The higher temperature required above the boiling point for removal of the coordinated DMF molecule reflects the additional energy required to break the coordination bond. A sharp mass loss at 300°C may be attributed to decarboxylation of (S)-L1 ligands, leading to thermal decomposition of 3.
The structure of 3 is similar to [Cu2(BDA)2(H2O)2]·2MeOH·4H2O (BDA = (S)-2,2′-dihydroxy-1,1′-binaphthalene-6,6′-dicarboxylic acid, CSD refcode: LABCOH)9 and [Cu2[(R)-L5]4(H2O)2]∙DMA (R)-L5 = (R)-2,2′-dimethoxy-1,10-binaphthalene-6,6′-dicarboxylic acid, CSD refcode: WAFPEB)13 in that all three CPs are 1-D looping chains containing a CuII paddlewheel motif. [Cu2(BDA)2(H2O)2]·2MeOH·4H2O and [Cu2[(R)-L5]4(H2O)2]∙DMA both feature a coordinated water molecule in the apical position of the CuII paddlewheel which contrasts with the coordinated DMF molecule in 3. The largest difference between the frameworks is in the extended packing of the 1-D chains within the crystal structure. The OH group from the 1,1′-binapthyl ligand in [Cu2(BDA)2(H2O)2]·2MeOH·4H2O is located on the edges of the 1-D chain pointing outwards, enabling the formation of an extensive hydrogen bonding network between adjacent chains yielding relatively tightly packed 1-D chains. The introduction of the methoxy group in [Cu2[(R)-L5]4(H2O)2]∙DMA still yields stacking of the 1-D chains, however, the CuII paddlewheels in the 1-D chains are now tilted at 69.2° in contrast with [Cu2(BDA)2(H2O)2]·2MeOH·4H2O where the CuII paddlewheels are orientated in the plane of the chain. The tilting of the 1-D chains and consequently the CuII paddlewheels is likely a result of accommodating the additional steric bulk of the methyl group and lack of hydrogen bonding ability of the methoxy group. The stacking of the 1-D chains in 3 can be attributed to the additional steric bulk afforded by the EtO substituent which consequently yields greater accessible void spaces of 33.8% when compared to 18% for [Cu2(BDA)2(H2O)2]·2MeOH·4H2O and 14.6% for Cu2[(R)-L5]4(H2O)2]∙DMA. The cylindrical voids present in [Cu2(BDA)2(H2O)2]·2MeOH·4H2O and Cu2[(R)-L5]4(H2O)2]∙DMA are retained in 3 where 1-D channels can be observed parallel to the a axis.
When a solution of (R)-L1 and LiOAc·2H2O in DMF/MeCN was allowed to slowly diffuse into an aqueous solution of AgNO3 at room temperature, colourless needles formed amid a brown gel after 3 days. Unfortunately, the formation of the crystals within the gel precluded the isolation of a bulk powder, and thus PXRD on the powder could not be performed. A solution was obtained in the orthorhombic space group I212121, which revealed a 1-D CP of composition [Ag((R)-L1)] (4) (Fig. 4). The structure contains AgI centres bound to the pyridyl nitrogen atoms of two (R)-L1 ligands. Each (R)-L1 adopts a bent conformation and links two AgI centres to form a chain (Fig. 4a). In contrast to 1–3, no looping 1-D chain structure is obtained due to the 2-c linear coordination of the AgI centre.
Crystal structure of [Ag((R)-L1)] (4) depicting (a) the 1-D zig-zag where both the AgI and (R)-L1 ligands act as two-connecting linkers, (b) the extended strips formed by half of the chains where chains belonging to adjacent layers are shown in red, green and blue and (c) the triple helix formed by half of the chains in C9, with the three chains shown in red, green and blue and the bent AgI centres represented in grey. (d) A view of the helix down the a axis. (e) A view along the a axis of the crystal packing. The extended strips are shown in blue and the triple helices in red.
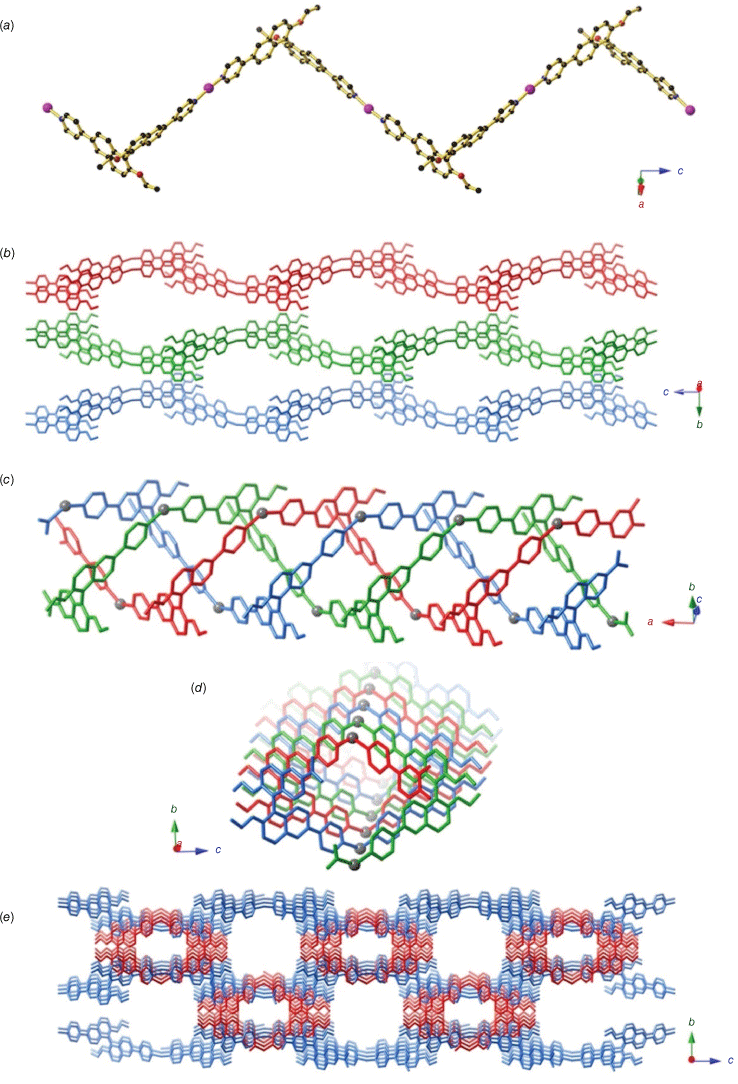
The structure contains two crystallographically distinct types of 1-D chains, which differ markedly in their packing arrangement. The first type consists of (R)-L1 ligands linking linear AgI centres to form extended strips that run parallel to the c axis. Adjacent chains pack along the a axis to form layers, which stack in an ABAB manner along the b axis (Fig. 4b). In the other half of the chains, the AgI surprisingly adopts a bent coordination geometry, resulting in the formation of a helical structure. Three separate chains are packed to form a triple helix, with the helical axis running parallel to the a axis (Fig. 4c, d). Extensive π–π interactions are present along the triple helix with the pyridyl rings stacked on top of naphthyl rings from an adjacent 1-D chain in a slightly offset manner. Cation–π interactions between the Ag+ ion and the pyridyl rings are present, where the Ag+ is sandwiched between two pyridyl rings. The helices partially occupy the spaces between adjacent layers, with the helical axis perpendicular to the direction of the zig-zag chains in each layer. This results in a structure with channels running along the a axis, which make up 23.6% of the structure (Fig. 4e).
Conclusion
Four 1-D chains containing bent 1,1′-binaphthyl ligands with dipyridyl and dicarboxylate coordinating groups were synthesised. Three NiII and CuII 1-D looping chains 1–3 were obtained containing 1,1′-binaphthyl ligands with both the dipyridyl, 1 and 2, and dicarboxylate in framework 3. The packing of the 1-D chains differed between the use of the dipyridyl and dicarboxylate ligands, where 1 and 2 pack in an AABB manner whereas 3 yields an ABAB packing. AgI yielded two 1-D chains in 4 – one a zig-zag chain and the other a bent helical chain. Relatively large accessible void volumes were present in frameworks 1–4 enabled by the introduction of an ethoxy group on the 1,1′-binaphthyl, highlighting how minimising opportunities for hydrogen bonding and increasing the steric bulk can help to prevent close packing of 1-D chains. Studies are ongoing with luminescent CPs containing 1,1′-binaphthyl for chiroptical fluorescence sensing.
Experimental
All chemicals and solvents were used as obtained and without further purification. 4,4′-Dibromo-2,2′-binaphthol,27 (S)-6,6′-dicarboxyl-2,2′-diethoxy-1,1′-binaphthalene (S)-L225,26 and Pd(PPh3)428 were synthesised according to literature procedures. (R)-4,4′-(2,2′-diethoxy-[1,1′-binaphthalene]-6,6′)dipyridine (R)-L1 was synthesised following a literature method29 with modification from (R)-6,6′-dibromo-2,2′-diethoxy-1,1′-binaphthalene. FT-IR spectra were measured on a Bruker Alpha spectrometer between 4000 and 400 cm−1 with 4 cm−1 resolution, using 32 scans and normalised as absorbance spectra. TGA was conducted on either a Mettler Toledo TGA/SDTA851 instrument using aluminium crucibles as sample holders or a PerkinElmer TGA 8000 instrument using ceramic sample holders under high-purity nitrogen. A typical analysis involved heating the sample to 450°C with a temperature increment of 4°C min−1. Microanalysis was performed at the Chemical Analysis Facility – Elemental Analysis Service in the Department of Chemistry and Biomolecular Science at Macquarie University, Australia.
Single crystal X-ray diffraction data were collected on either the MX1 or MX2 beamline at the Australian Synchrotron.30,31 In general, single crystals were transferred directly from the mother liquor into immersion oil and placed under a stream of nitrogen at 100 K. Crystal structures were solved by direct methods using the program SHELXT32 (ver. 2019/3, see https://shelx.uni-ac.gwdg.de/SHELX/) and refined using a full matrix least-squares procedure based on F2 (SHEXL),33 within the Olex234 GUI program (ver. 1.5, see https://www.olexsys.org/olex2/). In structures containing disordered solvent molecules that could not be satisfactorily modelled, the solvent mask routine within the Olex2 GUI was used.34
Ligand synthesis
(R)-6,6′-Dibromo-2,2′-diethoxy-1,1′-binaphthalene (4.62 g, 9.23 mmol), 4-pyridyl boronic acid (3.40 g, 27.7 mmol), Pd(PPh3)4 (533 mg, 0.461 mmol, 5 mol%) and potassium carbonate (6.38 g, 46.1 mmol) were dissolved in a mixture of degassed 1,4-dioxane (150 mL) and water (15 mL) to yield a pale yellow solution. The reaction mixture was heated at reflux under nitrogen for 40 h to yield a black suspension. The solvent was removed under vacuum and the residue dissolved in dichloromethane and filtered through celite. The filtrate was washed with water and the organic layer dried over MgSO4, filtered and the solvent removed to yield a yellow oil. The oil was crystallised by the dropwise addition of the crude product in the minimum amount of dichloromethane (~10 mL) into rapidly stirred hexane (300 mL) to yield the formation of a white solid (4.46 g, 97%). 1H NMR (CDCl3, 400 MHz) δ 8.67 (br s, 2H), 8.18 (d, J = 1.7 Hz, 1H), 8.06 (d, J = 9.0 Hz, 1H), 7.61 (d, J = 4.6 Hz, 2H), 7.50 (d, J = 9.0 Hz, 2H), 7.26 (d, J = 9.0 Hz, 2H), 4.11 (q, J = 7.0 Hz, 2H), 1.11 (t, J = 7.0 Hz, 3H) ppm. HRMS (ESI) m/z: [M + H]+ calculated for [C34H28N2O2 + H]+, 497.2229; found, 497.2446.
Framework synthesis
(R)-L1 (9.9 mg, 0.02 mmol) and Ni(NO3)2∙6H2O (5.8 mg, 0.02 mmol) were dissolved in DMF (1.0 mL) in a small vial. The vial was then placed within a larger vial and diethyl ether allowed to diffuse in as a vapour over 2 weeks to yield the formation of green plate crystals (4.0 mg, 18.4%). Elemental analysis (%): calculated for [C68H60N4NiO6]‧3.5H2O: C 70.96, H 5.87, N 4.87; found: C 70.96, H 5.93, N 4.87%.
A solution of (R)-L1 (9.9 mg, 0.02 mmol) in DMF (2.5 mL) was carefully layered over a solution of Cu(NO3)2∙3H2O (4.8 mg, 0.02 mmol) in H2O (2.5 mL) and allowed to diffuse at room temperature for 7 days to yield the formation of green block crystals (3.1 mg, 14%). Elemental analysis (%): calculated for [C68H60CuN4O6]‧2.9H2O: C 71.33, H 5.79, N 4.89; found: C 71.32, H 5.85, N 4.89%.
A mixture of (S)-L2 (6.45 mg, 0.015 mmol), Cu(NO3)2‧3H2O (7.25 mg, 0.03 mmol) and HCl (1 M, 100 µL) in DMF/H2O (2.0 mL, 3:1 v/v) was heated at 85°C for 2 days. The reaction mixture was cooled to room temperature before being washed with water and DMF, filtered and dried to afford bright green block crystals (2.7 mg, 15%). IR (ATR): 3060, 2928, 1665, 1622, 1467, 1401, 1344, 1230, 1138, 1092, 1045, 954, 814, 787, 766, 680, 651, 579, 497, 451 cm−1. Elemental analysis (%): calculated for [Cu2C58H54N2O14]‧2H2O: C 59.74, H 5.01, N 2.40; found: C 59.75, H 5.34, N 2.32%.
A solution of (R)-L1 (9.9 mg, 0.020 mmol) and LiOAc·2H2O (2.0 mg, 0.03 mmol) in DMF/MeCN (3 mL, 1:1 v/v) was allowed to slowly diffuse into a solution of AgNO3 (6.8 mg, 0.040 mmol) in H2O (2 mL) at room temperature. A brown viscous gel formed after 3 days, containing colourless needles suitable for single crystal X-ray diffration.
Supplementary material
Crystallographic and structural details, continuous shape analysis, PXRD, TGA and ATR-FTIR spectra are available in the Supplementary material. The crystallographic information files can be obtained from the Cambridge Structural Database: CCDC 2340981–2340984. Supplementary material is available online.
Data availability
The data that support this study will be shared upon reasonable request to the corresponding author.
Declaration of funding
This research was conducted by the Australian Research Council Centre of Excellence in Exciton Science (project number CE170100026) and funded by the Australian Government.
Acknowledgements
This research was undertaken in part at the Melbourne TrACEES Platform at the University of Melbourne and used the MX1 and MX2 beamlines at the Australian Synchrotron, part of ANSTO, making use of the Australian Cancer Research Foundation (ACRF) detector. This paper was contributed as an invited paper for the 2024 Awards Special Issue. Dr Hua was awarded the 2023 Alan Sargeson Lectureship of the RACI Inorganic Division.
References
1 Brunel JM. BINOL: a versatile chiral reagent. Chem Rev 2005; 105: 857-898.
| Crossref | Google Scholar |
2 Yu F, Chen Y, Jiang H, Wang X. Recent advances of BINOL-based sensors for enantioselective fluorescence recognition. Analyst 2020; 145: 6769-6812.
| Crossref | Google Scholar |
3 Tan JSJ, Paton RS. Frontier molecular orbital effects control the hole-catalyzed racemization of atropisomeric biaryls. Chem Sci 2019; 10: 2285-2289.
| Crossref | Google Scholar |
4 Pasini D, Nitti A. Recent advances in sensing using atropoisomeric molecular receptors. Chirality 2016; 28: 116-123.
| Crossref | Google Scholar |
5 Zhang X, Yin J, Yoon J. Recent advances in development of chiral fluorescent and colorimetric sensors. Chem Rev 2014; 114: 4918-4959.
| Crossref | Google Scholar |
6 Pu L. Enantioselective fluorescent sensors: a tale of BINOL. Acc Chem Res 2012; 45: 150-163.
| Crossref | Google Scholar |
7 Thoonen S, Hua C. Chiral detection with coordination polymers. Chem Asian J 2021; 16: 890-901.
| Crossref | Google Scholar |
8 Liang X, Liang W, Jin P, Wang H, Wu W, Yang C. Advances in chirality sensing with macrocyclic molecules. Chemosensors 2021; 9: 279-302.
| Crossref | Google Scholar |
9 Cui Y, Ngo HL, White PS, Lin W. Homochiral 3-D open frameworks assembled from 1- and 2-D coordination polymers. Chem Commun 2003; 2003: 994-995.
| Crossref | Google Scholar |
10 Cui Y, Ngo HL, White PS, Lin W. Hierarchical assembly of homochiral porous solids using coordination and hydrogen bonds. Inorg Chem 2003; 42: 652-654.
| Crossref | Google Scholar |
11 Deng W-T, Qu H, Huang Z-Y, Shi L, Tang Z-Y, Cao X-Y, Tao J. Facile synthesis of homochiral compounds integrating circularly polarized luminescence and two-photon excited fluorescence. Chem Commun 2019; 55: 2210-2213.
| Crossref | Google Scholar |
12 Sun X-P, Tang Z, Yao Z-S, Tao J. A homochiral 3D framework of mechanically interlocked 1D loops with solvent-dependent spin-state switching behaviors. Chem Commun 2020; 56: 133-136.
| Crossref | Google Scholar |
13 Tanaka K, Muraoka T, Otubo Y, Takahashi H, Ohnishi A. HPLC enantioseparation on a homochiral MOF–silica composite as a novel chiral stationary phase. RSC Adv 2016; 6: 21293-21301.
| Crossref | Google Scholar |
14 Wu C-D, Lin W. Highly porous, homochiral metal–organic frameworks: solvent-exchange-induced single-crystal to single-crystal transformations. Angew Chem Int Ed Engl 2005; 44: 1958-1961.
| Crossref | Google Scholar |
15 Zheng M, Liu Y, Wang C, Liu S, Lin W. Cavity-induced enantioselectivity reversal in a chiral metal–organic framework Brønsted acid catalyst. Chem Sci 2012; 3: 2623-2627.
| Crossref | Google Scholar |
16 Ma L, Wu C-D, Wanderley MM, Lin W. Single-crystal to single-crystal cross-linking of an interpenetrating chiral metal–organic framework and implications in asymmetric catalysis. Angew Chem Int Ed Engl 2010; 49: 8244-8248.
| Crossref | Google Scholar |
17 Wang Y, Gao K, Li J, Wang L, Wu J. Synthesis and characterization of a Cd compound for selectively sensing of nitro-explosives. Inorg Chem Commun 2018; 96: 189-193.
| Crossref | Google Scholar |
18 Wanderley MM, Wang C, Wu C-D, Lin W. A chiral porous metal–organic framework for highly sensitive and enantioselective fluorescence sensing of amino alcohols. J Am Chem Soc 2012; 134: 9050-9053.
| Crossref | Google Scholar |
19 Ma L, Mihalcik DJ, Lin W. Highly porous and robust 4,8-connected metal−organic frameworks for hydrogen storage. J Am Chem Soc 2009; 131: 4610-4612.
| Crossref | Google Scholar |
20 Thoonen S, Tay HM, Hua C. A chiral binaphthyl-based coordination polymer as an enantioselective fluorescence sensor. Chem Commun 2022; 58: 4512-4515.
| Crossref | Google Scholar |
21 Zhang Z, Ji YR, Wojtas L, Gao W-Y, Ma S, Zaworotko MJ, Antilla JC. Two homochiral organocatalytic metal organic materials with nanoscopic channels. Chem Commun 2013; 49: 7693-7695.
| Crossref | Google Scholar |
22 Ma L, Lin W. Chirality-controlled and solvent-templated catenation isomerism in metal–organic frameworks. J Am Chem Soc 2008; 130: 13834-13835.
| Crossref | Google Scholar |
23 Ma L, Falkowski JM, Abney C, Lin W. A series of isoreticular chiral metal–organic frameworks as a tunable platform for asymmetric catalysis. Nat Chem 2010; 2: 838-846.
| Crossref | Google Scholar |
24 Thoonen S, Siripanich P, Hua L, Tay HM, Ramkissoon P, Smith TA, Lessio M, Hua C. Enhancing enantioselectivity in chiral metal organic framework fluorescent sensors. Inorg. Chem. Front. 2024; [Advance Article, published 18 May 2024].
| Crossref | Google Scholar |
25 Seter M, Dakternieks D, Duthie A. Chiral rings from BINOL dicarboxylic acids and alkane ditin(IV) linkers. Main Group Met Chem 2012; 35: 73-80.
| Crossref | Google Scholar |
26 Alcázar V, Morán jR, Diederich F. Chiral molecular clefts for dicarboxylic acid complexation. Isr J Chem 1992; 32: 69-77.
| Crossref | Google Scholar |
27 MacLean MWA, Wood TK, Wu G, Lemieux RP, Crudden CM. Chiral periodic mesoporous organosilicas: probing chiral induction in the solid state. Chem Mater 2014; 26: 5852-5859.
| Crossref | Google Scholar |
28 Coulson DR, Satek LC, Grim SO. Tetrakis(triphenylphosphine)palladium(0). In: Cotton FA, editor. Inorganic Syntheses. Vol. 13. Wiley; 2007. pp. 121–124. [Online book] doi:10.1002/9780470132449.ch23
29 Zhang Q-W, Li D, Li X, White PB, Mecinović J, Ma X, Ågren H, Nolte RJM, Tian H. Multicolor photoluminescence including white-light emission by a single host–guest complex. J Am Chem Soc 2016; 138: 13541-13550.
| Crossref | Google Scholar |
30 McPhillips TM, McPhillips SE, Chiu HJ, Cohen AE, Deacon AM, Ellis PJ, Garman E, Gonzalez A, Sauter NK, Phizackerley RP, Soltis SM, Kuhn P. Blu-Ice and the Distributed Control System: software for data acquisition and instrument control at macromolecular crystallography beamlines. J Synchrotron Radiat 2002; 9: 401-406.
| Crossref | Google Scholar |
31 Aragão D, Aishima J, Cherukuvada H, Clarken R, Clift M, Cowieson NP, Ericsson DJ, Gee CL, Macedo S, Mudie N, Panjikar S, Price JR, Riboldi-Tunnicliffe A, Rostan R, Williamson R, Caradoc-Davies TT. MX2: a high-flux undulator microfocus beamline serving both the chemical and macromolecular crystallography communities at the Australian Synchrotron. J Synchrotron Radiat 2018; 25: 885-891.
| Crossref | Google Scholar |
32 Sheldrick GM. SHELXT – integrated space-group and crystal-structure determination. Acta Cryst A 2015; 71: 3-8.
| Crossref | Google Scholar |
33 Sheldrick GM. Crystal structure refinement with SHELXL. Acta Cryst C 2015; 71: 3-8.
| Crossref | Google Scholar |
34 Dolomanov OV, Bourhis LJ, Gildea RJ, Howard JAK, Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J Appl Cryst 2009; 42: 339-341.
| Crossref | Google Scholar |