

Compounds of amine-substituted cyclic amines: synthesis and structures
Neil F. Curtis A *
A *
A School of Chemical and Physical Sciences, Victoria University of Wellington, Box 600, Wellington 6140, New Zealand.

Neil Curtis was born in Auckland, New Zealand in 1931. He attended Auckland University, graduating with a PhD in Chemistry in 1955. After spending 2 years with (not yet Sir) Ron Nyholm in London, studying rhenium diarsine compounds, he was appointed as a lecturer at the Victoria University of Wellington. During his PhD studies he had been visited by the Princes of Serandip, who persuaded him to recrystallise some nickel(ii) ethanediamine compounds from acetone, which resulted in unexpected products with unusual and intriguing properties. In Wellington he resumed study of these compounds, eventually establishing that they had aza-macrocyclic ligand structures, reported in 1961. This started an extensive flurry of research into aza-macrocyclic ligand coordination chemistry. Much of Neil’s subsequent research has involved extensions of these reactions. During a period of sabbatical leave at MIT with Al Cotton in 1964 he returned to rhenium chemistry and was in the group reporting the rhenium–rhenium quadruple bond. During a later leave period with Alan Sargeson at ANU he developed new methods of synthesis of azamacrocycle ligand compounds, including those described in this Review. Neil has remained at the Victoria University of Wellington for his academic career. He was appointed as Professor of Chemistry in 1975, and after retiring in 1996 as Emeritus Professor of Chemistry. Neil was awarded a Fellowship of the Royal Society of New Zealand in 1975 and served on the Council of the Society in various roles, from 1982 to 1994, including terms as Hon. Treasurer and Vice-President. Neil has 45 publications in the Australian Journal of Chemistry and was included in the Journal’s list of ‘golden oldies of inorganic chemistry’, Aust. J. Chem., 1995, 48, 689. Goodness knows what that makes him now! |
Australian Journal of Chemistry 75(7) 439-466 https://doi.org/10.1071/CH22020
Submitted: 26 January 2022 Accepted: 12 April 2022 Published: 8 August 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
Cyclic tetra-amines with amine substituents on the central carbon atom of C3 ring segments are readily prepared by reduction of nitro-substituted aza-macrocycles, formed by reactions of metal-ion amine compounds with formaldehyde and a nitro-alkane. Reactions of bis-(ethane-1,2-diamine)-copper(ii) or -nickel(ii) cations with formaldehyde and nitroethane form the trans (anti) and cis (syn) isomers of (6,13-dimethyl-6,13-di-nitro-1,4,8,11-tetra-azacyclotetradecane)-copper(ii) or -nickel(ii)) cations, which are readily reduced to form the trans and cis-6,13-diamino cations, from which the trans- and cis-amine substituted cyclic tetra-amines can be isolated. Similar reactions for the (3,7-diaza-nonane-1,9-diamine)-copper(ii) or -nickel(ii) cations lead to 6-methyl-1,4,8,11-tetra-azacyclotetradecane-6-amine. Amine-substituted cyclic tetra-amines with different ring sizes or alkyl substituents can similarly be prepared by using different diamines or alkyl-nitro compounds. This review is primarily about compounds formed by trans-6,13-diamino and 6-amino-1,4,8,11-tetraazacyclotetradecanes with d-transition-metal ions. The amines react with transition-metal ions in much the same manner as 1,4,8,11-tetraazacyclotetradecane (cyclam), forming compounds with isomeric configurations arising from the four chiral nitrogen centres of coordinated cyclam, the cis- or trans-configurations of the amine substituents and a form of geometrical isomerism arising from the relationship between the chiral coordinated cyclam configuration and the orientation of the substituents. The amine-substituted cyclams coordinate by the four cyclam nitrogen atoms, in planar or folded arrangements, and also by one or both of the amine substituents. Non-coordinated amine substituents can be protonated. The amine substituents can be modified by methylation, amide formation and reaction with aldehydes to form imines. The imines formed with functionalised aldehydes can be reduced to form variously functionalised secondary amine substituents. This all leads to complex coordination chemistry and X-ray crystallographic structure determinations have been critical in understanding the configurations present. Structures in the Cambridge Crystallographic Data Base of all relevant compounds are listed.
Keywords: aldehyde, amine, azamacrocycles, coordinated ligands, coordination compounds, crystal structures, cyclic amines, isomerism, reactions of nitroalkanes.
Introduction
Cyclic tetra-amines with C-amine substituents are of interest because of the potentially increased denticity of the macrocyclic ligand and the variety of configurations and coordination modes possible. An amine substituent on the central C-atom of a six-membered chelate ring segment of a coordinated cyclic amine in the usual chair conformation cannot readily coordinate. If this chelate ring flexes to a boat conformation the amine can coordinate with relatively little additional ligand strain, resulting for cyclam-6,13-diamines in possible hexadentate coordination. For usually non-labile metal-ions, such as CoIII or CrIII, the coordinated amine substituents are more labile than the cyclam secondary amine groups and can be displaced by other ligands, forming cations with penta- and tetradentate coordination by the cyclic amine ligand. For metal ions which are usually labile to substitution, such as NiII, CuII or ZnII, the secondary amine groups of cyclic amines are much less labile than those of acyclic amine compounds because of their inclusion within the ring structure, but substituent amine donors remain labile, introducing additional aspects to the coordination chemistry.[1,2]
Amine substituted tetra-aza-macrocycles can be prepared by conventional syntheses, but metal-ion mediated condensation reactions give easier access to a variety of such ligands. These reactions generally include amine–carbonyl condensations involving imine formation steps. The NiII and CuII ions are usually the most effective ‘mediators’ for such reactions, as they form strong, but labile, metal–nitrogen bonds, combined with flexible coordination numbers and geometries, which facilitate such reactions. Reactions of bis-diamine compounds of CuII or NiII with formaldehyde and a nitro alkane, RNO2, form products with acyclic tetradentate ligands, with the diamines linked by a –NH–CH2–C(R,NO2)–CH2–NH– group. The formation of two such links leads to a tetraaza macrocyclic ligand with two nitro substituents. Similar reactions for acyclic tetraamine CuII or NiII cations lead to aza-macrocycles with one nitro-substituent.[3] Reduction of these nitro substituents forms cyclic tetra amine cations with amine substituents (see Scheme 1).
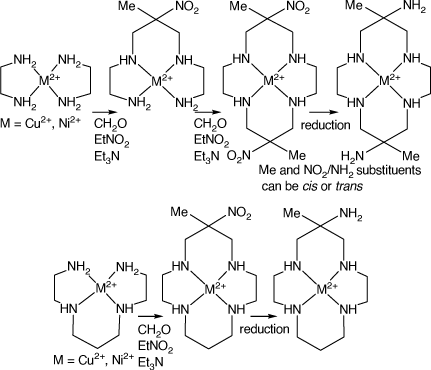
|
trans-6,13-Dimethyl-1,4,8,11-tetraazacyclotetradeca-6,13-diamine (diam) is probably the most-studied amine-substituted cyclic amine, largely because of its ready synthesis via reactions of bis(ethane-1,2-diamine)-CuII or -NiII with formaldehyde and nitroethane. Analogous compounds with larger ring sizes are similarly prepared from longer chain diamines and the alkyl substituent pattern can be changed by using substituted diamines or other nitro alkanes. Similar reactions of linear tetra-amine compounds of NiII or CuII lead to mono-(nitro, alkyl)-substituted macrocyclic tetra-amines, for which reduction of the nitro groups forms mono-(amino, alkyl)-substituted cyclic amines.
Compounds of these amino-substituted cyclic-amines, homologues and analogues, have been prepared with a variety of transition-metal ions, using methods appropriate for the amine chemistry of each metal ion. Compounds of diam have been reported for VIV, CrIII, FeII, FeIII, CoII, CoIII, NiII, CuII, ZnII, RhIII, PdII, PtII, PtIV, CdII, PbII and HgII.
This Review covers the preparations and properties of compounds of amine-substituted cyclic amines formed via nitroalkane-aldehyde condensations of metal-ion amine compounds, and such compounds with the amine substituents chemically modified. Structures of such compounds included in the Cambridge Crystallographic Database are listed and representative figures of the different structural types shown. In many cases compounds of the azamacrocycles discussed were synthesised for purposes specific to a research project, focussing on some aspect, such as electrochemistry, reaction kinetics or spectroscopy, but such aspects are not covered.
Historical background
Research into the preparations and properties of the compounds described in this Review began during a period of sabbatical leave which I spent with Alan Sargeson in the Chemistry Department of the Australian National University in Canberra, August 1980–May 1981.
Alan Sargeson and his group developed the facile synthesis of metal ion clathrochelate compounds by reactions of tris(ethane-1,2-diamine) compounds of MIII metal ions, with formaldehyde and ammonia, which form N(CH2–N–)3 capped clathrochelate ligands, and by their reaction with formaldehyde and nitromethane, which form C(NO2){(CH2–N–)}3 capped clathrochelate ligands[4–8], which could be reduced to form the amine substituted cations (see Scheme 2).
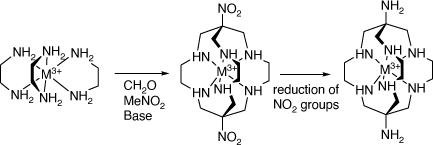
|
I attempted to extend these reactions to diamine compounds of the labile NiII and CuII ions. Facile reactions occurred, but gave complex and, as yet, uncharacterised products. Substituting the di-basic carbon acid nitroethane for tri-basic nitromethane was an obvious strategy change, with the potential of forming macrocyclic products by linking diamines with C(Me,NO2){(CH2–N–)}2 groups.
Reactions of [Ni(en)2]2+ and [Cu(en)2]2+ salts with nitroethane and formaldehyde, in refluxing methanol, with triethylamine as base, occurred rapidly, forming products which crystallised readily, and this encouraged continued studies. Compounds with the macrocyclic ligand with the two diamine residues doubly linked, [M(dino)]2+ (dino = 6,13-dimethyl-6,13-dinitro-1,4,8,11-tetraazacyclotetrdecane), were isolated (hereafter ‘1,4,8,11-tetraazacyclotetradecane’ will be represented in formulae by ‘cyclam’).
Bis(nitro,methyl) compounds of 16- and 18-ring membered macrocycles were similarly formed from propane-1,3-diamine and butane-1,4-diamine compounds, respectively, though yields were lower, with extensive polymer formation. Homologues were formed from C-methyl-substituted diamines or with other nitro alkanes. Similar reactions for CuII and NiII compounds of linear tetra-amines led to compounds of mono-(nitro,methyl)-substituted macrocyclic compounds. Copper(ii) is usually the most successful ‘mediating’ metal ion, although the reactions also occur for nickel(ii), palladium(ii), platinum(ii) and gold(iii) diamine compounds, with PdII most effective for the larger ring compounds.[3]
At this stage, I returned to the Victoria University of Wellington in New Zealand. Studies of these reactions continued in Canberra and Wellington. Geoffrey Lawrance moved to the University of Newcastle and continued the studies with PhD student Paul Bernhardt, who later moved to the University of Queensland, where he established a fourth group studying these compounds.
Synthesis of 6,13-dimethyl-6,13-dinitro-cyclam (dino) metal-ion cations
The dino macrocycle occurs as geometric isomers, with the two nitro-substituents oriented to opposite sides of the cyclam ring, anti- or trans-dino, or with them oriented to the same side, syn- or cis-dino. The reaction of [Cu(en)2]–(NO3)2 or –(ClO4) with nitroethane and paraformaldehyde (or aqueous formaldehyde added drop-wise) in refluxing methanol, made basic with triethylamine, forms acyclic [Cu(nelin)]2+ and macrocyclic [Cu(trans-dino)]2+ and [Cu(cis-dino)]2+ salts which co-crystallised. The nelin and trans/cis-dino salts can be separated by crystallisation or by chromatography. Relative yields of the acyclic and macrocyclic products depend on reaction conditions, which can be optimised to produce predominantly one or the other – with yields of up to 70% for [Cu(trans/cis-dino)](NO3)2. Chromatography of the [Cu(trans/cis-dino)]2+ ion showed two closely running bands which could not effectively be separated, indicating the presence, later confirmed, of both [Cu(trans-dino)]2+ and [Cu(cis-dino)]2+ isomers.[9]
Reaction of [Cu(en)2]2+ (or [Cu(pn)2]2+, [Cu(propane-1,3-diamine)2]2+ or [Cu(trans-cyclohexane-1,2-diamine)2]2+ with formaldehyde and a nitroalkane in the ionic liquid 1-butyl-3-methylimidazolium tetrafluoroborate form the same nitro-substituted cyclic tetraamines in similar yields to reaction in the usual alcohol solvents, with less acyclic by-products. Yields were higher for the 16-membered ring compound formed with propane-1,3-diamine.[10]
Similar reaction of [Ni(en)2]2+ salts, (usually [{Ni(en)2}2-µCl2]Cl2, ‘Ni(en)2Cl2’), with nitroethane, formaldehyde (or paraformaldehyde) and base in refluxing methanol, formed yellow singlet ground state (sgs) [Ni(dino)]Cl2 which crystallised in ~90% yield, with no indications of the formation of significant amounts of acyclic ligand compounds.[11,12]
Reduction of 6,13-dimethyl-6,13-dinitro-cyclam compounds
The possible reduction of the nitro groups to form amine-substituted cyclic tetra-amine compounds was an obvious extension of these studies. Reduction of the nitro-groups of the [M(trans-dino)]2+ or [M(cis-dino)]2+ cations form the trans- or cis-isomers of [M(6,13-dimethyl-cyclam-6,13-diamine)]2+ cations, labelled here as diam and cis-diam, respectively, elsewhere as diammac and cis-diammac.
Reduction of the nitro substituents of (6,13-dimethyl-6,13-dinitro-cyclam)-copper(ii) cations
The group in Canberra, led by Geoffrey Lawrance, focussing on the CuII compounds, found that the nitro groups of the mixture of products formed by reaction of [Cu(en)2](NO3)2 with nitroethane and formaldehyde were reduced by Zn, Zn/Hg or Sn in acid (usually by Zn/HCl) to form transient CuII amine cations, which demetalated to deposit Cu metal and leave protonated amines in solution. Addition of CuII nitrate and base to this resulting solution reformed the [Cu(amine)]2+ cations. Chromatography, using a Sephadex C25 column, eluting with NaCl solution, resulted in the isolation of five bands, the last two close-running. The first band contained unreacted starting material or readily hydrolysed compound(s). The second band gave crystals of aqua-(1,3-bis(2-aminoethyl)-5-ammonio-5-methyl-1,3-diazacyclohexane)copper(ii) perchlorate, labelled as [Cu(badcH)(OH2)](ClO4)3·3H2O, FANFIJ, and the third gave crystals of (5-ammonio-5-methyl-3,7-diazanonane-1,9-diamine)copper(ii) perchlorate hydrate, FASWAX, both derived from reduction of [Cu(nelin)]2+.
The fourth band gave crystals of trans-[Cu(H2diam-IIIaa-N4)(OClO3)2](ClO4)2·6H2O (labels defined below), FANFEF, in ~31% yield. Crystals isolated from the fifth band in ~15% yield were assigned as [Cu(cis-diam)](ClO4)2·2H2O, although isolation of the pure cis-isomer proved to be difficult[9] (see Scheme 3).
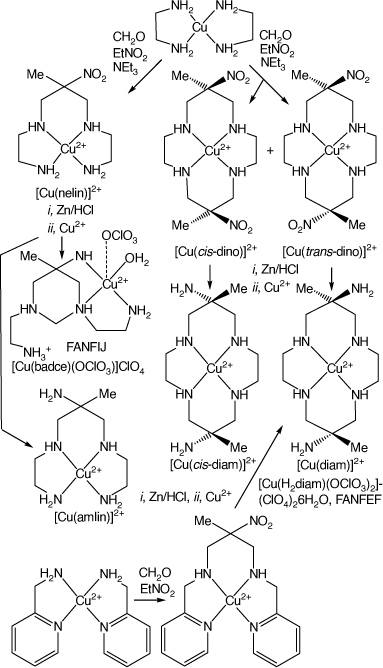
|
The [Cu(diam)]2+ ion was also formed by an unanticipated reaction when [Cu(2-methyl-2-nitro-N,N′-bis(pyridine-2-yl-methyl)propane-1,3-diamine)]2+ was reduced with Zn/HCl, the resulting amine reacted with Cu2+ and the product isolated as trans-[Cu(diam-IIIbb-N4)(OClO3)]ClO4, DUMGIB[13] (see Scheme 3).
Reduction of the nitro substituents of (6,13-dimethyl-6,13-dinitro-cyclam)nickel(ii) cations
Meanwhile, in Wellington, Asoskamali Siriwardena[11] and I focussed on the reduction of the [Ni(trans/cis-dino)]2+ cation.
The nitro groups of [Ni(trans/cis-dino)]Cl2 are not reduced by Zn/HCl and addition of NaBH4 to a solution causes precipitation of unreduced hydroborate salts. They are reduced by hydrogenation at ambient temperatures in water, using Raney nickel catalyst, typically taking ~8 h at 800 psi, or several days at 50 psi hydrogen pressure.[11,12]
Evaporation of the reduction solution resulted in a purple gum which was extracted with hot ethanol, giving a purple solution, and leaving residual blue-violet solid. Evaporation of the ethanol solution led to crystallisation of [Ni#(diam-III-N6)]Cl2·6H2O (the label Ni# indicating triplet ground state, tgs, NiII). Acidification of the ethanol extract with HCl caused crystallisation of trans-[Ni#(H2diam-III-N4)Cl(OH2)]Cl3·3H2O, KELVUT.[14] The residual solid crystallised from ice-water/propan-2-ol as trans-[Ni#(diam-I-N5)(OH2)]Cl2·0.5H2O, which recrystallised from dilute perchloric acid as grey–green trans-[Ni#(Hdiam-I-N5)Cl](ClO4)2·H2O, PEHXEF.[12]
Hydrogenation in water formed the [Ni(diam-I)]2+ and [Ni(diam-III)]2+ products in variable but approximately equal proportions. The proportion of the [Ni(diam-I)]2+ isomer formed decreased for reduction in acidic (1 mol dm−3 HCl) solution, or in aqueous ammonia solution, or with higher hydrogen pressures.[11]
Evaporation of the final filtrates from the preparation left a gum, from which a hygroscopic compound of composition [Ni(H2diam)]Cl4·xH2O was isolated by addition of propan-2-ol.[11] With hindsight this was probably the cis-diam isomer, formed by reduction of [Ni(cis-dino)]2+ (see discussion in Compounds of Nickel(ii) with cis-diam, below).
Isolation of trans- and cis-6,13-dimethyl-cyclam-6,13-diamine
Isolation from CuII compounds
Modifications of the procedure described in Reduction of the nitro substituents of (6,13-dimethyl-6,13-dinitro-cyclam)copper(ii) cations were used to isolate trans- and cis-diam. The mixed [Cu(trans/cis-dino)]2+ salt was reduced with Zn/HCl and the resulting solution of [H6(trans-diam]6+ and [H6(cis-diam]6+ reacted with Cu2+ and base. The resulting [Cu(trans-diam)]2+ and [Cu(cis-diam)]2+ ions were separated by chromatography into four bands, each with distinctive CuII d–d spectra. Each isolated band was reduced with Zn/HCl and the resulting amines isolated by chromatography, in overall quantitative yield. The amine isolated from bands 1, 2 and 4 was identified by 1H NMR spectra as diam and that from band 3 as cis-diam. The isomers were isolated as the salts [H6diam)]Cl6·2H2O and [H6-cis-diam)]Cl6·2.5H2O in ~3:1 proportions.[15,16]
Isolation from NiII diam compounds
The diam was released from trans-[Ni#(H2diam-III)(OH2)2]Cl4 by heating with conc. HCl in a sealed tube at 110°C, until the purple colour of the NiII compound completely faded, taking ~2 days. The [H6diam]Cl6·2H2O was filtered off and washed with conc. HCl/ethanol until free of Ni2+. Alternatively, the diam was released by reaction with 4 molar proportions of KCN in water at 100°C taking ~8 h for the purple colour of the [Ni#(diam)]2+ cation to completely fade. The K2[Ni(CN)4] was filtered off from the cold solution, the filtrate evaporated to dryness and the amine extracted from the residue with CH3Cl or CH2Cl2.[12]
Compounds of non-coordinated diam and cis-diam
The free amines were prepared by reacting the hexa-hydrochloride salts with NaOH in ethanol, filtering off the precipitated NaCl, evaporating the filtrate to dryness, extracting the amine from the residue with CH2Cl2 and isolating it as diam·3.5H2O[12] or cis-diam·2H2O.[15,16]
The amines were also prepared from the hydrochloride salts by using an anion exchange column, with the amine solution used directly, or evaporated and the amine extracted from the residue with CH2Cl2.
Hexa-, tetra- or di-protonated salts of diam crystallise from aqueous solution, depending on the acidity and anions present. See Table 1 for reported structures.[17–21]

|
Analytical results for samples of the amines and salts have indicated variable hydration numbers, which probably depend on crystallisation conditions or subsequent desiccation. Hydration numbers for the amines and salts will here generally be omitted in descriptions of preparations.
The protonation constants, log KH, of diam, measured by pH titration of [H6(diam)]6+ in 0.1 mol dm−3 NaClO4 at 25°C are: 11.2(1), 9.7(1), 6.2(1) and 5.3(1), with the remaining two constants too small to measure in water;[12] other reported values are: 11.0, 9.9, 6.3, 5.5[22] and 11.0, 10.0, 6.2, 5.5, ~1.5.[40] Values for cis-diam are: 10.6, 9.7, 6.3, 5.4, ~2.7.[40]
Coordination of diam and cis-diam
The four secondary amine groups of coordinated cyclam are chiral centres, which can occur in five configurations I–V,[53] see Scheme 4, and the same configurations occur for coordinated diam and cis-diam, with the 6,13-(methyl, amine) substituents giving rise to additional isomeric possibilities.
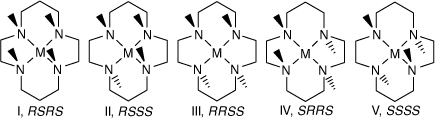
|
Diam (and cis-diam) can coordinate to metal ions by the four secondary-amine cyclam N-atoms, labelled here as diam-N4, or also by one (diam-N5) or both (diam-N6) primary-amine substituents. Uncoordinated amine substituents can be free or protonated, labelled as Hdiam+ or H2diam2+.
Isomerism of coordinated diam compounds arising from orientation of substituents
Distinct configurations are possible for coordinated diam, arising from the orientation of the substituents.[49] For a six-membered chelate ring of diam in the usual chair conformation, the amine substituent can be equatorially oriented (configuration a) or axially oriented (configuration b); neither is positioned suitably for coordination. If the chelate ring flexes into a boat conformation, a previously equatorially oriented amine substituent (a) becomes axially oriented and able to coordinate with minor distortion of the macrocycle conformation, while a previously axially oriented (b) amine becomes equatorially oriented and still unable to coordinate (see Scheme 5).
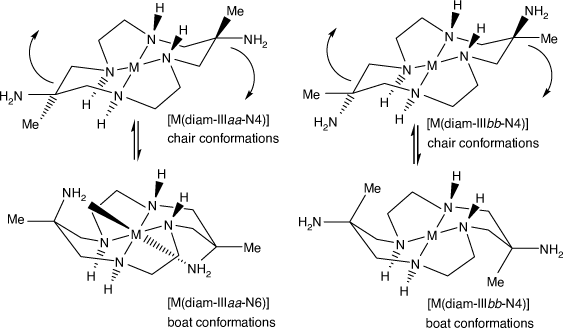
|
For diam coordinated in configuration IIIaa, both amine substituents coordinate for configuration diam-IIIaa-N6, one for diam-IIIaa-N5 or neither for diam-IIIaa-N4 – or variants with uncoordinated substituent amine groups protonated.
For configuration diam-IIIbb, neither substituent can coordinate, so only diam-IIIbb-N4 is possible. Since most diam-III compounds have the IIIaa configuration, the aa configuration will usually be assumed and the aa label omitted.
For cations with configurations diam-Iaa or diam Iab, one substituent amine can coordinate on the side of the N4 plane opposite to the four NH groups and for configuration diam-Ibb only diam-Ibb-N4 is possible. Since all reported structures have configuration Iab, the ab label will be assumed.
For cis-diam, all reported structures, other than two with CuII, have configuration cis-diam-Vaa-N6, so the aa label will be assumed.
The cis/trans geometrical configuration of diam is fixed during the [M(dino)]2+ synthesis. The cyclam chiral and a/b geometrical configurations of coordinated diam and cis-diam are determined during coordination. Interconversion of cyclam configurations requires inversion at cyclam NH centres, which can occur in basic solution, but is usually inhibited in acid solution. Interconversion of substituent aa/bb configurations requires inversion at all four cyclam NH centres and has not been reported. The IIIbb configuration for diam compounds has been observed only for CuII, PdII and PtII, but occurs frequently for compounds of diam with modified amine substituents. The IIIab configuration has been observed for compounds with modified amino substituents (see Compounds of diam, monam (and homologues) with modified amine substituents, below).
Metal-ion compounds of trans- and cis-6,13-dimethyl-cyclam-6,13-diamine, diam and cis-diam
Compounds are discussed in the order of metal ions: Zn–V, Cd–Mo, Pb–Hg.
Formation constants (log K) at 298 K in 0.05 mol dm−3 KCl or KNO3 for diam, cis-diam have been reported for ZnII; 15.0, 16.1, CdII; 10.6, 12.1 and HgII; 10.5, 12.2. PbII: 10.8, 11.8, with values for cis-diam consistently the higher.[22,40]
X-Ray crystallographic studies have been essential to disentangling the configurational isomerism of coordinated diam. Table 1 lists compounds of diam, cis-diam and homologues, with structures in the Cambridge Crystallographic Centre Data base. Distinguishing between Hdiam-IIIaa-N4 and Hdiam-IIIab-N4 or Hdiam-Iaa-N4 and Hdiam-Iab-N4 structures depends on assignment of CH3 and NH3+ groups, which may be difficult for disordered structures or for poor diffraction data sets.
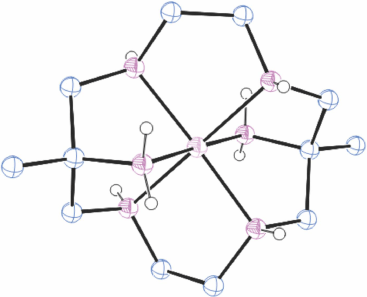
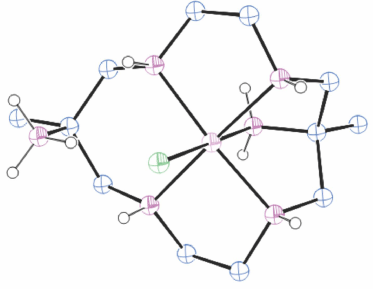
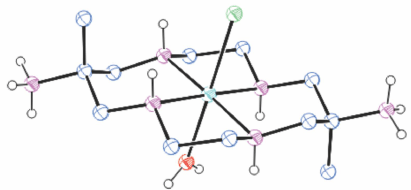
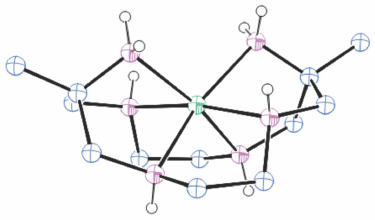
Structures of cations of compounds of different metal ions with the same coordination modes are generally similar, and protonation of uncoordinated amine substituents usually makes little difference to the overall structure of the cation. Figs 1–4 show representative examples of NiII cations in each of the most common coordination modes: [Ni#(diam-III-N6]2+, PEHWOO (Fig. 1), trans-[Ni#(Hdiam-I-N5)Cl]2+, PEHXEF (Fig. 2), trans-[Ni#(H2diam-III-N4)Cl(OH2)]Cl3·3H2O, KELVUT (Fig. 3) and cis-[Ni#(cis-diam-V-N6)]2+, MASRIH (Fig. 4). Figures shown for other cations display distinctive strutural features.
|
Compounds of zinc(ii) with diam and cis-diam
[Zn(OH2)6[(ClO4)2 reacts rapidly in water with diam, or a diam salt plus base, to form [Zn(diam-III-N6)]2+, which was isolated as [Zn(diam-III-N6)](ClO4)2, VISSUL.[22] [Zn(diam-III-N4)(OS2O5)2], KENPUO[19] crystallised from a concentrated aqueous solution of dithionate.
[Zn(diam-III-N4)(OH2)2]ZnCl4 crystallised when HCl and propan-2-ol were added to a solution of diam and ZnCl2 in 1:2 mole proportions. Mixing hot aqueous solutions of ZnCl2 and excess diam caused immediate precipitation of a compound of composition Zn3·diam·Cl5·OH, which recrystallised unchanged from dilute HCl.[11]
Titration of [Zn(diam)]2+ with acid enabled calculation of the protonation constants and distribution of protonated species as a function of pH (measurements in 0.5 mol dm−3 KCl at 298 K). The [Zn(diam)]2+ ion is slowly demetalated in dilute acid solution at ambient temperatures.[22]
The compound cis-[Zn(cis-diam-V-N6](ClO4)2, MASRON[50] (isomorphous with the NiII analogue, MASRIH) was prepared by reaction of cis-diam with Zn2+.
Compounds of copper(ii)
The formation kinetics of the reaction of diam with Cu2+ in water, between pH 0 and 5.5[54] and in strongly alkaline solution,[55] have been reported.
The acid dissociation rates for [Cu(H2diam-III-N4)]2+ were measured spectrophotometrically in HClO4/NaClO4, ionic strength 2 mol dm−3. The first order rate constants observed in 0.2, 0.6, 1.0 and 2.0 mol dm−3 HClO4 solutions were 1.3(5), 1.3(3), 1.3(3) and 1.2(6) × 10−9 s−1 at 50°C, respectively, and 7.3, 8.0, 7.2 and 7.5 × 10−9 s−1, respectively, at 70°C. The rate constant in HCl/NaCl (2 mol dm−3) at 70°C was 6.5 × 10−9 s−1. The independence of the rate on the H+ concentration indicates that the rate determining step is intramolecular for [Cu(H2diam)]2+.[11]
[H6diam]Cl6·2H2O in basic solution reacts rapidly with Cu2+ to form three isomeric [Cu(diam)]2+ ions which were isolated by chromatography in approximately equal proportions, as trans-[Cu(diam-IIIbb-N4)(OH2)2](ClO4)2, ROMWAR, Fig. 5 (which also crystallised as trans-[Cu(diam-IIIbb-N4)](ClO4)2, DUMGIB), [Cu(H2diam-IIIaa-N4)(OClO3)2](ClO4)2·2H2O, ROMWEV (which also crystallised as trans-[Cu(H2diam-IIIaa-N4)(OClO3)2](ClO4)2·6H2O, FANFEF), similar to KELVUT, Fig. 3, and trans-[Cu(diam-I-N5)(OClO3)]ClO4, ROMWIZ, Fig. 6.[49]
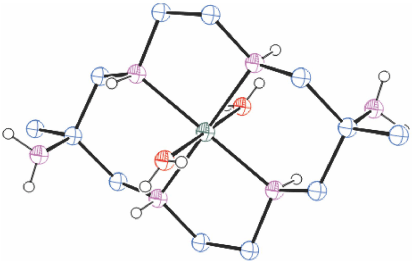
|
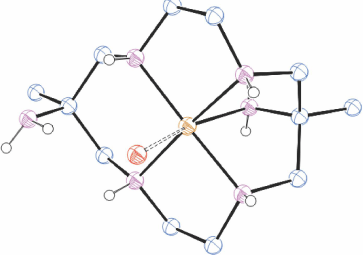
|
Other CuII compounds of diam prepared, but not structurally characterised, include [Cu(H2diam)]−Cl4·3H2O, −(ClO4)4, [Cu(diam)]−Cl2·4H2O, −(ClO4)2·2.5H2O and –(ClO4)2·4H2O.[11]
Reaction of Cu2+ with [cis-H6diam]Cl6·2.5H2O formed two cations, which were similarly isolated by chromatography in approximately equal proportions, as trans-[Cu(cis-diam-I-N5)(OClO3)]ClO4, ROMWOF, Fig. 7 and trans-[Cu(cis-H2diam-III-N4)(OClO3)2](ClO4)2, ROMWUL (with NH3+/CH3 disorder).[49]
All reported [Cu(diam)] structures have the four cyclam nitrogen atoms in planar coordination, crystallising from basic solution with the amine substituents unprotonated, or from acid solution with the substituents protonated. Reported structures usually have perchlorate ions or water molecules weakly coordinated in the tetragonal sites. The two CuII cis-diam compounds are the only structurally characterised cis-diam compounds with other than the cis-diam-Vaa-N6 configuration and [Cu(cis-diam-Iab-N5)(OClO3)]ClO4 is the only reported compound of cis-diam with N5 coordination (although the bond to the amine substituent is long at 2.323(4) Å).
Compounds of nickel(ii) with diam
Compounds of [N(diam-III)]2+
Reaction of a NiII salt with diam, or with a diam salt and base, rapidly forms [Ni#(diam-III-N6)]2+ and lilac coloured [Ni#(diam-III-N6)]2+ salts crystallise from basic solution. [Ni#(diam-III-N6)](ClO4)2·H2O, FEBZUH,[27] FEBZUH10[12], Ni#(diam-III-N6)]ZnCl4·1.5H2O, PEHWUU,[12] [Ni#(diam-III-N6)]Cl2·6H2O, PEHWOO[12] and [Ni#(diam-III-N6)]·Ni(CN)4[11] were prepared.
The protonation constants, pKa, for [Ni#(diam-III-N6)]2+ in 2 mol dm−3 NaClO4 at 25°C are 4.5(1), 4.0(1), so the amine substituents are protonated in acid solution.[12] In weakly acidic solution yellow sgs [Ni(H2diam-III-N4)]2+ and blue tgs trans-[Ni#(H2diam-III-N4)(OH)2]4+ cations exist in equilibrium, with the sgs form increasingly predominating with higher concentrations of electrolytes, particularly perchlorate or iodide. Sgs [Ni(H2diam-III-N4)](ClO4)4, PEHWII[12] and tgs trans-[Ni#(H2diam-III-N4)Cl(OH2)]Cl3·2H2O, KELVUT,[14] trans-[Ni#(H2diam-III-N4)(OH2)2]SO4·5H2O (and 2H2O) were prepared.[11] The amine substituents of [Ni#(diam-III-N6)]2+ are displaced by thiocyanate, with trans-[Ni#(diam-III-N4)(NCS)2], PEHXAB,[12] crystallising from neutral and trans-[Ni#(H2diam-III-N4)(NCS)2](NCS)2·0.5H2O from acid solution, and by cyanide, but not by azide, or amine ligands. A compound of unknown structure crystallised with oxalate and no reaction occurred with pentane-2,4-dionate.[12]
Compounds of [N(diam-I)]2+
The protonation constants, pKa, for trans-[Ni#(diam-I-N5)(OH2)]2+ in 2 mol dm−3 NaClO4 at 25°C are 4.94(2), 2.4(5), so cations are protonated in weakly acid solution.[12] Compounds prepared in neutral solutions were: trans-[Ni#(diam-I-N5)(OH2)]Cl2·0.5H2O, trans-[Ni#(diam-I-N5)(OH2)]Cl·ClO4, trans-[Ni#(diam-I-N5)(OH2)]ZnCl4·H2O, trans-[Ni#(diam-I-N5)(ONO)]ClO4·0.5H2O and trans-[Ni#(diam-I-N5)(CN)]Cl·0.5H2O. From weakly acidic solution trans-[Ni#(Hdiam-I-N5)Cl](ClO4)2·H2O, PEHXEF and from a strongly acidic solution [Ni#(H2diam-I-N4)](ClO4)4·2.5H2O crystallised. Most compounds crystallised poorly.[11,12]
In aqueous solution the [Ni(diam-I)]2+ cation slowly isomerises to [Ni(diam-III)]2+. The rate constant for isomerisation of trans-[Ni#(diam-I-N5)(OH2)]Cl2·0.5H2O, in 2 mol dm−3 aqueous NaCl at 25°C, at the self-established pH of 2.3, is 5.0(5) × 10−5 s−1, this rate increasing in both more acidic and more basic solution.[12]
The [Ni(diam-I)]2+ cation is also slowly demetalated in acid solution, the combined rate constant for loss of [Ni(H2diam-I)]4+ at 50°C, in 2 mol dm−3 NaCl, by isomerisation and demetalation is ~2.4 × 10−4·[H+] s−1.[12]
Compounds of nickel(ii) with cis-diam
Reaction of [Ni(OH2)6]Cl2 with [cis-H6diam)]Cl6 in basic solution forms the purple cis-octahedral cation, isolated as cis-[Ni#(cis-diam-V-N6)](ClO4)2, MASRIH (Fig. 6).[50]
Hygroscopic material of composition [Ni(H2diam)]Cl4·xH2O, isolated under acidic conditions from the residues from the hydrogenation of [Ni(dino)],[11] was probably a cis-diam compound, but as no structures have been reported it will be labelled as ‘diam?’.
Orange/yellow coloured (sgs) compounds of composition [Ni(H2diam?-N4)](ClO4)4, [Ni(H2diam?-N4)](ZnCl4)2 and brown [Ni#(H2diam?-N4)(NCS)2](NCS)2·3H2O (µef = 3.14 B.M. at 25°C) crystallised from solutions in propan-2-ol of the residual gum from the reduction of ‘[Ni(dino)]Cl2’ when HClO4, ZnCl2 or NaNCS were added. Recrystallisation of the thiocyanato compound from aqueous ammonia gave lustrous brown crystals of [Ni#(diam?-N4)(NCS)2]·0.5H2O.[11]
Compounds of cobalt(iii) with diam and cis-diam
[Co(diam-III-N6)]3+ compounds
Reaction of equimolar diam and Na3[Co(CO3)3] in solution rapidly forms yellow [Co(diam-III-N6)]3+ salts:[56] [Co(diam-III-N6)]Cl2·ClO4, FEBZOB,[27] FEBZOB10,[24] [Co(diam-III-N6)](ClO4)3, NOZHAL,[25] [Co(diam-III-N6)]Cl3·2H2O, RUPFOX,[26] [Co(diam-III-N6)]Cl3·3H2O, [Co(diam-III-N6)]Cl2·ClO4 and [Co(diam-III-N6)]Br2·ClO4. The mixed anion salts are much less soluble than the single anion salts.[56] Evaporation of a solution formed by reaction of [H6(diam)]Cl6 with Na3[Co(CO3)3]·3H2O, without acidification, gave yellow crystals of composition [Co(diam-III-N6)]8·(CO3)5·Cl14·56H2O, which also crystallised from an equimolar aqueous solution of [Co(diam-III-N6)]Cl3·3H2O and Na2CO3.[56]
When equimolar diam and [Co(OH2)6]Cl2 in dilute HCl solution[24] (or a solution of [Co(diam-III-N6)]3+ reduced with Zn,Hg/HCl[56]) was exposed to the air, green trans-[Co(H2diam-III-N4)]Cl2]3+, red trans-[Co(Hdiam-III-N5)]Cl]3+ and yellow [Co(diam-III-N6)]3+ formed, in proportions dependent on the acidity. The highest proportion of trans-[Co(Hdiam-III-N5)]Cl]3+ was formed at ~1 mol dm−3 HCl, with increasing amounts of the N6 cation formed as the HCl concentration decreased.[56] The trans-[Co(H2diam-III-N4)]Cl2]3+ and trans-[Co(Hdiam-III-N5)]Cl]3+ ions were separated by chromatography[24] or by crystallisation.[56] On evaporation of the solution, trans-[Co(H2diam-III-N4)]Cl2]Cl3·3H2O crystallised. On addition of perchlorate, trans-[Co(H2diam-III-N4)]Cl2](ClO4)3·3H2O, or on addition of ZnCl2 and ethanol, trans-[Co(H2diam-III-N4)Cl2)]ZnCl4·Cl·0.5H2O·C2H5OH, KUWBIO).[28] crystallised. Evaporation of the red filtrate, after crystallisation of green trans-[Co(H2diam-III-N4)]Cl2]Cl3·3H2O, gave red crystals of trans-[Co(Hdiam-III-N5)Cl]Cl3·3H2O (or, on addition of ZnCl2 and propan-2-ol, of trans-[Co(Hdiam-III-N5)Cl)]ZnCl4·Cl·0.5H2O·0.5C3H7OH, KUWBEK).[28] Substituting HBr for the HCl formed trans-[Co(H2diam-III-N4)Br2]Br3·H2O.[56]
When a solution of trans-[Co(H2diam-III-N4)Cl2]3+ in dilute HClO4 was heated, trans-[Co(H2diam-III-N4)(OH2)Cl)]4+ and trans-[Co(H2diam-III-N4)(OH2)2)]5+ slowly formed sequentially. When a solution of trans-[Co(H2diam-III-N4)Cl2]3+ in dilute HClO4 slowly evaporated, trans-[Co(diam-III-N4)(OH2)2)](ClO4)3·H2O crystallised.[56]
Heating trans-[Co(H2diam-III-N4)Cl2]Cl3 in water with NaNCS or Na(MeCO2) forms violet-brown trans-[Co(H2diam-III-N4)(NCS)2](NCS)3·2.5H2O or purple trans-[Co(H2diam-III-N4)(OCOMe)2]Cl3·5H2O, respectively.[56] Reaction with CN− forms yellow trans-[Co(diam-III-N4)(CN2)]ClO4·2H2O[25] (better prepared by reaction of trans-[Co(diam-I-N5)(OH)]2+ (below) with CN−[91]). Preparations of trans-[Co(diam-III-N4)(CN)2]·trans-[Co(H2diam-III-N4)(CN)2](ClO4)4·4H2O, FOMCIU and Na·trans-[Co(diam-III-N5)CN)](ClO4)3, FOMCEQ have been reported.[29]
[Co(diam-I)]3+ compounds
Dark brown peroxo compounds form when an equimolar solution of a Co2+ salt and diam in water or methanol is exposed to the air at ambient temperatures. When [Co(OH2)6](ClO4)2 was used, trans-[{Co(diam-I-N5)}2(µO2)](ClO4)4·H2O, KUWZEH,[28] crystallised. Reaction of diam with excess [Co(OH2)6]Cl2 (or -Br2) in air gave red-brown crystals formulated as trans-[{Co(diam-I-N5)]2(µO2)]CoCl4·2Cl·10H2O (or –CoBr4·2Br·8H2O), respectively.[56]
In basic aqueous solution trans-[{Co(diam-I-N5)}2(µO2)](ClO4)4 slowly forms [Co(diam-III-N6)]3+.
When an aqueous solution of trans-[{Co(diam-I-N5)}2(µO2)](ClO4)4 was heated, the colour changed to orange and orange trans-[Co(diam-I-N5)(OH)]ClO4)2·H2O crystallised. This compound was also formed by a rapid reaction of [Co(diam-III-N6)](ClO4)3 in 2 mol dm−3 NaOH solution. Reaction of trans-[Co(diam-I-N5)(OH)](ClO4)2·H2O with cold aqueous HCl formed orange coloured trans-[Co(Hdiam-I-N5)(OH2)]Cl3·ClO4, KUWZUX,[28] and with ZnCl2 formed red [Co(Hdiam-I-N5)Cl]ZnCl4·H2O, KUWZOR[28] while heating with conc. HCl formed red trans-[Co(Hdiam-II-N5)lCl3·3H2O, KUXBAG[28] (Fig. 8), with the uncommon cyclam configuration II. The configuration observed would be formed by inversion of one NH centre of diam-Iab, inversion of the second NH centre would form diam-IIIaa. It is probable that this is a metastable transient between configurations I and III, trapped by the strongly acidic solution.
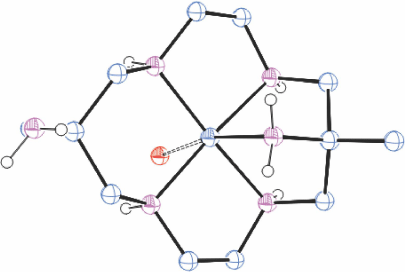
|
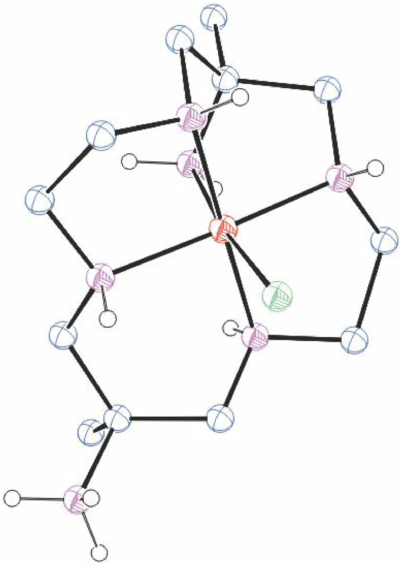
A compound of cobalt(iii) with cis-diam
The pH of an aqueous solution of cis-diam was adjusted to 7, [Co(OH2)6]Cl2 and charcoal were added and the solution aerated. The cis-[Co(cis-diam-V-N6)](ClO4)3·2H2O product, RUPFOX[26] was isolated by chromatography in ~30% yield.
Compounds of chromium(iii) with diam and cis-diam
Heating CrCl3 with diam in DMSO formed [Cr(diam-III-N6]Cl3·3H2O, which recrystallised from hot dilute HCl as trans-[Cr(Hdiam-III-N5)(OH2)]Cl4·3H2O. The amine substituent re-coordinated in basic solution.[11,30]
Chromatography of the solution formed by reaction of CrCl3 with [H6diam]Cl6/6NEt3 in anhydrous, de-oxygenated EtOH, using Sephadex-SP-C25 and eluting with NaClO4 solution, gave two close-running bands. The first yielded trans-[Cr(diam-III-N5)(OH)](ClO4)2·1.5H2O and the second cis-[Cr(diam-III-N5)(OH)](ClO4)2, with the configurations assigned after extensive analysis of the electronic and ESR spectra.[30] A third, slower running band yielded [Cr(diam-III-N6](ClO4)3, JEVCAO.[30]
Similar preparations using [H6cis-diam]Cl6 formed products isolated as cis-[Cr(cis-diam-V-N6)](ClO4)3·2H2O, HAHLOR and cis-[Cr(cis-diam-V-N6](ZnCl4)1.5·3H2O.[16]
Compounds of iron(ii) and iron(iii) with diam
[H6diam]Cl6 was reacted with [Fe(OH2)6]SO4·H2O for 1 day in de-oxygenated water at ambient temperature. Low-spin [Fe(diam-III-N6](ClO4)3, SANLAU[31] and [Fe(diam-III-N6](ClO4)2·Cl, SIZCEJ.[32] were isolated by chromatography.
Reaction of diam with [Fe(OH2)6]SO4·H2O in water under rigorously anaerobic conditions enabled isolation of blue, low-spin [Fe(diam-III-N6](PF6)2, PAZXAP.[33]
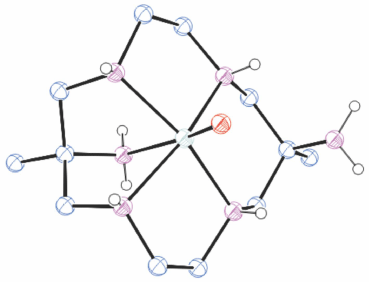
A vanadyl compound of diam, [VIVO]2+
Reaction of [V(acac)3] with [H6diam]Cl6/6Et3N in MeOH for 2 days formed a dark yellow solution. A purple coloured band was isolated by chromatography. When evaporated this changed to green and green crystals of trans-[VO(diam-I-N5)](ClO4)2, TAFPIZ[34] (Fig. 9) were isolated.
|
Compounds of cadmium(ii) with diam and cis-diam
Attempts to isolate salts of [Cd(diam-III-N6)]2+ from solutions containing diam and Cd2+ were unsuccessful.[15] When a solution of equimolar diam and CdCl2 in water was heated, fine white crystals of composition Cd·(diam)·Cl2·2H2O formed as the solution cooled.[11]
The compound cis-[Cd(cis-diam-V-N6)](ClO4)2, VOPLER,[15] was prepared as for the ZnII analogue.
Compounds of palladium(ii), platinum(ii) and platinum(iv) with diam
Compounds of PdII and PtII with diam were prepared by heating [PdCl4]2− or [PtCl4]2− salts with diam (or [H6diam]Cl6/6NEt3) in water. Isomeric PdII products were isolated as perchlorate salts by chromatography as [Pd(diam-IIIaa-N4)](ClO4)2·6H2O, TACVOI[37] and [Pd(diam-IIIbb-N4)](ClO4)2, NESXIU.[36] Compounds with the amine substituents protonated crystallised from acid solution; [Pt(H2diam-IIIbb-N4)](ClO4)4·6H2O, TACVUO.[37] NESXIU and TACVUO have the uncommon diam-IIIbb-N4 configuration.
Oxidation of the PtII compound with Cl2 formed trans-[Pt(H2diam-IIIaa-N4)Cl2](ClO4)2·Cl2·4H2O, VOZZAL and trans-[Pt(H2diam(NCl2)-IIIaa-N4)Cl2](ClO4)2 (diam(NCl2) = 6,13-dimethyl-cyclam-6,13-dichloro-amine), VOZYUE.[38]
Reaction of H2[PdCl4] with diam initially formed [Pd(H2diam-III-N4)]Cl3·0.5[PdCl4], which recrystallised from hot dilute HClO4 as [Pd(H2diam-III-N4)](ClO4)4·6H2O, or from aqueous NH3 as [Pd(diam-III-N4)](ClO4)2·0.5H2O.[11]
Compounds of rhodium(iii) with diam
Equimolar RhCl3 and diam were refluxed in water or methanol for ~10 h and two compounds isolated from the resulting solution by chromatography; yellow trans-[Rh(H2diam-III-N4)]Cl2](ClO4)2·2H2O and pale yellow trans-[Rh(Hdiam-III-N5)Cl](ClO4)3·3H2O. Heating the latter in NaOH solution and then evaporating with added ClO4− gave colourless crystals of [Rh(diam-III-N6)](ClO4)3, SERHUS.[35]
When RhCl3 and diam were refluxed in water or methanol for 10 h, and then NaClO4 and ZnCl2 added, [Rh(diam-III-N6)]3(ClO4}5·(ZnCl4)2·3H2O crystallised.[11,35]
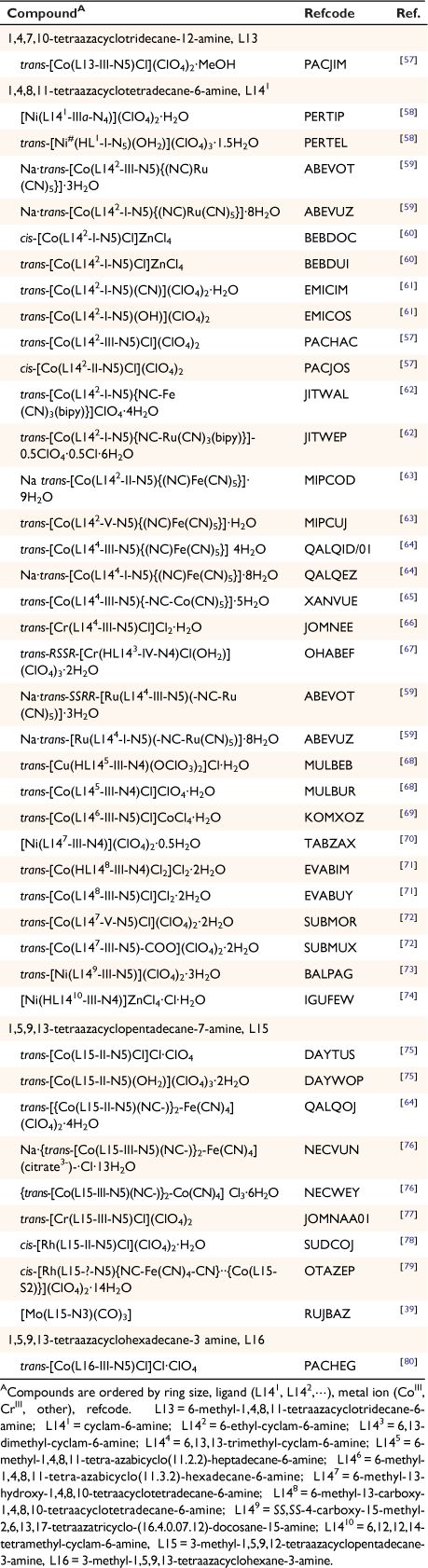
A compound of molybdenum with diam and carbon monoxide
Reaction of diam with molybdenum carbonyl formed [diam(Mo(CO4)2)]·DMF, RUJBED with two Mo(CO)4 groups each bonded to the diam by one amine substituent and an adjacent cyclam nitrogen in a bidentate and exo-cyclic manner[39] (see Table 2 for the related monam compound RUJBAZ).
|
Compounds of lead(ii) with diam and cis-diam
An aqueous solution of lead acetate was added dropwise to a solution of diam, with the pH adjusted to below seven with acetic acid (in final equimolar proportions). An aqueous solution of NaClO4 was then added dropwise until the solution became turbid. White crystals of composition Pb2+·(diamH+)3·(CH3CO2−)3·(ClO4−)2·(H2O)3.5 slowly formed. A similar preparation using lead nitrate formed crystals of composition Pb2+·(H3diam3+)5·(NO3−)7·H2O.[11]
When the pH of an equimolar aqueous solution of [H6cis-diam]Cl6 and Pb(NO3)2, plus NaClO4, was raised to 6 with NaOH and the solution evaporated, colourless crystals of cis-[Pb(cis-diam-V-N6)(OH2)](ClO4)2, WENVAM[40] formed over a period of weeks.
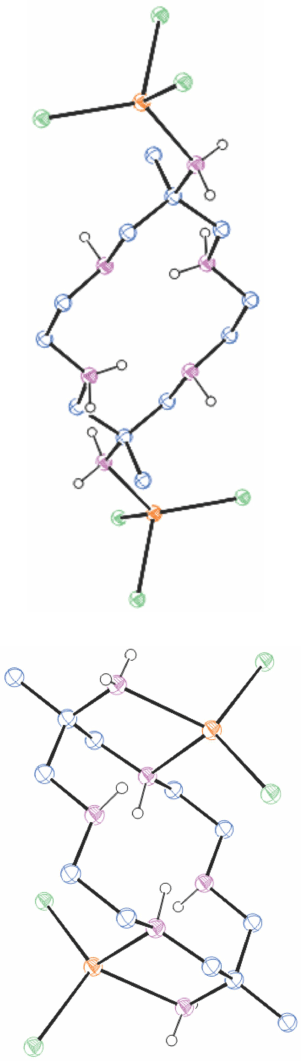
A compound of mercury(ii) with diam
When HgCl2 and NaClO4 were added to an aqueous solution of [H6diam]Cl6 and the pH raised to 6 with NaOH, colourless crystals of [(H2diam)(HgCl3)2]·[diam(HgCl2)2)]·2H2O, WENVIU[40] (Fig. 10) slowly crystallised. The diam is in exo-cyclic coordination to chloro-Hg2+ ions; in molecule 1 only by an amine substituent, in molecule 2 by an amine substituent and one ring N-atom.
|
Summary
Preparative methods for metal-ion compounds of diam and cis-diam are generally like those used for cyclam. Preparations for diam under basic conditions usually yield trans-[M(diam-IIIaa-N6]n+ products. All reported structures of diam compounds have the cyclam nitrogen atoms in planar coordination, but with folded, cis, coordination inferred from the d–d spectrum for one CrIII compound. Most metal ions react with diam to form products with the aa substituent configuration. Products with the bb configuration are formed by CuII, PdII and PtII. A CoIII compound with the uncommon cyclam configuration II was also prepared. Metastable configuration diam-I-N5 compounds are formed by reaction of Cu2+ with diam, hydrogenation of [Ni(trans-dino)]2+, by exposure of [Co(diam)]2+ to oxygen and by reaction of [Co(diam-III-N6)]3+ in 2 mol dm−3 NaOH solution. It is possible that compounds of these isomers are also formed by other elements, but not yet isolated.
All reported structures of compounds of cis-diam, other than with CuII, have folded macrocycle, cis-[M(cis-diam-V-N6)] configurations. Two compounds of CuII (and possibly compounds with NiII) have planar-macrocycle structures.
The cyclam ring secondary amine groups of coordinated diam are non-labile because of their inclusion in the ring-structure. The primary amine substituents of diam-III-N6 ions are more labile and can usually be protonated or substituted, rapidly for normally labile ions such as NiII, CuII or ZnII, slowly for normally non-labile ions, such as CoIII. [Ni(diam-III)]2+ and [Cu(diam-III)]2+ are extremely resistant to acid de-metalation, while [Zn(diam-III)]2+ and [Ni(diam-I)]2+ slowly hydrolyse in acid.
Most reported cis-diam compounds were prepared under basic conditions and have [M(cis-(cis-diam-Vaa-N6)] configurations. Reaction of Cu2+ with cis-diam forms trans-[Cu(cis-diam-I-N5)(OClO3]+ and trans-[cis-H2diam-IIIaa-N4)(OClO3)2]2+. It is likely that other cis-diam compounds with the amine substituents protonated, or displaced by other ligands, could be prepared, analogous to these and the putative [Ni(cis-diam)]2+ compounds mentioned in Compounds of Nickel(ii) with cis-diam.
Structures
Early in the studies of these ligands, structures of compounds of [Co(diam-III-N6)]3+, [Cr(diam-III-N6)]3+, [Ni(diam-III-N6)]2+ and [Cu(H2diam-III-N4)]4+ cations were determined by X-ray crystallography. The CoIII compound, in particular, raised interest because the Co–N bond lengths were shorter than those of other CoIII–N6 compounds such as [Co(seph)]3+ or [Co(en)3]3+. The d–d spectra, reduction potentials and CoIII/CoII self-exchange rates were also indicative of a higher ligand field strength for the [Co(diam-III-N6)]3+ cation. This led to speculation that for [diam-III-N6)]3+ the ligand might somehow be ‘constraining’ the CoIII ion.[27] For [Co(diam-III-N6)]3+ the Co–N distances to the cyclam nitrogen and amine substituent atoms are both unusually short. For the larger NiII and ZnII ions the Ni–N distances are more similar to those of other N6 type ligand compounds and the substituent Ni–N distances are longer than the cyclam Ni–N distances.
For any MN6 species the M–N distances will be determined by the intersection of two energy–distance relationship curves; firstly the bond energy of the MN6 group, determined by electronic interactions between the Mn+ ion and N donor-atoms and secondly the strain energy within the ligand(s) arising from positioning the N-atoms in their coordination sites, which will increase as the centre–N distances decreases. The short Co–N distances for [Co(diam-III-N6)]3+ suggest that the intra-ligand repulsive interactions are smaller for diam than other N6 ligands, probably because of fewer H–H repulsive interactions.
Strain energies versus centre–N distance were calculated for the diam molecule in the III-N6 configuration, with the N atoms constrained to lie on a sphere.[81] Three conformations for the [M(diam)]n+ ion were identified, differing principally by the relative conformations of the two five-membered chelate rings, conformations, 1 and 3 with centrosymmetrical δ,λ and chiral, 2 with δ,δ, λ,λ arrangements (also describable in terms of the relative axial or equatorial orientations of the C3, C4 and C9, C10-ring atoms.[48] These had strain energy versus distance relationships with minima at ~1.93, 2.00 and 2.1 Å, respectively. Observed structures for [Co(diam-III-N6)]3+ compounds (Co–N ~1.94 Å), and other small M3+ ions, have centrosymmetrical cations with the conformation 1, while structures for [Ni(diam-III-N6)]2+ (Ni–N ~2.07 Å for cyclam N-atoms, with longer Ni–N for the amine substituents, and also for other larger M2+ ions), are chiral, with the conformation 2. These conformational variations have been confirmed by NMR[16] and later molecular mechanics studies.[48,81,82]
Calculated strain energies increase steeply with increasing metal-ion size and [Zn(diam-III-N6)](ClO4)2 has the largest reported M–N distances of ~2.1 Å, attempts to prepare endo-cyclic compounds of Cd2+, Hg2+ or Pb2+ were unsuccessful.
Similar calculations for cis-[M(cis-diam-V-N6)]n+ suggest that the ligand strain energy minimum occurs at a larger M–N distance and increases less steeply with increasing M–N distance for cis-diam than for diam, and so the cis-isomer would be more compatible with larger metal ions.[16,81] cis-[Cd(cis-diam-V-N6)](ClO4)2, VOPLER, with Cd–N ~2.4 Å[16] and cis-[Pb(cis-diam-V-N6)(OH2](ClO4)2, WENVAM, with Pb–N ~2.8 Å have been prepared.[40] Variations of the structures and spectroscopic parameters of the cis-[M(cis-diam-V-N6)]n+ cations, M = CoIII, CrIII and NiII have been evaluated.[16]
Compounds of related di-amine-substituted cyclic tetraamines
6,13-Diethyl-cyclam-6,13-diamine compounds
Reaction of [Cu(en)2](NO3)2 with nitropropane, formaldehyde and NEt3 forms brown [Cu(Etdino)](NO3)2 (Etdino = 6,13-diethyl-6,13-dinitro-cyclam), which recrystallised from aqueous HClO4 as purple [Cu(Et2dino)](ClO4)2.[41] Similar reaction of [Ni(en)2]Cl2 produces [Ni(Etdino)]Cl2.[12]
The [Cu(Etdino)]2+ cation(s) were reduced with Zn/HCl and isomeric [Cu(Etdiam)]2+ (Etdiam = 6,13-diethyl-cyclam-6,13-diamine) ions, formed by reaction with Cu2+, were separated by chromatography into five bands with distinctive d–d spectra. These were each reduced with Zn/HCl, and the ligands characterised by NMR spectroscopy. Bands 1, 2, 3 and 5 contained the [Cu(trans-Etdiam)]2+ isomer and band 4 the [Cu(cis-Etdiam)]2+ isomer (cf. the reduction of [Cu(trans-dino)]2+. Compounds of Zinc(ii) with diam and cis-diam). [H4(trans-Etdiam)](ClO4)2·2H2O, ZUSCEN,[41] reacted with Na3[Co(CO3)3] to form trans-[Co(trans-Etdiam-IIIaa-N6)](ClO4)3·H2O, ZUSCIZ.[41]
2,9-meso-2,6,9,13-Tetramethyl- and 2,2,6,9,9,13-hexamethyl-cyclam-6,13-damine compounds
Reactions under the same conditions of [Cu(en)2]2+, [Cu(rac-pn)2]2+ or [Cu(2-methyl-pn)2]2+ with formaldehyde and nitroethane, form acyclic mono-nitro and macrocyclic dinitro products in 2:3, 10:1 and ~50:1 proportions, respectively. The acyclic products from rac-pn, REVQIS, and 2-methyl-pn, REVQOY,[42] have the diamine residue methyl substituents remote from the –CH2–C(Me, NO2)–CH2– link. The macrocyclic compound formed from pn was reduced with Zn/HCl and the hexa-hydrochloride salt of the cyclic amine product isolated and reacted with Cu2+ and base. The major product, isolated by chromatography, was trans-[Cu(2,9-meso-6,13-diamminium-2,6,9,13-tetramethyl-cyclam-IIIaa-N4)(OClO3)2](ClO4)2, NOPKUY,[83] with the C2 and C9-methyl substituents equatorially oriented.
Reaction of [Ni(rac-pn)2]Cl2 with nitroethane and formaldehyde forms [Ni(2,9-meso-2,6,9,13-tetramethyl-6,13-dinitro-cyclam)ICl2.[12]
5S,7S,12R,14R-5,7,12,14-Tetramethyl-cyclam-6,13-diamine compounds
Reactions of metal-ion diamine compounds with nitromethane and aldehydes generally produce extensive tars. Reaction of [Cu(en)2]2+ with four mole proportions of acetaldehyde, followed by reaction with nitromethane, forms [Cu(5,7,12,14-tetramethyl-6,13-dinitro-cyclam)]2+, which was reduced by Zn/HCl to form 5S,7S,12R,14R-5,7,12,14-tetramethyl-cyclam-6,13-diamine, L.[26,83] The compounds [H4L](ClO4)4·2H2O, NOCSUT, [Cu(H2L-IIIaa-N4)](ClO4)4·2H2O, NOCTAA.[83] NOCSUT and NOCTAA are isomorphous. trans-[Cu(H2L-IIIaa-N4)(ONO2)2]-(NO3)2·4H2O, QUXVAH[51] and trans-[Co(L-IIIaa-N6)]Cl2·ClO4, RUPFIR[26] have been structurally characterised (see Scheme 6).
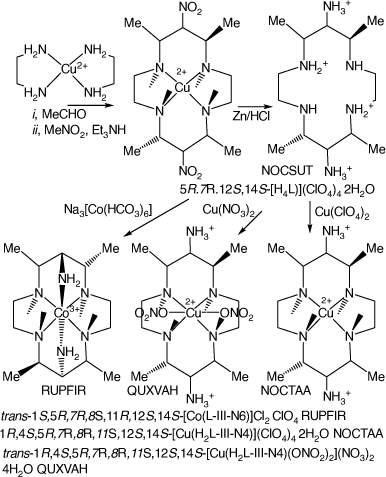
|
Protonation constants, pKas, for 5S,7S,12R,14R-5,7,12,14-tetramethyl-cyclam-6,13-diamine, at 298 K in 0.1 mol dm−3 Et4NClO4, are: 11.1(1), 10.3(1), 5.5(1), 5.2(1) 3.5(1). Molecular mechanics modelling of strain energies of the CoII and CoIII cations and their relationship to redox couples and self-exchange rates are discussed.
Mild reduction of the 6,13-dinitro(Cu) ion formed the 6,13-diketo oxime compound (L), isolated as [Cu(L-III-N4)Cl]Cl·7H2O, NOCTEE[83] and [Cu(L-III-N4)Cl]BF4·H2O, TASCOF,[84] which have the uncommon cyclam configuration II.
3,15-Dimethyl-1,5,9,13-tetraazacyclohexane-3,15-diamine compounds
Reaction of [M(propane-1,3-diamine)2]2+ with formaldehyde and nitroethane forms [M(3,15-dinitro-3,15-dimethyl-[16]ane)]2+, M = Cu2+ or Pd2+, with yields higher for Pd2+. Reduction by Zn/HCl forms the 3,15-cis-diamine. The overall yield of the amine was ~13% via the CuII compound and ~40% via the PdII compound. cis-[Cu(cis-[16]-diam-V-N6)](ClO4)2, CAHYEQ,[85] has a tetragonal structure, with significantly longer Cu–N bonds to one ring N-atom (2.35(4) Å) and one substituent N-atom (2.33(3) Å) which are in tetragonal positions.
4,15-Dimethyl-2,6,13,17-tetraazatricyclo[16.4.0.07,12]docane-4,15-diamine compounds
Reaction of [Cu(trans-(R,S)-cyclohexane-1,2-diamine)2]2+ or [Cu(cis-(R,R or S,S)-cyclohexane-1,2-diamine)2]2+, as nitrate or perchlorate salts, with nitroethane, formaldehyde and base, form the (mono-nitro) acyclic and (di-nitro) macrocyclic condensation products, which were isolated by chromatography. The nitro groups of one acyclic CuII compound were reduced with Zn/HCl, and [Co(RR,RR-1,3-bis(2′-aminocyclohexylamino)-2-methylpropane-2-amine-N5)Cl]Cl·ClO4·2H2O, WAPPIM, was prepared from the isolated amine salt.[86]
Compounds of (mono-amine)-substituted tetraaza-cyclic tetraamines
CuII and NiII compounds of linear tetraamines react with formaldehyde and nitroethane in basic solution to form tetraaza-macrocycle compounds with nitro and methyl substituents on the central carbon atom of one six-membered chelate ring. The CuII compounds have been reduced by Zn/HCl to form the methyl, amine substituted cyclic tetraamines, which have been isolated by ion exchange chromatography as the penta-hydrochloride salts, from which compounds of transition-metal ions have been prepared. The 3-methyl-3-nitro-1,5,9,13-tetraazacyclohexadecane (L16) compound was prepared in higher yield as the PdII compound (see Scheme 7). Compounds with reported structures are shown in Table 2.
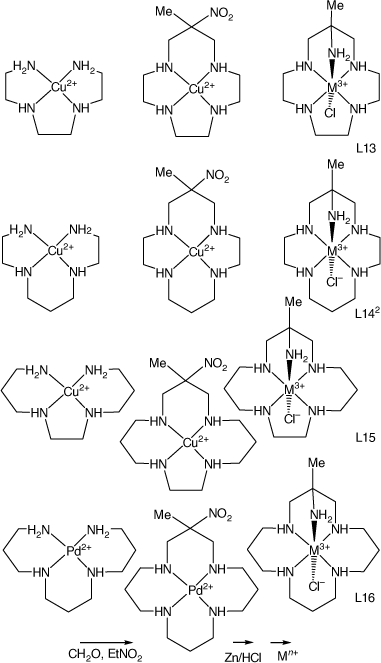
|
Some related compounds are included, including cyclam-6-amine (L141), which was synthesised by ‘non-templated’ reactions[58] (Scheme 8) and amine/methyl substituted cyclic tetraamines with other substituents, formed via reaction of variously substituted linear tetraamine CuII cations with EtNO2 and formaldehyde (Scheme 9).
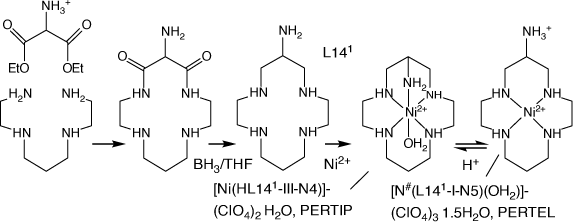
|
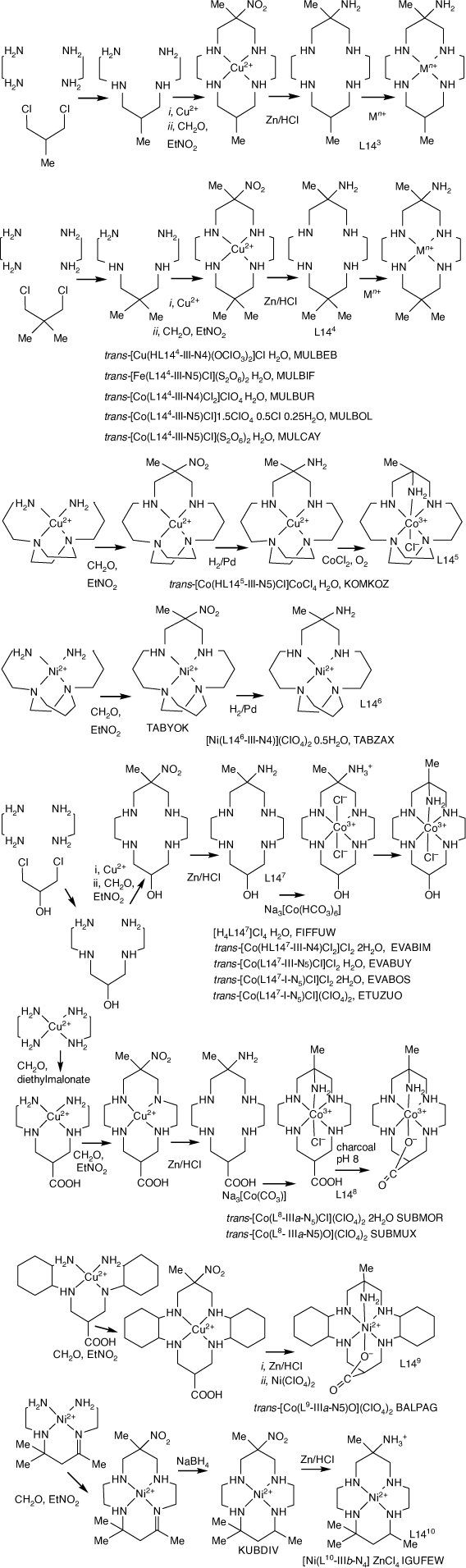
|
Most reported structures have octahedral trans-N5(X) coordination of the ligand to a M3+ metal ion, usually with configuration IIIa, occasionally with configurations Ia, many with cyano-linked di-nuclear structures. Compounds of a pentaamine with different configurations were either prepared by different procedures or isolated from mixed products by chromatography.
Dimensions of trans-[Co(ligand-III-N5)Cl]2+ cations with 13–16 ring-membered ligands, included in Table 2, show little variation in Co–N and Co–Cl distances with ring size or substituents.
Formation constants for metal ions with mono-methyl substituted cyclic tetraamines with a range of ring sizes have been reported.[87] Complexation kinetics for CuII and NiII with L15 have been reported.[88] Comparative basic hydrolysis rates for chlorocobalt(iii) compounds of L13, L141, L15 and L16 have been measured.[52]
Reaction of [Cu(3,6,9-triazaundecame-1,15-diamine)]2+ with nitroethane and formaldehyde forms [Cu(15-methyl-15-nitro-1,4,7,10,13-pentaazacyclohexadecane), which was reduced with Zn/HCl to form 15-methyl-1,4,7,10,13-pentaazacyclo-hexadecane-15-amine salts, which formed octahedral cations with CoIII, CrIII and FeIII. [Co(L16-N6)](ClO4)3, CAJBEV, has the amine substituent coordinated trans to the 7-aza group[89] (see Scheme 10).
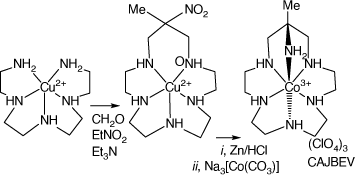
|
Compounds of diam, monam (and homologues) with modified amine substituents
Modifications of the amine substituents of diam, monam or homologues are shown in the following reaction schemes, which include CCDC refcodes of structures.
6,13-exo-Methylene-cyclam compounds
Reactions of [Cu(diam-III-N4)]2+ (and [Pd(diam-III-N4)]2+ with nitrous acid form exocyclic-6,13-bis(methylene)-cyclam compounds, with the structure of [Cu(L-III-N4)]2+)(ClO4)2, JUTXIF, reported[45,90] (see Scheme 11).
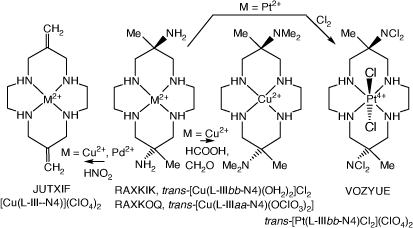
|
A 6,13-dimethyl-6,13-bis(dichloro-amine)-cyclam compound
Oxidation of trans-[Pt(H2diam-IIIbb-N4)Cl2]2+ with Cl2 forms the 6,13-dimethy-cyclam-6,13-bis(dichloro-amine), L, compound trans-[Pt(L)-IIIbb-N4)Cl2](ClO4)2, VOZYUE[38] (see Scheme 11).
6,13-Dimethyl-cyclam-6,13-bis(ethyl-amide) compounds
Reactions of trans-[Cu(diam-IIIaa-N4)(OClO3)2], [Ni(diam-IIIaa-N6)](ClO4)2 or trans-[Co(diam-IIIaa-N4)(CN)2]ClO4 with acetic or propionic anhydrides in DMF form 6,13-di-methyl-6,13-bis((m)ethyl-amido)-cyclam, L, compounds, [Cu(L-IIIbb-N4)(OClO3)2], DAPWOH[46] and trans-[Co(L-IIIbb-N4)(CN)2]ClO4·2H2O, BOHJOY.[47] The NiII cation forms sgs square-planar, and also tgs octahedral compounds with a variety of trans ligands. The structure of trans-[Ni#(L-IIIbb-N4)(OH2)]·trans-[Ni#(L-IIIbb-N4)(OH2)(OCOOH)]ClO4·HCO3, NAJZUU, which crystallised from NaHCO3 solution, was determined[91] (see Scheme 12).
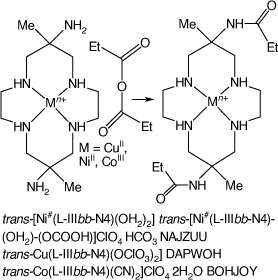
|
A 6,13-bis-(N,N-dimethylformamidinyl)-cyclam compound
A vigorous reaction of [Ni(diam-III-N6)](ClO4)2 dissolved in dimethylformamide with oxalyl chloride formed sparingly soluble, lilac coloured, trans-dichloro-(6,13-dimethyl-cyclam-6,13-bis-(N,N-dimethyl-formamidinyl))nickel(II) perchlorate dihydrate, UZAJEL[92] (see Scheme 13).
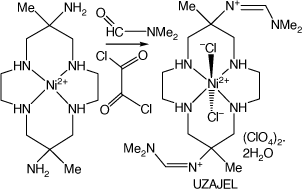
|
6,13-Dimethyl-cyclam-6,13-bis-(dimethylamine) compounds
Refluxing a solution of [Cu(diam)]2+ in formic acid/formaldehyde (a standard method for methylating amines[44,93]) for 2 days forms isomeric 6,13-bis(dimethylamino) CuII cations as the major products. Compounds with isomeric cations [Cu(L-IIIaa-N4)(OClO3)2], RAXKOQ and [Cu(L-IIIbb-N4)(OH2)2]Cl2, RAXKIK were isolated by chromatography, (cf. Compounds of Coppe(ii) with diam and cis-diam). The isomerism was investigated by molecular modelling[94] (see Scheme 11).
Compounds formed by reactions of diam compounds with aldehydes
Reaction of [Cu(diam)]2+ with formaldehyde and reaction of [H6diam)]6+ with pyridine-2-carbaldehyde form polycyclic products incorporating fused 1,3-diazetidene (diazacyclopentane) rings, which are tautomers of the methylene imines.
Compounds formed by reaction of [Cu(diam)]2+ with formaldehyde
Refluxing an aqueous solution of [Cu(OH2)4](NO3)2, [H6diam)]Cl6 and formaldehyde for 24 h formed a mixture of polycyclic products incorporating fused 1,3-diazetidene rings, tautomeric with methylene imines, which were separated by chromatography and the structures of three determined[95] (see Scheme 14).
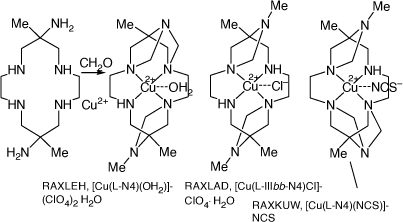
|
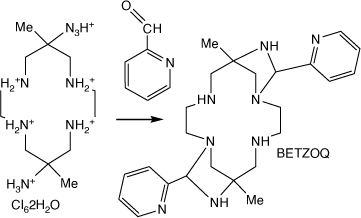
A compound formed by reaction of [H6diam]Cl6·2H2O with pyridine-2-carbaldehyde
Reaction of [H6diam]Cl6·2H2O with pyridine-2-carbaldehyde forms trans-6,14-dimethyl-8,16-bis(2-pyridyl-methyl)-1,4,7,9,12,15-hexa-azatricyclo(12.2.1.16,9)octadecane, BETZOQ, with 1,3-diazetidene rings[96] (see Scheme 15).
|
Compounds of diam and monam with secondary amine substituents, formed by reduction of imine functions formed by reaction with an aldehyde
Diam and monam react with aldehydes under anhydrous conditions to form imines (or 1,3-diazetidene tautomers), which are reduced by NaBH3CN or NaBH4 to form secondary amine substituents. Metal-ion compounds were prepared from the resultant amines. Alternatively, the metal ion (usually CuII) compound of diam or monam was reacted with the aldehyde and the resulting imine compound reduced, usually with NaBH3CN.
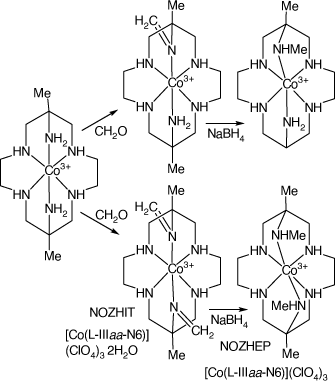
Compounds formed by reactions of [Co(diam-III-N6)](MeCO2)3 with formaldehyde
Reactions of [Co(diam-III-N6)](MeCO2)3 with formaldehyde in MeOH under anhydrous conditions form the 6-mono- and 6,13-bis-(methyl-imine), NOZHEP, cations, which were reduced with NaBH4 to form the 6-mono- and 6,13-bis-(methyl-amine), NOZHIT, cations, respectively[97] (see Scheme 16).
|
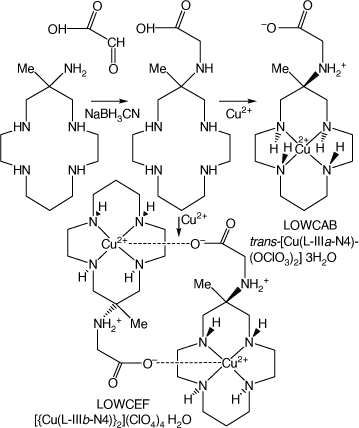
Compounds formed via reaction of monam with glyoxylic acid
Reaction of monam with glyoxylic acid, reduction of the resulting imine with NaBH3CN and reaction of the resulting N-carboxymethyl substituted macrocycle with Cu2+ forms isomeric cations, isolated as trans-[Cu(L-IIIa-N4)(OClO3)2]·3H2O, LOWCAB and [{Cu(L-IIIb-N4}2](ClO4)4·H2O, LOWCEF[98] (see Scheme 17).
|
CoIII compounds formed via reaction of diam with 4-carboxy-benzaldehyde
Reaction of [H6(diam)]Cl6 with 4-carboxy-benzaldehyde and NaBH3CN form the 6-(4-carboxy-phenyl-methyl-amine compound, which reacted with Na3[Co(CO3)3]3H2O to form the Co(III) compounds XASJUX and XASJOR[99] (see Scheme 18). The electrochemistry of XASJUX chemisorbed onto TiO2 was investigated.
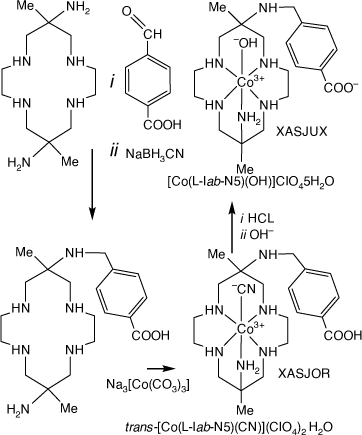
|
Copper(ii) compounds formed via reactions of monam with 2-, or 4-hydroxy-benzaldehyde
Reactions of monam with 4-carboxy-benzaldehydes and Cu2+ form 1,3-diazetidene type imine tautomers, LIZSUI and LIZTAP[100] which are reduced by NaBH4 to form the (4-carboxy-phenyl)-methyl) monam compound, WOHYEX. Reaction of 6-(2-or 4)-hydroxy-benzaldehyde with [Cu(monam)]2+ and NaBH4 form 6- (2- or 4)-hydroxy-phenyl)-methyl-monam) compounds, WOHYAT and WOHXUM[101] (see Scheme 19).
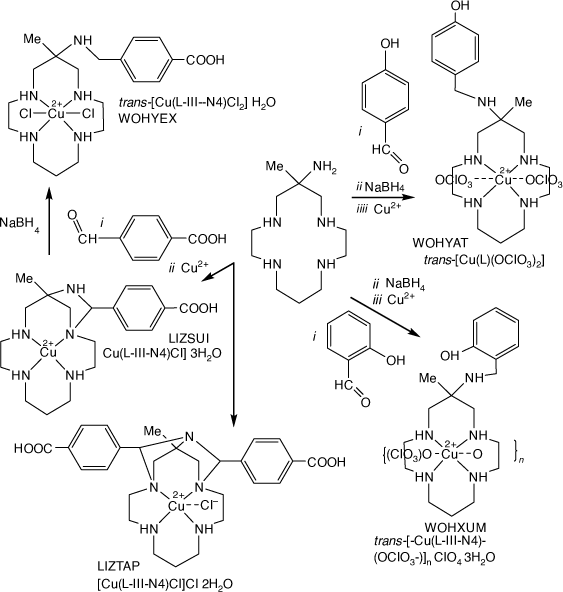
|
CoIII compounds of diam with N-methyl-(benzo cyclic ether) substituents
Reactions of trans-[Co(diam-N4)(OH2)2]3+ with a 4-formyl-benzo-(cyclic ether) and NaBH3CN form compounds with benzo-cyclic ethers N-appended to [Co(diam-III-N5)]3+. The cyclic ethers can host cations, MOGVEJ, MOGVIN, MOGVOT, MOGVUZ and MOGWAG[102] (see Scheme 20).
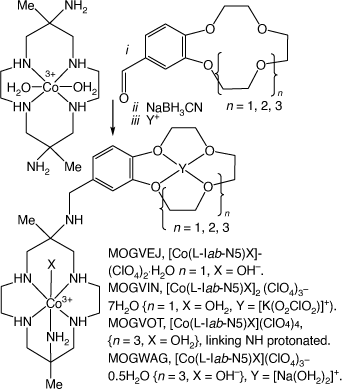
|
Compounds of diam, monam (and homologues with different ring sizes) with N-(methyl-aromatic) substituents
Reactions of diam, monam (and homologues with different ring sizes) with carboxy-methyl-aromatics, reduction of the resulting imine functions with NaBH3CN and reaction with metal ions form compounds with N-(methyl-aromatic) substituents. Compounds with methyl-phenyl, -naphthyl, and -anthacenyl aromatic groups and with mixed aromatic groups were formed to give XODTIT, XODTOZ and XODTUP from diam, and ECXIX, EXEXOD and ECXUJ from monam.[103] The ZnII compounds were used for photochemical studies of energy transfer between aromatic substituents, LESCOB.[104] Reaction with 4-dimethylamino-benzaldhyde forms the 6,13-bis-(4-dimethylamino-methyl-phenyl) compound, which reacts with 9-carboxymethyl-anthracene to substitute 6-anthraceneyl-methyl for one 4-dimethylamino-methyl-phenyl group, YEBZIP[105] (see Scheme 21).
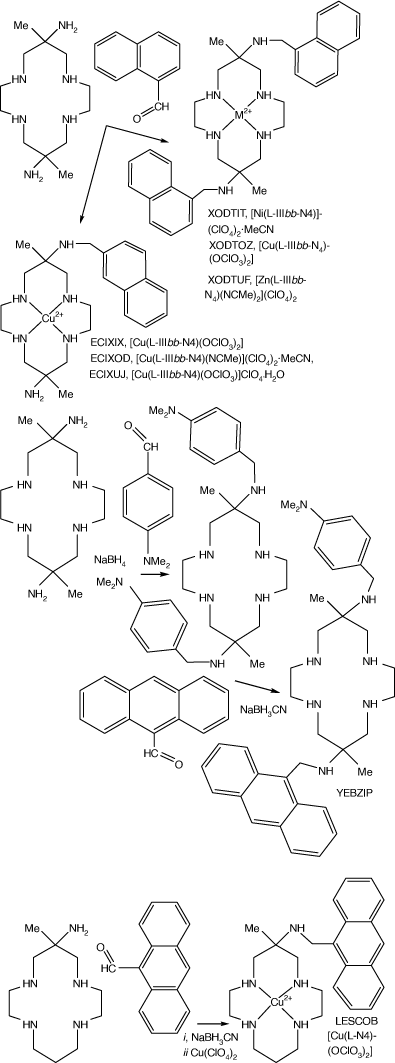
|
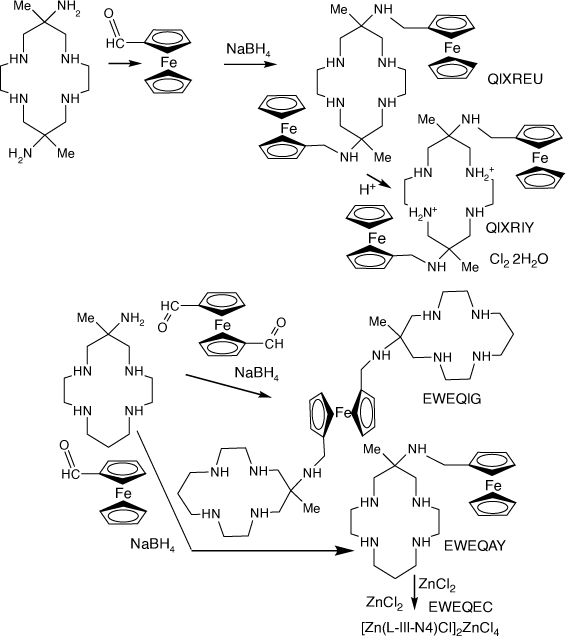
Compounds of diam and monam with N-(methyl-ferrocenyl) substituents
Compounds with N-(methyl-ferrocenyl) substituents were formed by reaction of diam or monam with carboxymethyl-ferrocene or bis(carboxymethyl)-ferrocene, reduction of the resulting imine compounds with NaBH4 and subsequent reaction with metal ions[106–108] (see Scheme 22).
|
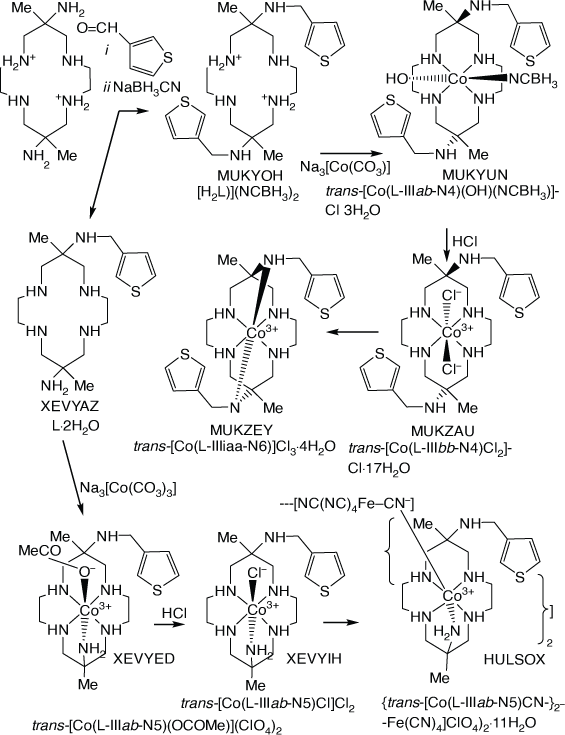
Cobalt(iii) compounds of diam with N-(3-methyl-thiophene) substituents
Compounds with 6- or 6,13-bis-(3-methyl-thiophene) substituents, were formed by reaction of [H2diam]2+ with 3-(carboxymethyl)-thiophene and NaBH3CN. CoIII compounds were formed by reaction with Na3[Co(CO3)3]·3H2O[109,110] (see Scheme 23).
|
Diam immobilised on cellulose
Epoxide activated cellulose was soaked in dilute HCl to hydrolyse any remaining epoxide groups, washed and then reacted with NaIO3 in water. Reaction ceased after a period of hours, when the oxidised cellulose was again washed. Diam dissolved in a KH2PO4/K2HPO4 buffer and NaBH3CN were added, the coupling reaction taking ~4 h at ambient temperatures. The washed and dried cellulose-diam had 4.2% N content, corresponding to a loading of 0.5 mmol of diam g–1 of cellulose-diam (Scheme 24).
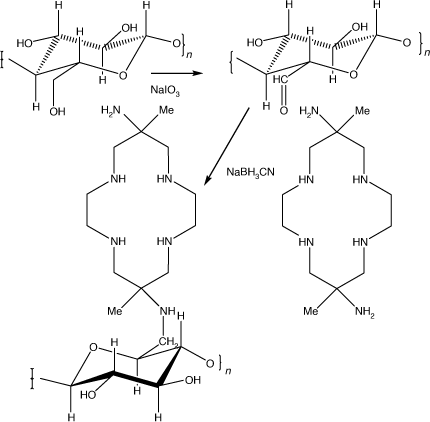
|
At pH 3 the cellulose-diam rapidly absorbed Cu2+, Ni2+, Co2+ and Fe2+, with residual solution concentrations of <0.02 ppm, with maximum loadings of ~1.2 mmol g–1 dry weight, significantly higher than the nominal diam loading. At pH 1 only Cu2+ was appreciably absorbed. No Cu2+ was leached from the Cu2+ loaded cellulose-diam at pH 1.[111]
Conclusions
Facile reactions of bis(ethane-1,2-diamine)-copper(ii) and -nickel(ii) cations with nitroethane and formaldehyde form (6,13-dimethyl-6,13-dinitro-4,8,11-tetraazacyclotetradecane)-copper(ii) or nickel(ii) cations. Reduction of the nitro functions of the CuII compound with Zn/HCl forms salts of isomeric 6,13-dimethyl-4,8,11-tetraazacyclotetradecane-6,13-diamine, with the amine substituents trans (anti), here labelled as ‘diam’, or cis (syn), here labelled as cis-diam, which can be separated by chromatography. Hydrogenation of the NiII cation forms [Ni(diam)]2+ (and probably also some [Ni(cis-diam)]2+, though this requires verification). Compounds of the isomeric amines with a wide range of transition metal ions have been prepared. The trans isomer, diam, forms metal-ion compounds with the cyclam nitrogen atoms in square-planar coordination, with both, one or neither of the amine substituents also coordinated. Most compounds formed by the cis-diam isomer have the cyclam nitrogen atoms in folded coordination, with the amine substituents coordinated cis. For the more intensively studied compounds of diam with NiII and CoIII, compounds with one or both of the amine substituents displaced by other ligands, or uncoordinated, free or protonated have been prepared. Compounds of CoIII and NiII have been prepared with cyclam configuration I, usually with N5 coordination. It is probable that related compounds can be prepared with cis-diam, but that requires verification. CuII, PdII and PtII react with diam to form isomeric products, distinguished by orientation of substituents relative to the cyclam secondary amine configuration.
Reactions of (3,7-diazanonane-1,9-diamine)-copper(ii) cations (and various C-substituted (3,7-diazanonane-1,9-diamine)-copper(ii) cations) with nitroethane and formaldehyde form (6-methyl-6-nitro-1,4,8,11-tetraazacyclotetradecane)-copper(ii) or nickel(ii) cations. Reduction of the nitro function of the CuII cations with Zn/HCl lead to variously C-substituted 6-methyl-cyclam-6-amines, from which compounds, mainly with CoIII or CrIII, with the cyclic amine penta-coordinated, have been prepared.
The amine substituents of diam have been modified by methylation and by conversion into amide functions. Reaction of diam and monam, or their metal-ion cations, with functionalised aldehydes form imines which can be reduced to form functionalised secondary amine substituents. Compounds with variously substituted methyl-benzene, methyl-anthacene (and poly-aromatic molecules), methyl-ferrocene, methyl-benzo-cyclic ethers (which can host other cations) and methyl-thiophene have been reported. Similar reactions, which could bind a cyclic tetraamine (or its metal-ion compounds) to significant biological molecules would appear to potentially have widespread applications.
Data availability
This is a review article. The data included was obtained from the primary publications listed as references. Secondary sources were American Chemical Society Chemical Abstracts and the Cambridge Crystallographic Data Centre database.
Conflicts of interests
The author declares that he has no known competing financial interests or personal relationships that could have, or appeared to have, influenced the content of this paper.
Declaration of funding
This work did not receive any external funding. The author does not have any links to commercial interests or funding agencies. The author is an Emeritus Professor at the Victoria University of Wellington, which provided office space and access to computer, internet and library services, but no financial support.
References
[1] GA Lawrance, PG Lye, Comments Inorg Chem 1994, 15, 339.| Crossref | GoogleScholarGoogle Scholar |
[2] GA Lawrance,, PV Bernhardt, Coord Chem Rev 1990, 104, 297.
| Crossref | GoogleScholarGoogle Scholar |
[3] NF Curtis, Coord Chem Rev 2018, 366, 3.
| Crossref | GoogleScholarGoogle Scholar |
[4] AM Sargeson, Chem Ber 1979, 15, 23.
[5] AM Sargeson, Pure and Appl Chem 1984, 56, 1603.
| Crossref | GoogleScholarGoogle Scholar |
[6] RJ Gene, TW Hambley, J Mac, B Harrowfield, AM Sargeson, MR Snow, J Am Chem Soc 1984, 106, 5478.
| Crossref | GoogleScholarGoogle Scholar |
[7] PV Bernhardt, AMT Bygott, RJ Geue, AJ Hendry, BR Korybut-Daszkiewicz, P Comba, II Creaser, LR Gahan, JM Harrowfield, GA Lawrance, LL Martin, AWH Mau, AM Sargeson, WHF Sasse, MR Snow, Inorg Chem 1986, 25, 384.
| Crossref | GoogleScholarGoogle Scholar |
[8] LR Gahan, JM Harrowfield, Polyhedron 2015, 94, 1.
| Crossref | GoogleScholarGoogle Scholar |
[9] P Comba, NF Curtis, GA Lawrance, AM Sargeson, BW Skelton, AH White, Inorg Chem 1986, 25, 4260., FANFEF, FANFIJ
| Crossref | GoogleScholarGoogle Scholar |
[10] J Zhang, S-X Luo, A McCluskey, GA Lawrance, Synth React Inorg, Met-Org, Nano-Met Chem 2013, 43, 1.
| Crossref | GoogleScholarGoogle Scholar |
[11] Siriwardena A. PhD Thesis, Compounds of 6,13-diamino-6,13-dimethyl-1,4,8,11-tetraazacyclotetradecane, Victoria University of Wellington, New Zealand, 1988.
[12] NF Curtis, A Siriwardena, GJ Gainsford, DC Weatherburn, Aust J Chem 1993, 46, 755. FEBZUH10, PEHWII, PEHWOO, PEHWUU, PEHXAB, PEHXEF
[13] GA Lawrance, BW Skelton, AH White, P Comba, Aust J Chem 1986, 39, 1101., DUMGIB
| Crossref | GoogleScholarGoogle Scholar |
[14] NF Curtis, R Pawley, WT Robinson, Acta Crystallogr, Sect C 2006, C62, m352., KELVUT
| Crossref | GoogleScholarGoogle Scholar |
[15] PV Bernhardt, P Comba, TW Hambley, GA Lawrance, K Varnagy, J Chem Soc, Dalton Trans 1992, 355, 355.VOPLER
[16] PV Bernhardt, P Comba, TW Hambley, Inorg Chem 1993, 32, 2804., HAHLOR
| Crossref | GoogleScholarGoogle Scholar |
[17] SL Jain, P Bhattacharyya, SA McLellan, DT Richens, P Lightfoot, AMZ Slawin, JD Woollins, J Chem Res 2004, 234., BETZIK
| Crossref | GoogleScholarGoogle Scholar |
[18] PV Bernhardt, GA Lawrance, BW Skelton, AH White, Z Anorg Allg Chem 2007, 633, 1036., GIFLUD, GIFMAK
| Crossref | GoogleScholarGoogle Scholar |
[19] PV Bernhardt,, TW Hambley, GA Lawrance, Aust J Chem 1990, 43, 699., KENPOI, KENPUO
| Crossref | GoogleScholarGoogle Scholar |
[20] PV Bernhardt, GA Lawrance, BW Skelton, AH White, Aust J Chem 1989, 42, 1035., VAZDIJ
| Crossref | GoogleScholarGoogle Scholar |
[21] AJ Lough, RM Gregson, G Ferguson, C Glidewell, Acta Crystallogr, Sect C 2000, 56, 859., MATGET
| Crossref | GoogleScholarGoogle Scholar |
[22] PV Bernhardt, GA Lawrance, M Maeder, M Rossignoli, TW Hambley, J Chem Soc, Dalton Trans 1991, 1167., VISSUL
| Crossref | GoogleScholarGoogle Scholar |
[23] LJ Farrugia, J Appl Crystallogr 2012, 45, 849.
| Crossref | GoogleScholarGoogle Scholar |
[24] PV Bernhardt, GA Lawrance, TW Hambley, J Chem Soc, Dalton Trans 1989, 1059., FEBZOB10
| Crossref | GoogleScholarGoogle Scholar |
[25] PV Bernhardt, LA Jones, PC Sharpe, J Chem Soc, Dalton Trans 1998, 1757., NOZHAL
| Crossref | GoogleScholarGoogle Scholar |
[26] PV Bernhardt, LA Jones, PC Sharpe, Inorg Chem 1997, 36, 2420., RUPFIR, RUPFOX
| Crossref | GoogleScholarGoogle Scholar |
[27] NF Curtis, GJ Gainsford, TW Hambley, GA Lawrance, KR Morgan, A Siriwardena, J Chem Soc Chem Commun 1987, 295., FEBZOB, FEBZUH
| Crossref | GoogleScholarGoogle Scholar |
[28] NF Curtis, WT Robinson, DC Weatherburn, Aust J Chem 1992, 45, 1683. KUWZEH, KUWZIL, KUWZOR, KUWZUX, KUWZBAG, KUWBEK, KUXBIO
[29] PV Bernhardt, NL Kilah, Acta Crystallogr, Sect C 2005, C61, m245., FOMCEQ, FOMCIU
| Crossref | GoogleScholarGoogle Scholar |
[30] PV Bernhardt, P Comba, NF Curtis, TW Hambley, GA Lawrance, M Maeder, A Siriwardena, Inorg Chem 1990, 29, 3208., JEVCAO
| Crossref | GoogleScholarGoogle Scholar |
[31] PV Bernhardt, T Hambley, GA Lawrance, Chem Commun 1989, 553., SANLAU
| Crossref | GoogleScholarGoogle Scholar |
[32] PV Bernhardt, P Comba, TW Hambley, GA Lawrance, Inorg Chem 1991, 30, 942., SIZCEJ
| Crossref | GoogleScholarGoogle Scholar |
[33] H Borzel, P Comba, H Pritzkow, AF Sickmuller, Inorg Chem 1998, 37, 3853., PAZXAP
| Crossref | GoogleScholarGoogle Scholar |
[34] PV Bernhardt, GA Lawrance, P Comba, LL Martin, TW Hambley, J Chem Soc, Dalton Trans 1990, 2859., TAFPIZ
| Crossref | GoogleScholarGoogle Scholar |
[35] GA Lawrance, PV Bernhardt, TW Hambley, J Chem Soc, Dalton Trans 1990, 983.SERHUS
[36] JK Clegg, JM Harrowfield, GA Lawrance. Yang Kim, A Madalan, P Thuery, A Woo, Aust J Chem 2012, 65, 734., NESXIU
| Crossref | GoogleScholarGoogle Scholar |
[37] PV Bernhardt, GA Lawrance, WC Patalinghug, AH White., BW Skelton, NF Curtis, A Siriwardena, J Chem Soc, Dalton Trans 1990, 2853., TACVOI, TACVUO
| Crossref | GoogleScholarGoogle Scholar |
[38] PV Bernhardt, GA Lawrance, TW Hambley, Inorg Chem 1992, 31, 631., VOZYUE, VOZZAL
| Crossref | GoogleScholarGoogle Scholar |
[39] M Rossignoli, PV Bernhardt, GA Lawrance, J Chem Soc, Dalton Trans 1997, 4247., RUJBAZ, RUJBED
| Crossref | GoogleScholarGoogle Scholar |
[40] PG Lye, GA Lawrance, M Maeder, BW Skelton, H Wen, AH White, J Chem Soc, Dalton Trans 1994, 793., WENVAM, WENVIU
| Crossref | GoogleScholarGoogle Scholar |
[41] PV Bernhardt, KA Byriel, CHL Kennard, PC Sharpe, Inorg Chem 1996, 35, 2045., ZUSCEN, ZUSCIZ
| Crossref | GoogleScholarGoogle Scholar |
[42] M Rossignoli, GA Lawrance, M Maeder, DCR Hockless, BW Skelton, Aust J Chem 1996, 49, 1307., REVQIS, REVQOY
| Crossref | GoogleScholarGoogle Scholar |
[43] M Rossignoli, PV Bernhardt, GA Lawrance, M Maeder, Aust J Chem 1997, 50, 529., NOPKUY
| Crossref | GoogleScholarGoogle Scholar |
[44] HT Clarke, HB Gillespie, SZ Weishaus, J Amer Chem Soc 1993, 55, 4571.
| Crossref | GoogleScholarGoogle Scholar |
[45] PV Bernhardt, P Comba, C Mitchell, Sp Publ Royal Soc Chem 1993, 131, 110.
[46] NF Curtis, H Puschman, Polyhedron 2006, 25, 1204., DAPWOH
| Crossref | GoogleScholarGoogle Scholar |
[47] NF Curtis, H Puschman, Polyhedron 2005, 24, 281., BOHJOY
| Crossref | GoogleScholarGoogle Scholar |
[48] PV Bernhardt, P Comba, Inorg Chem 1992, 31, 2638.
| Crossref | GoogleScholarGoogle Scholar |
[49] PV Bernhardt, LA Jones, PC Sharpe, J Chem Soc, Dalton Trans 1997, 1169., ROMWAR, ROMWEV, ROMWIZ, ROMWOF, ROMWUL
| Crossref | GoogleScholarGoogle Scholar |
[50] PV Bernhardt, Acta Crystallogr, Sect C 2000, 56, 744., MASRON, MASRIH
| Crossref | GoogleScholarGoogle Scholar |
[51] Y-Y Liu, H-Y Chu, Acta Crystallogr, Sect E 2010, E66, m837., QAXVAH
| Crossref | GoogleScholarGoogle Scholar |
[52] Y Baran, GA Lawrance, M Martinez, EN Wilkes, Inorg React Mech (Amsterdam) 2000, 1, 315.
[53] B Bosnich, CK Poon, ML Tobe, Inorg Chem 1965, 4, 1102.
| Crossref | GoogleScholarGoogle Scholar |
[54] PG Lye, GA Lawrance, M Maeder, J Chem Soc, Dalton Trans 2001, 2376.
| Crossref | GoogleScholarGoogle Scholar |
[55] PG Lye, GA Lawrance, M Maeder, Inorg React Mech (Amsterdam) 1999, 1, 153.
[56] NF Curtis, A Siriwardena, Aust J Chem 1991, 44, 1023.
| Crossref | GoogleScholarGoogle Scholar |
[57] GA Lawrance, TM Manning, M Maeder, M Martinez, MA O’Leary, WC Patalinghug, BW Skelton, AH White, J Chem Soc, Dalton Trans 1992, 1635., PACJIM, PACHAC, PACJOS
| Crossref | GoogleScholarGoogle Scholar |
[58] E Kimura, M Haruta, T Koike, M Shionoya, K Takenouchi, Y Iitaka, Inorg Chem 1993, 32, 2779., PERTEL, PERTIP
| Crossref | GoogleScholarGoogle Scholar |
[59] PV Bernhardt, F Bozoglian, BP Macpherson, M Martinez, Dalton Trans 2004, 2582., ABEVIN, ABEVOT, ABEVUN
| Crossref | GoogleScholarGoogle Scholar |
[60] PV Bernhardt, BP Macpherson, Acta Crystallogr, Sect C 2003, 59, m467., BEBDOC, BEBDUI
| Crossref | GoogleScholarGoogle Scholar |
[61] PV Bernhardt, BP Macpherson, Acta Crystallogr, Sect C 2003, 59, m533., EMICIM, EMICOS
| Crossref | GoogleScholarGoogle Scholar |
[62] PV Bernhatdt, F Bozoglian, M Font-Bardia, M Martinez, AP Meacham, B Sienra, X Solans, Eur J Inorg Chem 2007, 2007, 5270., JITWAL, JITWEP
| Crossref | GoogleScholarGoogle Scholar |
[63] PV Bernhardt, BP Macpherson, M Martinez, J Chem Soc, Dalton Trans 2002, 1435., MIPCOD, MIPCUJ
| Crossref | GoogleScholarGoogle Scholar |
[64] PV Bernhardt, BP Macpherson, M Martinez, Inorg Chem 2000, 39, 5203., QALQAV, QALQEZ, QALQID, QALQOJ
| Crossref | GoogleScholarGoogle Scholar |
[65] BP Macpherson, BM Alzoubi, PV Bernhardt, M Martinez, PA Tregloan, R van Eldik, Dalton Trans 2005, 1459., XANVUE
| Crossref | GoogleScholarGoogle Scholar |
[66] GA Lawrance, M Martinez, BW Skelton, AH White, J Chem Soc, Dalton Trans 1992, 823., JOMNEE
| Crossref | GoogleScholarGoogle Scholar |
[67] MG Basallote, PV Bernhardt, T Calvet, CE Castillo, M Font-Bardia, M Martinez, C Rodriguez, Dalton Trans 2009, 9567., OHABEF
| Crossref | GoogleScholarGoogle Scholar |
[68] JK Clegg, LF Lindoy, P Thuery, YH Lee, DKA Kusumohastuti, C Mora, HH Kim, JH Cho, Y Kim, J Inclusion, Phenom Macrocyclic Chem 2009, 65, 49., MULBEB, MULBUR
| Crossref | GoogleScholarGoogle Scholar |
[69] TN Mali, PW Wade, RD Hancock, J Chem Soc, Dalton Trans 1992, 67., KOMKOZ
| Crossref | GoogleScholarGoogle Scholar |
[70] RD Hancock, MP Ngwenya, PW Wade, JCA Boeyens, SM Dobson, Inorg Chim Acta 1989, 73, 164. TABZAX, TABYOK
[71] JM Harrowfield, Y Kim, GA Koutsantonis, YH Lee, P Thuery, Inorg Chem 2004, 43, 1689., EVABIM, EVABUY
| Crossref | GoogleScholarGoogle Scholar |
[72] TW Hambley, GA Lawrance, M Maeder, EN Wilkes, J Chem Soc, Dalton Trans 1992, 1283., SUBMOR, SUBMUX
| Crossref | GoogleScholarGoogle Scholar |
[73] G Wei, TW Hambley, GA Lawrance, M Maeder, Aust J Chem 2002, 55, 667., BALPAG
| Crossref | GoogleScholarGoogle Scholar |
[74] NF Curtis, H Puschman, WT Robinson, Polyhedron 2009, 28, 2927., IGUFEW
| Crossref | GoogleScholarGoogle Scholar |
[75] G Aullon, PV Bernhardt, F Bozoglian, M Font-Bardia, BP MacPherson, M Martinez, C Rodriguez, X Solans, Inorg Chem 2006, 45, 8551., DAYTUS, DAYWOP
| Crossref | GoogleScholarGoogle Scholar |
[76] PV Bernhardt, F Bozoglian, G Gonzalez, M Martinez, BP Macpherson, B Sienra, Inorg Chem 2006, 45, 74., NECVUN, NECWEY, NECWAU
| Crossref | GoogleScholarGoogle Scholar |
[77] GA Lawrance, M Martinez, BW Skelton, AH White, J Chem Soc, Dalton Trans 2009, 9567. JOMNAA01
[78] GA Lawrance, M Martinez, BW Skelton, R van Eldik, AH White, Aust J Chem 1992, 45, 351., SUDCOJ
| Crossref | GoogleScholarGoogle Scholar |
[79] PV Bernhardt, M Martinez, C Rodriguez, M Vazquez, Inorg Chem 2011, 50, 1429., OTAZEP
| Crossref | GoogleScholarGoogle Scholar |
[80] TW Hambley, GA Lawrance, M Martinez, BW Skelton, AH White, J Chem Soc, Dalton Trans 1992, 1643., PACHEG
| Crossref | GoogleScholarGoogle Scholar |
[81] PV Bernhardt, P Comba, Helv Chim Acta 1991, 74, 1834.
| Crossref | GoogleScholarGoogle Scholar |
[82] JE Bol, C Buning, P Comba, J Reedijk, M Strohle, J Comput Chem 1998, 19, 512.
| Crossref | GoogleScholarGoogle Scholar |
[83] PV Bernhardt, PC Sharpe, Inorg Chem 1998, 37, 1629., NOCTAA. NOCTEE, NOCSUT
| Crossref | GoogleScholarGoogle Scholar |
[84] PV Bernhardt, PC Sharpe, Chem Commun 1996, 1267., TASCOF
| Crossref | GoogleScholarGoogle Scholar |
[85] PV Bernhardt, GA Lawrance, S Luther, M Maeder, M Rossignoli, Inorg Chim Acta 2000, 306, 1., CAHYEQ
| Crossref | GoogleScholarGoogle Scholar |
[86] BL Elliott, TW Hambley, GA Lawrance, M Maeder, G Wei, J Chem Soc, Dalton Trans 1993, 1725., WAPPIM
| Crossref | GoogleScholarGoogle Scholar |
[87] Y Baran, A Bayada, GA Lawrance, PG Lye, M Maeder, EN Wilkes, Trans Metal Chem, (Dorrecht) 2003, 28, 460.
| Crossref | GoogleScholarGoogle Scholar |
[88] N McCann, GA Lawrance, Y-M Neuhold, M Maeder, Inorg Chem 2007, 46, 4002.
| Crossref | GoogleScholarGoogle Scholar |
[89] PV Bernhardt, GA Lawrance, M Napitupulu, G Wei, Inorg Chim Acta 2000, 300, 604., CAJBEV
| Crossref | GoogleScholarGoogle Scholar |
[90] PV Bernhardt, P Comba, Chem Commun 1993, 113., JUTXIF
| Crossref | GoogleScholarGoogle Scholar |
[91] NF Curtis, R Pawley, WT Robinson, Polyhedron 2008, 27, 3041., NAJZUU
| Crossref | GoogleScholarGoogle Scholar |
[92] NF Curtis, J Waikara, Inorg Chem Commun 2011, 14, 1344., UZAJEL
| Crossref | GoogleScholarGoogle Scholar |
[93] W Eschweiler, Chem Ber 1905, 38, 880.
| Crossref | GoogleScholarGoogle Scholar |
[94] PV Bernhardt, J Chem Soc, Dalton trans 1996, 4319., RAKIK, RAXJOQ
| Crossref | GoogleScholarGoogle Scholar |
[95] PV Bernhardt, JC Hetherington, LA Jones, J Chem Soc, Dalton Trans 1996, 4325., RAXKAW, RAXLAD, RAXLEH
| Crossref | GoogleScholarGoogle Scholar |
[96] SL Jain, P Bhattacharyya, SA McLellan, DT Richens, P Lightfoot, AMZ Slawin, JD Woollins, J Chem Res 2004, 234., BETZOQ
| Crossref | GoogleScholarGoogle Scholar |
[97] PV Bernhardt, LA Jones, J Chem Soc, Dalton Trans 1998, 1757., NOZHEP, NOZHIT
| Crossref | GoogleScholarGoogle Scholar |
[98] PV Bernhardt, PC Sharpe, Inorg Chem 2000, 39, 4994., LOWCAB, LOWCEF
| Crossref | GoogleScholarGoogle Scholar |
[99] PV Bernhardt, NL Kilah, AP Meacham, P Meredith, R Vogel, Dalton Trans 2005, 2508., XASJOR, XASJUX
| Crossref | GoogleScholarGoogle Scholar |
[100] PV Bernhardt, PC Sharpe, Inorg Chem 2000, 39, 2020., LIZSUI, LIZTAP, LIZTET
| Crossref | GoogleScholarGoogle Scholar |
[101] PV Bernhardt, PC Sharpe, Inorg Chem 2000, 39, 4123., WOHXUM, WOHYAT, WOHYEX
| Crossref | GoogleScholarGoogle Scholar |
[102] PV Bernhardt, EJ Hayes, Inorg Chem 2002, 41, 2892., MOGVEJ, MOGVIN, MOGVOT, MOGVUZ, MOGWAG
| Crossref | GoogleScholarGoogle Scholar |
[103] PV Bernhardt, EG Moore, MJ Riley, Inorg Chem 2002, 41, 3025., XODTIT, XODTIT, XODTOX, XODTUZ
| Crossref | GoogleScholarGoogle Scholar |
[104] PV Bernhardt, BM Flanagan, MJ Riley, J Chem Soc, Dalton Trans 1999, 3579., LESCOB
| Crossref | GoogleScholarGoogle Scholar |
[105] EG Moore, PV Bernhardt, A Fuerstenberg, MJ Riley, E Vauthey, J Phys Chem A 2005, 109, 11715., YEBZIP
| Crossref | GoogleScholarGoogle Scholar |
[106] PD Beer, PV Bernhardt, J Chem Soc, Dalton Trans 2001, 1428., QIXREU, QIXRIY
| Crossref | GoogleScholarGoogle Scholar |
[107] PV Bernhardt, NL Creevey, Dalton Trans 2004, 914.. EWEJAY, EWEQIG, EWEQEC
| Crossref | GoogleScholarGoogle Scholar |
[108] PV Bernhardt, NL Kilah, Polyhedron 2007, 26, 392., XEVYAZ, XEVYED, XEVYIH
| Crossref | GoogleScholarGoogle Scholar |
[109] S Hasan, PV Bernhardt, J Incl Phenom Macrocyclic Chem 2009, 65, 39., MUKYOH, MUKYON, MUKZEY, MUKZAU
| Crossref | GoogleScholarGoogle Scholar |
[110] S Hasan,, NL Kilah, M Martinez, PV Bernhardt, Aust J Chem 2009, 62, 1214., HULSOX
| Crossref | GoogleScholarGoogle Scholar |
[111] Siriwardena A, Levick S. Report RI 2848, Industrial Processing Division. Wellington, New Zealand: NZ DSIR; 1989.