

Photocycloadditions of benzene derivatives and their systematic application to organic synthesis
Arthur Desvals A and Norbert Hoffmann A *
A *
A CNRS, Université de Reims Champagne-Ardenne, ICMR, Equipe de Photochimie, UFR Sciences, B.P. 1039, 51687 Reims, France.

Arthur Desvals obtained his bachelor’s degree in 2017 from the University of Versailles Saint-Quentin-en-Yvelines and his master’s degree in 2019 from the University Paris-Saclay. He then received his PhD degree in 2023 under the supervision of Dr Norbert Hoffmann at the Reims Institute of Molecular Chemistry. His research focuses on photochemical reactions and valorisation of compounds derived from biomass. |

Norbert Hoffmann studied chemistry at the RWTH Aachen University, Germany and received his PhD degree in 1992 under the supervision of Hans-Dieter Scharf. In 1993, he obtained a permanent position (Chargé de Recherche) at the French National Centre for Scientific Research (CNRS) in Reims, France. In 2004, he was appointed Research Director in the CNRS. His main research interests are in the field of organic photochemistry: electron transfer, photoinduced radical reactions, stereoselective reactions, cycloadditions of aromatic compounds, reactions in photochemical continuous flow reactors and application of these reactions to organic synthesis. Further research interests concern the production of fine chemicals from biomass and the synthesis and characterisation of dyes (chemistry of polymethine and perylene derivatives). |
Australian Journal of Chemistry 76(3) 117-129 https://doi.org/10.1071/CH23029
Submitted: 10 February 2023 Accepted: 20 March 2023 Published: 4 May 2023
© 2023 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
Photocycloadditions of benzene derivatives with alkenes play an important role as key steps in organic synthesis. Intramolecular reactions have been most frequently studied in this context. Often, meta or [2 + 3] photocycloadditions take place in competition with ortho or [2 + 2] additions. The influence of the substitution pattern and the spin multiplicity of the excited state on the outcome of these reactions is discussed. A topological analysis permitting a systematic application of the [2 + 3] photocycloadditions to the total synthesis of natural products is presented and a selection of corresponding syntheses is discussed. More recently the [2 + 2] photocycloaddition and consecutive rearrangements on organic synthesis have been published. Some approaches in the context of asymmetric synthesis have also been reported.
Keywords: aromatic compounds, meta or [2 + 3] photocyloaddition, molecular complexity, natural products, ortho or [2 + 2] photocycloaddition, photocycloaddition, spin multiplicity, terpenes, total synthesis.
Introduction
Photochemical reactions of simple compounds can generate a high degree of molecular complexity and diversity which is of high interest, for example, for the search of new biologically active compounds.[1] The photochemical reactivity of organic compounds is significantly different from their ground state reactivity. This is due to the fact that photochemical or electronic excitation modifies the electronic configuration of such compounds.[2,3] New compounds or compound families become easily available that cannot, or only with difficulty, be synthesised by more conventional methods of organic synthesis. Consequently, these reactions have become an important tool in organic synthesis.[4–7] During recent years, photochemical reactions became also particularly attractive in chemical and pharmaceutical industries when searching for new biologically active compounds.[8,9] Photocycloadditions of alkenes with electronically excited benzene derivatives are typical examples. While ground state reactions of such compounds are often characterised by a high tendency to regenerate aromaticity in the products, photochemical reactions are frequently characterised by a high tendency to loose aromatic character in the products. In this context, it should also be mentioned that photochemical excitation significantly affects the aromatic or anti-aromatic character.[10–13] Thus benzene derivatives become anti-aromatic when they are photochemically excited. Consequently, the reactivity is significantly increased. For the same reason, product formation at the excited state potential energy surface is directed to structures which are less stable at the ground state potential energy surface. Thus, a high degree of molecular complexity is generated in one step with relatively simple starting compounds. In the context of sustainable chemistry, it should be pointed out that in these reactions the concept of the photon as a traceless reagent is realised.[14]
Three different modes of photocycloaddition are possible between ground state alkenes and electronically excited benzene type compounds (Scheme 1). They provide efficient access to complex non-aromatic polycyclic compounds. Ortho ([2 + 2]) photocycloadditions lead to bicyclo[4.2.0]octa-2,4-dienes, meta ([2 + 3]) photocycloadditions generate tricyclo[3.3.0.0]octa-3-enes and para ([2 + 4]) photocycloadditions give rise to bicyclo[2.2.2]octa-2,5-dienes.
Three modes of photocycloadditions of alkenes to electronically excited benzene derivatives.
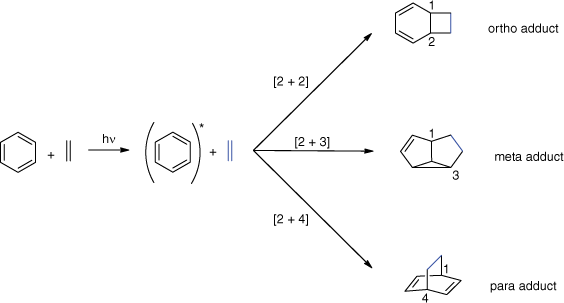
Despite promising recent studies,[15–17] para photocycloadditions are rarely observed for benzene-like aromatic compounds. Ortho and meta photocycloadditions have been studied much more intensively since their discoveries, respectively, in 1957[18,19] and 1966.[20,21] However, many aspects, such as the competition between these modes, still need intensive investigation. Also, the influence of substituents or the influence of the spin multiplicity on this competition are not yet well understood. These are important topics when the reactions are systematically applied to organic synthesis. Mechanisms based on the formation of an exciplex have been proposed to explain the selectivity.[22,23] The selectivity is strongly influenced by the nature of the substituents because they affect the electron-donating and accepting properties of the partners. A large electronic difference between the partners induced by the substituents directs the reaction towards the ortho photocycloaddition whereas a small difference favours the meta photocycloaddition.[22,24] The substitution pattern may also cause an exergonic photochemical electron transfer between the reaction partners leading to non-cyclisation products. A common intermediate for all three photocycloadditions was later found in a computational study.[25] It is easily accessible from the S1 state and corresponds to a conical intersection between the S1 and S0 surfaces. Over the years, numerous reviews have described many aspects of these reactions, ranging from experimental conditions and mechanistic descriptions to applications in total synthesis.[26–30] The present article gives an overview of historical and recent applications in total syntheses. Topological aspects are detailed as they are crucial for the planning of total syntheses. Furthermore, new insights on the competition between ortho and meta photocycloadditions at their singlet excited state are highlighted.
Meta photocycloaddition at the singlet state
It was early established that the meta photocycloaddition occurs at the singlet excited state, based on the observed retention of configuration of the alkene partner and quenching experiments.[31] The mechanism of the meta photocycloaddition is detailed for the intramolecular version in Scheme 2. The intramolecular [2 + 3] cycloaddition of the aromatic compound 1 excited to the singlet state to the alkene function can either occur at positions 1,3 or 2,6, to lead to intermediates 2 or 3 respectively. They also possess zwitterionic character due to the singlet multiplicity.[32] After this initial formation of two σ-bonds, the charge or radical combination leads to two different products for each intermediate depending on the generated cyclopropane. The more common intermediate 2 usually yields angular triquinanes 4 or linear triquinanes 5. Intermediate 3 gives structures 6 or 7. Due to the zwitterionic character of the intermediates, electronic properties of substituent X have an influence on the regioselectivity. If X is electron-donating, it directs the addition towards positions 1,3 to form intermediate 2, while positions 2,6 and intermediate 3 are preferred when X is electron-withdrawing.[20,33,34] In the case of the absence of a strong donor or acceptor substituent X, compelling evidence showed the biradical nature of these intermediates.[35,36]
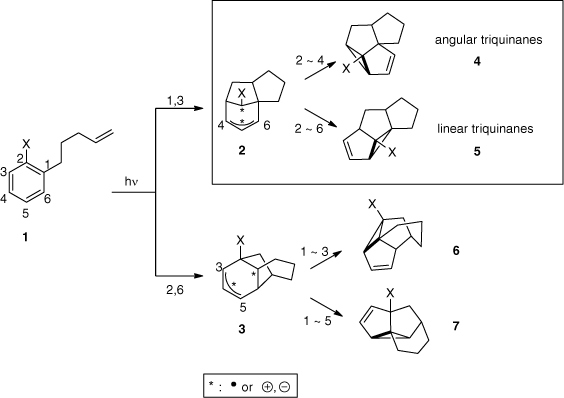
Applications of meta photocycloadditions in total syntheses
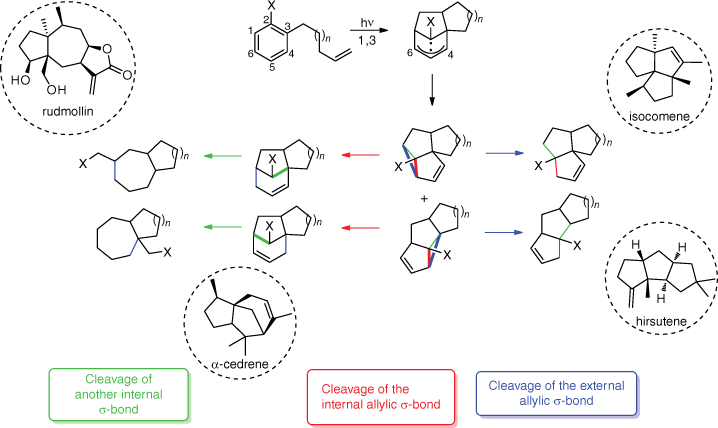
The meta photocycloaddition gives access to many different scaffolds depending on the opening of the cyclopropane ring (Scheme 3). A topological analysis is helpful for a targeted application to the synthesis of natural compounds. On the one hand, cleavage of the external allylic σ-bond (blue) leads to either angular triquinanes like isocomene, or linear triquinanes like hirsutene. On the other hand, cleavage of the internal allylic σ-bond (red) gives sesquiterpenoids like α-cedrene. From this type of product, another internal σ-bond (green) can be cleaved leading to other polycyclic structures like rudmollin.
The pioneer work of Wender and Howbert displayed many applications of the meta photocycloaddition in total syntheses of natural products.[37] In this first of a large series of articles, in 1981, they described their strategy considering the mode of addition (ortho, meta, para), regioselectivity, endo/exo selectivity and stereochemistry. First, they pointed out that the intramolecular reaction drastically reduces the number of possible cycloadducts compared to the intermolecular version. Then they adjusted the substituents and their electronic properties to favour the meta mode and to direct the regioselectivity. Finally, the endo/exo selectivity and stereochemistry were explained by looking at the conformations of the exciplexes. Some of these syntheses will now be briefly discussed focusing on the crucial steps of meta photocycloaddition and cyclopropane opening.
In the total synthesis of isocomene, the meta photocycloaddition of the aromatic compound 8 gives both the linear and angular cycloadducts 9 and 10 (Scheme 4).[38] It should be pointed out that the reaction was stereospecific with respect to the chiral centre in 8 which is explained by an allylic 1,3-strain effect.[39] This was observed in many similar reactions. The linear adduct 10 was transformed into the angular one (9) by a thermal vinyl cyclopropane rearrangement. Conveniently, this process also leads to the opening of the cyclopropane by a 1,5-hydrogen transfer to yield product 11. Isocomene was then obtained by a regioselective catalytic hydrogenation.
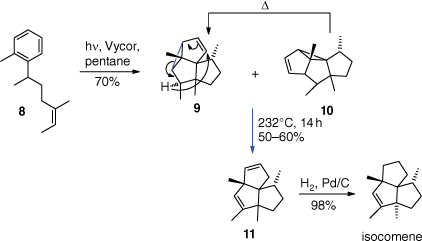
In the synthesis of hirsutene, the meta photocycloaddition was less efficient (Scheme 5).[40] It was improved by adding an acetyl function on the lateral chain of substrate 12. Treating the meta adduct 13 with 10-camphor-sulfonic acid then led to intermediate A with a tertiary allylic positive charge that was neutralised by an E1 elimination to yield product 14. Hirsutene was then obtained after four additional steps.
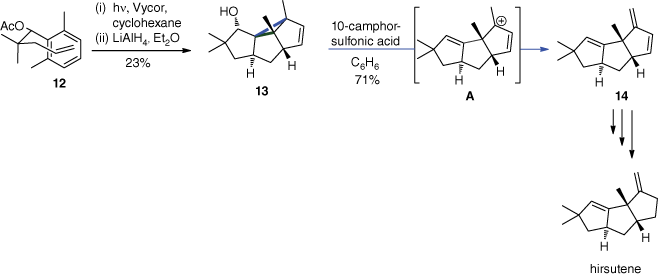
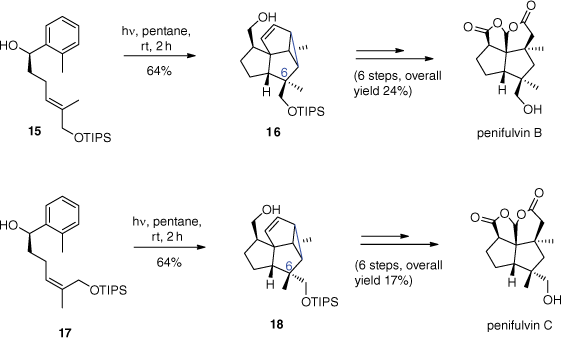
The enantioselective total syntheses of penifulvins A, B and C were developed more recently by Gaich and Mulzer using a similar methodology (Scheme 6).[41,42] The stereogenic quaternary carbon centres in position 6 of adducts 16 and 18 were set by the trisubstituted E/Z double bonds of substrates 15 and 17 respectively. Due to the fact that the photocycloadditions occur at the singlet excited state, no long living radical intermediates are generated enabling E/Z isomerisation. It should be pointed out that such reactions can be carried out with compounds possessing non-protected hydroxy functions.
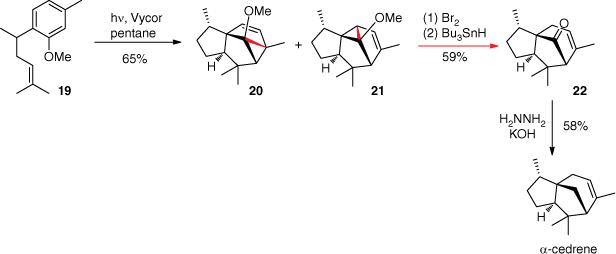
In the synthesis of α-cedrene, the meta photocycloaddition of compound 19 led to a mixture of products 20 and 21 (Scheme 7). These two adducts converged into product 22 when the internal allylic σ-bond was cleaved by an addition of Br2 followed by radical debromination using tributyltin hydride. α-Cedrene was finally obtained with 58% yield by a Wolff–Kishner reduction of product 22.
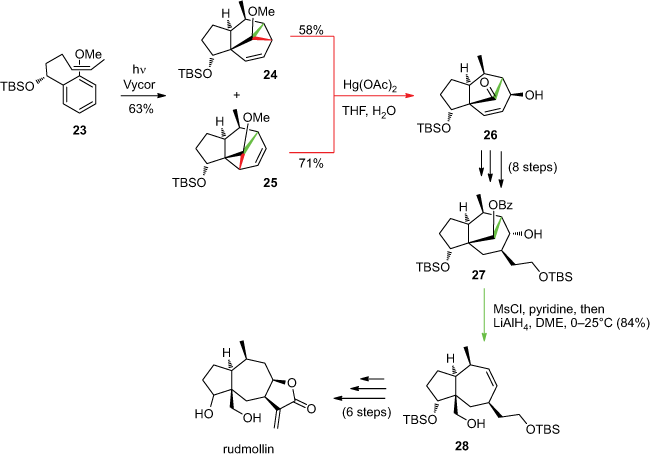
A similar strategy was applied to the synthesis of rudmollin. This synthesis required the cleavage of two σ-bonds of the cyclopropane (Scheme 8).[43] Irradiation of substrate 23 yielded a mixture of meta adducts 24 and 25. As for α-cedrene, the cleavage by electrophilic activation of the internal allylic σ-bond of the two products 24 and 25 led to a unique product 26. Compound 27 was obtained after eight additional steps; its geometry allowed the cleavage of another σ-bond originating from the initial cyclopropane. Mesylation of the alcohol, followed by addition of lithium aluminium hydride, induced fragmentation to give product 28. Rudmollin was then obtained after six additional steps.
Asymmetric induction of meta photocycloadditions
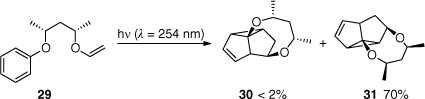
High diastereoselectivities were observed by Sugimura and co-workers by introducing a chiral tether (Scheme 9). Irradiation of substrate 29 yielded predominantly adduct 31 (70%) and traces of product 30 (<2%).[44,45]
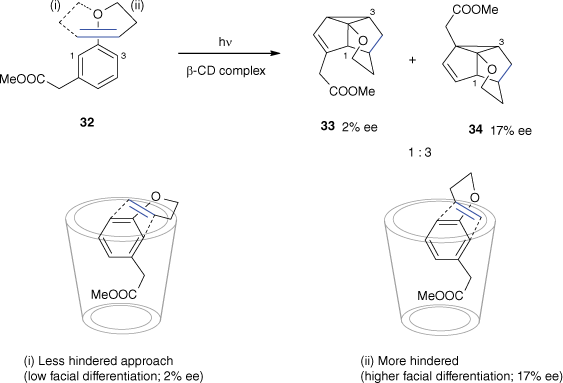
Several photochemical reactions have been carried out with cyclodextrin (CD) inclusion complexes in order to induce chirality by the host structure.[46,47] Moderate enantioselectivities were obtained by Vízvárdi and co-workers by complexation of the substrates in CDs (Scheme 10).[48] The irradiation of a β-CD complex of substrate 32 yields products 33 and 34 in a 1:3 ratio with an enantiomeric excess (ee) of 2 and 17% respectively. The difference of ee was explained by comparing the approaches leading to each product in the CD cavity. The stereodifferentiation in approach (ii) is more expressed than in approach (i), the facial enantioselectivity is thus higher for product 34 than for product 33.
Competition between [2 + 2] and [2 + 3] photocycloadditions at the singlet and triplet excited state
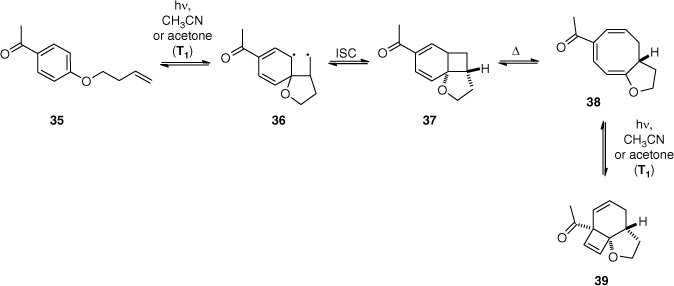
The intramolecular ortho photocycloaddition gives access to interesting tricyclic structures that often rearrange into more stable products. Selective ortho photocycloaddition is usually induced by placing an electron-withdrawing substituent on the aromatic ring. Furthermore, acetophenone derivatives give exclusively ortho photocycloadditions since they react at the triplet excited state. In accordance with the El Sayed rules,[2,49] this is due to the facile intersystem crossing from the n–π* singlet excited state to π–π* triplet excited state of the aromatic ketones.[50] In this case, the primary adduct 37 of ortho photocycloaddition is formed via a biradical intermediate[51] 36 that combines after intersystem crossing (Scheme 11). The initial ortho photocycloaddition adducts 37 then undergo a spontaneous thermal disrotatory ring opening. Finally, a photochemical disrotatory cyclisation of cyclooctatriene 38 leads to the stable product 39.[52]
Cosstick et al. observed the same consecutive rearrangements of the primary ortho adduct 41 when irradiating benzonitrile derivative 40 in acetonitrile (Scheme 12).[53] This study opened the question of singlet or triplet intermediates for these derivatives because they noticed that the reaction rate was not affected when irradiating solutions saturated by xenon.[54] In such conditions, xenon induces an increased spin-orbit coupling by a heavy atom effect. Later, the authors performed more quenching experiments.[55] First, they compared the intensity of fluorescence of compounds bearing an alkene on the side chain with the saturated analogues. The fluorescence was quenched by 50% for the derivatives with alkenes, proving that there is an interaction between the singlet excited state aromatic and ground state alkene. However, they noted that the quenching was less pronounced compared to aromatic compounds known to react at the singlet excited state. Then they observed that the formation of the cyclooctatriene 42 was quenched by 50% in the presence of a low concentration of 2,5-dimethylhexa-2,4-diene or 2,3-dimethylbuta-1,3-diene. As these dienes are triplet quenchers, the result may indicate that the first step of the ortho photocycloaddition occurs at the triplet state, but the possibility of side reactions with the dienes was not ruled out. Nevertheless, in this experiment the photocyclisation to form final product 43 was not affected, they thus concluded that the last step of photocyclisation of the cyclooctatriene takes place at the singlet excited state in acetonitrile as solvent.
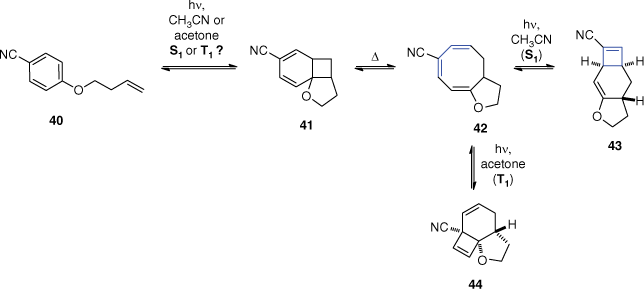
Following this work, Wagner et al. performed the irradiation of the same compound in acetone in a larger study investigating the effect of triplet sensitisation.[56] The triplet reaction in acetone yielded a different product 44. Triplet sensitisation did not impact the first step of the ortho photocycloaddition. However, it caused a different electrocyclisation of the cyclooctatriene 42, confirming that the product 43 is obtained from a photocyclisation at the singlet excited state.
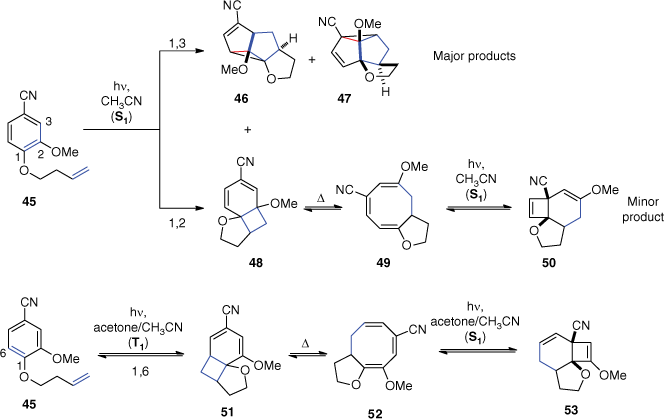
Our recent results give new insights on the nature of the excited state involved in the first step of ortho photocycloaddition of benzonitrile derivatives (Scheme 13).[57] The irradiation of benzonitrile derivative 45 bearing a methoxy substituent revealed that the singlet excited state prevails by direct absorption in acetonitrile. On the one hand, the major products 46 and 47 are the result of a singlet state meta photocycloaddition. Their formation is favoured by the methoxy substituent that stabilises the zwitterionic intermediate (see meta photocycloadditions section). Compound 46 was completely transformed into 47 by prolonged irradiation. On the other hand, the regioselectivity of the [2 + 2] addition served as a support that the ortho adduct 50 is formed at the singlet excited state. The irradiation in acetonitrile yielded product 50 resulting from the known rearrangements of adduct 48 which is formed by an ortho photocycloaddition on positions 1,2. After complete conversion of 45 and prolonged irradiation, its 15% portion of the product mixture didn’t change. In contrast, triplet sensitisation by irradiation in an acetonitrile/acetone mixture yielded product 53 resulting from an ortho photocycloaddition on positions 1,6. No product was isolated when irradiations were carried out in pure acetone, indicating that the photocyclisation of cyclooctatrienes 52 is not effective at the triplet state. The reaction is only possible in an acetonitrile/acetone mixture that permits the triplet ortho photocycloaddition by sensitisation of substrate 45 as well as the singlet photocyclisation by direct absorption of cyclooctatriene 52. A typical triplet-excited state reaction of a cyclooctatriene intermediate is reported in Ref. [58].
Applications of ortho photocycloaddition in total syntheses
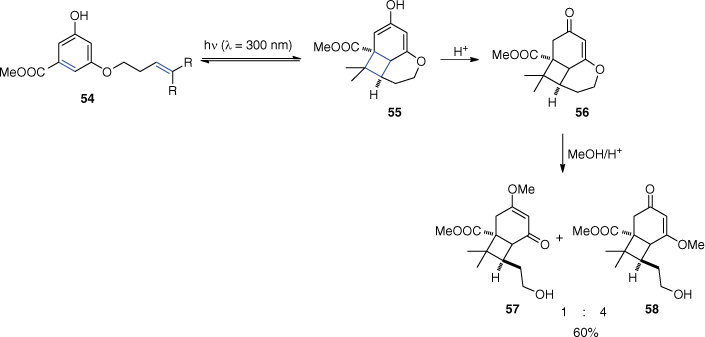
An early example of application of ortho photocycloadditions was the synthesis of compounds 57 and 58 (Scheme 14)[59,60] that are 5,5-dialkylcyclohexane-1,3-dione analogues of known herbicides.[61–64] The irradiation of the 3,5-dihydroxy derivative 54 gave product 56 via a tautomerisation of the primary ortho adduct 55. The final products 57 and 58 were obtained after a ring opening induced by methanol addition under acidic conditions.
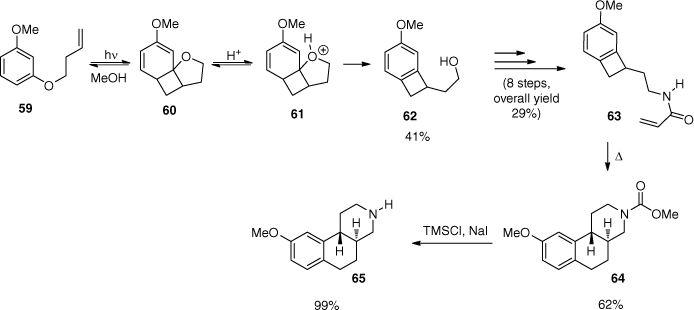
Resorcinol derivatives such as 59 possess low photo-reactivity when irradiated under standard conditions due to the reversibility of the reaction (Scheme 15). When irradiated in an acidic medium, product 62 was isolated.[65,66] Under these acidic conditions, the tetrahydrofuran moiety of the primary ortho adduct 60 is protonated. Subsequent opening of the tetrahydrofuran ring then leads to product 62. This type of benzocyclobutenes are valuable synthesis intermediates.[67] Product 63 was obtained after eight steps, and it was efficiently transformed into product 64 via a thermal rearrangement. Removal of the methyl carbamate protecting group finally gave benzooctahydroisoquinoline 65.[68,69] This compound is a rigidified structural analogue of dopamine and has a selective affinity to certain dopamine receptors.
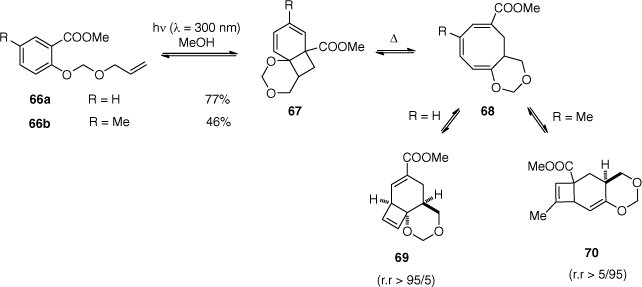
More recently, Zech and Bach have revisited the ortho photocycloadditions on salicylic acid derivatives.[70] In prevision of total syntheses,[71] gradual changes to the substrate structures were performed in order to study the selectivity of the reaction (Scheme 16).[72] For example, an additional oxygen atom in the side chain (66a) gave product 69 as expected, after the known rearrangements of primary adduct 67 and cyclooctatriene 68. However, a methyl substituent (66b, R═Me) yielded a different regioisomer (70) after photocyclisation of the cyclooctatriene 68.
In their strategy for the total synthesis of atlanticone C, this methyl group had to be introduced before the photochemical step so they replaced the ester function with a cyclic ketone (71) to force the formation of the desired regioisomer 72 (Scheme 17).[72] To prevent undesired photochemical reactivity of the α,β-unsaturated carbonyl function, an aqueous solution of Fe2(SO4)3 was used to absorb light below λ = 350 nm. Synthesis of atlanticone C from product 72 was completed by modifications of the oxidation state of several carbon atoms and introduction of a stereogenic centre. It was performed in nine steps with an overall yield of 6%.
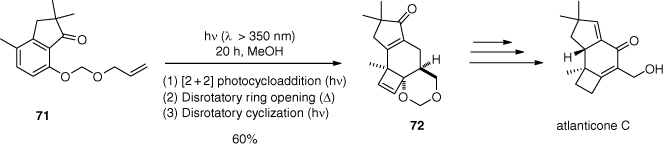
The research group later described the first enantioselective total synthesis of (+)-atlanticone C using a similar photochemical reaction cascade.[73] A catalytic chiral resolution of the racemic photoproduct was enabled by an enantioselective Corey–Bakshi–Shibata reduction. This time, (+)-atlanticone C was prepared in 10 steps with 18% yield from the enantiomerically pure photoproduct.
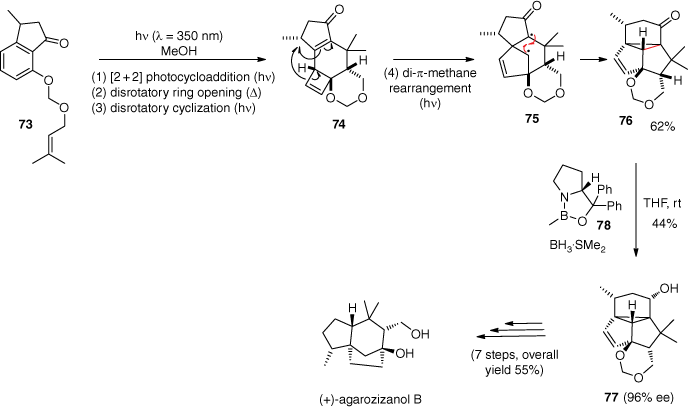
When similar compounds to 71 were irradiated at λ = 350 nm, a new reactivity was observed.[74] Rauscher and co-workers used this photochemical reaction cascade for the total synthesis of (+)-agarozizanol B (Scheme 18).[75] In this reaction, irradiation of substrate 73 yields product 74 after the classical sequence of ortho photocycloaddition, ring opening and photocyclisation. Compound 74 absorbs light under these conditions and undergoes a di-π-methane rearrangement to give product 76 via the biradical intermediate 75. The overall yield for these four successive reactions is 62%. Product 77 was then obtained with an ee of 96% with the same process used in the (+)-atlanticone C synthesis.[73] The oxazaborolidine 78 was used for a catalytic chiral resolution of the photoproduct 76 by a Corey–Bakshi–Shibata reduction.[76] Finally, (+)-agarozizanol B was obtained from compound 77 after seven steps with an overall yield of 55%.
The synthetic value of the ortho photocycloaddition is considerably increased when applied to hetero-aromatic substrates such a pentafluoropyridine,[77] oxazolopyridine,[78] benzotriazole,[79] or even polycyclic aromatics like cyanonaphthalene.[80] More complex alkene partners can also be considered like naphthalene[81,82] or 1-cyanonaphthalene.[83]
Chiral induction of ortho photocycloadditions
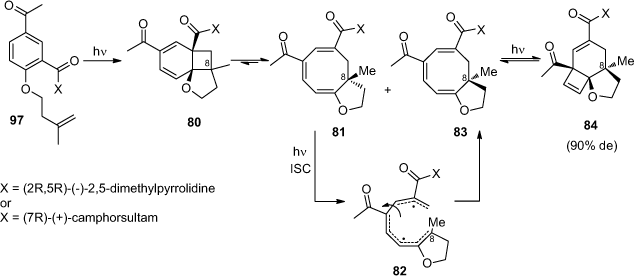
Wagner and McMahon performed diastereoselective ortho photocycloadditions by irradiating compounds bearing a chiral auxiliary (Scheme 19).[58] Using (2R,5R)-(–)-2,5-dimethylpyrrolidine or (7R)-(–)-camphorsultam attached to substrate 79, the final product 84 was isolated with a diastereoisomeric excess of 90%. The only permanent stereogenic centre C8 is created during the first step of the ortho photocycloaddition. Compound 80 is formed with a low diastereoselectivity. It is then increased by a photo-epimerisation of cyclooctatriene 81. The authors suggested that this kinetic resolution is due to the intersystem crossing to the triplet state facilitated by the acetyl substituent. It leads to ring opening of cyclooctatriene 81 to the tetraene biradical 82 where the rotation of the σ-bond between the allyl and pentadienyl moieties leads to an epimerisation during ring closing to cyclooctatriene 83.
Conclusion
Starting from relatively simple products, photocycloadditions of benzene derivatives with alkenes provide efficient access to compounds with a high degree of molecular complexity. Mainly [2 + 3] (meta) or [2 + 2] (ortho) photocycloaddition take place. In the latter case, photochemical and thermal rearrangements of the primary adducts are generally observed. The meta photocycloaddition occurs at the singlet excited state. The ortho addition, however, can take place either at the singlet or the triplet excited state. In this case, the spin multiplicity may have an influence on the regioselectivity of cycloaddition. In this context, more systematic and detailed studies of the influence of the substitution pattern on the regioselectivity and the cycloaddition mode is necessary. For example, recent findings suggest that the role of the singlet excited state in ortho photocycloadditions of nitrile and ester substituted aromatic compounds may have been overlooked.
The meta photocycloaddition has already proven to be a key step in many syntheses of natural products. A topological analysis is helpful in this regard. In this context, the ortho photocycloaddition was neglected in the past. However, recently, impressive syntheses have been published using this photocycloaddition as a key step. The application of photocycloadditions of benzene derivatives to organic synthesis can further be strengthened if challenges such as chiral induction or para photocycloadditions continue to be addressed.
References
[1] CJ Gerry, SL Schreiber, Chemical probes and drug leads from advances in synthetic planning and methodology. Nat Rev Drug Discov 2018, 17, 333.| Chemical probes and drug leads from advances in synthetic planning and methodology.Crossref | GoogleScholarGoogle Scholar |
[2] Klán P, Wirz J. Photochemistry of Organic Compounds. Chichester: Wiley; 2009.
[3] NJ Turro, G Schuster, Photochemical Reactions as a Tool in Organic Syntheses. Science 1975, 187, 303.
| Photochemical Reactions as a Tool in Organic Syntheses.Crossref | GoogleScholarGoogle Scholar |
[4] W Liu, C-J Li, Recent Synthetic Applications of Catalyst-Free Photochemistry. Synlett 2017, 28, 2714.
| Recent Synthetic Applications of Catalyst-Free Photochemistry.Crossref | GoogleScholarGoogle Scholar |
[5] N Hoffmann, Photochemical reactions as key steps in organic synthesis. Chem Rev 2008, 108, 1052.
| Photochemical reactions as key steps in organic synthesis.Crossref | GoogleScholarGoogle Scholar |
[6] T Bach, JP Hehn, Photochemical reactions as key steps in Natural Product Synthesis. Angew Chem Int Ed 2011, 50, 1000.
| Photochemical reactions as key steps in Natural Product Synthesis.Crossref | GoogleScholarGoogle Scholar |
[7] MD Kärkäs, JA Porco, CRJ Stephenson, Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis. Chem Rev 2016, 116, 9683.
| Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis.Crossref | GoogleScholarGoogle Scholar |
[8] HE Bonfield, T Knauber, F Lévesque, EG Moschetta, F Susanne, LJ Edwards, Photons as a 21st century reagent. Nat Commun 2020, 11, 804.
| Photons as a 21st century reagent.Crossref | GoogleScholarGoogle Scholar |
[9] M Oelgemöller, Green Photochemical Processes and Technologies for Research & Development, Scale-up and Chemical Production. J Chin Chem Soc 2014, 61, 743.
| Green Photochemical Processes and Technologies for Research & Development, Scale-up and Chemical Production.Crossref | GoogleScholarGoogle Scholar |
[10] Solà M, Boldyrev AI, Cyrański MK, Krygowski TM, Merino G. Aromaticity and Antiaromaticity. Chichester: Wiley; 2023.
[11] M Rosenberg, C Dahlstrand, K Kilså, H Ottosson, Excited State Aromaticity and Antiaromaticity: Opportunities for Photophysical and Photochemical Rationalizations. Chem Rev 2014, 114, 5379.
| Excited State Aromaticity and Antiaromaticity: Opportunities for Photophysical and Photochemical Rationalizations.Crossref | GoogleScholarGoogle Scholar |
[12] R Papadakis, H Ottosson, The excited state antiaromatic benzene ring: a molecular Mr Hyde? Chem Soc Rev 2015, 44, 6472.
| The excited state antiaromatic benzene ring: a molecular Mr Hyde?Crossref | GoogleScholarGoogle Scholar |
[13] J Yan, T Slanina, J Bergman, H Ottosson, Photochemistry Driven by Excited-State Aromaticity Gain or Antiaromaticity Relief. Chem Eur J 2023, 29, e202203748.
| Photochemistry Driven by Excited-State Aromaticity Gain or Antiaromaticity Relief.Crossref | GoogleScholarGoogle Scholar |
[14] N Hoffmann, Photochemical reactions of aromatic compounds and the concept of the photon as a traceless reagent. Photochem Photobiol Sci 2012, 11, 1613.
| Photochemical reactions of aromatic compounds and the concept of the photon as a traceless reagent.Crossref | GoogleScholarGoogle Scholar |
[15] F Birbaum, A Neels, CG Bochet, Photochemistry of Allenyl Salicylaldehydes. Org Lett 2008, 10, 3175.
| Photochemistry of Allenyl Salicylaldehydes.Crossref | GoogleScholarGoogle Scholar |
[16] U Streit, F Birbaum, A Quattropani, CG Bochet, Photocycloaddition of Arenes and Allenes. J Org Chem 2013, 78, 6890.
| Photocycloaddition of Arenes and Allenes.Crossref | GoogleScholarGoogle Scholar |
[17] Z Zhang, Y-j Zhou, X-W Liang, Total synthesis of natural products using photocycloaddition reactions of arenes. Org Biomol Chem 2020, 18, 5558.
| Total synthesis of natural products using photocycloaddition reactions of arenes.Crossref | GoogleScholarGoogle Scholar |
[18] DE Ayer, GH Buchi, US Patent 2 805 242. 1957. Chem Abstr 1958, 52, 2904.
[19] HJF Angus, D Bryce-Smith, Addition of Maleic Anhydride to Benzene. Proc Chem Soc 1959, 326.
[20] KE Wilzbach, L Kaplan, A Photochemical 1,3 Cycloaddition of Olefins to Benzene. J Am Chem Soc 1966, 88, 2066.
| A Photochemical 1,3 Cycloaddition of Olefins to Benzene.Crossref | GoogleScholarGoogle Scholar |
[21] D Bryce-Smith, A Gilbert, BH Orger, Photochemical 1,3-Cycloaddition of Olefins to Aromatic Compounds. Chem Commun 1966, 1966, 512.
| Photochemical 1,3-Cycloaddition of Olefins to Aromatic Compounds.Crossref | GoogleScholarGoogle Scholar |
[22] J Mattay, Selectivity and charge transfer in photoreactions of arenes with olefins: 2, Mode of cycloaddition. Tetrahedron 1985, 41, 2405.
| Selectivity and charge transfer in photoreactions of arenes with olefins: 2, Mode of cycloaddition.Crossref | GoogleScholarGoogle Scholar |
[23] J Mattay, T Rumbach, J Runsink, Radical ions and photochemical charge transfer phenomena. 27. Molecular recognition phenomena in the excited state: the meta photocycloaddition of cis- and trans-2,5-dihydro-2,5-dimethoxyfuran and cis-1,4-dimethoxycyclopent-2-ene to anisole. J Org Chem 1990, 55, 5691.
| Radical ions and photochemical charge transfer phenomena. 27. Molecular recognition phenomena in the excited state: the meta photocycloaddition of cis- and trans-2,5-dihydro-2,5-dimethoxyfuran and cis-1,4-dimethoxycyclopent-2-ene to anisole.Crossref | GoogleScholarGoogle Scholar |
[24] F Müller, J Mattay, Photocycloadditions: control by energy and electron transfer. Chem Rev 1993, 93, 99.
| Photocycloadditions: control by energy and electron transfer.Crossref | GoogleScholarGoogle Scholar |
[25] S Clifford, MJ Bearpark, F Bernardi, M Olivucci, MA Robb, BR Smith, Conical Intersection Pathways in the Photocycloaddition of ethene and Benzene: A CASSCF Study with MMVB Dynamics. J Am Chem Soc 1996, 118, 7353.
| Conical Intersection Pathways in the Photocycloaddition of ethene and Benzene: A CASSCF Study with MMVB Dynamics.Crossref | GoogleScholarGoogle Scholar |
[26] U Streit, CG Bochet, The arene-alkene photocycloaddition. Beilstein J Org Chem 2011, 7, 525.
| The arene-alkene photocycloaddition.Crossref | GoogleScholarGoogle Scholar |
[27] R Remy, CG Bochet, Arene-Alkene Cycloaddition. Chem Rev 2016, 116, 9816.
| Arene-Alkene Cycloaddition.Crossref | GoogleScholarGoogle Scholar |
[28] J Cornelisse, The meta photocycloaddition of arenes to alkenes. Chem Rev 1993, 93, 615.
| The meta photocycloaddition of arenes to alkenes.Crossref | GoogleScholarGoogle Scholar |
[29] D Chappell, AT Russell, From α-cedrene to crinipellin B and onward: 25 years of the alkene-arene meta-photocycloaddition reaction in natural product synthesis. Org Biomol Chem 2006, 4, 4409.
| From α-cedrene to crinipellin B and onward: 25 years of the alkene-arene meta-photocycloaddition reaction in natural product synthesis.Crossref | GoogleScholarGoogle Scholar |
[30] N Hoffmann, Photochemical Cycloaddition between Benzene Derivatives and Alkenes. Synthesis 2004, 2004, 481.
| Photochemical Cycloaddition between Benzene Derivatives and Alkenes.Crossref | GoogleScholarGoogle Scholar |
[31] A Morikawa, S Brownstein, RJ Cvetanovic, Kinetics of the cycloaddition of photoexcited benzene to 2-butene in the gas phase. J Am Chem Soc 1970, 92, 1471.
| Kinetics of the cycloaddition of photoexcited benzene to 2-butene in the gas phase.Crossref | GoogleScholarGoogle Scholar |
[32] PW Peterson, RK Mohamed, IV Alabugin, How to Lose a Bond in Two Ways — The Diradical/Zwitterion Dichotomy in Cycloaromatization Reactions. Eur J Org Chem 2013, 2013, 2505.
| How to Lose a Bond in Two Ways — The Diradical/Zwitterion Dichotomy in Cycloaromatization Reactions.Crossref | GoogleScholarGoogle Scholar |
[33] J Mattay, J Runsink, JA Piccirilli, AWH Jens, J Cornelisse, Photochemical cycloadditions of 1,3-dioxoles to anisole. J Chem Soc Perkin Trans 1 1987, 15.
| Photochemical cycloadditions of 1,3-dioxoles to anisole.Crossref | GoogleScholarGoogle Scholar |
[34] J Mattay, J Runsink, J Gersdorf, T Rumbach, C Ly, Selectivity and Charge Transfer in Photoreactions of α,α,αTrifluorotoluene with Olefins. Helv Chim Acta 1986, 69, 442.
| Selectivity and Charge Transfer in Photoreactions of α,α,αTrifluorotoluene with Olefins.Crossref | GoogleScholarGoogle Scholar |
[35] RS Sheridan, Azo precursors to a putative biradical in arene metal photoadditions: evidence for a novel concerted nitrogen expulsion. J Am Chem Soc 1983, 105, 5140.
| Azo precursors to a putative biradical in arene metal photoadditions: evidence for a novel concerted nitrogen expulsion.Crossref | GoogleScholarGoogle Scholar |
[36] DE Reedich, RS Sheridan, Independent generation of arene meta photoaddition biradicals. J Am Chem Soc 1985, 107, 3360.
| Independent generation of arene meta photoaddition biradicals.Crossref | GoogleScholarGoogle Scholar |
[37] PA Wender, JJ Howbert, Synthetic studies on arene-olefin cycloadditions: total synthesis of (.+-.)-.alpha.-cedrene. J Am Chem Soc 1981, 103, 688.
| Synthetic studies on arene-olefin cycloadditions: total synthesis of (.+-.)-.alpha.-cedrene.Crossref | GoogleScholarGoogle Scholar |
[38] PA Wender, GB Dreyer, Synthetic studies on arene-olefin cycloadditions. ii. total synthesis of (±)-isocomene. Tetrahedron 1981, 37, 4445.
| Synthetic studies on arene-olefin cycloadditions. ii. total synthesis of (±)-isocomene.Crossref | GoogleScholarGoogle Scholar |
[39] RW Hoffmann, Allylic 1,3-strain as a controlling factor in stereoselective transformation. Chem Rev 1989, 89, 1841.
| Allylic 1,3-strain as a controlling factor in stereoselective transformation.Crossref | GoogleScholarGoogle Scholar |
[40] PA Wender, JJ Howbert, Synthetic studies on arene-olefin cycloadditions -III- total synthesis of (±)-hirsutene. Tetrahedron Lett 1982, 23, 3983.
| Synthetic studies on arene-olefin cycloadditions -III- total synthesis of (±)-hirsutene.Crossref | GoogleScholarGoogle Scholar |
[41] T Gaich, J Mulzer, Total Synthesis of (−)-Penifulvin A, an Insecticide with a Dioxafenestrane Skeleton. J Am Chem Soc 2009, 131, 452.
| Total Synthesis of (−)-Penifulvin A, an Insecticide with a Dioxafenestrane Skeleton.Crossref | GoogleScholarGoogle Scholar |
[42] T Gaich, J Mulzer, From Silphinenes to Penifulvins: A Biomimetic Approach to Penifulvins B and C. Org Lett 2010, 12, 272.
| From Silphinenes to Penifulvins: A Biomimetic Approach to Penifulvins B and C.Crossref | GoogleScholarGoogle Scholar |
[43] PA Wender, K Fisher, Seven-Membered Ring Synthesis Based on Arene Olefin Cycloadditions: The Total Synthesis of (±)-Rudmollin. Tetrahedron Lett 1986, 27, 1857.
| Seven-Membered Ring Synthesis Based on Arene Olefin Cycloadditions: The Total Synthesis of (±)-Rudmollin.Crossref | GoogleScholarGoogle Scholar |
[44] K Hagiya, A Yamasaki, T Okuyama, T Sugimura, Asymmetric meta-arene-alkene photocycloaddition controlled by a 2,4-pentanediol tether. Tetrahedron Asymmetry 2004, 15, 1409.
| Asymmetric meta-arene-alkene photocycloaddition controlled by a 2,4-pentanediol tether.Crossref | GoogleScholarGoogle Scholar |
[45] T Sugimura, A Yamasaki, T Okuyama, Stereocontrolled intramolecular meta-arene–alkene photocycloaddition reactions using chiral tethers: efficiency of the tether derived from 2,4-pentanediol. Tetrahedron Asymmetry 2005, 16, 675.
| Stereocontrolled intramolecular meta-arene–alkene photocycloaddition reactions using chiral tethers: efficiency of the tether derived from 2,4-pentanediol.Crossref | GoogleScholarGoogle Scholar |
[46] C Yang, Y Inoue, Supramolecular photochirogenesis. Chem Soc Rev 2014, 43, 4123.
| Supramolecular photochirogenesis.Crossref | GoogleScholarGoogle Scholar |
[47] V Ramamurthy, J Sivaguru, Supramolecular Photochemistry as a Potential Synthetic Tool: Photocycloaddition. Chem Rev 2016, 116, 9914.
| Supramolecular Photochemistry as a Potential Synthetic Tool: Photocycloaddition.Crossref | GoogleScholarGoogle Scholar |
[48] K Vízvárdi, K Desmet, I Luyten, P Sandra, G Hoornaert, E Van der Eycken, Asymmetric Induction in Intramolecular meta Photocycloaddition: Cyclodextrin-Mediated Solid-Phase Photochemistry of Various Phenoxyalkenes. Org Lett 2001, 3, 1173.
| Asymmetric Induction in Intramolecular meta Photocycloaddition: Cyclodextrin-Mediated Solid-Phase Photochemistry of Various Phenoxyalkenes.Crossref | GoogleScholarGoogle Scholar |
[49] MA El-Sayed, Vanishing First- and Second-Order Intramolecular Heavy-Atom Effects on the (π*→n) Phosphorescence in Carbonyls. J Chem Phys 1964, 41, 2462.
| Vanishing First- and Second-Order Intramolecular Heavy-Atom Effects on the (π*→n) Phosphorescence in Carbonyls.Crossref | GoogleScholarGoogle Scholar |
[50] PJ Wagner, K Nahm, Regiospecific intramolecular reaction of an alkene group with the benzene ring of a triplet ketone. J Am Chem Soc 1987, 109, 4404.
| Regiospecific intramolecular reaction of an alkene group with the benzene ring of a triplet ketone.Crossref | GoogleScholarGoogle Scholar |
[51] K-L Cheng, PJ Wagner, Biradical Rearrangements during Intramolecular Cycloaddition of Double Bonds to Triplet Benzenes. J Am Chem Soc 1994, 116, 7945.
| Biradical Rearrangements during Intramolecular Cycloaddition of Double Bonds to Triplet Benzenes.Crossref | GoogleScholarGoogle Scholar |
[52] PJ Wagner, K Nahm, Interconversion of Bicyclooctadienes and Cyclooctatrienes Formed by Intramolecular Photocycloaddition of Phenyl Ketones Containing Remote Double Bonds. J Am Chem Soc 1987, 109, 6528.
| Interconversion of Bicyclooctadienes and Cyclooctatrienes Formed by Intramolecular Photocycloaddition of Phenyl Ketones Containing Remote Double Bonds.Crossref | GoogleScholarGoogle Scholar |
[53] KB Cosstick, MGB Drew, A Gillbert, Specific intramolecular ortho photocycloaddition of substituted 4-phenoxybut-1-enes. J Chem Soc Chem Commun 1987, 1987, 1867.
| Specific intramolecular ortho photocycloaddition of substituted 4-phenoxybut-1-enes.Crossref | GoogleScholarGoogle Scholar |
[54] H Morrison, A Miller, An Analysis of Heavy-Atom Perturbation of Intersystem Crossing as a Mechanistic Tool in Photochemistry. Tetrahedron 1981, 37, 3405.
| An Analysis of Heavy-Atom Perturbation of Intersystem Crossing as a Mechanistic Tool in Photochemistry.Crossref | GoogleScholarGoogle Scholar |
[55] SY Al-Qaradawi, KB Cosstick, A Gilbert, Intramolecular photocycloaddition of 4-phenoxybut-1-enes: a convenient access to the 4-oxatricyclo[7.2.0.0]undeca-2,10-diene skeleton. J Chem Soc Perkin Trans 1 1992, 1145.
| Intramolecular photocycloaddition of 4-phenoxybut-1-enes: a convenient access to the 4-oxatricyclo[7.2.0.0]undeca-2,10-diene skeleton.Crossref | GoogleScholarGoogle Scholar |
[56] PJ Wagner, RP Smart, Acetone Sensitized Intramolecular ortho Photocyclization of Substituted 4-Phenoxybut-1-enes and 5-Phenoxypent-1-enes. Tetrahedron Lett 1995, 36, 5135.
| Acetone Sensitized Intramolecular ortho Photocyclization of Substituted 4-Phenoxybut-1-enes and 5-Phenoxypent-1-enes.Crossref | GoogleScholarGoogle Scholar |
[57] A Desvals, SA Baudron, V Bulach, N Hoffmann, Photocycloadditions of Arenes Derived from Lignin. J Org Chem 2021, 86, 13310.
| Photocycloadditions of Arenes Derived from Lignin.Crossref | GoogleScholarGoogle Scholar |
[58] PJ Wagner, K McMahon, Chiral Auxiliaries Promote both Diastereoselective Cycloaddition and Kinetic Resolution of Products in the Ortho Photocycloaddition of Double Bonds to Benzene Rings. J Am Chem Soc 1994, 116, 10827.
| Chiral Auxiliaries Promote both Diastereoselective Cycloaddition and Kinetic Resolution of Products in the Ortho Photocycloaddition of Double Bonds to Benzene Rings.Crossref | GoogleScholarGoogle Scholar |
[59] N Hoffmann, J-P Pete, Acid catalyzed intramolecular [2+2] photocycloaddition of 3,5-dihydroxybenzoic acid derivatives. Tetrahedron Lett 1998, 39, 5027.
| Acid catalyzed intramolecular [2+2] photocycloaddition of 3,5-dihydroxybenzoic acid derivatives.Crossref | GoogleScholarGoogle Scholar |
[60] N Hoffmann, J-P Pete, Intramolecular [2+2] Photocycloaddition of Bichromophoric Derivatives of 3,5-Dihydroxybenzoic Acid and 3,5-Dihydroxybenzonitrile. Synthesis 2001, 1236.
| Intramolecular [2+2] Photocycloaddition of Bichromophoric Derivatives of 3,5-Dihydroxybenzoic Acid and 3,5-Dihydroxybenzonitrile.Crossref | GoogleScholarGoogle Scholar |
[61] A Maier, A Golz, HK Lichtenthaler, N Meyer, G Retzlaff, Studies on the Effect of Different Cyclohexane-1,3-diones on de-novo Fatty Acid Biosynthesis in Poaceae. Pestic Sci 1994, 42, 153.
| Studies on the Effect of Different Cyclohexane-1,3-diones on de-novo Fatty Acid Biosynthesis in Poaceae.Crossref | GoogleScholarGoogle Scholar |
[62] AJ Liepa, JS Wilkie, DA Winkler, KN Winzenberg, Preparation of Bicyclic Herbicide Precursors by Intramolecular Stork-Danheiser Kinetic Alkylation Reactions of Methyl 1-(Haloalkyl)-3-methoxy-5-oxocyclohex-3-ene-1-carboxylate Derivatives. Aust J Chem 1992, 45, 759.
| Preparation of Bicyclic Herbicide Precursors by Intramolecular Stork-Danheiser Kinetic Alkylation Reactions of Methyl 1-(Haloalkyl)-3-methoxy-5-oxocyclohex-3-ene-1-carboxylate Derivatives.Crossref | GoogleScholarGoogle Scholar |
[63] JD Burton, JW Gronwald, DA Somers, JA Connelly, BG Gengenbach, DL Wyse, Inhibition of plant acetyl-coenzyme A carboxylase by the herbicides sethoxydim and haloxyfop. Biochem Biophys Res Commun 1987, 148, 1039.
| Inhibition of plant acetyl-coenzyme A carboxylase by the herbicides sethoxydim and haloxyfop.Crossref | GoogleScholarGoogle Scholar |
[64] AR Rendina, JM Felts, Cyclohexanedione Herbicides Are Selective and Potent Inhibitors of Acetyl-CoA Carboxylase from Grasses. Plant Physiol 1988, 86, 983.
| Cyclohexanedione Herbicides Are Selective and Potent Inhibitors of Acetyl-CoA Carboxylase from Grasses.Crossref | GoogleScholarGoogle Scholar |
[65] N Hoffmann, J-P Pete, Acid catalyzed intramolecular photochemical reactions of 3-alkenyloxyphenols. Tetrahedron Lett 1996, 37, 2027.
| Acid catalyzed intramolecular photochemical reactions of 3-alkenyloxyphenols.Crossref | GoogleScholarGoogle Scholar |
[66] N Hoffmann, J-P Pete, Intramolecular Photochemical Reactions of Bichromophoric 3-(Alkenyloxy)phenols and 1-(Alkenyloxy)-3-(alkyloxy)benzene Derivatives. Acid-Catalyzed Transformations of the Primary Cycloadducts. J Org Chem 1997, 62, 6952.
| Intramolecular Photochemical Reactions of Bichromophoric 3-(Alkenyloxy)phenols and 1-(Alkenyloxy)-3-(alkyloxy)benzene Derivatives. Acid-Catalyzed Transformations of the Primary Cycloadducts.Crossref | GoogleScholarGoogle Scholar |
[67] AK Sadana, RK Saini, WE Billups, Cyclobutarenes and Related Compounds. Chem Rev 2003, 103, 1539.
| Cyclobutarenes and Related Compounds.Crossref | GoogleScholarGoogle Scholar |
[68] C Verrat, N Hoffmann, J-P Pete, An Easy Access to Benzo[f]isoquinoline Derivatives Using Benzocyclobutenes Derived from Resorcinol. Synlett 2000, 166.
| An Easy Access to Benzo[f]isoquinoline Derivatives Using Benzocyclobutenes Derived from Resorcinol.Crossref | GoogleScholarGoogle Scholar |
[69] Verrat C. Photocycloadditions [2+2] intramoléculaires d’éthers de polyphénols: accès au squelette de produits naturels hétérocycliques, PhD Thesis, Université de Reims Champagne-Ardenne, Reims, France; 2000.
[70] A Zech, T Bach, Photochemical Reaction Cascade from O-Pent-4-enyl-Substituted Salicylates to Complex Multifunctional Scaffolds. J Org Chem 2018, 83, 3069.
| Photochemical Reaction Cascade from O-Pent-4-enyl-Substituted Salicylates to Complex Multifunctional Scaffolds.Crossref | GoogleScholarGoogle Scholar |
[71] A Gilbert, T Bach, The Awakening of a Sleeping Beauty: The ortho Photocycloaddition in the Total Synthesis of Protoilludane- and Prezizaene-Type-Sesquiterpenes. Synlett 2022,
| The Awakening of a Sleeping Beauty: The ortho Photocycloaddition in the Total Synthesis of Protoilludane- and Prezizaene-Type-Sesquiterpenes.Crossref | GoogleScholarGoogle Scholar |
[72] A Zech, C Jandl, T Bach, Concise Access to the Skeleton of Protoilludane Sesquiterpenes through a Photochemical Reaction Cascade: Total Synthesis of Atlanticone C. Angew Chem Int Ed 2019, 58, 14629.
| Concise Access to the Skeleton of Protoilludane Sesquiterpenes through a Photochemical Reaction Cascade: Total Synthesis of Atlanticone C.Crossref | GoogleScholarGoogle Scholar |
[73] J Proessdorf, A Zech, C Jandl, T Bach, Concise Synthesis of (+)-Atlaticone C. Synlett 2020, 31, 1598.
| Concise Synthesis of (+)-Atlaticone C.Crossref | GoogleScholarGoogle Scholar |
[74] L Næsborg, C Jandl, A Zech, T Bach, Complex Carbocyclic Skeletons from Aryl Ketones through a Three‐Photon Cascade Reaction. Angew Chem Int Ed 2020, 59, 5656.
| Complex Carbocyclic Skeletons from Aryl Ketones through a Three‐Photon Cascade Reaction.Crossref | GoogleScholarGoogle Scholar |
[75] N Rauscher, L Næsborg, C Jandl, T Bach, Concise Total Synthesis of Agarozizanol B via a Strained Photocascade Intermediate. Angew Chem Int Ed 2021, 60, 24039.
| Concise Total Synthesis of Agarozizanol B via a Strained Photocascade Intermediate.Crossref | GoogleScholarGoogle Scholar |
[76] EJ Corey, CJ Helal, Reduction of Carbonyl Compounds with Chiral Oxazaborolidine Catalysts: A New Paradigm for Enantioselective Catalysis and a Powerful New Synthetic Method. Angew Chem Int Ed 1998, 37, 1986.
| Reduction of Carbonyl Compounds with Chiral Oxazaborolidine Catalysts: A New Paradigm for Enantioselective Catalysis and a Powerful New Synthetic Method.Crossref | GoogleScholarGoogle Scholar |
[77] MG Barlow, DE Brown, RN Haszeldine, Photochemical Addition of Ethylene to Pentafluoropyridine: Formation of 1:1- and 2:1-Adducts. J Chem Soc Chem Commun 1977, 669.
| Photochemical Addition of Ethylene to Pentafluoropyridine: Formation of 1:1- and 2:1-Adducts.Crossref | GoogleScholarGoogle Scholar |
[78] D Donati, S Fusi, F Ponticelli, Photocycloaddition on 2-Methyloxazolo[5,4-b]pyridine: a route to the oxazolo[5,4-b]azocine system. Tetrahedron Lett 1996, 37, 5783.
| Photocycloaddition on 2-Methyloxazolo[5,4-b]pyridine: a route to the oxazolo[5,4-b]azocine system.Crossref | GoogleScholarGoogle Scholar |
[79] KI Booker-Milburn, PM Wood, RF Dainty, MW Urquhart, AJ White, HJ Lyon, JPH Charmant, Photochemistry of Benzotriazole: An Unprecedented Tautomer-Selective Intermolecular [2+2] Photocycloaddition. Org Lett 2002, 4, 1487.
| Photochemistry of Benzotriazole: An Unprecedented Tautomer-Selective Intermolecular [2+2] Photocycloaddition.Crossref | GoogleScholarGoogle Scholar |
[80] T Noh, D Kim, Y-J Kim, Photocycloaddition of Cyanonaphthalenes to 1,3-Cyclohexadiene. J Org Chem 1998, 63, 1212.
| Photocycloaddition of Cyanonaphthalenes to 1,3-Cyclohexadiene.Crossref | GoogleScholarGoogle Scholar |
[81] N Hoffmann, J-P Pete, Y Inoue, T Mori, Novel [2 + 2] Photocycloaddition-Induced Rearrangement of Bichromophoric Naphthalene-Tethered Resorcinol Ethers. J Org Chem 2002, 67, 2315.
| Novel [2 + 2] Photocycloaddition-Induced Rearrangement of Bichromophoric Naphthalene-Tethered Resorcinol Ethers.Crossref | GoogleScholarGoogle Scholar |
[82] N Hoffmann, New photochemical rearrangements and extrusion reactions of aromatic compounds induced by an intramolecular [2+2] photocycloaddition between a naphthalene and a resorcinol moiety. Tetrahedron 2002, 58, 7933.
| New photochemical rearrangements and extrusion reactions of aromatic compounds induced by an intramolecular [2+2] photocycloaddition between a naphthalene and a resorcinol moiety.Crossref | GoogleScholarGoogle Scholar |
[83] M Sakamoto, T Yagi, T Mino, K Yamaguchi, T Fujita, A Novel Photochemical Cycloaddition of 1-Cyanonaphthalene to Substituted Pyridines. J Am Chem Soc 2000, 122, 8141.
| A Novel Photochemical Cycloaddition of 1-Cyanonaphthalene to Substituted Pyridines.Crossref | GoogleScholarGoogle Scholar |