

Electronic structure study of H3BXH3 (X═B, N and P) as hydrogen storage materials using calculated NMR and XPS spectra
Feng Wang A * and Delano P. Chong B
A * and Delano P. Chong B
A
B
Abstract
Boron-based materials have been used for hydrogen storage applications owing to their high volumetric and gravimetric hydrogen density. The present study quantum mechanically investigates the electronic structures of three compounds: diborane (DB, B2H6), ammonia borane (AB, H3BNH3) and phosphine borane (PB, H3BPH3). The exploration is facilitated using calculated nuclear magnetic resonance (NMR) chemical shifts, together with outer valence ionisation potentials (IP) and core electron binding energy (CEBE). The findings show a distinct electronic structure for diborane, differing notably from AB and PB, which exhibit certain similarities. Noteworthy dissimilarities are observed in the chemical environments of the bridge hydrogens and terminal hydrogens in diborane, resulting in a substantial chemical shift difference of up to 5.31 ppm. Conversely, in AB and PB, two distinct sets of hydrogens emerge: protic hydrogens (Hp–N and Hp–P) and hydridic hydrogens (Hh–B). This leads to chemical shifts as small as 0.42 ppm in AB and as significant as 3.0 ppm in PB. The absolute isotropic NMR shielding constant (σB) of 11B in DB is 85.40 ppm, in contrast to 126.21 ppm in AB and 151.46 ppm in PB. This discrepancy indicates that boron in PB has the most robust chemical environment among the boranes. This assertion finds support in the calculated CEBE for B 1s of 196.53, 194.01 and 193.93 eV for DB, AB and PB respectively. It is clear that boron in PB is the most reactive atom. Ultimately, understanding the chemical environment of the boranes is pivotal in the context of dehydrogenation processes for boron-based hydrogen storage materials.
Keywords: borane compounds, CEBE, core electron binding energy, DFT calculations, 1H proton and 11B NMR chemical shift–shielding constant, role of hydrogens and boron in boranes, valence ionisation energy spectrum.
Introduction
New momentum has emerged globally in the study of energy storage materials, catalysed by two decades of intensive research and development across both academic and industrial domains.1 Despite these advances, there remains a need to address persisting scientific, technological and economic hurdles associated with the hydrogen life cycle. This includes production, storage, transportation, distribution and utilisation of hydrogen. Hydrogen storage and transportation have become significant for hydrogen energy export countries such as Australia.2 As a result, the processes of dehydrogenation and hydrogenation are important in chemical hydrogen storage using hydrogen carriers. The storage issue is particularly challenging,1 given its implications in transportation such as on board shipping. Among hydrogen storage techniques, chemical hydrogen storage, using chemical binding (chemisorption), has been more attractive than conventional physical techniques, including the cryogenic liquefaction of hydrogen at −253°C. Hydrogen carriers such as ammonia borane (AB) exhibit notable advantages. AB boasts a high hydrogen content of 19.6 wt-%, remarkable stability under ambient conditions, non-toxicity, and high solubility in common solvents.2,3 AB holds potential as both a liquid-state hydrogen carrier and a solid-state hydrogen storage material.1 When dissolved, AB functions as a liquid-state hydrogen carrier (LHC).3 This property aligns well with pre-existing energy infrastructure, allowing reuse with minimum modification.2 Nevertheless, challenges persist with AB, primarily revolving around the dehydrogenation–hydrogenation process and the establishment of economically viable chemical pathways for these operations. Compounds like B–N and P–B adducts, such as AB and phosphine borane (PB, H3BPH3), stand out as efficient and lightweight materials for hydrogen storage.
Ammonia borane may be the simplest amine borane, yet its history has been anything but simple.4 As a solid hydrogen storage material that is both efficient and safe, AB has gained significant attention in recent years. In a comprehensive review by Demirci1 summarising the literature up to June 2020, a range of hydrogen chemical storage compounds containing boron were examined. These include borohydride, lithium borohydride, sodium borohydride, potassium borohydride, borane, AB, diborane (DB), amidoboranes, hydrazine borane, triborane and dodecaborane. The hydrogen generating capacity of AB, essentially its role as a hydrogen carrier, has been recognised for a considerable period.1 Although AB possesses characteristics associated with propellant and energetic materials, its primary identity remains that of a hydrogen carrier. The hydrogen released from AB is used for electricity generation through conversion in fuel cells.1
Phosphine borane, which can dissolve in liquid ammonia, has been comparatively less explored as an LHC. The behaviour or property of PB involves the thermal or catalytical elimination of H2, yielding cyclic and polymeric phosphinoboranes.5 Mechanistic insights into the reversible activation of H2 in this context indicate an interaction with the Lewis acidic boron centre, followed by intramolecular proton migration to phoshorus.5 Notably, the arrangement of cations and anions positions the BH and PH units facing each other, with a BH⋯HP proximity of 2.75 Å, significantly surpassing typical intermolecular H-bonding distances. In the domain of synthesis and industry, PB plays a pivotal role in facilitating efficient dehydrocoupling of secondary PB adducts, leading to the formation of the corresponding polymers.6 Although discussions touch on the nature of the coordinate bond between phosphines and the borane group, comprehensive and definitive information concerning the precise nature of the P–B bond remains to be firmly established.7
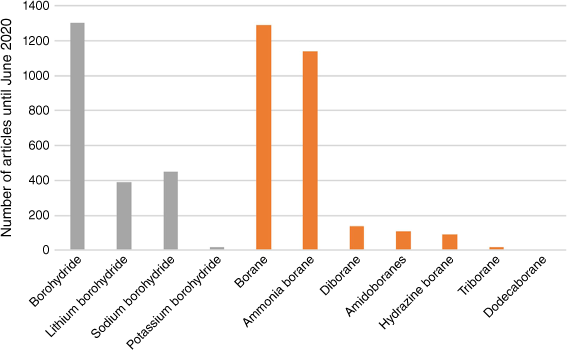
Diborane stands out as a clear precursor for AB and PB; however, it poses challenges due to its pyrophoric and toxic nature.8 The amine and phosphine adducts of borane exhibit improved stability and diminished reactivity compared with B2H6, thereby reducing safety concerns. AB and PB both offer substantial promise as pivotal elements in hydrogen storage fuels, primarily owing to their stability and high gravimetric hydrogen content. Fig. 1 illustrates that DB has undergone considerable investigation,1 not only for its applications in energy materials but also owing to its fundamental chemical bonding, notably the unique B–H–B three-centre–two-electron (3c–2e) bonding character.9 An extensive overview encompassing several decades of research in the application of AB in chemical hydrogen storage can be found in Demirci.1,10
Number of articles from a search on the Web of Science for boron-containing hydrogen chemical storage materials to June 2020.1 Grey bars are borohydrides and derivatives, and orange bars are borane and its derivatives.
PB, the simplest adduct of its kind, was synthesised as ‘diborane diphosphine’ and later was found to be the monomer H3BPH3,11,12
Phosphine borane tends to undergo substantial dissociation, resulting in the formation of B2H6 and the corresponding phosphorus ligand.13
The direct addition of H2 to unsaturated bonds is typically symmetry-forbidden, necessitating the involvement of catalysts, which can be either heterogeneous or homogeneous transition metal catalysts, for hydrogenation reactions. However, the activation of hydrogen by main group compounds under mild conditions remained unexplored until a significant breakthrough was reported in 2005.14 Power and co-workers, particularly through their work on digermyne, achieved the first directed addition of hydrogen to a closed-shell unsaturated main group compound under ambient conditions.14 This transition metal-like behaviour is demonstrated by a variety of main group element species, offering intriguing possibilities for applications in homogeneous catalysis. The emergence of transition metal-free reactions highlights the potential of reactive main group compounds and underscores the significance of molecular design, particularly the strategic selection of substituents at the main group element, for both isolation and practical applications.15 This avenue not only holds the promise of cost reduction but also addresses environmental concerns, which are of great importance to the industry.
Crucial to this achievement is an understanding of kinetic stabilisation, thermodynamic stabilisation and the use of ligands possessing the requisite electronic properties for the reactive species.15 To stabilise low-valent and electron-deficient compounds, strong s- and p-donors have proved to be suitable, with examples encompassing N-heterocyclic imines and their carbon and phosphorus derivatives.15 The hydrogen release of AB is exothermic and thus non-reversible.16 Consequently, understanding the electronic properties of boranes, including ionisation potentials, becomes pivotal for gaining deeper insights into the thermodynamics of hydrogen storage materials.17 The study of suppressing the thermal dehydrogenation of AB11,18 involves an exploration of the added nucleophile’s role in inhibiting dehydrogenation. This aspect is further supported by the observation of facile phosphine–ammonia exchanges.10
The consideration of B–N and B–P adducts as hydrogen storage materials stems from their potential to influence hydrogen storage capacity and release characteristics. Their structural properties can significantly affect their performance in this regard. For example, AB displays a high theoretical hydrogen capacity of 19.6 wt-%, releasing hydrogen at ~6.5 wt-% below 385 K but up to 15.4 wt-% under certain conditions.16 By contrast, PB boasts a moderate theoretical hydrogen capacity of 14.5 wt-% and can release hydrogen in the range of 8–10 wt-%. Diborane, with a theoretical hydrogen capacity of 18.5 wt-%, can release hydrogen up to 6.3 wt-%.19 AB and PB represent examples of Lewis acid–Lewis base adducts, where the stability of the central dative bond plays a significant role. Describing the factors influencing the strength of B–N and B–P bonds is instrumental in understanding this phenomenon.20 Although this approach provides a general estimate of the dative bond’s strength, the dissociation energy of the dative bond is not solely linked to the strengths of the Lewis acid or Lewis base. As a result, the Lewis acidity of boron is primarily influenced by substituent size and electronegativity.20
The investigation of the electronic structures of compounds involving BH3 bonding with BH3, NH3, and PH3 is significant when studying the release of hydrogen. Numerous theoretical studies have been conducted on AB and its dehydrogenated products, as well as small boron-nitrogen hydrides, yielding insights into the hydrogen release process.21,22 This understanding of B–N and B–P adducts electronic structures in the gas phase is crucial for understanding their behaviour in hydrogenation and dehydrogenation processes. Although various gas-phase studies for DB, AB and PB are available, including our recent research on AB and its dehydrogenated borane hydrides,22,23 fewer investigations have delved into the nature of protic N–H and hydridic B–H hydrogen in AB and PB. Furthermore, these studies rarely explore how the electronic structures, especially chemical bonding characterised by valence ionisation energies, influence the behaviour of protic and hydridic hydrogens in AB and PB compared with DB. In an ongoing study, well-established quantum mechanical methods were employed to elucidate the roles of hydrogens in AB, PB and DB. We recently combined O 1s X-ray photoelectron spectroscopy (XPS) and 1H NMR chemical shifts to study the intramolecular hydrogen bonding of salicylic acid.24 The present analysis incorporates 1H NMR spectroscopy with valence ionisation potentials (IP) as well as B 1s XPS to shed light on the functions of protic and hydridic hydrogens in these borane compounds. By combining these techniques, a comprehensive understanding of the interplay between electronic structure, chemical bonding and hydrogen behaviour within these compounds can be obtained.
Computational details
The details of the quantum mechanical calculations are the same as mentioned in our previous study.22 Briefly, the geometries of DB, AB and PB are optimised in the gas phase, based on coupled-cluster theory (CCSD(T)) with the cc-pVTZ basis set, i.e. the CCSD(T)/aug-cc-pVQZ level of theory. NMR calculations are conducted using density functional theory (DFT) B3PW91/aug-cc-pVTZ level of theory, and the valence vertical ionisation energies (VIEs) are calculated using the ΔPBE0(SAOP)/et-pVQZ method developed by Segala and Chong.25 The CEBE calculations use the Δ(PW86-PW91/et-pVQZ) + Crel method.26 All calculations were performed using the Gaussian 16 computational chemistry package27 and Amsterdam Density Functional (ADF) suites of programs.28
Results and discussion
Major structural differences among the boranes
The primary distinguishing structural feature among DB, AB and PB compounds lies in the types of atoms and groups attached to the central boron atom. DB only has hydrogen atoms bonded to the boron atoms, whereas AB and PB respectively have nitrogen and phosphorus atoms connected to the boron atom.9 These distinct bonding arrangements and molecular architectures lead to variations in the physical and chemical characteristics of these compounds, including their hydrogen storage capacity and reactivity. In Fig. 2, the structural differences are evident. Diborane has two boron atoms linked by two bridging hydrogen atoms (Hb), with each boron atom forming bonds with two additional hydrogen atoms. In AB, the boron atom is connected to an amino (NH3) group and three boron–hydrogen (B–H) bonds. Similarly, in PB, the boron atom is attached to a phosphine (PH3) group and three B–H bonds. Each of these compounds harbours two categories of hydrogen atoms: bridge (Hb) and terminal (Ht) hydrogens in diborane (DB),9 and protic (Hp) hydrogens in AB and PB originating from the N–H and P–H bonds. These hydrogens possess partial positive charges (δ+). Additionally, there are hydridic (Hh) hydrogens in AB and PB within B–H bonds, carrying partial negative charges (δ−).3 This intricate arrangement of atoms and hydrogen groups forms the foundation for the distinctive properties and behaviours of these compounds.
Chemical structures of diborane (DB), ammonia borane (AB) and phosphine borane (PB) and their point group symmetry. In DB, the hydrogens are labelled as bridge hydrogens (Hb) and terminal hydrogens (Ht); in AB and PH, the hydrogens are labelled as protic (N–H and P–H) hydrogens (Hp) and hydridic (B–H) hydrogens (Hh).
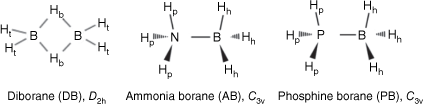
The structural differences between DB and both AB and PB are quite pronounced. In DB, two boron atoms are linked through bridge hydrogens, presenting a unique arrangement. By contrast, AB and PB feature a single B–X bond (where X═N or P), which allows rotations around this bond. The energy barriers associated with these rotations are defined as the energy difference between eclipsed and staggered conformers, analogous to the behaviour observed in molecules like ethane.29 Specifically, the calculated rotational energy barriers around the B–X bond for H3B–NH3 and H3B–PH3 have been reported as 2.48 and 2.62 kcal mol–1 (1 kcal mol–1 = 4.186 kJ mol–1) for AB and 2.47 and 2.55 kcal mol–1 for PB, calculated using the MP2/6-31++G(d,p) and DFT-based B3LYP/6-31++G(d,p) levels of theory respectively.29 For comparison, the rotational energy barrier of ethane (CH3–CH3) is 2.875 kcal mol–1.30 As a result, the present study focused solely on eclipsed AB and PB for the C3v point group symmetry. This choice aligns with the low-temperature dynamics study of AB, which also adopted the eclipsed structure.31 Importantly, the energy barriers observed for AB and PB are approximately half of the earlier estimated 4–6 kcal mol–1 barrier that restricts internal rotation in DB.32 This difference underscores the varying rotational dynamics between these compounds.
Table 1 compares properties among the three compounds DB, AB and PB. Notably, the B–X bond lengths shows different values: 1.762 Å for B–B (or B⋯B distance) in DB, 1.655 Å for B–N in AB and 1.949 Å for B–P in PB. This trend in bond lengths aligns with expectations, as the van der Waals radii33 for B, N, and P are 1.92, 1.55 and 1.80 Å respectively. The reported complexation energy (Ec) reveals that the stability of PB is −21.10 kcal mol–1, comparatively less stable than AB with an Ec of −25.97 kcal mol–1.33 Examining the structural parameters of H3BXH3 in Table 1, it is evident that the HBN angle in AB (104.88°) increases to 117.82° for the HBP angle in PB, whereas the bond lengths of hydridic hydrogens (Hh–B) in the boranes remain quite similar (1.188 Å for H–B in DB, 1.210 Å for H–B in AB and 1.207 Å for H–B in PB). The lengths of protic hydrogen bonds (Hp–N and Hp–P) exhibit significant differences. Specifically, the Hp–N bond length in AB is notably shorter at 1.015 Å, whereas the Hp–P bond length in PB is significantly longer at 1.405 Å compared with their corresponding hydridic bonds (Hh–B). The H–X bond lengths in AB and PB closely resemble the bond lengths of H–N in ammonia (NH3), at 1.017 Å, and H–P in phosphine (PH3), at 1.421 Å.34 In DB, the bridge Hb–B bond length (1.315 Å) is significantly longer than the terminal Ht–B bond length (1.188 Å), indicating a distinct bonding nature. This set of structural parameters contributes to the unique characteristics and behaviours of these boron compounds.
| Property | H2B2H4 | H3BNH3 | H3BPH3 | |
|---|---|---|---|---|
| Symmetry | D2h | C3v | C3v | |
| Etot (Eh) | ‒53.143253 | ‒83.063511 | ‒369.267118 | |
| Ec (kcal mol–1) | – | −25.97 A | −21.10 A | |
| RB–X (Å) | 1.762 | 1.655 (1.66) 10, 35 | 1.949 | |
| ∠HXB (°) | t, 122.24 (121.5) B | 111.06 | 117.82 (116.9) 36 | |
| ∠HBX (°) | b, 95.85 (95.8) | 104.88 | 103.42 (103.6)38 | |
| RH–X (Å) | 1.315 (1.314),b B | 1.015 (1.020) A | 1.405 (1.399, 36 1.404 A) | |
| RH–B (Å) | 1.188 (1.184),t B | 1.210 | 1.207 (1.212) 36 | |
| μ (D) | 0 | 5.40 (5.64) C | 4.35 (4.36) C | |
| A (MHz) | 80 404 (79 616) D | 73 821 | 57 188 | |
| B (MHz) | 18 344 (18 181) D | 17 713 | 10 476 (10 591) E | |
| C (MHz) | 16 848 (16 707) D | 17 713 | 10 476 (10 591) E |
All the data except for those in parentheses are calculated using the method in CCSD(T)/cc-pVTZ level of theory. Etot, total electronic energy of the compound; A, B and C, rotational constants; μ, dipole moment; b, bridge H; t, terminal H in DB.
The nature of the N–B bond in AB and P–B bond in PB is predominantly polar. The interaction between the electron lone pair of phosphines and boranes leads to increased acidity of the P–H bond, whereas the P–B bond decreases electron density on the phosphorus atom. As a result, PB displays a longer P–B bond length of 1.949 Å and exhibits a weaker adjacent P–H bond in comparison with AB. In PB, the bond length of P–Hh is 1.416 Å, considerably longer than the N–Hh bond length of 1.015 Å. Owing to its high symmetry (D2h), DB is a non-polar compound with no permanent dipole moment. By contrast, the dipole moment of AB is 5.40 D, surpassing PB’s dipole moment of 4.35 D. This divergence in dipole moments can be attributed to the larger dipole moment of the Lewis base NH3 (1.55 D for NH3 using MP2/6-311+G(3df,2p)) in comparison with PH3 (0.58 D).37 Furthermore, the interaction energy within the bond formed by PH3 must result from the distortion of the PH3 electron pair due to the positive field of the Lewis acid (BH3). This intricate interplay of polarities and interactions contributes to the varied characteristics and behaviours observed in these compounds.
NMR chemical shifts of the boranes
Further advances in understanding the structure and reactivity of these borane compounds will not only contribute to tackling challenges in the field of hydrogen storage but also to other domains of boron chemistry.38 NMR spectroscopy is a powerful tool for unravelling molecular structures. The structural information provided by the NMR technique is multifaceted, site‐specific, remarkably stable and reliably reproducible over time.39 Our investigation looked into the role of hydrogen atoms within DB, AB and PB through the lens of 1H NMR chemical shifts. To accomplish this, the NMR absolute isotropic shielding constants (chemical shifts)7 for the boranes were calculated. The absolute isotropic shielding constants (σ, ppm)7 were calculated using the gauge-independent atomic orbital (GIAO) method40–43 in conjunction with the B3PW91 functional. This functional has demonstrated accuracy in predicting NMR shield tensor (chemical shifts) for molecules43 and compounds containing boron.44
The chemical shift (δA, ppm) of atom A in the boranes is given by:
where σA0 is the absolute isotropic shielding constant of same atom A in a reference compound. Usually, the reference standards for σH0 and σC0 are the shielding constants of 1H and 13C in tetramethylsilane (TMS) respectively, whereas the reference standard for σN0 is 15N in NH3 and for σB0 is 11B in BF3·OEt2 (boron trifluoride diethyl etherate).43 In the G16 computational chemistry package, σH0 = 31.8821 ppm in TMS, σB0 = 83.6 ppm in B2H6, σN0 = 258.4 ppm in NH3, all calculated using B3LYP/6-311+G(2d,p) (GIAO).
Table 2 reports the calculated absolute isotropic shielding constants (σB) for DB, AB and PB. The marked differences in these σB constants arise from the diverse chemical environments of these compounds. Specifically, boron in DB features the smallest shielding constant (σB = 85.40 ppm), whereas boron in PB exhibits the largest shielding constant (σB = 151.46 ppm). AB falls between DB and PB, with a shielding constant of σB = 126.21 ppm. The shielding constants of hydrogens (σH) within DB, AB and PB are reported in Fig. 2. A standardised absolute shielding scale for boron is established with reference to the shielding constant in liquid BF3·OEt2, which is 110.9 ppm for 11B.45 In the present study, utilising the absolute isotropic shielding constant (σ) directly rather than the chemical shift (δ) serves to minimise discrepancies stemming from different methods. For example, σB0 = 83.6 ppm of B2H6 is calculated using the B3LYP/6-311+G(2d,p) (GIAO) and 85.40 ppm using B3PW91/aug-cc-pVTZ. It is important to acknowledge that, although the accuracy of an atom’s absolute isotropic shielding constant in a specific chemical environment hinges on the chosen level of theory,43 employing the same level of theory across a series of compounds can help mitigate certain systematic errors. NMR techniques have been leveraged to investigate B–N and B–P coupling for AB46 and PB.7
| Molecules | Structure | σ(11B) (δB) (ppm) | σ(X) (δX) (ppm) | σ(Hb (HB)) (δH,h) (ppm) | σ(Ht (HX)) (δH,p) (ppm) | Δδ (ppm) | |
|---|---|---|---|---|---|---|---|
| B2H6 (DB) | 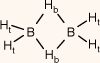 | 85.40 (53.32) A | 85.4 A, B | 32.32 (−0.44) B | 27.01 (4.87) B | 5.31 | |
| H3BNH3 (AB) | 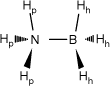 | 126.21 (94.13) A | 231.39 (249.122) A B C | 29.47 (2.41) B | 29.05 (2.83) B | 0.42 | |
| H3BPH3 (PB) |  | 151.46 (119.38) A | 454.37 A B D | 30.59 (1.28) B | 27.60 (4.28) B | 3.0 |
The experimental chemical shifts of 11B in BH3 and H3BNH3 are 86 and −16.6 ppm.44 Unless results are associated with a reference citation, data are calculated using the method in B3PW91/aug-cc-pVTZ (ppm). No reference – absolute shielding (σ).
The calculated chemical shifts δ(11B) of AB and PB are referenced against δ(11B) of DB, which is 85.40 ppm. Experimentally, δ(11B) for AB is observed at −21.6 ppm relative to the IUPAC standard BF3·OEt2 (δ = 0 ppm) at 20°C. Calculated δ(11B) values for AB show some variability depending on the methods employed, yielding values of −24.7, −23.7 and −27.1 ppm with respect to the IUPAC standard BF3·OEt2.50 As reported in Table 2, the distinct chemical environments of boron in DB, AB and PB are clearly apparent, as evidenced by their NMR chemical shifts of δ(11B). The shifts in δ(11B) across the boranes are quite significant. Specifically, δ(11B) for AB and PB shifts by 40.80 and 66.06 ppm respectively, relative to δ(11B) of DB. The trend is δ(11B) (PB) > δ(11B) (AB) > δ(11B) (DB). This order reveals the principle that the stronger the chemical environment of boron, the larger its chemical shift.
The hydrogens within the Lewis acid BH3 and Lewis base XH3 (X═N and P) of AB and PB exhibit notable differences from the hydrogens in DB. In the staggered configurations, the chemical shifts of the three-fold degenerate protic hydrogens (Hp–X) are 2.83 ppm in AB and 4.28 ppm in PB. Conversely, the chemical shift of three-fold degenerate hydridic hydrogens (Hh–B) in the BH3 fragment follows the reverse order: 2.47 ppm in AB and 1.28 ppm in PB. Moving to DB, the chemical shifts of the bridge hydrogens (Hbs) and terminal hydrogens (Hts) are −0.44 and 4.87 ppm respectively. As a result, the two distinct groups of hydrogens in DB, AB and PB exhibit chemical shifts of 5.31, 0.42 and 3.0 ppm respectively. The most substantial difference is observed in the Hbs and Hts of DB, followed by the Hhs and Hps of PB. By contrast, the differences between the Hhs and Hps of AB are the smallest. However, both AB and PB show the same trend: protic hydrogens have a stronger chemical environment than hydridic hydrogens. Importantly, the calculated chemical shifts align with findings from other NMR studies of AB in diverse mediums, including solutions.46 This consensus indicates the robustness and validity of the trends obtained in chemical shifts, which reflect the distinct chemical environments and bonding characteristics within these borane compounds.
Fig. 3 compares the NMR hydrogen shielding constants (σH) of the boranes. The boranes studied here feature two distinct groups of hydrogen atoms, each with markedly different roles and bonding characteristics. The hydrogen chemical shifts (δH) of the boranes are relatively modest when compared with the reference methyl hydrogens (–CH3). The σH value for TMS is 31.88 ppm, indicating that σH(boranes) is generally lower than or comparable with σH(TMS). The borane compounds host two distinct types of hydrogen atoms, Hb and Ht in DB, and Hp and Hh in AB and PB. As shown in Fig. 3, the difference between hydrogen groups is most pronounced (δH = 5.31 ppm) in DB, whereas the difference is least marked (δH = 0.42 ppm) in AB. In PB, the hydrogen shielding constant difference is substantial, with a large splitting of 3.0 ppm.
Comparison of the NMR absolute isotropic hydrogen and boron shielding constants σ for DB, AB and PB calculated using B3PW91/aug-cc-pVTZ (ppm). The hydrogen chemical shifts of the boranes are in blue for hydridic hydrogens (Hhs), red for protic hydrogens (Hps) and green for hydrogens in DB.
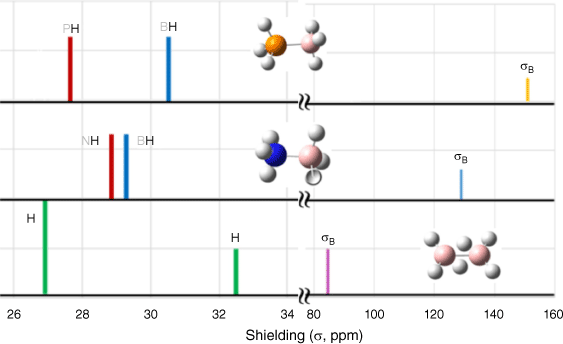
A notable point is the nuanced role of the hydrogens in DB. It can be argued that DB may be more appropriately represented as H2B2H4, rather than B2H6, as the six hydrogens in DB do not all play identical roles. Specifically, the bridge Hbs in DB, formed through three-centre-two-electron bonds, exhibit stronger chemical characteristics compared with the terminal Hts in DB.9 For both AB and PB, the hydridic hydrogens (Hhs) (represented in blue in Fig. 3) within the hydrogens in the H–B bonds demonstrate larger shielding constants than their protic hydrogen (Hps) counterparts (in red) within the same compound.
Ionisation potentials of the boranes
To further understand the borane compounds, we calculated their IPs.23 It is important to note that the IP of a compound includes both the valence IP and core IP, which are measured using photoelectron spectroscopy (PES) and XPS respectively. The complete valence IP of DB is readily available,9 whereas the availability of data on the experimental valence IPs for AB and PB are quite limited. The first (valence) IP of AB was measured as 10.58 eV for the VIP and 9.44 eV for the adiabatic ionisation potential (AIP) using mass spectrometry.51 The AIP value agrees with 9.26 ± 0.03 eV determined by a recent photo-electron–photoion coincidence (PEPICO) spectroscopic measurement.23 The measured IP value of a compound depends on experimental conditions, spectroscopic techniques, resolution and the spectral analyses, and the measurement process.52
Accurate theoretical calculations are essential for molecular spectroscopy, guiding the interpretation of experimental results and enabling more accurate determination of key properties.52,53 Table 3 compares the calculated valence IPs for the three borane compounds. The agreement between calculated valence IPs and measurements9 for DB suggests that the ΔPBE0(SAOP)/et-pVQZ method in the present study is accurate. Therefore, the same method was used to calculated the valence IPs for both AB22 and PB. The complete valence IPs of AB were confirmed by recent gas-phase measurements at the Elettra synchrotron in Italy (please see ‘In proof’ section at the end of this article). This recent measurement of AB increases our confidence in the calculated valence IPs of PB, although no available IPs for PB exist.
| Molecule | Valence configuration | First | Second | Third | Fourth | Fifth | Sixth | |
|---|---|---|---|---|---|---|---|---|
| B2H6 D2h | (2ag)2(2b3u)2(1b1u)2 (1b2u)2(3ag)2 (1b 1g) 2 | 1b1g 11.80 (11.89) A | 3ag 13.01 (13.30) A | 1b2u 13.49 (13.91) A | 1b1u 14.69 (14.75) A | 2b3u 15.52 (16.11) A | 2ag 21.56 (22.33) B | |
| H3BNH3 C3v | (3a1)2(1e 1) 4(4a 1) 2(5a1)2(2e1)4 | 2e1: 10.57 C (10.58) D, E | 5a1: 12.58 C | 4a1: 16.95 C | 1e1: 18.56 C | 3a1: 29.88 C | ||
| H3BPH3 C 3v | (5a1)2(6a 1) 2(2e 1) 4 (7a1)2(3e 1) 4 | 3e1: 11.06 | 7a1: 11.28 | 2e1: 14.96 | 6a1: 16.54 | 5a1: 22.33 |
The HOMO is highlighted in bold. The symmetries of orbitals underlined are swapped positions in AB and PB.
The IPs of DB diverge significantly from those of AB and PB complexes, which can be attributed to DB’s distinct D2h point group symmetry and different structural characteristics. AB and PB share the C3v point group symmetry in the form of Lewis acid–Lewis base complexes. This commonality allows AB and PB to share the same character table and irreducible representations with orbitals of a1 and e1 symmetries. However, the ground state configurations of AB and PB diverge owing to the variations in the valence electronic structures stemming from the nitrogen and phosphorus atoms within the complexes.
Fig. 4 compares the simulated valence IP spectra of AB and PB in the energy region of 5–35 eV. Four major IP bands in PB (top) but five major bands in AB (bottom) appear in the spectra. Although some similarities exist between the spectra of PB and AB, there are notable differences. The lowest five valence IPs of AB span a wider energy range (~11–30 eV) compared with PB (~11–22 eV). The valence IPs and their assignment exhibit an interesting pattern for the a1 orbitals and the doubly degenerate e1 orbitals in both AB and PB. There is a small energy gap (ΔIP 0.22 eV) in PB between the outer valence highest occupied molecular orbital (HOMO), 3e1, a doubly degenerate orbital and the next HOMO, HOMO-1, 7a1. As a result, the first IP band at approximately 11 eV, which consists of three closely lying states, leads to a single band so that there are four distinct IP bands in the outer valence IP spectrum of PB. However, the IP energy gap between HOMO and HOMO-1 of AB is larger, measuring 2.01 eV, which is likely sufficient for spectrometer resolution, resulting in five IP bands in the spectrum of AB. The IPs of AB agree with a previous calculation using the B3LYP/6-31++G(d,p)//MP/6-31++G(d,p) method,29 which however, does not include orbital 3a1 at 29.88 eV.
Calculated valence ionisation spectra of AB (bottom) and PB (top). The ionisation energies (degeneracy is considered) in Table 3 are convoluted using a Gaussian broadening function with full width at half maximum (FWHM) at 0.05 eV. The related valence states (orbitals) are also indicated in the spectra. Purple spectrum (top) is for PB and blue spectrum (bottom) is for AB.
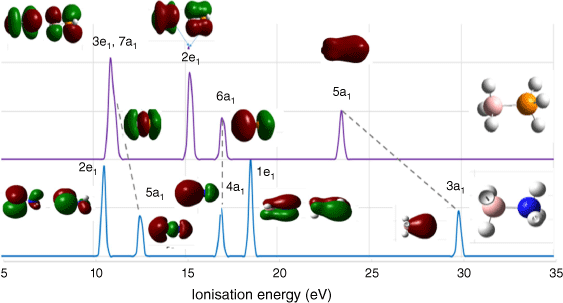
Further information using energy decomposition analysis (EDA) reveals that in AB,29 the charge transfer (CT) is largely governed by the π(HOMO) orbital with an N → B CT profile. Mallajosyula et al.29 suggested that in PB, the dominant contribution is from the σ(6a1) orbital with a marginal contribution from the HOMO (3e1). However, the present study reveals that the outer valence orbitals, such as π(3e1, 2e1) and σ(7a1), do not exhibit distinct CT character, whereas the σ(6a1) orbital displays a B ← P CT character, and the σ(5a1) orbital exhibits a B → P CT character. As a result, the IP spectra of AB and PB exhibit similarities in their σ(a1) states with CT characteristics, but their π(e1) orbitals are notably different. The π(e1) orbitals of AB displays CT character, whereas the π(e1) orbitals of PB exhibit a more covalent character.
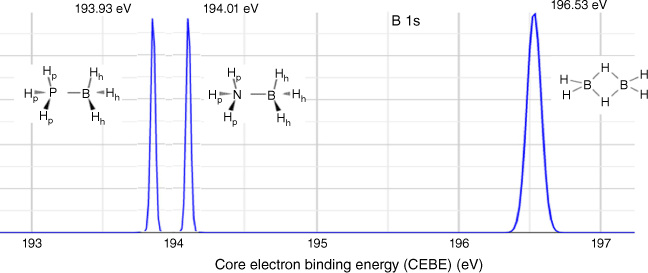
Core electron binding energies provide valuable information about the chemical environments of atoms within compounds.57 The comparison of calculated CEBE for boron (B 1s) in the different borane compounds – DB, AB, and PB – can reveal important insights into their local electronic structures. As shown in Fig. 5, B 1s CEBE values for DB, AB and PB are 196.53, 194.01 and 193.93 eV respectively. The B 1s CEBE values for DB and AB agree well with available experimental values of 196.5 eV for DB58 and 193.73(4) eV for AB,59 whereas no experimental B 1s CEBE for PB is available. The trend of B 1s CEBE values among the compounds is as follows: B 1s (PB) < B 1s (AB) < B 1s (DB). This implies that the core electrons of boron in PB require less energy to be ionised (oxidised) compared with those in AB, whereas the core electrons of boron in DB require more energy to be ionised. Lower CEBE values indicate a more chemically reactive environment, which corresponds to a more active chemical environment. This trend aligns with the larger 11B NMR chemical shifts reported in Fig. 3. It is important to note that whereas XPS and NMR techniques have different mechanisms, they both provide valuable information about the chemical and electronic properties of a compound, local to a specific atom.
Conclusion
The present study elucidates the electronic structures of two sets of hydrogens in diborane (H2B2H4), ammonia borane (H3B–NH3) and phosphine borane (H3B–PH3). The electronic structure and bonding nature of DB markedly differ from the donor–acceptor borane complexes of AB and PB. Although all three boranes process two distinct hydrogen groups, the bridge hydrogens (Hbs) and terminal hydrogens (Hts) in diborane exhibit a significant chemical shift difference of up to 5.31 ppm in the 1H NMR spectra. AB and PB share a common bonding pattern involving protic hydrogens (H–N and H–P) (Hps) and hydridic hydrogens (H–B) (Hhs), linked by the B ← X (X═N and P) bond. However, the chemical shift difference between Hps and Hhs in AB is small at ΔδH = 0.42 ppm, and it increases to 3.0 ppm for PB. The NMR chemical environment of 11B was calculated as 85.40 ppm in diborane, 126.21 ppm in AB, and 151.46 ppm in PB, consistent with the calculated B 1s CEBE of 196.53, 194.01 and 193.93 eV for DB, AB and PB respectively. Both 11B NMR and B 1s XPS spectroscopy clearly reveal that the chemical environments of boron in AB and PB are more similar compared with that in DB. It is always worth considering novel approaches and combining existing techniques to obtain a more profound understanding of systems under investigation, despite the significantly different mechanisms of NMR and XPS. Exploring the interplay between NMR and XPS could represent an interesting and potentially valuable research avenue for investigating novel compounds, including those relevant to energy storage materials. Finally, the outer valence orbitals of AB and PB reflect their differing bonding characteristics, with AB exhibiting slightly greater susceptibility to oxidation due to its slightly lower first ionisation potential (10.57 eV) compared with PB (11.07 eV).
Confirmation
We are pleased to announce the successful completion of gas-phase synchrotron-sourced photoemission and X-ray absorption measurements for ammonia borane at Elettra Sincrotrone Trieste in Italy. The outcomes of these experiments show exceptional agreement with our valence IP and CEBE calculations. Specifically, we precisely determined the complete valence IP spectrum, which encompasses the calculated band at 29.99 eV (3a1). The calculated B 1s energy, at 194.01 eV, has been confirmed. We are currently in the final stages of manuscript preparation, describing these compelling findings resulting from a decade collaboration between theoretical and experimental approaches.
Acknowledgements
F. Wang acknowledges Swinburne Supercomputer Facilities for providing computing facilities for this work.
References
1 Demirci UB. Ammonia borane: an extensively studied, though not yet implemented, hydrogen carrier. Energies 2020; 13(12): 3071.
| Crossref | Google Scholar |
2 Wang F, Swinbourn R, Li C. Shipping Australian sunshine: liquid renewable green fuel export. Int J Hydrogen Energy 2023; 48: 14763-14784.
| Crossref | Google Scholar |
3 Akbayrak S, Özkar S. Ammonia borane as hydrogen storage materials. Int J Hydrogen Energy 2018; 43(40): 18592-18606.
| Crossref | Google Scholar |
4 Li H, Yang Q, Chen X, Shore SG. Ammonia borane, past as prolog. J Organomet Chem 2014; 751: 60-66.
| Crossref | Google Scholar |
5 Welch GC, Stephan DW. Facile heterolytic cleavage of dihydrogen by phosphines and boranes. J Am Chem Soc 2007; 129(7): 1880-1881.
| Crossref | Google Scholar | PubMed |
6 Oldroyd NL, Chitnis SS, Annibale VT, Arz MI, Sparkes HA, Manners I. Metal-free dehydropolymerisation of phosphine-boranes using cyclic (alkyl)(amino)carbenes as hydrogen acceptors. Nat Commun 2019; 10(1): 1370.
| Crossref | Google Scholar | PubMed |
7 Cowley AH, Damasco MC. Donor–acceptor bond in phosphine–borane complexes. J Am Chem Soc 1971; 93(25): 6815-6821.
| Crossref | Google Scholar |
8 Sakai S. Theoretical study of the chemical reactions of B2H6 with Lewis bases (NH3, PH3, H2O, and H2S). J Phys Chem 1995; 99(22): 9080-9086.
| Crossref | Google Scholar |
9 Wang F, Pang W, Huang M. Valence space electron momentum spectroscopy of diborane. J Electron Spectrosc Relat Phenom 2006; 151(3): 215-223.
| Crossref | Google Scholar |
10 Demirci UB. Ammonia borane, a material with exceptional properties for chemical hydrogen storage. Int J Hydrogen Energy 2017; 42(15): 9978-10013.
| Crossref | Google Scholar |
11 Nordman CE, Reimann C. The molecular and crystal structures of ammonia–triborane. J Am Chem Soc 1959; 81(14): 3538-3543.
| Crossref | Google Scholar |
12 Nguyen VS, Matus MH, Nguyen MT, Dixon DA. Reactions of diborane with ammonia and ammonia borane: catalytic effects for multiple pathways for hydrogen release. J Phys Chem A 2008; 112(40): 9946-9954.
| Crossref | Google Scholar | PubMed |
13 Rudolph RW, Parry RW. Fluorophosphine ligands. IV. The apparent base strengths of difluorophosphine, trifluorophosphine, and phosphine toward the Lewis acid borane. J Am Chem Soc 1967; 89(7): 1621-1625.
| Crossref | Google Scholar |
14 Spikes GH, Fettinger JC, Power PP. Facile activation of dihydrogen by an unsaturated heavier main group compound. J Am Chem Soc 2005; 127(35): 12232-12233.
| Crossref | Google Scholar | PubMed |
15 Sarbajna A, Swamy V, Gessner VH. Phosphorus ylides: powerful substituents for the stabilization of reactive main group compounds. Chem Sci 2020; 12(6): 2016-2024.
| Crossref | Google Scholar | PubMed |
16 Wolf G, Baumann J, Baitalow F, Hoffmann FP. Calorimetric process monitoring of thermal decomposition of B–N–H compounds. Thermochim Acta 2000; 343(1–2): 19-25.
| Crossref | Google Scholar |
17 Nielsen TK, Besenbacher F, Jensen TR. Nanoconfined hydrides for energy storage. Nanoscale 2011; 3(5): 2086-2098.
| Crossref | Google Scholar | PubMed |
18 Ramachandran PV, Drolet MP, Kulkarni AS. A non-dissociative open-flask hydroboration with ammonia borane: ready synthesis of ammonia–trialkylboranes and aminodialkylboranes. Chem Commun 2016; 52(80): 11897-11900.
| Crossref | Google Scholar | PubMed |
19 Züttel A. Materials for hydrogen storage. Mater Today 2003; 6(9): 24-33.
| Crossref | Google Scholar |
20 Staubitz A, Robertson AP, Sloan ME, Manners I. Amine− and phosphine−borane adducts: new interest in old molecules. Chem Rev 2010; 110(7): 4023-4078.
| Crossref | Google Scholar | PubMed |
21 Dixon DA, Gutowski M. Thermodynamic properties of molecular borane amines and the [BH4–][NH4+] salt for chemical hydrogen storage systems from ab initio electronic structure theory. J Phys Chem A 2005; 109(23): 5129-5135.
| Crossref | Google Scholar | PubMed |
22 Chong DP, Wang F. Dehydrogenation of ammonia borane impacts valence and core electrons: a photoemission spectroscopic study. ACS Omega 2022; 7(40): 35924-35932.
| Crossref | Google Scholar | PubMed |
23 Schleier D, Gerlach M, Pratim Mukhopadhyay D, Karaev E, Schaffner D, Hemberger P, Fischer I. Ammonia borane, NH3BH3: a threshold photoelectron–photoion coincidence study of a potential hydrogen-storage material. Chem Eur J 28(42): e202201378.
| Crossref | Google Scholar |
24 Hill A, Wang F. Intramolecular O···H hydrogen bonding of salicylic acid: further insights from O 1s XPS and 1H NMR spectra using DFT calculations. J Phys Chem A 2023; 127(12): 2705-2716.
| Crossref | Google Scholar | PubMed |
25 Segala M, Chong DP. An evaluation of exchange-correlation functionals for the calculations of the ionization energies for atoms and molecules. J Electron Spectrosc Relat Phenom 2009; 171(1): 18-23.
| Crossref | Google Scholar |
26 Chong DP. Density functional calculation of core-electron binding energies of glycine conformers. Can J Chem 1996; 74(6): 1005-1007.
| Crossref | Google Scholar |
29 Mallajosyula SS, Datta A, Pati SK. Conformational preference in heteroatomic analogues of ethane, H3X−YH3 (X = B, AL; Y = N, P): implicationsof charge transfer. J Phys Chem A 2006; 110(15): 5156-5163.
| Crossref | Google Scholar | PubMed |
30 Mo Y, Wu W, Song L, Lin M, Zhang Q, Gao J. The magnitude of hyperconjugation in ethane: a perspective from ab initio valence bond theory. Angew Chem Int Ed Engl 2004; 43(15): 1986-1990.
| Crossref | Google Scholar | PubMed |
31 Cho H, Shaw WJ, Parvanov V, Schenter GK, Karkamkar A, Hess NJ, Mundy C, Kathmann S, Sears J, Lipton AS, et al. Molecular structure and dynamics in the low temperature (orthorhombic) phase of NH3BH3. J Phys Chem A 2008; 112(18): 4277-4283.
| Crossref | Google Scholar | PubMed |
32 Stitt F. The gaseous heat capacity and restricted internal rotation of diborane. J Chem Phys 1940; 8(12): 981-986.
| Crossref | Google Scholar |
33 Anane H, El Houssame S, El Guerraze A, Jarid A, Boutalib A, Nebot-Gil I, Tomás F. Ab initio molecular orbital study of the substituent effect on ammonia and phosphine–borane complexes. J Mol Struct 2004; 709(1): 103-107.
| Crossref | Google Scholar |
35 Matus MH, Grant DJ, Nguyen MT, Dixon DA. Fundamental thermochemical properties of ammonia borane and dehydrogenated derivatives (BNHn, n = 0−6). J Phys Chem C 2009; 113(37): 16553-16560.
| Crossref | Google Scholar |
36 Durig JR, Li YS, Carreira IA, Odom JD. Microwave spectrum, structure, dipole moment, and barrier to internal rotation of phosphine–borane. J Am Chem Soc 1973; 95(8): 2491-2496.
| Crossref | Google Scholar |
37 Burrus CA. Stark effect from 1.1 to 2.6 millimeters wavelength: PH3, PD3, DI, and CO. J Chem Phys 1958; 28(3): 427-429.
| Crossref | Google Scholar |
38 Roy B, Pal U, Bishnoi A, O’Dell LA, Sharma P. Exploring the homopolar dehydrocoupling of ammonia borane by solid-state multinuclear NMR spectroscopy. Chem Commun 2021; 57(15): 1887-1890.
| Crossref | Google Scholar | PubMed |
39 Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL. How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov 2010; 9(3): 203-214.
| Crossref | Google Scholar | PubMed |
40 McWeeny R. Perturbation theory for the Fock–Dirac density matrix. Phys Rev 1962; 126(3): 1028-1034.
| Crossref | Google Scholar |
41 Ditchfield R. Self-consistent perturbation theory of diamagnetism. Mol Phys 1974; 27(4): 789-807.
| Crossref | Google Scholar |
42 Wolinski K, Hinton JF, Pulay P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J Am Chem Soc 1990; 112(23): 8251-8260.
| Crossref | Google Scholar |
43 Cheeseman JR, Trucks GW, Keith TA, Frisch MJ. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J Chem Phys 1996; 104(14): 5497-5509.
| Crossref | Google Scholar |
44 Gao P, Wang X, Huang Z, Yu H. 11B NMR chemical shift predictions via density functional theory and gauge-including atomic orbital approach: applications to structural elucidations of boron-containing molecules. ACS Omega 2019; 4(7): 12385-12392.
| Crossref | Google Scholar | PubMed |
45 Jackowski K, Makulski W, Szyprowska A, Antusek A, Jaszuński M, Jusélius J. NMR shielding constants in BF3 and magnetic dipole moments of 11B and 10B nuclei. J Chem Phys 2009; 130(4): 044309.
| Crossref | Google Scholar | PubMed |
46 Staubitz A, Robertson APM, Manners I. Ammonia–borane and related compounds as dihydrogen sources. Chem Rev 2010; 110(7): 4079-4124.
| Crossref | Google Scholar | PubMed |
47 Shaw WJ, Linehan JC, Szymczak NK, Heldebrant DJ, Yonker C, Camaioni DM, Baker RT, Autrey T. In situ multinuclear NMR spectroscopic studies of the thermal decomposition of ammonia borane in solution. Angew Chem Int Ed Engl 2008; 47(39): 7493-7496.
| Crossref | Google Scholar | PubMed |
48 Sundholm D, Gauss J, Schäfer A. Rovibrationally averaged nuclear magnetic shielding tensors calculated at the coupled‐cluster level. J Chem Phys 1996; 105(24): 11051-11059.
| Crossref | Google Scholar |
49 Lantto P, Jackowski K, Makulski W, Olejniczak M, Jaszuński M. NMR shielding constants in PH3, absolute shielding scale, and the nuclear magnetic moment of 31P. J Phys Chem A 2011; 115(38): 10617-10623.
| Crossref | Google Scholar | PubMed |
50 Rzepa HS, Arkhipenko S, Wan E, Sabatini MT, Karaluka V, Whiting A, Sheppard TD. An accessible method for DFT calculation of 11B nmr shifts of organoboron compounds. J Org Chem 2018; 83(15): 8020-8025.
| Crossref | Google Scholar | PubMed |
51 Yuan B, Shin J-W, Bernstein ER. Dynamics and fragmentation of van der Waals and hydrogen bonded cluster cations: (NH3)n and (NH3BH3)n ionized at 10.51 eV. J Chem Phys 2016; 144(14): 144315.
| Crossref | Google Scholar | PubMed |
52 Carniato S, Millié P. Accurate core electron binding energy calculations using small 6-31G and TZV core hole optimized basis sets. J Chem Phys 2002; 116(9): 3521-3532.
| Crossref | Google Scholar |
53 Wang F. Future of computational molecular spectroscopy – from supporting interpretation to leading the innovation. Phys Chem Chem Phys 2023; 25: 7090-7105.
| Crossref | Google Scholar | PubMed |
55 Cederbaum LS. One-body Green’s function for atoms and molecules: theory and application. J Phys B 1975; 8(2): 290-303.
| Crossref | Google Scholar |
56 Lloyd DR, Lynaugh N. Photoelectron studies of boron compounds. Part 3.– Complexes of borane with Lewis bases. J Chem Soc Faraday Trans 2 1972; 68(0): 947-958.
| Crossref | Google Scholar |
57 Jolly WL, Finn P, Pearson RK, Hollander JM. Chemical shifts in core electron binding energies for some gaseous nitrogen compounds. Inorg Chem 1971; 10(2): 378-381.
| Crossref | Google Scholar |
58 Allison DA, Johansson G, Allan CJ, Gelius U, Siegbahn H, Allison J, Siegbahn K. Molecular spectroscopy by means of ESCA: V. Boron compounds. J Electron Spectrosc Relat Phenom 1972; 1(3): 269-283.
| Crossref | Google Scholar |
59 Beach DB, Jolly WL. Photoelectron spectroscopic study of the bonding in borane adducts. Inorg Chem 1985; 24(4): 567-570.
| Crossref | Google Scholar |