

Impact of the 2Fe2P core geometry on the reduction chemistry of phosphido-bridged diiron hexacarbonyl compounds†
Odi Th. E. Selan A B , Mun Hon Cheah A C * , Brendan F. Abrahams A , Robert W. Gable A and Stephen P. Best A *
A C * , Brendan F. Abrahams A , Robert W. Gable A and Stephen P. Best A *
A School of Chemistry, The University of Melbourne, Parkville, Melbourne, 3010, Vic., Australia.
B Department of Chemistry, Faculty of Science and Engineering, Nusa Cendana University, Kupang – NTT, 85001, Indonesia.
C Department of Chemistry, Molecular Biometics, Ångström Laboratory, Uppsala University, SE 75237 Uppsala, Sweden.
Handling Editor: George Koutsantonis
Australian Journal of Chemistry 75(9) 649-659 https://doi.org/10.1071/CH21309
Submitted: 30 November 2021 Accepted: 19 January 2022 Published: 26 February 2022
© 2022 The Author(s) (or their employer(s)). Published by CSIRO Publishing. This is an open access article distributed under the Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND)
Abstract
The effect of core geometry constraints of hydrogenase H-cluster analogues on reduction chemistry have been explored by a combination of structural, electrochemical and IR spectroelectrochemical (IR-SEC) studies. A series of phosphido-bridged diiron hexacarbonyl complexes, Fe2(µ2-PPh2(CH2)xPPh2)(CO)6, x = 2 (2P) and 4 (4P) and previously reported with x = 3 (3P) and the unlinked bis-diphenylphosphido (DP) analogues were investigated. The X-ray structures of the neutral complexes demonstrate the effect of the linking group on the Fe2P2 core geometry with P–Fe–Fe–P torsion angles of 95 (2P), 101 (3P), 108 (4P) and 109° (DP) and a twisting of the Fe(CO)3 fragments from an eclipsed geometry (2P, 3P and DP) for 4P. For all four compounds the primary reduction process involves two close-spaced one-electron reactions (E1 and E2) with a systematic trend to more negative reduction potentials with a shorter link between the bridging phosphorus atoms. This reflects the greater constraint that the bridging group places on the adoption of a planar 2Fe2P geometry. The sensitivity of the core geometry is greater for E2 than E1 and this impacts the stability of the monoanion with respect to disproportion (Kdisp(298 K) = 0.02 (2P), 2.4 (3P) and 3540 (4P and DP)). 4P has a stable dianion and gives reversible cyclic voltammetry at 298 K and is quasi-reversible at 253 K, whereas the response of 2P is irreversible at 298 K, with two distinct daughter products, but becomes quasi-reversible at 253 K. IR-SEC measurements enabled elucidation of the spectra and time evolution of the reduction products. These results are consistent with a bimolecular reaction giving a distinct reduced product modelled as a dimeric, 4Fe species. The sensitivity of the reduction chemistry of the bridged diiron compounds underpins their utility as catalytic proton reduction catalysts and the systematic trends delineated in this investigation provide the framework for charting the path of their redox-coupled chemical reactions.
Keywords: [Fe–Fe]-hydrogenase H cluster, bridged diiron carbonyl compounds, electrochemistry of transition metal complexes, IR spectroelectrochemistry, IR spectroscopy, phosphido-bridged diiron compounds, redox-coupled chemical reactions, transition metal carbonyl compounds.
Introduction
Both economic and environmental imperatives have driven the spectacular rise in investment in so-called green hydrogen and this, in turn, has energised the search for efficient catalysts of proton reduction and dihydrogen oxidation based on earth-abundant elements. The evolutionary solution, hydrogenases, outperform laboratory-based catalysts but are large (ca. 60 kDa) complex molecules susceptible to denaturation and are unlikely to be suitable for applications which have a significant power density requirement. Crystallographic characterisation of the metal complexes at the active site of [FeFe]-hydrogenase has underpinned the intense focus on dithiolato/azadithiolato-bridged diiron carbonyl chemistry[1,2] and while those complexes exhibit hydrogenase chemistry (mostly proton reduction),[3] the abiological complexes are uncompetitive in terms of reaction rate and efficiency (high overpotential). Those studies allow delineation of the impact of the steric and electronic properties of the complex on electrocatalytic proton reduction at the diiron centre. Elucidation of that chemistry has relied heavily on the combination of experimental observations[1,4] (voltammetry and spectroelectrochemistry) with theory[5,6] (ab initio calculations of the reaction path) and simulation of the voltammetric response. While a clear link between the chemistry of dithiolato-bridged complexes and specific phosphido-bridged analogues has been established,[7,8] differences between electronic and stereochemical characteristics of the sulfur and phosphorus donor atoms impact on the chemistry and can be used to tune the properties of the catalyst.
It has previously been shown that the identity of the bridging atom of the bridged-diiron carbonyl compounds has systematic effect on the relative stabilities and geometry of the FeIFeI and Fe0Fe0 redox states,[7,8] where the reduced form of the sulfido-bridged compounds retains a butterfly geometry 2Fe02S core[9] but a planar structure is obtained for analogous P-bridged compounds.[10] The geometry of the 2Fe02P core can be constrained to non-planar by linking the two P atoms together, where a C3 linker is calculated to have a non-planar 2Fe02P core geometry and is observed to undergo catalytic proton reduction at the 2e−, 2H+ level, albeit with a smaller rate constant than is observed for the analogous sulfido-bridged compound. The calculated geometry for the respective diprotonated 2Fe02X intermediates has a shorter H–H contact for the sulfido-bridged compound and that has been interpreted as the key factor underlying the difference in rate. While the argument for such a causal relationship is attractive, it needs to be recognised that there may be intrinsic differences in reactivity between the S- and P-bridged compounds that may complicate the comparison. Moreover, bridgehead isomer effects have been shown to have a significant impact on electrocatalytic proton reduction by related phosphido-bridged diiron carbonyl compounds.[11,12] In those studies electrocatalytic proton reduction was reported at the 2e−2H level for unlinked phosphido bridging ligands, e.g. PHPh−. For these reasons we have investigated the reduction chemistry of the complexes Fe2(μ, μ-PhP(CH2)xPPh)(CO)6, x = 2 (2P) and 4 (4P), which when considered together with unlinked and 3-C linked complex (x = 3, 3P) allows evaluation of the importance of the 2Fe2P core geometry on the reduction chemistry.
Results and discussion
Influence of the linker on the X-ray structures of the 2Fe2P forms
The X-ray structures of 2P and 4P have not previously been published and these, together with 3P,[13] illustrate the importance of the bridging ligand on the conformation of the two Fe(CO)3 fragments. When viewed parallel to the Fe–Fe bond, the two Fe(CO)3 fragments are eclipsed for both 2P and 3P but there is a significant twist about the Fe–Fe bond for 4P (Fig. 1). For 4P the X-ray structure has two crystallographically-distinct molecules in the asymmetric unit where the C–Fe–Fe–C torsion angle of the two molecules take values of 13.1° (4Pa) and 31.4° (4Pb) for the apical CO groups, with corresponding values of 5.9° and 8.3° for the basal CO groups (average). These values are much larger than those of 2P (8.7° apical, 1.8° basal) and 3P (6.7° apical, 2.0° basal). Associated with a greater twist, there is more asymmetry of the two P–Fe distances for each bridging phosphorus atom in 4Pb than 4Pa (Δ(P–Fe)av = 0.010, 4Pa, 0.026 Å 4Pb, Supplementary Table S3) and there is a small difference in Fe–Fe distance (Δ(Fe–Fe)av = 0.003 Å) consistent with a weaker Fe–Fe bond for the more twisted form. The longer Fe–Fe bond of the more distorted form of 4P, relative to the other bridged forms suggests a weakening of the Fe–Fe bonding in response to distortion of the 2P2Fe core. Clearly for 4P, the barrier to rotation of the Fe(CO)3 fragments about the Fe–Fe bond is not high and may provide more ready access to a rotated structure. This is proposed to be important for the rearrangement reactions and electrocatalytic proton reduction.[3,8,14–21]
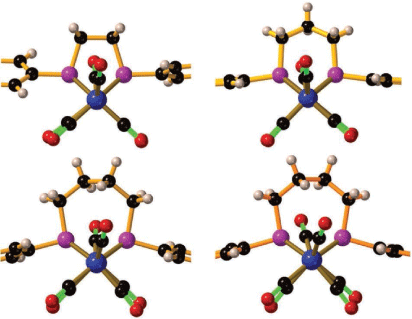
|
Reduction chemistry of 2P and 4P
Electrochemistry
Reduction of the phosphide-bridged diiron hexacarbonyl compounds DP (Fe2(μ-PPh2)2(CO)6) and 3P have been shown to proceed by a quasi-reversible, metal-based, two-electron ECE (electron-chemical-electron) process[22,23], where the chemical (C) step corresponds to a flattening of the 2Fe2P core with consequential loss of the Fe–Fe bonding contact. The relative stabilities of the one- and two-electron reduced products are closely linked to the geometry of the 2Fe2P core where, in the absence of a linking group between the phosphorus atoms, the one-electron reduced product is unstable with respect to disproportionation (Kdisp > 50 based on unobservable concentrations of [DP]1− during reduction of DP)[23]. This reaction becomes less pronounced for 3P which has a Kdisp value of ca. 13.[22] Consistent with a less constrained 2Fe2P core geometry, 4P gives a well-defined two-electron quasi-reversible reduction (Fig. 2). On lowering the temperature there is a marked increase in ΔEp (175–348 mV for ν = 100 mV s−1) and is consistent with an E1CE2 path, E2 > E1, where the chemical step is interconversion between the planar and butterfly forms of the 2Fe2P core. The general features of the electrochemistry are adequately represented by Scheme 1a, where the details of the simulation parameters used to generate Fig. 2b are given in the Supplementary Table S4. The values of E1 and E2 are most clearly delineated by the voltametric response at lower temperatures (Fig. 2c, d) and the value of E2−E1 (0.27 V) corresponds to Kdisp = 3.7 × 104 under standard conditions. Delineation of the relationship between the identity of the bridging phosphido ligand and the reduction potential requires careful attention to the calibration of the potential of the reference electrode. For non-aqueous solvents a common approach is to measure the voltammetry of the solute of interest with an approximately equimolar concentration of ferrocene. Measurements of this sort, combined with electrochemical simulation, for 2P, 3P, 4P and DP are shown in the Supplementary material (Supplementary Fig. S5, Supplementary Table S5). There are clear trends in the first and second reduction potentials, with E(2P) < E(3P) < E(4P) < E(DP) for both E1 and E2. This effect is readily explained in terms of the facility of the bridge to accommodate the more flattened 2Fe2P core of the reduced species.[7,10,22] The sensitivity of the reduction potential to the bridge is approximately twice as large for E2 as for E1 and this leads to increasing stability of the monoanion with respect to disproportionation.
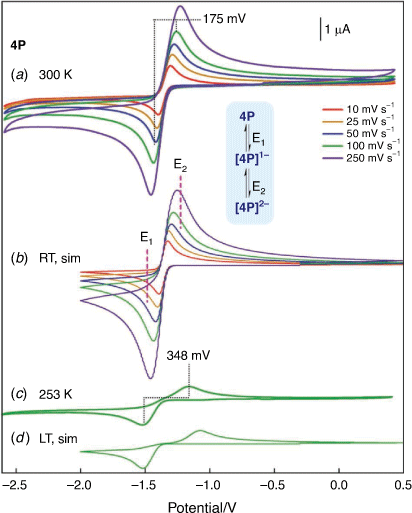
|
|
A cyclic voltammogram of 2P in 1,2-difluorobenzene was included in the supporting information of a previous publication[24] and over the limited scan range (−1.6 to −2.7 V vs Fc+/Fc) are consistent with measurements conducted in THF and CH3CN. When measured over a wider potential range it is clear that the reduction chemistry of 2P is complicated by the generation of at least two daughter products, with anodic waves for processes attributed to E3 (Epa = −1.39 V) and E4 (Epa = −0.67 V) prominent at RT voltammetry (Fig. 3a). The primary reduction involves closely spaced one-electron reductions, with E1 > E2, which are irreversible at room temperature but partly reversible at low temperature (Fig. 3c). The alternate assignment of the anodic wave at −1.39 V in the RT voltammetry to E1 can be excluded based on the very large value of ΔEP (280 mV) when compared to the value obtained at low temperature (115 mV). The value of ΔEP is most commonly larger for low temperature measurements owing to increases in uncompensated resistance. It is noted that the value of ΔEP for E1 at 253 K is in the range obtained for the Fc+/Fc couple when measured under analogous experimental conditions and supports assignment to a diffusion-controlled partly reversible process at low temperatures.
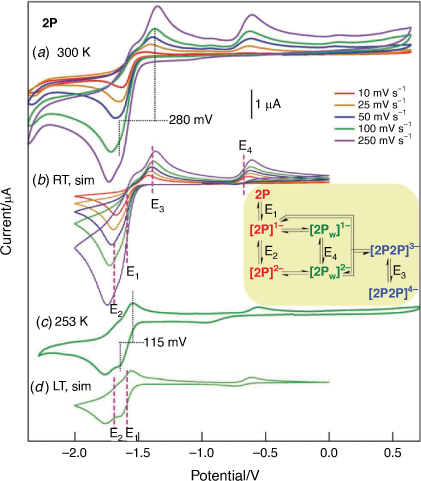
|
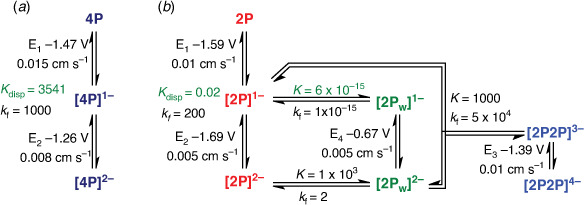
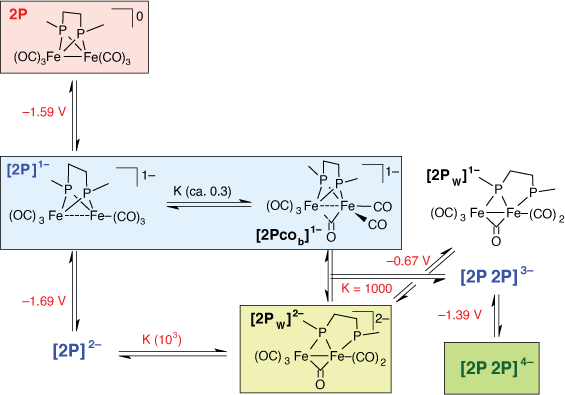
Simulation of the voltametric response of 2P can be achieved using reaction schemes which either involve an intermediate species formed en route to a more stable rearranged two-electron reduced product or where the second daughter product results from reactions following two-electron reduction. The former reaction scheme corresponds to a six-membered double square scheme[25,26] and satisfactory reproduction of the cyclic voltammetry (CV) can be obtained using this approach (Supplementary Scheme S1, Supplementary Fig. S1). Alternatively, rearrangement of the complex occurs after two-electron reduction to give a comparatively stable species ([2Pw]2−) and additional species result from the association of [2Pw]2− and reduced 2P species ([2P2P]n−, Scheme 1b). Simulations shown in Fig. 2b, d are based on the latter approach where the key thermodynamic and kinetic parameters are given in Scheme 1. The rationale for adoption of the latter, more complicated, approach is based observations of the time evolution of the species during thin-layer electrochemical experiments discussed in the following section.
IR spectroelectrochemistry (IR-SEC) of 2P and 4P
Complementary methods are necessary to identify the molecular details of the species implied by electrochemical analysis. Previous work has demonstrated that a combination of IR spectroscopy and ab initio DFT (density functional theory) calculations present an extremely effective strategy for the identification of transiently stable species generated during redox reactions[7,9,22] and provide the basis for interpretation of the reduction chemistry of 2P and 4P.
Consistent with the CV, reduction of 4P and the subsequent re-oxidation take the form of simple interconversions without the generation of observable concentrations of intermediate products (Fig. 4). Based on wavenumbers of the ν(CO) modes the reduction can be assigned to a two-electron process giving [4P]2−. Whereas the wavenumbers and profiles of the 1917, 1888, 1832 and 1805-cm−1 bands match closely that of [3P]2−, an additional weak band is observed at 1933 cm−1 which shares the same potential-dependent growth and depletion response and is also assigned to [4P]2−. Different conformations of the seven-membered rings formed by the bridging phosphido ligands evident from the crystalline form of 4P (Fig. 1) may lead to lower symmetry of the resulting dianion and/or lead to distinct conformers which are in rapid equilibrium. Either outcome would account for the presence of an additional band. A rapid equilibrium of this sort will neither impact on the chemical reversibility of the system nor, in most cases, give an observable signature in cyclic voltametric experiments. Finally, the high stability of [4P]2− and [DP]2−[23] facilitates collection of well-defined spectra of the highly reduced species and interrogation of their sensitivity to the planarity of the 2Fe2P core. The shift of the ν(CO) band wavenumbers of [DP]2− relative to those of [4P]2− of 9–11 cm−1 reflects changes to the electron richness at the metal centre, this being modified by the substituents on the bridging phosphido ligand. The deviation of the 2Fe2P core geometry from planar is manifest by the ν(CO) band profile of the lowest wavenumber band (1824 cm−1 [DP]2−, Fig. 4), this being split into two components for [4P]2− and [3P]2−.
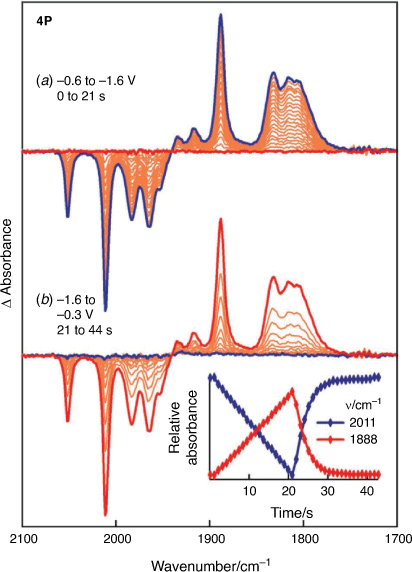
|
The CV of 2P indicates that reduction, while chemically reversible, is accompanied by rapid chemical change and the generation of two electroactive daughter products (Fig. 3). The time and potential dependence of the spectral changes in IR-SEC experiments of the reduction of 2P in THF indicate the presence of minor and major products which are oxidised at potentials of −0.8 and 0.2 V, respectively (Supplementary Fig. S2). The build-up of minor products is more pronounced for experiments conducted in CH3CN (Fig. 5) and the species identified by deconvolution of the spectra are labelled with reference to Scheme 1b – e.g. the species re-oxidised at ca. −1.39 and −0.67 V are attributed to [2P2P]4− and [2Pw]2−, respectively. Reduction of 2P is accompanied by the generation of three species where the generation of significant, though transient, concentrations [2P]1− is expected owing to the favourable comproportionation equilibrium. It is surprising that the growth of [2P2P]4− requires a significant concentration of [2Pw]2−, and the rate of growth of [2Pw]2− slows more rapidly than the rate of disappearance of 2P (6–10 s, Fig. 5 inset) and rapid growth of [2P2P]4− occurs when there are significant concentrations of both [2P]1− and [2Pw]2− (Fig. 5 inset). Further, reoxidation of [2P2P]4− is accompanied by transient increases in the concentrations of [2P]1− and [2Pw]2−. These observations exclude assignment of this species to a simple dianion of 2P and are most straightforwardly explained with the dimeric (4Fe) formulation of [2P2P]4−. The model for simulation of the cyclic voltametric response of 2P which incorporates the dimeric species (Scheme 1, Fig. 3) can be used to calculate the surface concentrations of the different species. These calculations generate trends (Supplementary Fig S4) consistent with those observed in the thin-layer IR-SEC experiments (Fig. 5). Finally, the slow depletion of [2Pw]2− at potentials intermediate between E3 and E4 may be explained by an additional equilibrium involving [2Pw]2− and [2P2P]4−.
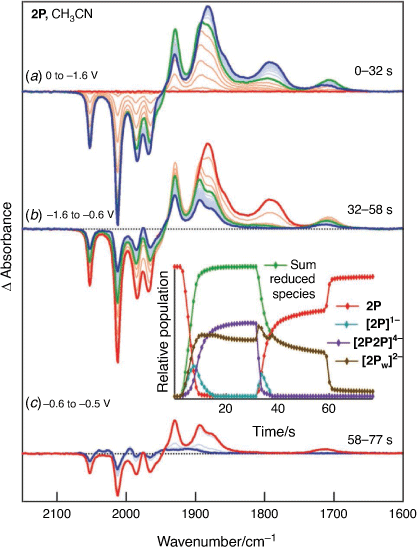
|
The ν(CO) band profile of the different reduction products can be extracted from the IR-SEC spectra by using an independently measured spectrum of the starting complex. The reliability of the spectra depends on the complexity of the product distribution and where there is simple interconversion between the oxidised and reduced components of the couple, e.g. for 4P, then well-defined spectra of the reduction product can be obtained. Despite the complexity of the reduction chemistry of 2P, during the reoxidation at times between 42 and 58 s (Fig. 5) there are significant concentrations of 2P and [2Pw]2− and this allows extraction of the spectrum of [2Pw]2−. The spectrum of [2P2P]4− can then be deduced (t = 20–30 s) and finally the spectrum of [2P]1− (t = 5–7 s). The spectra of the species identified in the IR-SEC experiments of 2P and 4P, together with the published spectra of the corresponding species derived from DP[23] and 3P[22] are brought together in Fig. 6. Despite subtle differences in the 2Fe2P core geometry of the neutral complexes there is a high level of consistency in the wavenumber and band profile of the ν(CO) modes (Fig. 6a). Structure assignment based on the agreement between the spectral profiles is straightforward for the dianion of 4P (Fig. 6c) and the rearranged two-electron reduction product of 2P (Fig. 6d). In both of these cases the ν(CO) band profiles match closely the corresponding species of 3P which, in turn, can be related to the reduction chemistry of DP. Together with the X-ray structure of [DP]2−,[10] the structures proposed for the reduction products of DP and 3P were supported by EXAFS analysis and DFT calculations[22,23] and earlier NMR measurements.[27]
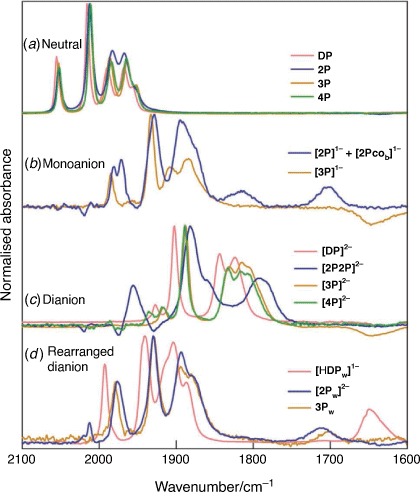
|
The spectra of the monoanion and dianion forms of 2P differ markedly from those of the reference compounds (Fig. 6b, c). Electrochemical simulation based on Scheme 1 suggests that there is significant, transient formation of [2P]1− near to the electrode and the time evolution of that product is consistent with the multicomponent analysis shown in Fig. 5. The band profile however contains more bands than can be accommodated by a single diiron species and the major component has a band profile which can be matched onto that of [3P]1−. The minor product has well-defined bands near 1710 and 1820 cm−1 together with bands overlapping those attributed to [2P]1−. The 1710-cm−1 band is indicative of a bridging CO group and this together with the spread of bands would be consistent with a structure having differentiated Fe centres as might be formed by rotation of one of the Fe(CO)3 fragments of 2P to give a bridging CO group and two terminally-bridging CO groups on one Fe centre and three on the other (Scheme 2, species [2P]1− and [2Pcob]1−). The energies of the corresponding isomeric forms of the neutral species (ΔE = 6.3 kcal) have previously been reported as part of an investigation into substituted forms of 2P and the dithiolato-bridged analogues[24] and an equilibrium closer to 1 for the corresponding isomers of the monoanion would account for the observed spectra. It is noted that the reduction potentials for the [2P]1−/2− and [2Pw]1−/2− and equilibrium constant for interconversion between [2P]2− and [2Pw]2− exclude significant contribution of [2Pw]2− to the spectrum of the monoanion forms of 2P.
|
The spectrum of the dianion form of 2P has a value of the average ν(CO) wavenumber compatible with assignment to a species reduced by two electrons per diiron fragment, but a band profile which indicates a structure significantly different from that of the DP, 3P or 4P analogues. Electrochemical simulation (Scheme 1) and the product evolution during IR-SEC experiments (Fig. 5) indicate a bimolecular reaction involving the 1e reduced form of 2P and [2Pw]2− with the product(s) reduced at potentials more negative than 1 V. Reoxidation appears to proceed with transient recovery of both [2P]1− and [2Pw]2− (Fig. 5). The structure of [2Pcob]1− includes a five-coordinate Fe centre which would be susceptible to attack by the terminally-bound phosphido ligand of [2Pw]2− which is, by analogy to [HDP]1−, Lewis basic in character. Reduction and rearrangement, including a change in binding mode of the bridging CO ligand of [2Pw]2−, gives the species designated as [2P2P]4− in Scheme 2. Preliminary DFT calculations to explore possible geometric structure of [2P2P]4− suggest that the dimer is formed by sharing of bridging phosphide ligand between two diiron subunits (Supplementary Fig. S7). We defer in depth DFT calculation exploring multiple conformations of [2P2P]4− and reduction pathway of 2P to a later publication owing to the complexity of the reduction chemistry.
The current investigation demonstrates the sensitivity of the reduction chemistry of diphosphido-bridged diiron carbonyl compounds to steric effects introduced by changing the bridge linking the phosphorus atoms. The trend in reduction potentials and relative stability of the one-electron reduced product can be linked directly to the accessibility of the reduced species to a planar 2Fe2P core geometry. This effect can be quantified in silico where P–Fe–Fe–P torsion angles of [DP]2− (180.0°), [4P]2− (147.2°), [3P]2− (140.8°) and [2P]2− (134.0°) were obtained from DFT calculations. It is interesting to consider these results in the context of earlier reports on the impact of substitution effects on the bridgehead phosphorus atom on the reduction chemistry and proton reduction catalysis.[11,12] Whereas bis(phosphido)-bridged complexes would be expected generally to accommodate a planar 2Fe2P core and have reduction chemistry analogous to DP, asymmetric substitution of the P atom (Me and CH2–Fc) leads to partly reversible or irreversible two-electron reduction. The CV response was explained in terms of the barrier to rearrangement of the planar 2Fe2P core and slow redox equilibria between the mono and dianion forms.[12] IR spectra recorded during reduction in an OTTLE (optically transparent thin-layer electrode) cell are consistent with the presence of a two-electron reduced species analogous to [DP]2− together with an ill-defined product, with spectral features and reoxidation potential analogous to [2Pw]2−. Accordingly, we suggest that the weak anodic wave designated a E8 in ref. 12 is more likely to be due to a two-electron reduced, rearranged form of the complex analogous to 3Pw.[22]
The electrochemistry of cases involving 1°-phosphido bridges, e.g. Fe2(PH(CH2Fc))2(CO)6, present more interesting electrochemical behaviour.[11,12] Whereas the primary reduction wave in those cases has a potential in line with expectations based on the phosphido-bridged compounds considered herein, the current response indicates net one-electron reduction. Based on the accessibility of a planar 2Fe2P core, the one-electron reduced species would be expected to be unstable with respect to disproportionation, i.e. a second reduction is expected at more positive potentials. While chemical reaction could in principle outrun the following reduction as is possibly implied in the earlier study[11,12] – this scenario is unlikely as the proposed reaction involves dihydrogen generation which most sensibly proceeds through the reaction of two one-electron reduced species. Simulations based on an alternate reaction path involving two-electron reduction with E1 < E2 (as observed for DP, 4P and 3P) and inclusion of bimolecular reaction between the two-electron reduced and neutral species giving dihydrogen and two equivalents of the phosphinidene bridged species [Fe2(P(CH2Fc))(PH(CH2Fc))(CO)6]1− can give results consistent with the electrochemistry and the reduction chemistry of related diiron compounds (Supplementary Fig. S6).
Conclusions
Differences in the reduction chemistry of phosphido-bridged diiron carbonyl compounds highlighted here by studies of DP and 3P have been put in context by extension to analogues with shorter (2P) and longer (4P) linking groups between the bridging phosphido donors. Distortion of the geometry of the neutral complex by the butylene group of 4P both make accessible conformers with different rotation of the Fe(CO)3 fragments about the Fe–Fe bond and accommodate a more planar 2Fe2P core for the dianion. For compounds of this class, reduction proceeds through two close-lying one-electron steps (E1 and E2) with the potentials more clearly defined by simulating the CV measurements at room and low temperature. The values of E1 and E2 are highly sensitive to the planarity of the 2Fe2P core of the dianion. DP and 4P have similar values with E1 < E2, 3P and 2P have increasingly negative values with E1 close to E2 for 3P and E1 > E2 for 2P. Contrary to reports of the modelling of voltammetry of 1°-phosphido bridged diiron carbonyl compounds, which imply one-electron reduction of the parent complex,[12] unlinked phosphido-bridged compounds undergo two-electron reduction with E1 < E2.
Both DP and 4P give monoanions that are unstable with respect to disproportionation (Kdisp = 3.7 × 104 at RT) and dianions which are stable on the timescale of CV and IR-SEC experiments. The lowest wavenumber ν(CO) band of the dianion is sensitive to the planarity of the 2Fe2P core, where a single band is obtained for [DP]2− which is split for [4P]2− and [3P]2−. The reduction chemistry of 2P is marked by the formation of two daughter products which have well-defined voltametric and spectroscopic signatures. Reduction proceeds through sequential one-electron reductions, with [2P]1− stable with respect to disproportionation (Kdisp = 2 × 10−2). Reduction to [2P]2− is irreversible at RT, but partly reversible at lower temperatures with an observable anodic wave associated with [2P]1−. There are two main daughter products formed following reduction, [2P2P]4− and [2Pw]2−.[2P2P]4− is oxidised at less negative potentials but evolves more slowly than [2Pw]2−. These observations suggest that [2P2P]4− forms from reactions involving [2Pw]2−. The electrochemical response is well modelled by inclusion of the reaction between [2Pw]2− and [2P]1−. The IR-SEC measurements show three reduction products with different time and potential dependent behaviour, which map onto the species deduced from the CV. Proposed structures for these species are based on reduction products obtained for 3P and DP in the presence of acid and are supported by the calculated band profile. The structure proposed for the dimer is speculative and is unprecedented in the chemistry of the phosphido-bridged diiron chemistry. Equilibria between 2Fe2S and 4Fe4S cores is important in iron–sulfur chemistry and a dimer has been inferred from IR-SEC measurements from related dithiolato-bridged compounds,[28] with the structure ultimately confirmed by independent chemical synthesis and X-ray characterisation.[29] The 4Fe4P species, [2P2P]4−, has a distinct structure which contains only terminally-bound CO groups and remains the subject of further investigation. The sensitivity of the reduction chemistry of the bridged diiron compounds to the constraints imposed by the linking group underpins their potential as catalytic proton reduction catalysts and the systematic trends delineated in this investigation provide the framework for charting the path of their redox-coupled chemical reactions. The extension of this work to the electrocatalytic reduction chemistry of these compounds, complemented with computational methods, will provide more nuanced insights into catalysed proton reduction by bridged diiron carbonyl compounds.
Experimental
Chemicals were purchased from Aldrich and used without further purification. Solvents were dried using standard procedures[30] and distilled under an atmosphere of dinitrogen immediately prior to use or transferred to the Vacuum Atmospheres glove box. Solutions used for electrochemical analysis were prepared under a dinitrogen atmosphere either using standard Schlenk techniques or with the aid of a glove box. The tetra-n-butylammonium hexafluorophosphate ([NBu4][PF6], TBAPF6) used for electrochemical measurements was synthesised and purified using published procedures.[31]
Synthesis
The phosphido bridging ligands were prepared by slight adaption of literature procedures.[3] This involved the cleavage of a phosphorous phenyl bond of triphenylphosphine by lithium followed by reaction with an appropriate dihaloalkane followed by protonation by THF/water. On the scale of the reaction, final purification of the air-sensitive product by vacuum distillation was, in our hands, inefficient and the crude product was reacted with an excess of Fe3(CO)12. Specifically, Fe2(PhP(CH2)4PPh)(CO)6, 4P, was prepared by the reaction of a solution of Fe3(CO)12 (2.398 g, 5.056 mmol) in dry toluene (75 mL) with a toluene solution of PHPh(CH2)4PHPh (1.4 g, ca. 5 mmol, 10 mL). The reaction mixture was stirred for 3 h at 100 °C. The solvent was removed under vacuum and the resulting residue extracted in dichloromethane (2 × 5 mL). The product was purified by column chromatography (silica using a 30:70 mixture of dichloromethane:hexane as eluent). The first yellow band was collected, the solvent removed and the product obtained as a yellow crystalline solid following recrystallisation from THF. Yield (19%), IR (THF): νCO 2051, 2011, 1983, 1964, 1952 cm−1.[31] P[1H] NMR (CDCl3): δ 131.6 (s). 1H NMR (CD2Cl2): δ 7.83−7.40 (m, C6H5, 10H), 2.00 (m, PCH2, 4H), 2.09 (m, PCH2CH2CH2CH2P, 2H). Samples of Fe2(PhP(CH2)2PPh)(CO)6, 2P were obtained using a procedure analogous to that previously reported.[24]
Electrochemistry
An autolab PGSTAT30 potentiostat with GPES software (Autolab) was used for cyclic voltammetric experiments which were conducted in a purpose-build gas-tight glass electrochemical cell. The solutions were prepared, and cell was assembled in a Vacuum Atmospheres glove box. The electrodes comprised a 1 mm disc working (Pt or vitreous carbon), Pt wire counter and a Ag wire jacketed with solvent/supporting electrolyte served as a pseudo-reference electrode. The disc electrodes were polished using alumina (0.3, 3 and 9 μm) impregnated discs (Buehler). The potential of the pseudo-reference electrode was referenced against the Fc+/Fc couple. All potentials are given with reference to the standard hydrogen electrode (SHE), this being calibrated against ferrocene (E°(Fc+/Fc). Similar rates of heterogeneous electron transfer for 2P, 4P and Fc were needed to simulate the cyclic voltametric response of mixtures of Fc and the respective solute species (Supplementary Fig. S5). Measurements of the voltammetric response at low temperatures were achieved using a jacketed electrochemical cell with water:ethylene glycol (50:50) coolant and a Kent refrigerated circulating water bath.
Electrochemical simulations were conducted using DigiElch[32] – Professional build version 8.225. Simulations employed a planar electrode geometry (area = 0.007 cm2) and for simplicity the uncompensated resistance and capacitance were set to zero. Diffusion was treated using a semi-infinite 1D model.
Infrared spectroelectrochemistry (IR-SEC)
The high-pressure IR-SEC cell has previously been described.[33] The cell operates in reflection/absorption mode with a flat and highly polished 3 mm vitreous carbon disc working electrode. Platinum ring counter and a Ag pseudo-reference electrode complete the electrochemical cell. Owing to practical constraints the potential of the reference electrode was not calibrated during IR-SEC experiments. The potential of the Ag electrode was typically +0.4 V vs SHE. The cell was assembled then transferred to the glove box, filled with 0.5–1 mL of the solution under investigation, sealed and removed from the glove box. A gas line is then connected to the cell and a high-purity N2 atmosphere of 200 kPa maintained throughout the experiment. A Bio-Rad FTS 175C spectrophotometer (glowbar source, a Ge/KBr beam splitter and liquid-nitrogen cooled MCT detector). The solution under investigation comprised a thin film of solution (10–20 μm) trapped between the working electrode and the CaF2 window. The thickness of the film could be adjusted under micrometer control.
X-ray crystal structures
A crystal of 2P was mounted on a Siemens SMART Apex CCD diffractometer[34] in a stream of cooled nitrogen. Data were collected on a yellow plate (0.20 × 0.20 × 0.10 mm) at 130 K using MoKα radiation (λ = 0.71073 Å). The structure was solved using direct methods (SHELXS-86)[35] and refined using a full matrix least squares procedure based upon F2 (SHELXL).[36] Absorption corrections were applied using SADABS.[34] All hydrogens were included in the refinement, constrained to geometrical estimates, with an isotropic displacement parameter of 1.2 times the displacement parameter of the parent carbon atom. Crystal data and refinement details: C20H14Fe2O6P2, monoclinic, P21/n; a = 12.2037(8), b = 11.7826(8) (1), c = 15.1800(10) Å, β = 102.5500(10)°, V = 2130.6(2) Å3; Z = 4; μ = 15.46 cm−1, max. and min. transmission factors, 0.864, 0.748; 2θ range: 2.2–28.2°; 13 304 reflections measured of which 5022 were independent, all 5022 reflections were employed in the refinement; goodness of fit = 1.033; wR2 (all data based on F2) = 0.0695; R (I < 2σ(I) = 0.0282).
A crystal of 4P was transferred directly from the mother liquor to a viscous protective oil before being mounted on an Oxford Diffraction SuperNova diffractometer in a stream of cooled nitrogen. Data were collected on a yellow block-shaped crystal (0.20 ×0.35 × 0.39 mm) at 130 K using CuKα radiation (λ = 1.5418 Å). The structure was solved using direct methods (SHELXS) and refined using a full matrix least squares procedure based upon F2 (SHELXL-97).[37] Crystal data and refinement details: C44H36Fe4O12P4 (formula for two crystallographically distinct molecules in the asymmetric unit); monoclinic, P21/n; a = 16.0952(1), b = 13.8062(1), c = 20.8322(2) Å, β = 100.628(1)°, V = 4549.78(6) Å3; Z = 4; μ = 11.882 cm−1, max. and min. transmission factors, 0.233, 0.051; 2θ range: 6.4–134.3°; 29 437 reflections measured of which 8132 were independent, all 8132 reflections were employed in the refinement; goodness of fit = 1.031; wR2 (all data based on F2) = 0.1489; R (I < 2σ(I) = 0.0556).
CCDC 2124850 and 2124851 contain supplementary X-ray crystallographic data for 2P and 4P respectively. This data can be obtained free of charge via http://www.ccdc.cam.ac.uk/structures/, or from the Cambridge Crystallographic Data Centre, Union Road, Cambridge, CB2 1EZ; fax(+44) 1223–336–033 or email: deposit@ccdc.cam.ac.uk.
DFT calculations
DFT geometry optimisations were performed on the Tetralith cluster, National Supercomputer Centre performed using ORCA 5.0.1 software package[38] with TPSS functional[39] and def2-TZVP basis sets.[40] Solvation effects were included using the CPCM model with parameters for THF.[41] The resolution of identity approximation (RI-J)[42] was applied and all phenyl groups in [2P2P]4−were replaced by methyl groups so as to reduce the computational time needed for convergence.
Data availability
The crystallographic data can be obtained free of charge via http://www.ccdc.cam.ac.uk/structures/, or from the Cambridge Crystallographic Data Centre, Union Road, Cambridge, CB2 1EZ; fax(+44) 1223–336–033 or email: deposit@ccdc.cam.ac.uk. The remaining data that support this study will be shared upon reasonable request to the corresponding author.
Conflicts of interest
The authors declare no conflict of interest in relation to any aspect of this publication.
Declaration of funding
The crystallographic component of the research was supported by a research grant (LE100100109) from the Australian Research Council. MHC gratefully acknowledges funding support from the Swedish Energy Agency through grant no. 50529-1 and computation resources provided by SNIC through the National Supercomputer Centre at Linköping University (Tetralith) under projects SNIC2021/5-474 and SNIC2021/5-305. No other external funding was directly associated with the research or preparation of manuscript.
Supplementary material
Supplementary material is available online.
Acknowledgements
SPB thanks the Australian Research Council for the support which enabled establishment of the electrochemical and IR-SEC facilities at the University of Melbourne. SPB thanks Alcoa Australia for the donation of the Vacuum Atmospheres glove box. OTES thanks the Australian government for the award of an Australia Awards Scholarship (AAS) Indonesia.
References
[1] Apfel U-P, Petillon FY, Schollhammer P, Talarmin J, Weigand W. [FeFe] hydrogenase models: an overview. In: Weigand W, Schollhammer P, editors. Bioinspired Catalysis: Metal-Sulfur Complexes. Wiley-VCH Verlag GmbH & Co. KgaA; 2015. pp. 79–103.[2] W Lubitz, H Ogata, O Rudiger, E Reijerse, Hydrogenases. Chem Rev 2014, 114, 4081.
| Hydrogenases.Crossref | GoogleScholarGoogle Scholar | 24655035PubMed |
[3] F Wittkamp, M Senger, ST Stripp, UP Apfel, [FeFe]-Hydrogenases: recent developments and future perspectives. Chem Commun 2018, 54, 5934.
| [FeFe]-Hydrogenases: recent developments and future perspectives.Crossref | GoogleScholarGoogle Scholar |
[4] J-F Capon, F Gloaguen, P Schollhammer, J Talarmin, Catalysis of the electrochemical H2 evolution by di-iron sub-site models. Coordination Chemistry Reviews 2005, 249, 1664.
| Catalysis of the electrochemical H2 evolution by di-iron sub-site models.Crossref | GoogleScholarGoogle Scholar |
[5] Greco C, De Gioia L, DFT investigation of models related to the active site of hydrogenases. In: Weigand W, Schollhammer P, editors. Bioinspired Catalysis: Metal-Sulfur Complexes. Wiley-VCH Verlag GmbH & Co. KgaA; 2015. pp. 137–160.
[6] D Chouffai, J-F Capon, L De Gioia, FY Petillon, P Schollhammer, J Talarmin, G Zampella, A Diferrous Dithiolate as a Model of the Elusive Hinactox State of the [FeFe] Hydrogenases: An Electrochemical and Theoretical Dissection of Its Redox Chemistry. Inorg Chem 2015, 54, 299.
| A Diferrous Dithiolate as a Model of the Elusive Hinactox State of the [FeFe] Hydrogenases: An Electrochemical and Theoretical Dissection of Its Redox Chemistry.Crossref | GoogleScholarGoogle Scholar | 25496017PubMed |
[7] MH Cheah, SP Best, XAFS and DFT Characterisation of Protonated Reduced Fe Hydrogenase Analogues and Their Implications for Electrocatalytic Proton Reduction. Eur J Inorg Chem 2011, 7, 1128.
[8] MH Cheah, C Tard, SJ Borg, X Liu, SK Ibrahim, CJ Pickett, SP Best, Modeling [Fe-Fe] Hydrogenase: Evidence for Bridging Carbonyl and Distal Iron Coordination Vacancy in an Electrocatalytically Competent Proton Reduction by an Iron Thiolate Assembly That Operates through Fe(0)-Fe(II) Levels. J Am Chem Soc 2007, 129, 11085.
| Modeling [Fe-Fe] Hydrogenase: Evidence for Bridging Carbonyl and Distal Iron Coordination Vacancy in an Electrocatalytically Competent Proton Reduction by an Iron Thiolate Assembly That Operates through Fe(0)-Fe(II) Levels.Crossref | GoogleScholarGoogle Scholar | 17705475PubMed |
[9] SJ Borg, JW Tye, MB Hall, SP Best, Assignment of molecular structures to the electrochemical reduction products of diiron compounds related to [Fe-Fe] hydrogenase: a combined experimental and density functional theory study. Inorg Chem 2007, 46, 384.
| Assignment of molecular structures to the electrochemical reduction products of diiron compounds related to [Fe-Fe] hydrogenase: a combined experimental and density functional theory study.Crossref | GoogleScholarGoogle Scholar | 17279816PubMed |
[10] RE Ginsburg, RK Rothrock, RG Finke, JP Collman, LF Dahl, The (metal-metal)-nonbonding[Fe2(CO)6(μ2-PPh2)2]2- dianion. Synthesis, structural analysis of its unusual dimeric geometry, and stereochemical-bonding implications. J Am Chem Soc 1979, 101, 6550.
| The (metal-metal)-nonbonding[Fe2(CO)6(μ2-PPh2)2]2- dianion. Synthesis, structural analysis of its unusual dimeric geometry, and stereochemical-bonding implications.Crossref | GoogleScholarGoogle Scholar |
[11] A Rahaman, C Gimbert-Surinach, A Ficks, GE Ball, M Bhadbhade, M Haukka, L Higham, E Nordlander, SB Colbran, Bridgehead isomer effects in bis(phosphido)-bridged diiron hexacarbonyl proton reduction electrocatalysts. Dalton Trans 2017, 46, 3207.
| Bridgehead isomer effects in bis(phosphido)-bridged diiron hexacarbonyl proton reduction electrocatalysts.Crossref | GoogleScholarGoogle Scholar | 28221379PubMed |
[12] C Gimbert-Surinach, M Bhadbhade, SB Colbran, Bridgehead Hydrogen Atoms Are Important: Unusual Electrochemistry and Proton Reduction at Iron Dimers with Ferrocenyl-Substituted Phosphido Bridges. Organometallics 2012, 31, 3480.
| Bridgehead Hydrogen Atoms Are Important: Unusual Electrochemistry and Proton Reduction at Iron Dimers with Ferrocenyl-Substituted Phosphido Bridges.Crossref | GoogleScholarGoogle Scholar |
[13] AL Reingold, Structure of Fe2(μ-PhPprPPh)(CO)6. Acta Crystallogr, Sect C: Cryst Struct Commun 1985, 41, 1043.
[14] C Greco, M Bruschi, P Fantucci, L De Gioia, Relation between coordination geometry and stereoelectronic properties in DFT models of the CO-inhibited [FeFe]-hydrogenase cofactor. J Organomet Chem 2009, 694, 2846.
| Relation between coordination geometry and stereoelectronic properties in DFT models of the CO-inhibited [FeFe]-hydrogenase cofactor.Crossref | GoogleScholarGoogle Scholar |
[15] C Tard, CJ Pickett, Structural and Functional Analogues of the Active Sites of the [Fe]-, [NiFe]-, and [FeFe]-Hydrogenases. Chem Rev 2009, 109, 2245.
| Structural and Functional Analogues of the Active Sites of the [Fe]-, [NiFe]-, and [FeFe]-Hydrogenases.Crossref | GoogleScholarGoogle Scholar | 19438209PubMed |
[16] R Goy, L Bertini, C Elleouet, H Goerls, G Zampella, J Talarmin, L De Gioia, P Schollhammer, U-P Apfel, W Weigand, A sterically stabilized FeI-FeI semi-rotated conformation of [FeFe] hydrogenase subsite model. Dalton Trans 2015, 44, 1690.
| A sterically stabilized FeI-FeI semi-rotated conformation of [FeFe] hydrogenase subsite model.Crossref | GoogleScholarGoogle Scholar | 25436832PubMed |
[17] MK Harb, A Daraosheh, H Goerls, ER Smith, GJ Meyer, MT Swenson, T Sakamoto, RS Glass, DL Lichtenberger, DH Evans, M El-khateeb, W Weigand, Effects of Alkane Linker Length and Chalcogen Character in [FeFe]-Hydrogenase Inspired Compounds. Heteroat Chem 2014, 25, 592.
| Effects of Alkane Linker Length and Chalcogen Character in [FeFe]-Hydrogenase Inspired Compounds.Crossref | GoogleScholarGoogle Scholar |
[18] W Wang, TB Rauchfuss, CE Moore, AL Rheingold, L De Gioia, G Zampella, Crystallographic Characterization of a Fully Rotated, Basic Diiron Dithiolate: Model for the Hred State? Chem - Eur J 2013, 19, 15476.
| Crystallographic Characterization of a Fully Rotated, Basic Diiron Dithiolate: Model for the Hred State?Crossref | GoogleScholarGoogle Scholar | 24130068PubMed |
[19] S Munery, J-F Capon, L De Gioia, C Elleouet, C Greco, FY Petillon, P Schollhammer, J Talarmin, G Zampella, New FeI-FeI Complex Featuring a Rotated Conformation Related to [2Fe]H Subsite of the [Fe-Fe] Hydrogenase. Chem - Eur J 2013, 19, 15458.
| New FeI-FeI Complex Featuring a Rotated Conformation Related to [2Fe]H Subsite of the [Fe-Fe] Hydrogenase.Crossref | GoogleScholarGoogle Scholar | 24127299PubMed |
[20] C-H Hsieh, OF Erdem, SD Harman, ML Singleton, E Reijerse, W Lubitz, CV Popescu, JH Reibenspies, SM Brothers, MB Hall, MY Darensbourg, Structural and Spectroscopic Features of Mixed Valent FeIIFeI Complexes and Factors Related to the Rotated Configuration of Diiron Hydrogenase. J Am Chem Soc 2012, 134, 13089.
| Structural and Spectroscopic Features of Mixed Valent FeIIFeI Complexes and Factors Related to the Rotated Configuration of Diiron Hydrogenase.Crossref | GoogleScholarGoogle Scholar | 22774845PubMed |
[21] T Liu, MY Darensbourg, A Mixed-Valent, Fe(II)Fe(I), Diiron Complex Reproduces the Unique Rotated State of the [FeFe]Hydrogenase Active Site. J Am Chem Soc 2007, 129, 7008.
| Fe(II)Fe(I), Diiron Complex Reproduces the Unique Rotated State of the [FeFe]Hydrogenase Active Site.Crossref | GoogleScholarGoogle Scholar | 17497786PubMed |
[22] MH Cheah, SJ Borg, SP Best, Steps along the Path to Dihydrogen Activation at [FeFe] Hydrogenase Structural Models: Dependence of the Core Geometry on Electrocatalytic Proton Reduction. Inorg Chem 2007, 46, 1741.
| Steps along the Path to Dihydrogen Activation at [FeFe] Hydrogenase Structural Models: Dependence of the Core Geometry on Electrocatalytic Proton Reduction.Crossref | GoogleScholarGoogle Scholar | 17256930PubMed |
[23] MH Cheah, SJ Borg, MI Bondin, SP Best, Electrocatalytic Proton Reduction by Phosphido-Bridged Diiron Carbonyl Compounds: Distant Relations to the H-Cluster? Inorg Chem 2004, 43, 5635.
| Electrocatalytic Proton Reduction by Phosphido-Bridged Diiron Carbonyl Compounds: Distant Relations to the H-Cluster?Crossref | GoogleScholarGoogle Scholar | 15332815PubMed |
[24] R Zaffaroni, TB Rauchfuss, A Fuller, L De Gioia, G Zampella, Contrasting Protonation Behavior of Diphosphido vs Dithiolato Diiron(I) Carbonyl Complexes. Organometallics 2013, 32, 232.
| Contrasting Protonation Behavior of Diphosphido vs Dithiolato Diiron(I) Carbonyl Complexes.Crossref | GoogleScholarGoogle Scholar |
[25] AJL Pombeiro, Guedes da Silva, MFC Lemos, MANDA , Electron-transfer induced isomerizations of coordination compounds. Coord Chem Rev 2001, 219–221, 53.
| Electron-transfer induced isomerizations of coordination compounds.Crossref | GoogleScholarGoogle Scholar |
[26] S-X Guo, AWA Mariotti, C Schlipf, AM Bond, AG Wedd, Investigation of the Pronounced Medium Effects Observed in the Voltammetry of the Highly Charged Lacunary Anions [α-SiW11O39]8- and [α-PW11O39]7-. Inorg Chem 2006, 21, 8563.
| Investigation of the Pronounced Medium Effects Observed in the Voltammetry of the Highly Charged Lacunary Anions [α-SiW11O39]8- and [α-PW11O39]7-.Crossref | GoogleScholarGoogle Scholar | 17029367PubMed |
[27] YF Yu, J Gallucci, A Wojcicki, Novel mode of reduction of phosphido-bridged, metal-metal-bonded binuclear complexes. Synthesis and reactivity of an unsymmetrical anion from bis(μ-diphenylphosphido)hexacarbonyldiiron [Fe2(CO)6(μ-PPh2)2]. J Am Chem Soc 1983, 105, 4826.
| Novel mode of reduction of phosphido-bridged, metal-metal-bonded binuclear complexes. Synthesis and reactivity of an unsymmetrical anion from bis(μ-diphenylphosphido)hexacarbonyldiiron [Fe2(CO)6(μ-PPh2)2].Crossref | GoogleScholarGoogle Scholar |
[28] SJ Borg, T Behrsing, SP Best, M Razavet, X Liu, CJ Pickett, Electron Transfer at a Dithiolate-Bridged Diiron Assembly: Electrocatalytic Hydrogen Evolution. J Am Chem Soc 2004, 126, 16988.
| Electron Transfer at a Dithiolate-Bridged Diiron Assembly: Electrocatalytic Hydrogen Evolution.Crossref | GoogleScholarGoogle Scholar | 15612737PubMed |
[29] I Aguirre de Carcer, A DiPasquale, AL Rheingold, DM Heinekey, Active-Site Models for Iron Hydrogenases: Reduction Chemistry of Dinuclear Iron Complexes. Inorganic Chemistry 2006, 45, 8000.
| Active-Site Models for Iron Hydrogenases: Reduction Chemistry of Dinuclear Iron Complexes.Crossref | GoogleScholarGoogle Scholar | 16999394PubMed |
[30] Errington RJ. Guide to Practical Inorganic and Organo-Metallic Chemistry. London: Blackie Academic & Professional; 1997. p. 256.
[31] Sawyer DT, Sobkowiak A, Roberts JJL. Electrochemistry for Chemists, 2nd edn. New York, NY: Wiley-Interscience; 1995. p, 505.
[32] M Rudolph, DP Reddy, SW Feldberg, A Simulator for Cyclic Voltammetric Responses. Anal Chem 1994, 66, 589A.
| A Simulator for Cyclic Voltammetric Responses.Crossref | GoogleScholarGoogle Scholar |
[33] SJ Borg, SP Best, Spectroelectrochemical cell for the study of interactions between redox-activated species and moderate pressures of gaseous substrates. J Electroanal Chem 2002, 535, 57.
| Spectroelectrochemical cell for the study of interactions between redox-activated species and moderate pressures of gaseous substrates.Crossref | GoogleScholarGoogle Scholar |
[34] Bruker SMART, SAINT & SADABS; Bruker AXS Inc.: Madison, Wisconsin, USA, 2000.
[35] GM Sheldrick, Acta Cryst A 1990, 46, 467.
| Crossref | GoogleScholarGoogle Scholar |
[36] GM Sheldrick, Crystal structure refinement with SHELXL. Acta Crystallogr C 2015, 71, 3.
| Crystal structure refinement with SHELXL.Crossref | GoogleScholarGoogle Scholar |
[37] GM Sheldrick, A short history of SHELX. Acta Crystallogr A 2008, 64, 112.
| A short history of SHELX.Crossref | GoogleScholarGoogle Scholar | 18156677PubMed |
[38] F Neese, Software update: the ORCA program system, version 4.0. WIREs Computational Molecular Science 2018, 8, e1327.
| Software update: the ORCA program system, version 4.0.Crossref | GoogleScholarGoogle Scholar |
[39] J Tao, JP Perdew, VN Staroverov, GE Scuseria, Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids. Phys Rev Lett 2003, 91, 146401.
| Climbing the density functional ladder: nonempirical meta-generalized gradient approximation designed for molecules and solids.Crossref | GoogleScholarGoogle Scholar | 14611541PubMed |
[40] J Zheng, X Xu, DG Truhlar, Minimally augmented Karlsruhe basis sets. Theor Chem Acc 2011, 128, 295.
| Minimally augmented Karlsruhe basis sets.Crossref | GoogleScholarGoogle Scholar |
[41] M Garcia-Rates, F Neese, Effect of the Solute Cavity on the Solvation Energy and its Derivatives within the Framework of the Gaussian Charge Scheme. J Comput Chem 2020, 41, 922.
| Effect of the Solute Cavity on the Solvation Energy and its Derivatives within the Framework of the Gaussian Charge Scheme.Crossref | GoogleScholarGoogle Scholar | 31889331PubMed |
[42] F Neese, An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix. J Comput Chem 2003, 24, 1740.
| An improvement of the resolution of the identity approximation for the formation of the Coulomb matrix.Crossref | GoogleScholarGoogle Scholar | 12964192PubMed |
† This paper is dedicated to Professor Glen Deacon, in appreciation of his contributions to the Inorganic Chemistry community.