

The Future of Retrosynthesis and Synthetic Planning: Algorithmic, Humanistic or the Interplay?
Craig M. Williams A B and Madeleine A. Dallaston A
A B and Madeleine A. Dallaston A
A School of Chemistry and Molecular Biosciences, University of Queensland, Brisbane, Qld 4072, Australia.
B Corresponding author. Email: c.williams3@uq.edu.au

Craig M. Williams (C.M.W.) was born in Adelaide, Australia. He received his B.Sc. (Hons) degree in chemistry in 1994 from Flinders University. In 1997, he was awarded his Ph.D. in organic chemistry from the same institution under the supervision of Professor Rolf H. Prager. He undertook post-doctoral studies as an Alexander von Humboldt Fellow working with Professor Armin de Meijere at the Georg-August-Universität, Göttingen, Germany, from 1997 to 1999. In early 1999, he accepted a second post-doctoral fellowship at the Australian National University with Professor Lewis N. Mander. Professor Williams has held an academic position at the University of Queensland since 2000, and during this time has won a number of awards including a Thieme Chemistry Journals Award in 2007, an Australian Research Council Future Fellowship award in 2011, and the Award for Outstanding Contribution to Research (SCMB, UQ, 2019). The Williams research group explores numerous interests within the discipline of organic chemistry (e.g. medicinal chemistry, fundamental molecules, natural product isolation, microelectronics, drug and agrichemical development, impact sensitive molecules) enabled by organic synthesis refined through the construction of biologically active complex natural products (diterpenes, polyketides, alkaloids), and designs synthetic methodology to assist in this endeavour (synthetic transformations and reagents). Professor Williams especially enjoys teaching whole molecule retrosynthesis to undergraduate and post-graduate students. |

Madeleine A. Dallaston (M.A.D.) was born in Leicester, England, and grew up in Brisbane, Australia. In 2016, she received her B.Sc. from Griffith University, earning the RACI Queensland Branch Prize for that year. In 2017, she undertook a B.Sc. (Hons) at the University of Queensland in collaboration with Defense Science and Technology, working on energetic materials for countermeasures development. She then commenced her Ph.D. in organic chemistry at the University of Queensland under the supervision of Professor Williams and G. Paul Savage in 2018, focusing on the synthesis of novel heterocycles for application as bioisosteres. |
Australian Journal of Chemistry 74(5) 291-326 https://doi.org/10.1071/CH20371
Submitted: 21 December 2020 Accepted: 25 March 2021 Published: 12 May 2021
Journal Compilation © CSIRO 2021 Open Access CC BY
Abstract
The practice of deploying and teaching retrosynthesis is on the cusp of considerable change, which in turn forces practitioners and educators to contemplate whether this impending change will advance or erode the efficiency and elegance of organic synthesis in the future. A short treatise is presented herein that covers the concept of retrosynthesis, along with exemplified methods and theories, and an attempt to comprehend the impact of artificial intelligence in an era when freely and commercially available retrosynthetic and forward synthesis planning programs are increasingly prevalent. Will the computer ever compete with human retrosynthetic design and the art of organic synthesis?
Keywords: retrosynthesis, biomimetic synthesis, natural products, computer assisted synthesis, total synthesis, computer assisted retrosynthesis, artificial intelligence, antithetic analysis.
Introduction
Retrosynthesis (or retrosynthetic analysis or antithetic analysis) is defined as ‘a problem-solving technique for transforming the structure of a synthetic target molecule to a sequence of progressively simpler structures along a pathway which ultimately leads to simple or commercially available starting materials for a chemical synthesis’.[1] This definition was formulated and underpinned by extensive research endeavours in heuristic chemical synthesis performed by the research group led by Elias James Corey, who won the 1990 Nobel Prize in Chemistry ‘for his development of the theory and methodology of organic synthesis’.[2] It should be recognised, however, that many talented and famous scientists performing organic synthesis were already utilising some form of innate retrosynthesis before the term was coined. From an educational perspective, Warren masterfully described synthetic planning using the disconnection approach,[3] which is an intuitive method that likely embodied early thinking. Overall, however, it is hard to ascertain whether the very early methods preceding Corey’s antithetic analysis focussed on bond breaking (disconnection), or more on literature-inspired bond formation (i.e. literature knowledge of chemical reaction methodology), or both,[4] but in modern times, the situation is rapidly changing. For example, reviews covering synthetic achievements by subject (e.g. drug synthesis[5] and natural product synthesis[6–8]) are generating highly refined retrosynthetic approaches, as are reaction popularity assessments (e.g. medicinal chemistry[9]) and modern methodology-focussed retrosynthetic planning (e.g. C–H activation[10] and photochemistry[11]). The most evolutionary change currently being confronted by the synthetic community, however, is the realisation that computer-aided retrosynthesis,[12] and the accompanying artificial intelligence (AI) that proposes forward syntheses,[13] may very soon become commonly available to educators and practitioners. It is this verge of revolutionary retrosynthesis that has inspired the present review, and as such the authors will attempt to shed light on a subject that has been in motion since the late 1960s, but not in the past widely adopted. To assist in achieving the goal of this perspective, classical and historical methods and practices of retrosynthesis will be briefly overviewed (i.e. using examples and experiences well known to the authors), along with an assessment of currently available retrosynthesis programs. Combined, this assessment will hopefully generate philosophical debate around the pros and cons of AI introduction and contemplation on the future of organic synthesis design.
Retrosynthetic Methods and Practices
There are essentially two forms of retrosynthesis, and these are highly integrated. The first is functional group interconversion (FGI) or functional group exchanges, and the second is whole-molecule (or target-orientated) retrosynthesis.
FGI plays a role in both, whereby the first mode is the ability to navigate through a range of functional groups in a linear fashion to arrive at an alternative functional group (this also includes protecting groups). An example to highlight this situation is the stereoselective synthesis of squalene (1) in 1959, which was performed by husband-and-wife team Drs John and Rita Cornforth and, presumably, their joint student K. Mathew.[14] At this time, the term retrosynthesis did not exist; however, conceptual thinking around construction of the target was discussed. Interestingly, the approach presented focussed on the number of carbon subunits required to achieve the 30-carbon framework (i.e. in terms of numerical values entered into an equation[15]), and that a reported olefin synthesis method would be used. This is a clear example of deploying a literature-inspired bond formation strategy, and to achieve this goal a large number of FGIs were undertaken (Scheme 1). To exemplify this point, homogeranyl lithium (2) was required to add to dichlorodiketone (3) in the final stages of the synthesis, and this was derived from homogeranyl bromide (4). Bromide 4 was in turn obtained from alcohol 5, which was prepared via reduction of homogeranic acid (6). Finally, base hydrolysis of geranyl cyanide (7) afforded 6 (Scheme 1). Overall, five FGIs were deployed to transform 7 into 2, but the homogeranyl carbon framework went unchanged, i.e. only the end functional group is different.
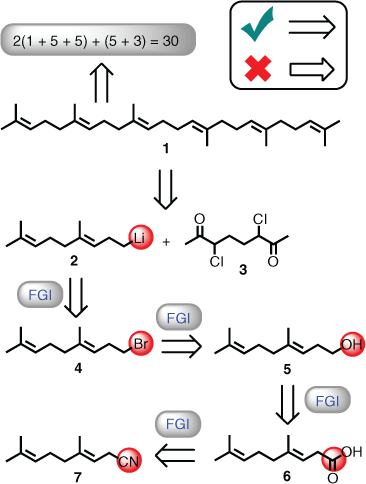
|
Although arguably FGI can also be considered target-orientated synthesis, the latter term is really reserved for targets that require as part of their retrosynthetic planning a combination of carbon framework manipulations and FGIs. Examples that fit this category include small, large, simple, and complex molecular targets, and thus are only limited by academic imagination and industrial desires. Some (of the many) areas that have benefitted from these endeavours include flavours, fragrances, pharmaceuticals, agrichemicals, materials, natural products, and environmental and fundamental molecules.
With all these drivers and inspiration to make such molecules, members of the synthetic community have resorted to numerous retrosynthetic approaches to achieve successful completion of their proposed targets (see below).[17a] Interestingly, the majority of these approaches were developed through the process of performing natural product total synthesis, mainly because natural products were the first on the scene during the emergence of the discipline of organic chemistry.[17b] Following isolation and subsequent elucidation of a natural product, there was an immediate structural challenge, often augmented by intriguing biology activity, that provided a substantial synthetic challenge. This is not to say that non-natural targets have not contributed to the art, but the fact that Mother Nature has for eternity been generating a vast library of molecules has presented a ‘spoiled for choice’ situation for synthetic enthusiasts and educationalists.
The various retrosynthetic approach concepts are outlined below using select examples well known to the authors.
Biomimetic Synthesis
Biomimetic, or biogenetically inspired, synthesis continues to be a regularly deployed method to construct complex natural products. In essence, this area evolved from natural product isolation and synthetic chemists postulating how organisms might produce secondary metabolites through biosynthesis and/or accompanying metabolic processes. The retrosynthetic planning process therefore adopts synthetic transformations, and/or key intermediates, that mimic a postulated biological process (e.g. enzymatic oxidation) undertaken by the organism.[18] It should be noted that domino,[19] tandem,[20] and cascade[21] reactions are prevalent, but importantly inspired by nature.
An example that embraces both biomimetic and domino-based retrosynthetic planning is the total synthesis of diterpenes in the vibsane series of natural products.[22] Interestingly, the isolation of this diterpene class was inspired by reports that the scrub Viburnum awabuki (Caplifoliaceae) was used as a fish poison by Japanese villagers (i.e. piscicidal activity).[23] Since that time, other viburnum species (i.e. V. odoratissimum and V. suspensum) have yielded a wide variety of different structure types all proposed to be biosynthetically related, and many displaying pronounced neurite outgrowth activity.[24] Of relevance to this section are the neovibsanins, which Fukuyama[25] proposed were biosynthetically derived from vibsanin B (8), via key intermediate 9 (Scheme 2). Intermediate 9 was then proposed to undergo a series of cyclisations and rearrangements, the first being tetrahydrofuran formation to give 10, which is poised to undergo further cyclisation to neovibsanin A (11, via path A), or provide carbocation 12 via path B. Carbocation 12 can then be attacked either intermolecularly by water to give neovibsanin H (13, via path C), or intramolecularly by the pendant homoisoprenyl alkene to give the bicyclic cation 14 (via path D). Cation 14 is then open to either nucleophilic attack by water, resulting in the formation of neovibsanin F (15, via path E), or elimination via path F to afford neovibsanin G (16) (Scheme 2).
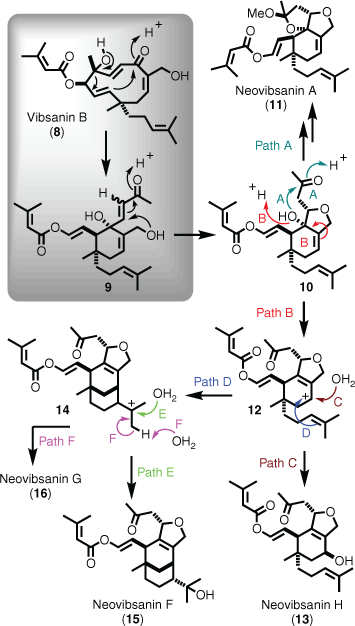
|
Digestion of this extensive biosynthetic proposal suggested that if a candidate similar to intermediate 9 was entertained during retrosynthetic analysis, then via multiple acid-catalysed steps, it might be possible to access the entire neovibsanin class of natural products. Equally, it could be considered that total synthesis of vibsanin B (8) might be a viable starting point, but approaches to construct the 11-membered ring are daunting in comparison with developing strategies for decorating a six-membered ring of the type seen in 9. With these considerations front of mind, the following retrosynthesis was formulated (Scheme 3). It was opted to install the enol ester side chain last, giving rise to 17, which although carrying the requisite retrons was better equated as 18 to hold oxygenation at the doubly allylic position. Access to 18 would likely arise from a combination of alkylation and addition to cyclohexenone 19, with the overall sequence starting from methylcyclohexenone 20 (Scheme 3).
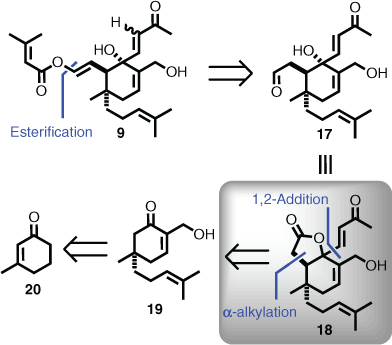
|
Although commercially available at the time, it was more convenient to synthesise methylcyclohexenone 20 in-house on large scale from ethyl acetylacetate (21).[26] Addition of homoprenyl magnesium bromide (22) to 20 could be achieved using standard cuprate conditions on reasonable scale in the racemic series (i.e. 23),[27] but also asymmetrically using the chiral ligand 24, affording the 11R-isomer 25 (Scheme 4). Dehydrogenation using IBX·NMO (26; IBX = o-iodoxybenzoic acid, NMO = N-methylmorpholine N-oxide)[28] gave 27, setting the stage for introduction of the important methylene hydroxy group (i.e. 28) via a Morita–Baylis–Hillman reaction. Ultimately, this was achieved by developing a green chemistry method using the common surfactant sodium dodecyl sulfate (SDS) in water, which could be performed at scale.[29] Silyl protection of the pendant hydroxyl group enabled enolate-mediated alkylation of ketone 28 with ethyl iodoacetate, which gave a mixture of diastereomers 29 and 30. The major diastereomer 30 was then reacted with the acyl anion equivalent,[30] lithium dithiazide (31)[31] to give lactone 32. Unmasking the aldehyde was performed using mercury acetate, and the final methyl enone was installed using ylid 33, to afford 34 (Scheme 4).[32]
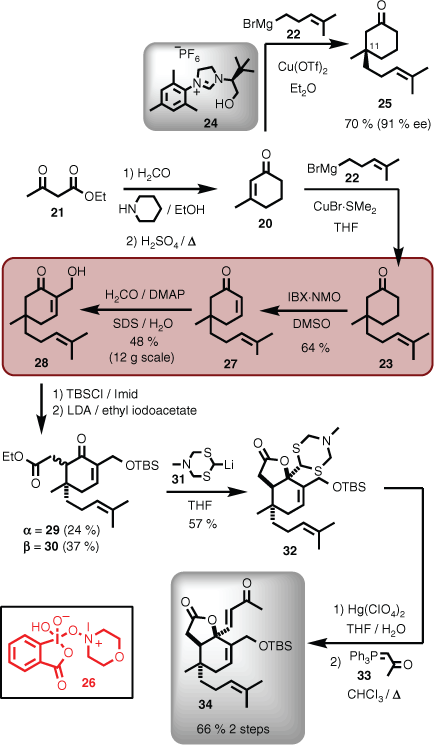
|
Having access to an advanced intermediate (i.e. 34) that most closely matched that proposed biosynthetically (i.e. intermediate 9) enabled the next phase of the biomimetic synthesis to be investigated. An important aspect in moving forward in this regard was the fact that many neovibsanin family members contained methoxy groups, presumably from methylation of the corresponding hydroxyl group via plant methyl transferases.[33] Therefore, instead of attempting to emulate an aqueous environment or conditions, methanol could be used as both the solvent and nucleophile. Accordingly, 34 was dissolved in methanol and treated with concentrated sulfuric acid at room temperature overnight. The strongly acidic conditions promoted several cascading reactions, the first of which was deprotection of the t-butyldimethylsilyl (TBS) group to unmask the primary alcohol seen in 35. Oxy-Michael addition formed the tetrahydrofuran ring observed in 36, which underwent epimerisation (i.e. 37), before solvolysis and attack by the solvent (path A) to give carboxylic acid 38. Under the reaction conditions, the acid undergoes Fischer esterification to afford the methyl ester (39). Global reduction followed by global oxidation provided aldehyde 40, which was immediately subjected to conditions developed by Davies to install the elaborate enol ester sidechain. This final manoeuvre delivered (±)-2-O-methylneovibsanin H (41) in 12 synthetic operations, whereby an acid-catalysed, one-pot, four-step sequence was key to achieving a concise total synthesis based on a biosynthetic proposal (Scheme 5).[32]

|
Based on these biogenetically inspired results, a similar domino reaction, albeit combining Brønsted and Lewis acid promotion, successfully led to the asymmetric total synthesis of (–)-neovibsanin G (42) starting from 25 (Scheme 5).[34] However, it is worthwhile to note that even the most well-considered retrosynthetic planning can come unstuck, and this occurred when attempting the total synthesis of neovibsanins A and B.[35] It was thought that the proposed advanced intermediate 34 was suitably flexible to follow the proposed biosynthetic route, but one fault resided within 34, which amounted to a single stereocentre that was opposite to that proposed for biosynthetic intermediate 9 (Scheme 5, red highlight). On treatment of 34 with concentrated sulfuric acid, although this time at 4°C overnight (room temperature (r.t.) resulted in decomposition), the resulting alcohol (43) did not undergo solvolysis, but instead acted as a nucleophile, giving rise to ester 44. Ketalisation then afforded the 4,5-bis-epi-esters 45 and 46, which were carried through via the respective aldehydes (47 and 48) to the non-natural isomers 4,5-bis-epi-neovibsanin A (49) and B (50) (Scheme 5).[35] Of note is that the biological activity of 49 and 50 was very similar to that of neovibsanins A and B.[35] Furthermore, a related outcome was experienced when spirovibsanin A was targeted, in that (±)-5,14-bis-epi-spirovibsanin A (51) was obtained (Scheme 5).[36]
Although retrosynthetic planning based on biosynthesis postulates rarely utilises enzymatic methods for performing forward synthesis, such synthetic transformations mediated by enzymatic control have found wide application in all other areas of organic synthesis.[37] That said, they are very substrate-dependant.[38] Lastly, from a philosophical perspective, adopting a retrosynthetic plan based on naturally occurring biosynthetic considerations could be considered plagiarism from Mother Nature. A similar argument could be mounted for AI, which presents a similar philosophical dilemma (see further below).
Linear Synthesis
Linear (or consecutive) syntheses are the most common outcomes of retrosynthetic planning, because they are by design iterative, and thus cater for both small and large target molecules. Like all synthetic approaches to a target molecule, linear syntheses have their place; however, generally they are avoided from both an elegance perspective (e.g. ideal syntheses)[39] and efficiency point of view (e.g. step economy),[40] i.e. convergence approaches are preferred when possible (see below).
A landmark contribution of tactical linear retrosynthesis deployment is that of the cubane skeleton (i.e. cubane-1,4-dicarboxylic acid (52) and the corresponding dimethyl ester (53)) reported in 1964 by Philip Eaton, and coworker Thomas Cole.[41] The sequence was later modified by the same authors for conversion into the Platonic solid cubane (54), a seminal contribution to the field of strained hydrocarbons.[42] Although a retrosynthetic approach was not discussed, as was the convention in that era, the clear inspiration for the route taken hinged on the fact that cyclopentadienone (55) underwent regioselective [4+2]-dimerisation.[43] However, at that time the endo stereoselective nature of these reactions was not defined, but was thought to be endo as drawn by Hafner and Goliasch in 1961.[44] Interestingly, Eaton independently confirmed the endo assignment via the known hydrocarbon 56, but the route was not verified with either experimental procedures or cited.[41] Apparently, this was achieved by reduction of dicyclopentadien-1,8-dione (57) to the diketone (58), followed by conversion to the bisthioketal (59), and final reduction via Raney nickel to endo-tetrahydrodicyclopentadiene (56). These key pieces of knowledge regarding the endo-stereochemistry, combined with reports that chlorinated cyclopentadienones also undergo [4+2]-dimerisation,[43,45] enabled Eaton to consider the following approach. Gaining access to a suitably substituted cyclopentadienone (60) would provide a dihalogenated endo-dicyclopentadien-1,8-dione (61). Although, given the photochemically sensitive bridgehead bromide seen in 61, a daring regioselective photolysis was postulated to drive a [2+2]-cycloaddition to form the precursor cage (62), a subsequent double ring contraction was then envisaged to give the 1,4-substituted cubane framework (i.e. 63), enabled by the Favorskii rearrangement (Scheme 6).
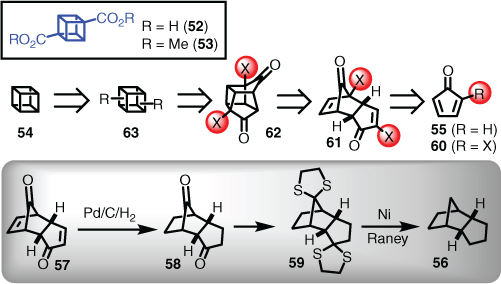
|
In the forward direction, the synthesis started by treating cyclopentenone (64) with N-bromosuccinimide, which gave the monobrominated material 65 en route to the tribromide 66. Exposing 66 to ethylamine in diethyl ether at low temperature generated 2-bromo-cyclopentadienone (67), which underwent immediate dimerisation to afford the diketone dimer (68) in 40 % yield over three steps. Subsequent UV irradiation effected the [2+2]-cycloaddition to give the cage precursor 69, which was treated with potassium hydroxide to mediate the double Favorskii, and reveal cubane-1,4-dicarboxylic acid (52). The corresponding dimethyl ester (53) could be readily obtained through classical Fischer esterification conditions (i.e. methanol and catalytic amounts of sulfuric acid). Although there are now numerous methods to access cubane itself (i.e. 54), a more expedient route consists of double decarboxylation of the diacid (52),[46] using modified Barton conditions that use chloroform as the H atom donor (Scheme 7).[47]
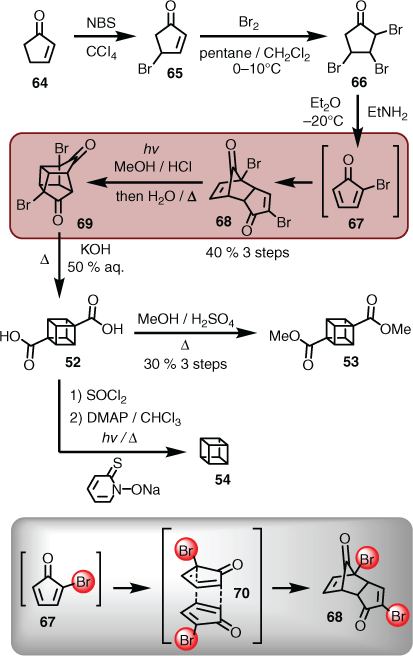
|
Given the original questions around endo-selectivity concerning the cyclopentadienone dimerisation, Eaton justified this stereochemical configuration on the grounds that ‘interactions of like dipoles should be minimized in the geometry of the transition state [i.e. 70] for Diels–Alder dimerization’ (Scheme 7).[41]
Since Eaton’s reports, there has been a great deal of activity in terms of both broadening the understanding of and developing synthetic methods for the cubane system.[48] A large-scale industrial process was developed for the synthesis of dimethyl cubane-1,4-dicarboxylate (53),[49] which was found not to be impact-sensitive, unlike the corresponding diacid (52).[50] Furthermore, cubane has more recently become known as a phenyl ring isostere, and has been deployed as a bioisostere in medicinal and agricultural chemistry.[51]
Convergent Synthesis
Convergent syntheses are based on a branched retrosynthetic pathway such that the individual branches lead to smaller fragments (synthons) that are more synthetically tractable, and that can be rapidly assembled in a convergent manner. This approach saves considerable synthetic steps (and or manipulations) compared with other methods, in particular linear sequences (see above).[52]
The Australian rainforest polyketide EBC-23 (71), isolated from Cinnamomum laubatii (family Lauraceae) in northern Queensland, is an example of a three-component convergent synthesis.[53] The inspiration to undertake this collaborative industry project included encouraging biological activity (i.e. growth inhibitor activity of the androgen-independent prostate tumour cell line DU145), and the necessity to confirm both the proposed relative and absolute stereochemical assignment. Ancillary drivers included undertaking a total synthesis campaign under project management milestone conditions, and being mindful that the synthesis should be designed such that it was process chemistry transferrable for potential large-scale production.
The retrosynthetic approach for EBC-23 started by disconnection of the EE-configured spiroketal unit, because it was plausible that this was occurring biosynthetically (i.e. selective oxidation of the polyol chain to a single keto moiety facilitating thermodynamic cyclisation via the anomeric effect), and it was also highly conceivable synthetically.[54] The corresponding ketone (72) initiated the first point of convergence, which was facilitated by considering acyl anion equivalent chemistry (i.e. 73),[30] which provided a left- (74) and right- (75) hand fragment. The left-hand fragment (74) could be further dissected using the same acyl anion approach to give epoxides 76 and 77, whereas the right-hand fragment (75) was perfectly poised for a ring-closing metathesis-styled disconnection leading to 78, which could be accessed from a functionalised syn-diol of the type seen in 79. The convergent approach also provided several built-in fallback strategies associated with the left- (74) and right- (75) hand fragments. For example, the left could be accessed via traditional asymmetric aldol methods (e.g. sequentially coupling 80 to aldehydes 81 and 82), whereas the right hand could materialise from the chiral pool (e.g. deoxy sugar 83) (Scheme 8).

|
In the forward direction, the left-hand fragment 74 was tackled first, as it was a chance to explore and select the appropriate acyl anion chemistry for the final coupling of both halves. In this regard, Tietze[55]–Smith[56] linchpin methodology was chosen, which is founded on the Brook rearrangement of silylated dithiane anions that facilitate an anion relay reaction involving non-symmetrical epoxides. On that basis, epoxide 84 was first prepared. This was achieved in two steps (via 85) using the method reported by Lepoittevin,[57] which provided the desired material in high enantiomeric excess (ee) and in large quantities, starting from commercial epi-chlorohydrin (86). TMS-dithiane (87) was prepared by treating dithiane (88) with n-butyl lithium (nBuLi) and quenching the resulting anion with trimethylsilyl chloride (TMSCl). Subsequent treatment of TMS-dithiane (87) with nBuLi provided the TMS-dithiane anion (89), which when reacted with epoxide 84, followed by addition of epi-chlorohydrin (86), afforded the desired disubstituted dithiane (90). Interestingly, unmasking the acyl anion equivalent (i.e. dithiane moiety seen in 90) by oxidative removal with mercury perchlorate not only provided the ketone function, but also mediated TMS deprotection, giving the hydroxyketone 91. The unstable hydroxyketone (91) was immediately subjected to a Prasad syn-selective reduction using diethylmethoxyborane and sodium borohydride,[58] and the resulting diol protected as an acetonide (i.e. 92). All transformations proceeded in high yields with high diastereomeric (de) selection and enantiomeric excess (Scheme 9). To effect the second acyl anion coupling, a second epoxide was required that contained syn-1,2-diol attributes (i.e. 79). Access to this unit (i.e. 93) arose from deployment of an enantiotopic group and diastereotopic face-selective Sharpless epoxidation reported by Schreiber and others[59] starting from divinyl carbinol (94). However, a stereochemical inversion of the first formed product (i.e. 95) was required to obtain the desired stereochemistry, and this was achieved using Mitsunobu conditions, which gave ester 96. The ester was then cleaved, and TBS protection installed using t-butyldimethylsilyl chloride (TBSCl). Although the TMS-dithiane (87) was used initially, the TBS-dithiane anion (97) (i.e. treatment of TBS-dithiane (98) with nBuLi) was found to provide superior coupling in terms of delivering the advanced intermediate 99 (Scheme 9).

|
Completing the synthesis required considerable troubleshooting in terms of finding ring-closing metathesis (RCM) conditions, and a workable sequence of deprotection and cyclisation through to the target.[53,60,61] The final sequence proceeded as follows. Addition of the acryloyl group to 99 proceeded smoothly to afford 100, but the ensuing RCM reaction required both harsh conditions and a catalyst that could maintain sustained activity. In this regard, the second-generation Hoveyda–Grubbs (101) catalyst[62] delivered in spades, in what is believed to be overcoming severe steric encumbrance and likely counterproductive co-ordination to the dithiane sulfur atoms. The desired product (102) was then stripped of silyl protection using hydrogen fluoride (HF) to give the polyol (103), setting the stage for final spiroketalisation. Cerium ammonium nitrate (CAN) was found to be critical for this final step in that it not only facilitated the unmasking of the keto functionality, but also metal-templated the cyclisation, as observed previously by Evans.[63] This last manoeuvre delivered EBC-23 (71) concisely (i.e. two degrees of convergence with nine total linear steps in 8 % overall yield), which enabled confirmation of structure and further biological studies (Scheme 10).
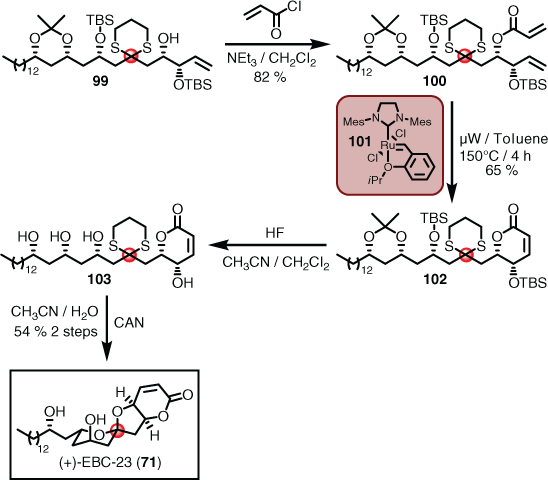
|
Chemical Degradation
Chemical degradation covers an aspect of retrosynthetic thinking that was only really made possible owing to the then-emerging field of natural product isolation and elucidation (i.e. before modern methods involving NMR spectroscopy).[64] The main approach in the early era was to perform a multitude of chemical degradation studies on the unknown isolate looking for functional group and skeletal clues (e.g. via melting point, boiling point, density, refractive indices, optical rotation) to assist in solving the structure.[64] Elucidation methodology of this type took a great leap forward, however, when in 1948 Woodward combined the evolving mechanistic understanding of chemical reactions with natural product elucidation (i.e. understanding degradation pathways from a mechanistic standpoint).[65] By this time, of course, chemical reactions were becoming better understood through the adoption of reaction arrows; however, although having been introduced 25 years prior,[66] these concepts were slow to be adopted in the natural product isolation field.[67] To highlight the point of mechanistic insight, Woodward chose the santonin (104) and santonic acid (105) story as an exemplar case.[67] For 100 years, many organic chemists had been trying to solve the structure of santonin (104),[68] which had been isolated from wormseed (Artemisia santonica) in 1830.[69] Although the flat structure of santonin (104) was solved at the end of this 100-year period, one of the unexplained products of degradation was santonic acid (105).[67] Interestingly, this Holy Grail of the time was obtained by vigorous boiling of 104 with concentrated alkali.[67] Woodward set about demonstrating that by evaluating the various base-induced degradation products and intermediates from a mechanistic view point, a proposed structure could be rationally determined, and then verified experimentally.[65,67] Woodward focussed on the functionality that would be likely susceptible to treatment with hydroxide, which first involved ring-opening of the lactone in santonin (104) to give alcohol 106. Enolisation of the conjugated γ-enone in 106, via 107, revealed the possibility of a ketone (i.e. 108) in the C-ring. The C-ring ketone could then undergo enolisation and reach over and attack the enone in the A-ring (i.e. 109). It was this critical thinking that led to the notion that santonic acid (105) was a cage bicyclic system (i.e. quite the novelty system in the day) (Scheme 11). Note it took many more decades for the absolute stereochemistry of both (–)-α-santonin (110)[70] and santonic acid (111)[71] to be solved.
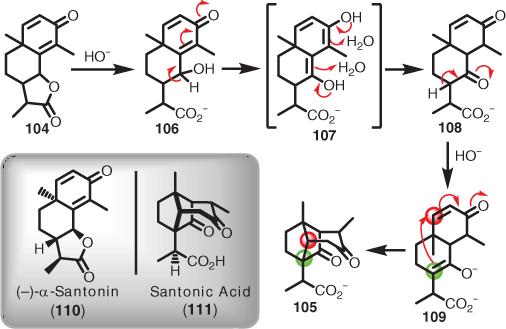
|
Although santonic acid (105) was not a natural product in its own right, this endeavour married the emerging field of reaction mechanism understanding with the field of natural product chemistry. The outcome of this merger produced several key discipline shifts: (1) it added a new dimension to the elucidation of natural products (and degradation products); (2) it elevated the notion that natural products could be transformed into related natural products and/or systems; (3) it assisted in laying the foundation for partial and relay syntheses (see below); and (4) it demonstrated that new chemical transformations could be incorporated into retrosynthetic thinking.
A further example to highlight these points are the Galbulimima alkaloids, which also integrated biosynthetic understanding. These unique alkaloids, which now number ~35 members in four separate classes, have so far been isolated from the bark of the tropical rainforest trees Galbulimima belgraveana and Galbulimima baccata.[72–75] These evergreen species are found in northern Australia, Indonesia, and Papua New Guinea, and have been used extensively by native tribes owing to their hallucinogenic and medicinal properties.[72–75]
Himgaline (112) is an interesting case in point.[72,76,77] It was solved using classical chemical degradation reinforced by semi-synthesis, with the final stages of the structure being proved using oxidative elimination of 112 using 70 % nitric acid to give GB13 (113). The overall conversion was represented in several simplified schemes showing the starting alcohol moiety (114) giving the eliminated enone function (i.e. 115) and the fragmented dialkylamine (116).[78,79] Furthermore, it was reported that catalytic hydrogenation of GB13 (113) in the presence of acid reconstituted himgaline (112), both installing the last nitrogen-containing ring (via an aza-Michael addition) giving oxohimgaline (not shown), and hydrogenating the resulting ketone to an alcohol (i.e. himgaline).[78,79] In light of this information, it was clear that any total synthesis of himgaline would be best achieved via GB13 (Scheme 12).
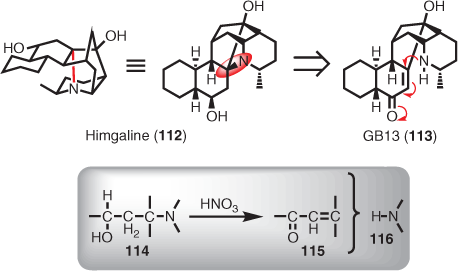
|
Mander and McLachlan were the first to report a synthesis of (±)-GB13, via a sequence that involved multiple dearomatisation steps, ring contraction/cleavage/annulation, and a Diels–Alder cycloaddition.[80] The details of this synthesis, which showcases the utility of benzenoid synthons,[81] started with a Birch reduction[82] alkylation protocol.[83] The dianion of 2,5-dimethoxybenzoic acid (117) was generated via lithium–ammonia reduction, and then quenched with 3-methoxybenzyl bromide (118) to give the dearomatised bis-enol ether 119. Double protonolysis and cyclisation of 119 with sulfuric acid gave a benzobicyclo[3.3.1]nonane (120), which underwent decarboxylation followed by methoxymethyl (MOM) protection of the bridgehead hydroxyl (i.e. 121). Installation of the adjacent diazo function with p-nitrobenzenesulfonyl azide gave ketone 122, setting the stage for a Wolff rearrangement-mediated ring contraction. Irradiation of 122 in the presence of hexamethyldisilazane resulted in the intermediate ketene being intercepted, and on protonolysis afforded the amide (123). Even though a mixture of diastereomers eventuated, this was inconsequential given that the next step aimed to install a double bond for the envisaged Diels–Alder reaction. This was achieved first by conversion of the amide function into a cyano group, and then via deprotonation and trapping with diphenylselenide, providing the option to install the bridge double bond (i.e. 124) by syn-elimination of the phenylselenyloxide. A normal demand Diels–Alder reaction involving the nitrile (124) and silyl enol ether 125 proceeded smoothly albeit slowly under Lewis acid catalysis (126) to produce the desired adduct (i.e. 127). Interestingly, this reaction was performed neat but using a large excess of the silyl enol ether, i.e. the diene acted as both reagent and solvent. TBS deprotection, reduction, and protection afforded 128, which was subjected to a second dissolving metal reduction (keto and cyano) to unmask the enone (129) via liberation of the intermediate enol ether. Stepwise epoxidation of 129 provided epoxyketone 130, which underwent Eschenmoser fragmentation in high yield to give 131. Both the acetylenic unit and the keto function were then converted into oximes (i.e. 132), and then reduced and cyclised as an annulation tactic to install the last ring of the GB13 skeleton (i.e. 133). Skeletal manipulations and protecting group dancing afforded ketone 134, which after Saegusa oxidation created the necessary enone moiety seen in 135. Double deprotection of the piperidine ring nitrogen and the bridgehead hydroxy group provided GB13 (113) in a total of 29 steps (Scheme 13).

|
At the completion of the synthesis, Mander and McLachlan stated that the synthesis of GB13 laid a foundation for the preparation of himgaline (112).[80] They further commented that intramolecular Michael addition of the nitrogen to the enone in GB13 followed by reduction of the ensuing saturated cyclic ketone should lead to himgaline, according to the degradation studies reported by Mander and Prager et al. in 1967.[78,79] Subsequent work by Chackalamannil,[84] and Evans,[85] achieved the total synthesis of himgaline (112) via different routes, although the final stages of both syntheses were based on a retrosynthetic analysis involving GB13 (113) as the penultimate step before Mander’s predicted Michael addition reduction sequence. For other Galbulimima alkaloid syntheses (e.g. (–)-GB17), see Thomson et al.[86]
Additional examples that are recommended to the interested reader include the total syntheses of bleomycin A2 (136),[87] based on degradation-inspired retrosynthesis, and xylopinine (137),[88] based on a mass spectrometry degradative fragmentation-inspired retrosynthesis. However, it should be noted that there are instances where complex unknown substances have resisted elucidation by natural product degradation, e.g. phorbol (138) (Fig. 1),[89] and in turn attracted considerable synthetic attention.[90]
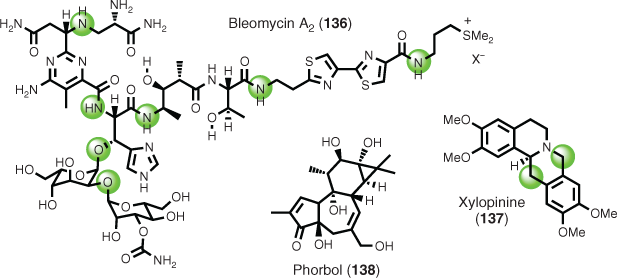
|
Partial Synthesis
Partial synthesis (or semi-synthesis) relates to natural product synthesis using a mixture of biosynthetic, degradation, and FGI-inspired retrosynthetic concepts. This often means that the skeleton is already (or mostly) intact and that FGI chemistry is then deployed to achieve the synthesis of the target. In some cases, the starting point (i.e. starting material) can be non-natural, but ultimately it is also derived from a nature source (e.g. via degradation). A wonderful example where the wide utility of semi-syntheses can contribute to an area of science is that of the gibberellin plant hormones.[91] A specific example is the conversion of gibberellic acid (139) to gibberellin A19 (GA19, 140).[92] At the time, GA19 had only ever been isolated once in workable analytical quantities from bamboo shoots (i.e. 14 mg from 44 tons), and was required for biosynthetic studies.[92] First, gibberellic acid (139) was esterified with diazomethane followed by protection of the bridgehead hydroxyl (via MOM) to give 141. Dissolving metal reduction cleaved the lactone bridge, removed the A ring oxygenation, and isomerised the double bond to afford 142, which was converted into the diazoketone (143). Cyclopropanation mediated by copper was the key synthetic manoeuvre to introduce the desired carbon bridge, and as expected, the cyclopropylketone (144) was obtained in high yield. A second lithium in ammonia reduction cleaved the cyclopropane bond that made up a six-membered ring, in turn leaving a five-membered ring ketone (145). Oxidative cleavage of the resulting five-membered ring was challenging, but it eventually yielded to a two-step process first involving treatment with potassium hydride (KH) and quenching with methyl iodide to afford an enol ether (146), followed by exposure to ozone. Reductive workup with dimethylsulfide (DMS) provided the desired tertiary aldehyde and C4 carboxylic acid functional groups (i.e. 147). Sequential basic and acidic hydrolytic conditions revealed the target (Scheme 14).

|
Pharmaceutically relevant examples include the semi-synthesis of both Taxol® (148)[93] and artemether (149) (Fig. 2).[94]
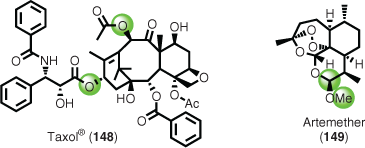
|
Relay Synthesis
Relay synthesis is a term used for the conversion of one natural product into another so as to intercept and supply an advanced intermediate for a total synthesis campaign. For retrosynthetic thinking, a route that employs a relay overcomes access to advanced intermediates in meagre quantities via chemical synthesis by using partial synthesis. A very early example was provided by Robinson for the synthesis of cholesterol (150),[95] with more recent examples reported by Shi, Tan, and Gin for the synthesis of the C18-bisnorditerpene alkaloid neofinaconitine (151),[96–98] and Deslongchamps et al. for ouabagenin (152).[99] The latter is a particularly instructive example whereby advanced intermediate 155 was achieved via the planned Diels–Alder based retrosynthetic route (i.e. reaction of 153 with 154 to give 155), but additional material (i.e. 156) was produced, enabled by a relay sequence obtained from degradation of 152 (via 157). The ability to inject a vital advanced intermediate into the total synthesis campaign was critical to the completion of the planned route, which involved re-establishment of the furanone ring via 158 and 159 (Scheme 15).

|
Chemoenzymatic Synthesis
Building on statements made above concerning biomimetic syntheses, it was noted that enzymes are an important set of ‘reagents’ that can be integrated solely or partly into retrosynthetic route design. A prime example is enzyme-mediated hydroxylation of hydrocarbon scaffolds using cytochromes P450 (P450s).[100] To highlight this point, Shen and Renata took commonly available diterpenes and converted them into high-value and rare natural products to demonstrate the power of combining chemical and enzymatic oxidation methods via a chemoenzymatic platform.[101] Among the numerous examples, steviol (160) was first oxidised with PtmO6, which was better overproduced on expression in Escherichia coli, followed by a traditional pyridinium dichromate (PDC) oxidation to give the C7 ketone (161). Not only was the yield high over two steps, but the selectivity was remarkable.
Note: regioselective C7 oxidation of terpenoid substrates is challenging, as exemplified by attempts to construct the ABC ring system (i.e. 162) of gedunin (163) via the devised retrosynthetic plan involving advanced intermediate 164. After considerable investigation, the synthesis of 162 was achieved in 17 steps starting from 165 (Scheme 16).[102]

|
Continuation of the chemoenzymatic synthesis involved another enzymatic oxidation, but this time using PtmO5, which is a class I P450 that requires a separate reductase partner (e.g. RhFRed) to support function and turnover. The adoption of this enzyme installed a β-OH at C11 with absolute stereospecificity to give 166. Acylation of 166 and deployment of the Appel reaction gave amide 167, which underwent double hydride reduction to give 168. Subjecting 168 to selenium dioxide gave the bridge allylic alcohol (i.e. C15) that was selectively oxidised with IBX[103] to afford rosthornin C (169) (Scheme 16).
Formal Synthesis
A formal synthesis is achieved when a practitioner performs a synthesis of a known target molecule using a different approach to that reported, but instead of completing the entire synthesis, the sequence stops at an advanced intermediate intersecting the previously disclosed synthetic route. For the purposes of retrosynthetic thinking, it is not the target itself that warrants a full disconnection evaluation, and associated forward planning, but the advanced intermediate of choice becomes the synthetic target. These indirect targets are usually embedded in the final stages of a prior reported synthetic route.
Formal syntheses are numerous and widespread. An instructive example is that of the marine natural product 7,20-diisocyanoadociane (170), which became a popular target because of considerable antimalarial activity shown by this and related systems.[104] Corey was the first to complete a total synthesis of 170 in 26 steps, via the dione advanced intermediate 171, starting from glutaric anhydride (172).[105] Mander was the first to establish the stereochemical relationships at C7 and C20, by completing the first formal synthesis of 170 starting from the anisole derivative 173.[106] The intersecting target in this formal synthesis was the diamine (174), which Simpson and Garson had previously reported could be converted in two steps to 170 (Scheme 17).[107] However, numerous groups focussed on targeting Corey’s dione (i.e. 171) as the point of establishing their formal syntheses. The first was that by Miyaoka,[108] which took 29 steps from (S)-4-methyltetrahydro-2H-pyran-2-one (175), followed by Vanderwal in 21 steps from perillaldehyde (176),[109] and then Thomson with a shattering 12-step synthesis of the dione (171) originating from desmethylcarvone (177).[110] Vanderwal ended up finishing this entertaining fight with a knockout blow constituting a 10-step total synthesis (not formal) of 170 starting from dehydrocryptone (178) (Scheme 17).[111]
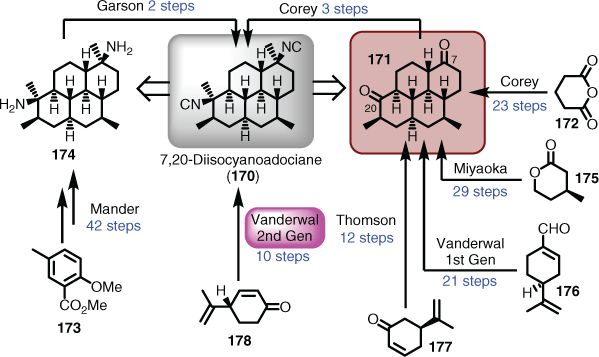
|
Methodology-Driven Synthesis
Many target-orientated syntheses are retrosynthetically designed around a certain type of synthetic methodology. This choice can be inspired by a multitude of reasons, some of which include: (1) testing newly discovered methodology designed in-house (e.g. C–H activation);[10] (2) long-standing experience with certain reaction types (e.g. Claisen rearrangement);[112] and (3) trust (or even distrust) of certain synthetic methodologies (Suzuki cross-couplings).[9] Overall, a key piece of methodology is identified as the focal point of the synthetic route design, and this in turn impacts the retrosynthetic thinking process, as the prime task then becomes strategic implementation (and designing accommodation) to reach the target.
A synthetic campaign that encapsulated aspects of methodology-driven synthesis concepts was that of vibsanin E (179)[113] and 5-epi-vibsanin (180),[114] which are two rarely occurring complex cage-containing diterpenes isolated from Viburnum awabuki in Japan.[115] Initially, the synthetic methods of choice combined an acid-catalysed cyclisation of 181, followed by ring expansion of 182, to give an advanced intermediate containing features seen in 183.[27] However, although this work provided substantial experience for the neovibsanins (see Schemes 3 and 4), it was not possible to convert the advanced intermediate into either of the targets.[116] Therefore, a more modern methodology was employed. However, it is always a concern when adopting new methods as many often fail when applied to complex systems. In this case, however, the Davies group had shown that donor–acceptor diazo functions (e.g. 184) undergo rhodium-catalysed [4+3]-cycloaddition with dienes (e.g. 185) (a cyclopropanation Cope rearrangement sequence) to give seven-membered rings (e.g. 186). In addition, they had applied this protocol to the total synthesis of (±)-5,10-bis-epi-vibsanin E (187), which was a non-natural product.[117] Clearly, combining the [4+3] and [4+2]-cycloaddition methodology was very powerful, but the end game was compromised. This opened an opportunity for the Williams and Davies groups to collaborate and pool experiences to complete the synthesis, by adopting the Davies protocol to access an advanced intermediate of type 183, and then deploying Williams end-game know-how (Scheme 18).[118]
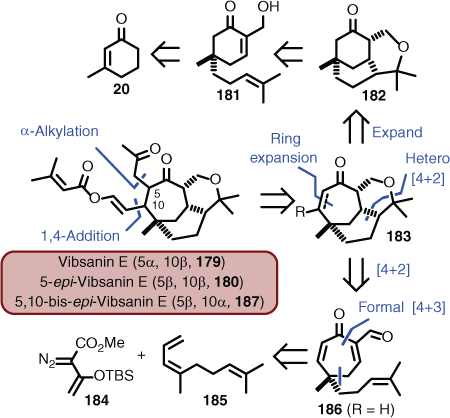
|
Reducing this approach to practice, the initial seven-membered ring (i.e. 188) was obtained in 65 % yield using the chiral catalyst known as PTAD (189), which required deoxygenation using in part Comin’s reagent (190) to afford a suitably substituted ring (i.e. 191) poised for the [4+2] hetero cycloaddition. The Diels–Alder reaction proceeded as planned and the resulting tricyclic ring system (192) was converted to the key advanced intermediate 193 in three steps. Conjugate addition using the lithiated MOM derivative took place both in high yield and stereoselectively. Introducing TMSCl was critical to facilitating the reaction and providing the desired material (194). Transmetallation and quenching with allyl bromide gave the allyl ether (195), and subsequent Claisen rearrangement afforded a mixture of syn- (196) and anti-isomers (197). The syn-isomer (196) was taken forward through a sequence of deprotection, alcohol oxidation, and Wacker oxidation to unmask the penultimate tricarbonyl 198. The last, and perhaps hopeful, step was heavily reliant on in-house-optimised Anders–Wittig chemistry (Anders–Gaßner reaction),[119] which delivered the target (i.e. 180 as the corresponding enantiomer 200), albeit in a low yield of 26 % using the Wittig reagent 199 (Scheme 19).[114]

|
Although the Claisen rearrangement gave a mixture of syn- and anti-isomers, it was possible to convert the anti-isomer into vibsanin E (179) (Fig. 3), although this was only undertaken in the racemic series.[113] However, although a well-devised synthetic route was in hand for these systems, it was not guaranteed that other members of the vibsane family could be conquered, and a spectacular failure was endured with attempts to synthesise 3-hydroxyvibsanin E (201). The synthetic route above that was arduously devised for vibsanin E and 5-epi-vibsanin E came completely unstuck in the penultimate step in the campaign to 3-hydroxyvibsanin E, when t-butyldimethylsilyl deprotection could not be achieved, giving 3-OTBS-vibsanin E (202) (Fig. 3).[120]
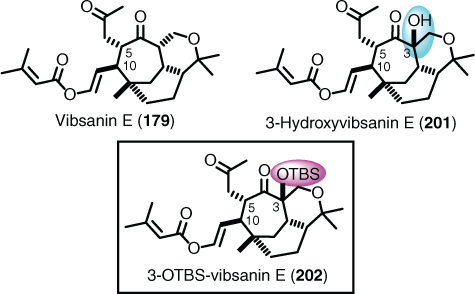
|
Collective Total Synthesis
The term collective total synthesis is a fairly new concept that was first put forward by MacMillan in 2011, when his group achieved the synthesis of six alkaloids from the same advanced intermediate.[121] MacMillan argued that although syntheses targeting an advanced core structure applicable to the synthesis of closely related natural products within a family had been previously reported, it was much less common to find examples where the preparation of an advanced intermediate with appropriate functionality was amenable to the construction of structurally diverse natural products in different families, the latter constituting the definition of the term collective total synthesis, which has analogies with a biosynthetic pathway providing collections of metabolites.[121] Since that time, further examples using different natural product classes have been added,[122] but the term is somewhat contentious, because there is an element of false pretences (i.e. the deck can be stacked by making a judicious choice of targets to match the key advanced intermediate). Alternatively, a collective total synthesis can only be achieved if enough natural product elucidation has been performed such that there is sufficient choice to undertake such an endeavour. Overall, however, the concept has substantial merit in that it aims to make one advanced intermediate multitask for multiple target syntheses, which has several important advantages. For example, providing a wide array of targets for biological testing saves time and effort designing individual synthetic routes, and is green chemistry-orientated (i.e. limiting waste streams).
The tetranortriterpenes (limonoids) are such a natural product class that fit the collective total synthesis criteria, in that a huge amount of structure elucidation has been undertaken on many different species within the meliaceae and rutaceae families.[123] A case of serendipitous collective total synthesis is that of the bicyclononanolides (e.g. mexicanolide (203)), whereby the retrosynthetic analysis aimed to access azedaralide (204) as the key advanced intermediate to achieve a total synthesis of mexicanolide (203), via a pre-established proposed biosynthetic route (i.e. via 205). Entry to azedaralide (204) was envisioned to be directly achievable from pyroangolensolide (206), a known degradation product that had been synthesised several times previously, and could be obtained starting from 2,6-dimethylcyclohexenone (207) (Scheme 20). In this case not only were two limonoid natural products synthesised (i.e. 203 and 204), but an additional three were also achieved (i.e. 208–210).
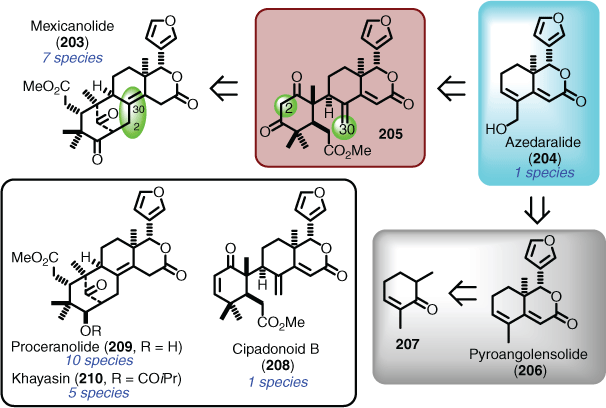
|
In the forward direction, the asymmetric synthesis of (+)-azedaralide (216) followed the racemic synthetic route (i.e. 204).[38] First, cyclohexenone 211 was subjected to in-house optimised Morita–Baylis–Hillman conditions,[29] which afforded the extended ketone (212) after TBS protection. Methylation (i.e. 213), followed by a (−)-diisopinocampheylchloroborane- (see 215 for (+)-DIP-Cl isomer; DIP = diisopinocampheylborane) mediated asymmetric diastereoselective aldol, provided the furylalcohol (214). The alcohol function was then acetylated to facilitate cyclisation, followed by TBS deprotection, to give the first natural product as the advanced intermediate (i.e. (+)-azedaralide (216)). A rarely utilised ketal-Claisen rearrangement was used to attach the A and B ring components of the targets to azedaralide. For this retrosynthetic manoeuvre to even be considered, a suitably functionalised cyclohexenane ring, matching the features seen in 217, was required. This goal was achieved in both racemic and asymmetric form using the following sequence, the only difference being that in the racemic procedure, a Mukiyama aldol was deployed, whereas the enantioselective route employed a boron-mediated aldol using (+)-DIP-Cl (215). In brief, treating aldehyde 218 with the boron enolate of 2-butanone, followed by potassium hydride-induced cyclisation gave the desired cyclohexanone (219), which was easily converted into the required enol ether (217) poised for condensation and rearrangement. Heating a mixture of 216 and 217 in the presence of anhydrous tosic acid afforded, via the extended enol ether 220, the second natural product, cipadonoid B (208), as the major product.[124] Epoxidation of cipadonoid B to introduce the additional ring oxygenation proceeded smoothly. Interestingly, it was not possible to reduce the epoxide to access the proposed intermediate 205 (Scheme 20) using traditional reagents. However, treatment of the epoxide (221) with aluminium amalgam not only regioselectively reduced the epoxide ring, but the resulting enolate subsequently underwent a 1,6-conjugate addition to afford the third natural product, proceranolide (209). This fortuitous result opened the opportunity to synthesise the fourth natural product, khayasin (210), using a peptide coupling protocol to install the hindered isopropyl ester. In addition, the original target and the fifth natural product, mexicanolide (203), could also be synthesised from proceranolide (209) using the Jones oxidation protocol (Scheme 21).[125]

|
Chemical Database-Aided Synthesis
Several searchable chemical databases are available to the practitioner. Reaxys and SciFinder are the main staple in this regard. Key features include the capability to find specific compounds and substructures, general and structure specific reactions, physical properties (experimental and calculated), and spectroscopic data (experimentally determined and calculated).
In the context of database searching as applied to retrosynthesis, D’Angelo and Smith put forward the notion that retrosynthesis should be used to initially find a product from undertaking a disconnection, which then enables a computer search to generate the remainder of the retrosynthesis. They termed this process as ‘a hybrid retrosynthesis approach’, because the concept utilises the basics of retrosynthetic analysis to identify a working intermediate that is subsequently searched for in a database using a computer.[126]
Computer-Aided Synthesis and Artificial Intelligence
The use of computers in chemistry has been around nearly as long as personal computers themselves,[127] and their possible application to planning synthetic routes has been realised for nearly as long.[128] There have been several comprehensive reviews on the topic,[13c,129] and so here we will only cover the topic briefly and highlight a few key programs and ideas. A selection of past and present programs is listed in Table 1, which by no means covers every program in the literature.

|
One of the first challenges to overcome in the realm of computers in chemistry was to develop a method to code chemical structures and atom connectivity in a way that was machine-readable. The most commonly used method is molecular graph theory, in which the molecule is represented by nodes and edges. Feature matrices are then used to encode various properties of the atoms (i.e. nodes) and relationships or bonds between them (i.e. edges), meaning that a 2D graph can encode 3D information such as stereochemistry.[130] Graph theory has been particularly useful in describing, and exploring, the small-molecule chemical universe.[131]
At its heart, retrosynthetic analysis can be seen as a pattern recognition exercise, an activity that computers are exceptionally good at. A set (albeit an ever-expanding set) of fairly simple rules can be used to solve even incredibly complex problems. In this regard, the parallels between retrosynthetic analysis and chess have been discussed before.[129a] In 1997, a computer called Deep Blue famously beat the world chess champion in a six-game match after making a move that has variously been attributed to a bug in the software and to superior intelligence.[132] Whether this move was a software bug or deliberately executed (and indeed whether a computer can be said to ‘deliberately’ do anything) remains up for debate. Deep Blue relied on human-programmed rules and strategies, and could not learn new tactics simply by observing other players during the game. In this way, Deep Blue was analogous to the early retrosynthesis programs such as LHASA and can be described as a ‘brute-force’ player, defeating opponents through sheer knowledge and ability to quickly evaluate every known (to the computer) option. More recently, board game-playing computers have used deep neural networks and are capable of not only playing the games, but also of independently developing new strategies.[133] These neural network computers display a game style described as more ‘human’ and ‘elegant’ than the earlier computers such as Deep Blue, and are regularly used as training tools by today’s competitors. There is also a great deal of research into using neural networks for chemical synthesis and retrosynthesis (see below). While neural networks show great promise for this type of problem, it is important to note that they are not without their issues, often being described as ‘brittle’, i.e. incredibly strong until they are faced with something unknown, carelessly entered, or deliberately edited, when they can break in spectacular fashion.[134]
Historically, two general methods of building computer-aided retrosynthesis programs have emerged: (1) human-coded rules combined with a heuristic search; and (2) machine learning techniques that automatically generate rules from a provided reaction database. Human-coded rule programs dominated up until the mid-2000s, starting with Corey’s pioneering work in the late 1960s. OCSS (Organic Chemical Simulation of Synthesis),[12a] which evolved to become LHASA (Logic and Heuristics Applied to Synthetic Analysis),[135] is considered the first true computer-assisted retrosynthesis planning tool. It was designed to be interactive to allow the chemist to choose which branches of the synthesis tree were worth exploring based on their own knowledge, favoured reactions, and experience in the laboratory. This concept of being able to explore a synthesis tree has been common to nearly all programs developed since, although the tree has sprouted considerably more branches (Fig. 4).

|
Even though the methods employed by the programs can be described by umbrella terms (e.g. hand-coded rule set, machine learning, a combination of both), often the authors of a program would select an overarching approach that differentiated it from other contemporary programs. For example, SYNCHEM was unusual in that it first considered only the carbon skeleton of the target molecule and systematically broke C–C bonds to find the most efficient (convergent) route from a database of starting materials; once starting materials were found, SYNCHEM generated intermediates by adding all possible functionalities to the carbon skeletons and mechanistically testing the reactions and deleting non-viable ones, whereas other programs would start from considering the target molecule as a whole.[137] For forward prediction programs, EROS considered reactions as bond- and electron-shifting processes, and then implemented evaluation processes to select only chemically viable reactions, whereas many other prediction programs focus on functional groups and their transformations as the basis of chemical reactions.[138]
Although it has been proposed that the synthesis of longifolene (222) was Corey’s inspiration for LHASA, and likely the birthplace of the term retrosynthesis, it was not subjected to LHASA.[67] That said, it did form its ground rules.[1c] The synthetic approach also utilised as the key step the santonic acid degradation performed by Woodward (see Scheme 11 above).[67] Therefore, based on this premise, it is worthwhile discussing the longifolene synthesis in detail. The initial retrosynthetic manoeuvre consisted of introducing carbonyls adjacent to the gem-dimethyl group, and in place of the exocyclic double bond (i.e. 223). These key carbonyl insertions provided retro-conjugate addition synthons to be formulated, which resulted in the enone (224) being proposed. Rationalising the three-dimensional structure in two dimensions reveals 225, which could be derived from the Wieland–Miescher ketone (226), accessed via a Robinson annulation of methylcyclohexandione (227) with methylvinylketone (228) (Scheme 22).[2b]
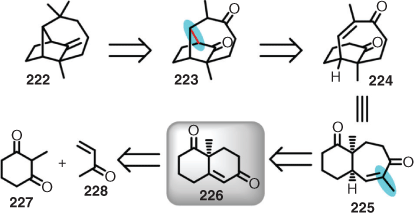
|
Starting from the Wieland–Miescher ketone (229), the A-ring carbonyl was protected as a ketal (230), which enabled the enone to be chain-extended (i.e. 231), thus setting the stage for a pinacol rearrangement via diol 232. The six- to seven-membered ring enlargement was the first key hurdle in the synthetic approach, and although proceeding smoothly in the end, it did require activating conditions to promote the pinacol rearrangement and afford the desired ring system (i.e. 234), albeit a different isomer to that proposed in the retrosynthesis (i.e. 225). The second key hurdle followed directly after in that conditions were required to mediate cage bicyclic formation. This was eventually achieved using triethylamine at high temperature, and only produced material (i.e. 223) in the order of 12 % yield. Having surmounted these hurdles, the remaining part of the synthetic campaign amounted to a single skeleton embellishment, and several FGIs, for example, completion of the gem-dimethyl function (i.e. 235), followed by removal of the adjacent ketone to give 237 via reduction of a dithiane (i.e. 236). Final oxidation of alcohol 237 and subsequent methylenation over two steps gave the long-awaited and seminal target longifolene (222)[160] (Scheme 23).

|
Interestingly, the Corey group also spawned many of the next generation of programs when those that worked on OCSS and LHASA moved on to their own independent careers. Jorgensen, who worked on the LHASA project as a graduate student in Corey’s laboratory, went on to develop CAMEO (Computer-Assisted Mechanistic Evaluation of Organic Reactions).[139,161] Instead of retrosynthetic analysis, this was a forward prediction program, which relied on human-programmed mechanistic rules to predict the outcome of a reaction. In total, 26 papers detailing the development and capabilities of CAMEO were published; the last appeared in 1995.[162] It seems that ceasing work on CAMEO was a conscious though difficult decision for Jorgensen, as he discussed in his autobiography in 2015.[163] Wipke also worked on OCSS as a postdoctoral researcher and went on to develop both SST[141] and SECS.[13b] It is likely that many early programs were abandoned because upkeep and expansion of the rule set became too time-consuming, and the results were not promising enough to warrant the commitment.
Since the 2000s, advances in machine learning and computing power have opened the door to more complex machine learning approaches. In terms of synthetic chemistry, three learning modes are used: (1) supervised learning, where both the input (reactants, reagents, conditions) and output (products, yield) are provided to the computer in the form of a training set; (2) unsupervised learning, in which only the input data are provided to the computer; and (3) reinforcement learning, where the computer aims to find the optimal path towards the goal and is given a reward for each step.[164] The method or mode of learning is independent of the end-goal of the program, i.e. a supervised learning mode can be utilised in a retrosynthesis program, a forward prediction program, or any other end-goal.
In machine learning, the programs are trained using databases of chemical reactions, and the quality of data (e.g. experimental details, stereochemistry, regioselective information) varies greatly between different databases. Often the information is extracted automatically and this can result in errors or missed information, which would have otherwise been identified by a human, especially when the information is being extracted from prose (written experimental procedures, patents) rather than drawn reaction schemes (although drawn reaction schemes often omit crucial information such as temperature and time).[155b] In a recent interview about a new collaboration, MIT researchers stated that they hoped the quality of data in the CAS (Chemical Abstracts Service) database would improve the predictive power of their machine learning algorithm that has previously only been trained on freely available datasets.[165] While it seems intuitive that an algorithm trained on data rich in, for example, stereochemical information would be better at predicting stereochemical outcomes of reactions, there is some evidence that the dataset an algorithm is trained on may have little effect on the quality of its predictions.[166]
A final consideration for both industry and academic chemists is that confidentiality and intellectual property is of utmost importance. Many of the browser-based free programs (e.g. IBM RXN) explicitly state that any information submitted through their website (i.e. into the retrosynthesis program) is visible to the developers. IBM RXN go on to state that by providing information, the user is granting IBM unrestricted use rights.[155] Some commercial services (e.g. SpayaAI) also make the search input data visible to the developers.[158] Others (e.g. ICSynth) can be installed locally and therefore the data are only visible to the user.[150] For these subscription services, there is also the issue of what happens to the data on termination of the licence or agreement. As the capabilities and availability of computer-assisted synthesis programs expand, the status of the ownership of the input and output data will continue to evolve.
Chiral Pool
The chiral pool, which has already been mentioned above (see for example Scheme 17), is the collection of optically active starting materials that are available from natural sources, such as amino acids, carbohydrates, and terpenes. They are highly utilised in synthesis, because of their exceptional enantiopurity, and thus can be incorporated into the final structure of the target or as chiral auxiliaries. Furthermore, the chiral pool presents an interesting opportunity for the use of computers to recognise structural relationships between targets and starting materials.
As a prime example, the chiral pool approach was used by Corey for the synthesis of helminthosporal (239).[167] Starting with S-(+)-carvone (240), a natural product readily available from caraway seeds, provided both a chiral centre and the isopropyl side chain, which inserted the correct configuration of the isopropyl group in the target. In brief, hydrogenation of 240 gave the saturated ketone (241), which was converted into a sacrificial 1,3-dicarbonyl (i.e. 242) to induce smooth albeit slow conjugate addition to methylvinylketone (228). Tricarbonyl 243 was then decarbonylated (i.e. 244), and subsequently cyclised to the bicyclo[3.3.1] system (i.e. 245) through treatment with the Lewis acid boron trifluoride. Wittig-mediated chain extension installed the masked aldehyde (246), which underwent a transprotection (i.e. direct interconversion of protecting groups[168]) with ethylene glycol to give the ketal (247) (note no change in oxidation state converting an enol ether to a ketal). Dihydroxylation gave the diol (248), which was oxidatively cleaved to give the dicarbonyl (249) as the key step in setting up the ring-contraction strategy towards the bicyclo[3.2.1] system, the latter being achieved via a Claisen condensation affording the cage bicyclic enal (250). The final step entailed a simple acid-mediated deprotection to unmask the remaining aldehyde in helminthosporal (239) (Scheme 24).

|
Such a synthesis is described as taking a chiron approach to retrosynthetic planning. Often, however, the relationship between the structure of the target and the chiral pool is not immediately obvious, and in the case of sugars can result in much of the initial functionality being removed (e.g. to access the carbon framework) to achieve synthesis of the target (e.g. leukotriene B5 (251) from tri-O-acetyl-d-glucal (252)[169]). In the helminthosporal example, a substantial clue to considering a chiron approach, beyond the importation of stereochemistry, was the isopropyl group and the cyclohexane moiety that S-(+)-carvone provides. Furthermore, when the hydrogenated material (i.e. 241) is redrawn in the chair configuration, the relationship between the starting material and target is more apparent, and now lends more weight to the adopted retrosynthetic design (Scheme 25).
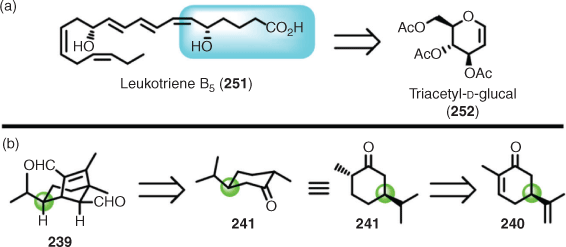
|
Actually, the term chiron (chiral synthon) was coined in the early 1980s by Hanessian,[170] who also developed a computer program specifically to search for chiral starting materials given a target molecule.[140a] CHIRON was an interactive program that projected different perspectives of a drawn molecule, allowing the chemist to ‘see’ the compound in new ways. The program also mapped stereochemical information onto the target and searched its hand-assembled starting material database for materials with maximal overlap in functional and stereochemical features. The aim was to find starting materials that would require the least transformation to reach the product, specifically focussing on the carbon framework and rings.
Currently Available Programs
Reaction Predictor,[151] Seq2Seq,[154] IBM RXN for Chemistry,[155] LillyMol,[157] SpayaAI,[158] AutoSynRoute,[12e] and AiZynthFinder[159] are all currently freely available, either as browser-based applications or as code downloadable from GitHub. They all rely on machine learning methods for training, and do not include any hand-coded reaction rules. ICSynth, a commercial machine learning-based program, has seen use in the pharmaceutical industry for route design.[150]
Arguably the most advanced computer-aided synthesis tool today is Synthia (formerly Chematica) developed by Grzybowski et al. and recently licenced by Merck. From its conceptual beginnings in 2001 at the MIT Bridge Club[35a] to being validated by in-laboratory results in 2018, the road to this autonomous synthesis planning tool was certainly a long one.[136] The technical details of how Synthia works are covered in detail elsewhere, and so here only a few differences and similarities between this program and others that have come before will be mentioned.
First, despite the rapid advances and interest in machine learning of late, at the core of Synthia’s workings is a massive database of over 50000 human-coded reaction rules. These rules were compiled by chemists over many years and, much like the early now-abandoned programs that relied on this type of database, will need to be kept up to date for the program to continue to be of use as new chemistry is discovered. Second, Synthia has a library of algorithms that it can draw on. The algorithms can, for example, augment the reaction rules to improve regio- and stereo-selectivity predictions, allow the program to plan over several steps, or combine reactions that proceed under the same conditions (i.e. tandem reactions) into one step. These algorithms set it apart from other programs, which can only plan one step at a time.
This hybrid hand-coded-rules-and-algorithm approach means that Synthia approaches retrosynthetic problems in a seemingly more ‘human’ way than other programs, which can only apply hand-coded rules strictly in the way they were written, or may miss an important reaction that is not covered in the database it was trained on. Synthia has probably the largest chemical reaction rule knowledge set of any program thus far (both as a consequence of the effort put in to coding the rules and by virtue of there simply having been more chemistry known now than in the past).
In 2018, verification of Chematica’s capabilities was published, with the in-laboratory confirmation of several routes devised by the program (Fig. 5).[136a] The Chematica-devised syntheses all provided improvements on the literature procedures in terms of yield and/or number of steps (253–257). In two examples (i.e. 259 and 260), Chematica was used to specifically avoid patented routes and provided comparable or significantly improved yield. One was a natural product (i.e. 258) that had not previously been synthesised. This verification clearly showed that there has been great improvement in the synthesis planning capabilities of computer programs over the last 50 years; however, the commercial aspect of the more capable programs (Synthia and ICSynth) means that their impact on the wider research community will be limited. Synthia also risks encountering the same fate as earlier hand-coded programs, abandoned owing to the time required and difficulty in keeping them up to date. Additionally, there is a great amount of research being carried out in the area of automatic data extraction,[155b] neural networks,[171] natural language sequence to sequence processing,[129d,154] and machine learning,[172] which negates the need for manual extraction of data and updating reaction rules.
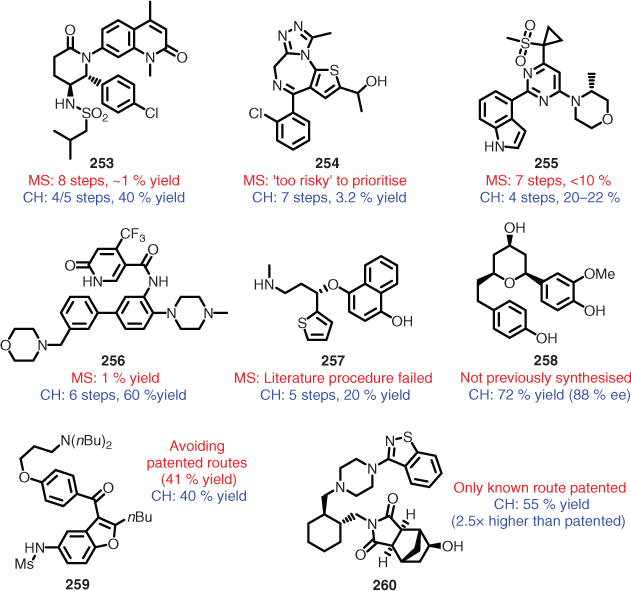
|
Discussion
Having canvassed both humanistic and computer-assisted retrosynthetic methods and practices in detail, it is important that with this knowledge, questions be posed and considered to better understand a probable direction this art form will take in the future, i.e. in the context of a computation era attempting to revolutionise the discipline of chemical synthesis through both retrosynthetic design and forward synthetic planning.
Is the Target Real?
Both natural and synthetic products are regularly assigned incorrect structures,[173] and it is often not until practitioners attempt their synthesis for the first time (e.g. trunkamide A (261)),[174] or repeat a synthesis (e.g. tetrahydrooxazepine (262)),[175] that the error is detected and corrected. Compounding this problem, in some cases even achieving a synthesis of the proposed target may not offer any additional information on the proposed structure (e.g. afzeliindanone (263)).[176] Another dimension to this problem is that some structures are extremely difficult to solve (e.g. EBC-232 (264)),[177] or an outright solution is not possible (e.g. a flat structure lacking relative and/or absolute stereochemistry). Therefore, the question in this context is whether AI development will in the future attempt to verify the input target structure has been correctly elucidated, which is especially important for those suspicious structure proposals that defy physical organic concepts such as aromaticity (e.g. 265)[178] and Bredt’s rule (e.g. 266)[179,180] (Fig. 6). Indeed, when describing an earlier incarnation of Chematica (pre 2016), Grzybowski et al. stated that ‘In full disregard of Bredt’s rules, it installed double bonds at bridgehead atoms’, and described this as a ‘creative’ and ‘non-existent or improbable’ structural feature.[152a] The structures synthesised via Chematica suggested routes in 2018 (Fig. 5) do not contain rare or unusually strained motifs, so it remains to be seen how Synthia would deal with such a structure now.
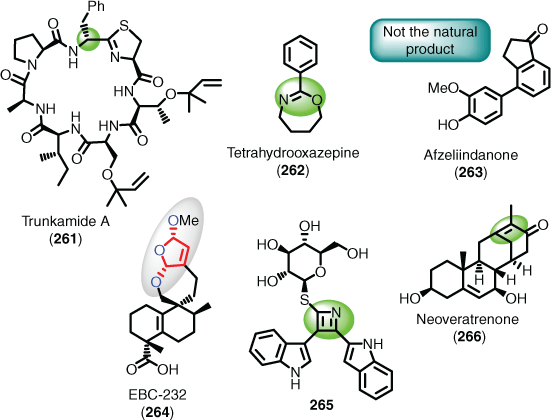
|
It is certainly conceivable that AI will indeed adopt an elucidation aspect, because it is currently possible through modern Computer-Assisted Structural Elucidation (CASE)[177,181] to screen target structures for validity. More importantly, this feature can be undertaken rapidly with current density functional theory (DFT) and associated methods[182] to give surprisingly accurate re-assignments, for example, the proposed structure for botryosphaerihydrofuran (267) and its revised structure 268 (Fig. 7).[183] Assigning absolute stereochemistry is a developing possibility with such methods.[184]
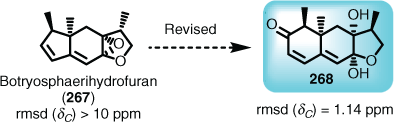
|
With that said, there are risks associated with automated NMR-based structural elucidation in terms of DFT energy miscalculations and subsequent erroneous consequences.[185]
Will AI Consider Model Systems?
Model systems (or model studies) in organic synthesis are loosely defined as testing a ‘key step’ in a proposed synthesis without probing the real system. Such studies have been widely used, and in some cases with great success, for example, establishing whether ring-closing metathesis was a viable option for the construction of the D ring seen in manzamine A (269) (Fig. 8).[186] However, the general view of the community is that a ‘model is a model, and real is real’,[7] meaning that the model system may or may not work, and that outcome is not a true reflection of whether the proposed transformation would work (or not) in the real system. Therefore, it could be justified that model systems be deemed obsolete if the practitioner has embraced full AI assistance.
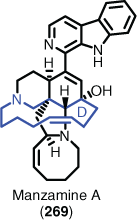
|
Will AI Discount Fundamental Molecules?
Fundamental molecules, such as strained hydrocarbons (e.g. cubane (54)[48] and [1.1.1]propellane (270)[187]), have fascinated theoretical and synthetic chemists since the discipline of organic chemistry began. However, the issue with many of these systems is undesirable physical properties, for example, explosivity and impact sensitivity. Therefore, will future AI retrosynthesis and synthetic planning programs take into account these details, which are a potential safety concern if not handled with practical knowledge and expertise? Cubanes and bicyclo[1.1.1]pentanes are such examples, but their impact sensitivity depends solely on the functional group appended to the parent skeleton (Fig. 9). For example, iodinated systems (e.g. 271 and 272) are quite sensitive, but not methyl esters (e.g. 273),[50b] although some carboxylic acids are strong candidates (e.g. 274).[50a] Hence, will AI search the literature for this know-how or calculate strain energy before making such synthetic design proposals? With available literature, and the ability to calculate strain energy and impact sensitivity,[50a] it should be feasible to introduce these features. Information such as impact sensitivity, however, is often only reported in the written text and has no standardised reporting protocol, meaning that this information is very difficult to automatically extract from the literature. Overall, however, these intriguing molecule types are playing an increasingly important role in bioactive molecule discovery,[51] and thus cannot be eliminated from consideration.
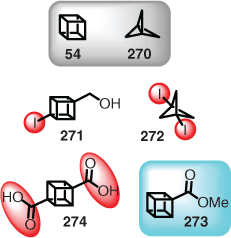
|
Will AI Learn from Practical Synthetic Problems?
It is often the case with synthetic practice that retrosynthesis design is very much considered a rough plan for the forward synthesis, which in turn becomes a trial-and-error process to achieve the target. This process has been elegantly described as ‘dead ends and detours’.[188] Given this aspect is an inherent process of chemical synthesis, it is likely that AI will by default follow a similar path, but in collaboration with the practitioner to determine what works in reality. This essential feedback loop provides further information on unexpected failures arising from the initial AI retrosynthetic plan and projected synthetic approach. This process will ultimately achieve the stated goal, but it needs to be determined whether this approach is any different for what is currently undertaken based on solely human design. Efforts to evaluate such AI performance are ongoing; see for example Fig. 5.
Another aspect for consideration under this section is that of reaction yields. The main issue is that reported yields vary in accuracy owing to a range of factors (e.g. scale of reaction). To complicate matters further, academic laboratories do not determine yield values to the same level of accuracy being undertaken by industry.[189] Hence, this can be considerably misleading when coding software for predictive forward syntheses. This complication is unlikely to change anytime soon.
How will Protecting Groups be Interpreted?
The famous opening chapter to ‘Protecting Groups’ by Kocieński, states up front ‘Death, Taxes and Protecting Groups’. This very statement sets the stage for the necessary evil that comes with performing chemical synthesis, which is only exacerbated by an increasingly complex target that demands an extended synthetic route. Kocieński follows by introducing seven tactical considerations to define how effectively a protecting group should perform.[190] In essence, protection group chemistry is a very large and complicated component of performing chemical synthesis, and must be very well understood for AI adoption.
Synthia is currently the only program that fully engages and utilises the concept of protecting groups, because it is the only software package that considers strategies over more than one synthetic step. Previous versions of Synthia indicated to the user when protecting groups were required for a particular intermediate or step, and this information came from the hand-coded reaction rules. It has recently been reported that the latest version of Synthia will now explicitly include all protection and deprotection steps in the proposed syntheses, so this is clearly still an area under development.[191] At this point in time, it is also reliant on human choice to decide on whether a synthetic step can actually be performed without the use of a protecting group. There are numerous syntheses that have been completed without the use of a single protection or deprotection, and this is a preferred situation as it limits synthetic steps, is economical, and limits waste.[192] However, there is no denying that syntheses without protecting group deployment are difficult, and if the synthetic route to the target (or the target itself) is not conducive to it, then the effort involved in attempting to achieve this goal may end up using many more resources (e.g. labour) than adopting a protecting group or two. That is, protecting group-free synthesis is an admirable goal, but must be viewed in realistic terms.
Can Domino and Cascade Reactions be Readily Predicted?
Domino and cascade reactions are always a welcome opportunity for the synthetic practitioner to implement because of numerous benefits, for example increased efficiency and waste minimisation of solvents, reagents, adsorbents, energy, and labour. Therefore, such reactions facilitate ecologically and economically favourable production and manufacture, and would greatly assist meeting Mulzer’s 25-step limit for any complex synthesis.[193]
The terms domino, cascade, tandem, and one-pot are used apparently interchangeably in much of the literature, but they are used to describe very different reaction procedures. A domino reaction, as defined by Tietze,[19] forms several bonds in one sequence without isolating the intermediates, changing the reaction conditions, or adding reagents. Importantly, in a domino reaction, each bond-forming reaction depends on the previous step to set up the required functionality. A one-pot reaction is similar in that it is a sequence of transformations taking place without isolation of the intermediates; however, further reagents are added to effect each step and reaction conditions can be changed,[21] whereas a tandem reaction has been defined by Nicolaou et al. as two or more reactions occurring on the same substrate and under the same conditions, but essentially in isolation of one another.[21] Tandem is commonly used to describe reactions that would be more accurately defined as one-pot or sequential;[194] however, tandem reactions are not time-resolved and so this usage is not preferred as justified by Tietze.[19] According to Nicolaou et al., cascade can be used as an umbrella term for all domino, one-pot, and tandem reactions,[21] Tietze argues that while cascade is often used to describe domino reactions, it is ambiguous and the latter term is preferred.[19]
As much as these reactions are highly desirable, their design is by no means straightforward, and is generally associated with natural product syntheses due to inspiration provided by Mother Nature (see the Biomimetic Synthesis section above). In terms of AI, this places a significant demand on programming. Synthia will identify reactions that can take place under the same reaction conditions,[195] which addresses simultaneous or tandem reactions, using the definition of Nicolaou et al.[21] Tactical combinations, as coined by Corey,[1a] can be defined as strategic retrosynthetic transformation sequences, the first of which does not immediately or obviously produce any benefit and often increases molecular complexity, but sets the scene for the second transformation, which results in molecular simplification. The term can in theory be applied to any combination of two transformations that fit the criteria; however, previously only ~500 such combinations had been explicitly identified in the literature and catalogued. Grzybowski et al. used Chematica to discover ~4.85 million tactical combinations and subsequently explicitly coded them into the program (along with the ~500 ‘known’ tactical combinations).[152b] This is part of the reason why Synthia can plan over multiple steps, but currently only two-step combinations have been included.[152b] These tactical combinations are not necessarily suitable for domino reactions, however, because there is no requirement for them to preclude the isolation of intermediates.
Overall, it is possible that Synthia would identify a route that would, in practice, result in a domino process. However, it seems that the user would have to manually review the retrosynthetic tree and identify that each reaction step would take place sequentially and under the exact same reaction conditions, unless the domino sequence was already known and was coded in as a reaction rule. Therefore, this implementation could be some time away and is far from being perfected.
Assessing Molecular Complexity
In light of what we know about computer-assisted and AI methods for devising retrosyntheses and route planning, there is a requirement for the computer to understand complexity. Complexity is two-fold in this context, although related: (1) molecular or target complexity, and (2) synthetic design or route complexity. Note that the molecular complexity of the target may or may not be directly proportional to the number of steps required to synthesise that target. Therefore, should there be some consideration as to whether this impacts AI performance in undertaking retrosynthetic design and predicting forward syntheses more efficiently? Obviously, AI will inherently attempt to provide the ‘best’ synthetic approach following consideration of the proposed retrosynthetic plan.
How is complexity perceived? Although molecular complexity is difficult for the practitioner to define, it is generally intuitively understood. For example, taking into account the target’s carbon skeleton, number of appendages, and functional groups, stereochemistry and conformation,[196] one can quickly ascertain a level of molecular complexity, and subsequently synthetic difficulty. As mentioned above, the latter is not necessarily determined by molecular complexity, i.e. complex molecules can be readily synthesised, but it is all dependent on retrosynthetic planning. When coding tactical combinations, molecular complexity had to be considered by the Chematica/Synthia team, and was quantified by the number of rings, the number of stereocentres, and the length of the SMILES string code of the molecule.[152b] Some targets, such as Taxol® (148) are currently ‘too complex’ for Synthia, and the route search times out (i.e. exceeds the available RAM).[191]
In an effort to conceptualise retrosynthetic planning based on complexity, there are three general parameters that have been used by the lead author over the years to categorise synthetic approaches to targets. These three categories, which are essentially a ranking system of synthetic difficulty, are best imagined as analogies to proton NMR splitting patterns, which include first-order,[197] second-order,[198] and non-first-order[197] interpretations.
A good first-order example is the total synthesis of EBC-329 (275),[199] as it is mostly first-order (n + 1) retrosynthetic bond breaking. When further simplified by adopting a chiral pool approach, as suggested by the cyclopropyl-gem-dimethyl residue, the conjugated enlactone is disconnected in the first instance to give an enyne (276) as conjured from metal coupling. Further disconnection of the enyne to a methyl ketone (i.e. 277) is inspired by Wittig chemistry, as is the inspiration for the subsequent disconnect to the dicarbonyl (278). Two carbonyls distanced by six carbon units imply an oxidative ring cleavage strategy fitting carene (279) (Scheme 26).
|
Based on this first-order retrosynthesis, the forward synthesis almost became pedestrian, albeit with one minor hurdle. Oxidative cleavage performed best over two steps to give 278, which reacted smoothly with the phosphorane (280) to afford 277. Some experimentation was required to procure the second olefination, but this was eventually achieved using Horner–Wadsworth–Emmons (i.e. 281) methodology, although a mixture of E- and Z-isomers was obtained (i.e. 282). The final step consisted of installing the γ-alkylidenebutenolide function, which was accomplished in one step via a copper-mediated reaction with iodoacid 283 (Scheme 26). HPLC purification then afforded the pure natural product, establishing the absolute stereochemistry in a total of seven steps (Scheme 27).

|
Second-order considerations include multiple bond disconnections at once, which would equate to strategies consisting of skeletal rearrangements (e.g. Scheme 21), cascade reactions (e.g. Scheme 5), and cycloadditions (e.g. Scheme 13) as prime examples in this category.
Non-first-order retrosynthesis (akin to aromatic ring-splitting patterns) encompasses irrational bond breaking and forming retrosynthetic manoeuvres that generally only become clear once viewed. A seminal contribution to this category was the total synthesis of nominine (284) by Peese and Gin.[200] The retrosynthesis boldly aimed to build four bonds using a convergent dual-cycloaddition approach via key intermediate 285 (Scheme 28).
|
The elegant forward synthesis is immediately daring by not only starting with an unstable dimethyl acetal (i.e. 286), but performing an ortho-lithiation on a substrate where multiple positions could have in principle been lithiated. The product (288) was converted into a functionally dense azide (289), which was converged with a cyclohexylaldehyde (290) as obtained from enone 20 via triflate 291. The audacious Staudinger reaction afforded the adduct (292) in high yield, which on treatment with acid gave the desired zwitterion (293), setting the stage for the first key 1,3-dipolar cycloaddition. Astoundingly, heating 293 at 180°C gave in one step the majority of the target skeleton (i.e. 294). FGI gave rise to the second cycloaddition precursor, alkene 297, via intermediates 295 and 296. Enamine formation derived from 297 facilitated the second cycloaddition to give 298 in 78 % yield. Installation of the exocyclic double bond followed by allylic oxidation gave nominine (284) (Scheme 29). This bold and exemplary case reduced a previously 40-step synthesis to just 15 steps.

|
Given practitioners can intuitively categorise both forms of complexity (molecular and synthetic), an in silico complexity index has been developed to rank both molecule and synthetic complexity,[201] which opens the door for AI adoption if a requirement is justified. The overall message being conveyed, however, is that complexity is much better understood by the practitioner than the computer at this point in time.
Mechanism
Understanding the mechanism of a chemical process is a fundamental part of the practitioner’s toolbox, as it enables further discovery and innovation. However, is this process important for retrosynthesis and forward synthesis if it merely comes down to bond breaking and bond forming from a graph theory or AI perspective? This aspect is difficult to judge because some chemical processes take decades to elucidate, although not hindering their use and adoption by the community. The Ley–Griffith oxidant, tetra-n-propylammonium perruthenate (TPAP), is a good example that was routinely used for mild and controlled oxidation of alcohols and many other substrates, but whose mechanism of oxidation was only recently elucidated, having stood unknown for almost four decades.[202] In the eyes of the computer, it is likely going to be a case that if it has literature precedent as working, then it can be adopted irrespective if the mechanism is understood or not.
Synthetic Methodology
From the perspective of AI coding, and current day AI performance (e.g. Synthia), has sufficient methodology already been discovered? If the paradigm shifts introduced by ring-closing metathesis[203] and C–H activation[10] are any indication, then methodological development must continue, whether by rational design or accidental discovery. However, what must not happen is the removal of old methodology from consideration as new methodology is developed, i.e. this is not a case of replacement, but bolstering the repertoire.
Will AI Develop a Chemical Mind or Brain Equivalent?
Perhaps the most important, albeit emotionally confronting, question for practitioners of synthetic chemistry is whether they will become obsolete owing to AI thinking and robotics (e.g. performing chemical reactions).[204] In fact, this is an area of debate for humankind in general and across all facets of life.
As early as 1950, the question ‘Can machines think?’ was proposed by Turing, who also embarked on defining the term ‘think’.[205] Some 30 years later, Searle proposed essentially the same question, ‘Could a machine think?’ but attempted to address the mind, brain, and program interface using the argument of intentionality in human beings.[206] He then went on to differentiate the brain–mind relationship (dualism), often called the mind–body problem, by considering the brain as a digital computer, and the mind as a computer program.[207] More recently, Signorelli considered the idea of machines overcoming humans and, if so, that they must be intrinsically related to conscious machines.[208]
The Turing test, originally called the imitation game by Turing, essentially asks whether an interrogator would be able to tell the difference between two participants, one of which is human and one a machine.[205] Importantly, the Turing test examines behaviour (output) rather than mechanism or working (process). There have been many variations of the Turing test, and the subject matter expert or Feigenbaum test is particularly applicable to chemical synthesis programs.[209] Chematica was recently subjected to the Feigenbaum or Turing test by having 18 experts grade the likelihood of 40 (20 from the literature and 20 from Chematica) synthetic routes to be human- or computer-designed. Overall, the experts were unable to differentiate between the human- and computer-designed routes, and even scored the computer-designed routes as being slightly more ‘elegant’ than the human-designed routes.[191]
With that said, all these considerations are philosophical debate, and are in the process of being translated, tested, and determined in silico. Therefore, it seems a long way off before synthetic chemists undergo complete morphological change into scientific generalists, i.e. when a ‘scientist’ of the future acquires molecules (based on society drivers) using the push of a button.
Conclusion
There is no doubt that synthetic chemistry has been and will continue to be a substantial enabling foundation discipline for humankind. The past and present output of pharmaceuticals, agrochemicals, and materials (e.g. plastics, microelectronics) is a reflection of the magnitude of this discipline being deployed by industry.[210]
However, even though this branch of science is highly sophisticated, it is by no means trouble-free, as Cornforth flagged back in 1993,[211] likely with Seebach’s 1990 proposal of ‘Organic synthesis – where now?’[212] front of mind. If we accept the answer to Seebach’s question is ‘everywhere’ and that Cornforth’s conjecture remains correct, then evolution of the discipline is inevitable, and in modern times that requires integration of algorithmic methods to assist in prompting human decision-making. Therefore, with the emergence of programs such as Synthia, the real quandary is whether the art of organic synthesis will be maintained, i.e. will the syntheses be elegant, and meet all social related desires of efficiency and environmental impact? Beyond that point, will computer-assisted methods and AI truly be revolutionary for the discipline, and importantly should they be relied on completely as Judson intimates?[213]
‘The GPS navigation device may render paper maps redundant – but not the driver of the car.’[214]
Lastly, the present article expresses certain thoughts and philosophies on retrosynthesis and forward synthetic planning that are designed to generate discussion among students and colleagues alike. It is fully realised that this important and ever-evolving area comes with many individual views and experiences that constantly drive synthetic organic chemistry thinking, and Sir John Warcup Cornforth[215] is one of several iconic individuals who have made significant contributions to the evolution of chemical synthesis.
Conflicts of Interest
The authors declare no conflicts of interest.
Declaration of Funding
This research did not receive any specific funding. The University of Queensland provided all the resources to enable the preparation of this manuscript.
Acknowledgements
C.M.W. is eternally grateful to the many bright and talented students and post-doctoral researchers who have passed through his laboratories, bringing culture, philosophy, and humour to the Williams research group enterprise. C.M.W. is profoundly grateful for financial and collaborative support over the last 20 years from the Australian Research Council (ARC), National Health and Medical Research Council (NHMRC), State and Federal Government departments (e.g. DPI, DAWE), Defence Science Technology Group (DSTG), Commonwealth Scientific and Industrial Research Organisation (CSIRO), and local and international industry (e.g. EcoBiotics Ltd, Bayer, Dow, DuPont). C.M.W. is eternally grateful for the many wonderful scientific collaborations that have developed over time, and in turn allowed his research to blossom in a multitude of unexpected directions. Such relationships with both academic and industrial collaborators are dearly cherished. M.A.D gratefully acknowledges the Australian Government Research Training Program Scholarship.
C.M.W. is especially grateful to Emeritus Professor Curt Wentrup (University of Queensland) for the invitation to write an Australian Journal of Chemistry Sir John Cornforth review article. Beyond the invitation itself, C.M.W. is especially honoured to contribute to this series having had the fortunate opportunity to meet Sir John W. Cornforth Jr. during the course of being interviewed for a Lectureship in 1999 at the University of Sussex, Brighton, UK. Anecdotally, Sir John Cornforth sat in the front row of the audience about 1 m away from the presenting interviewees, and at the end of C.M.W’s lecture, asked (shouted owing to deafness) a stereochemistry question regarding the proposed research program, which was not likely answered satisfactorily. At the end of the lecture C.M.W. asked his host Professor Phil Parsons (now at Imperial College London), ‘Who was that?’, to which he laughed and replied, ‘That was John Cornforth!’ Perhaps it was no surprise that Associate Professor Gareth Rowlands (now at Massey University, NZ) ended up getting the position.
Lastly, the authors are most grateful to those that have provided feedback on this topic over the years and the ensuing manuscript.
References
[1] (a) E. J. Corey, X.-M. Cheng, The Logic of Chemical Synthesis 1995 (Wiley: New York, NY).(b) E. J. Corey, Chem. Soc. Rev. 1988, 17, 111.
| Crossref | GoogleScholarGoogle Scholar |
(c) E. J. Corey, Pure Appl. Chem. 1967, 14, 19.
| Crossref | GoogleScholarGoogle Scholar |
[2] (a) The Nobel Prize, The Nobel Prize in Chemistry 1990. Nobel Media AB 2020. Available at: https://www.nobelprize.org/prizes/chemistry/1990/summary (accessed 11 Jun 2020)
(b) E. J. Corey, Angew. Chem. Int. Ed. Engl. 1991, 30, 455.
| Crossref | GoogleScholarGoogle Scholar |
[3] (a) S. Warren, Designing Organic Syntheses: A Programmed Introduction to the Synthon Approach 1982 (Wiley: New York, NY).
(b) S. Warren, P. Wyatt, Organic Synthesis: The Disconnection Approach, 2nd Edn 2008 (John Wiley & Sons: New York, NY).
[4] M. B. Smith, J. Chem. Educ. 1990, 67, 848.
| Crossref | GoogleScholarGoogle Scholar |
[5] (a) J. Saunders, Top Drugs: Top Synthetic Routes 2000 (Oxford University Press: Oxford).
(b) T. P. Stockdale, C. M. Williams, Chem. Soc. Rev. 2015, 44, 7737.
| Crossref | GoogleScholarGoogle Scholar |
[6] K. C. Nicolaou, E. J. Sorensen, Classics in Total Synthesis: Targets, Strategies, Methods 1996 (Wiley-VCH: Weinheim).
[7] K. C. Nicolaou, S. A. Snyder, Classics in Total Synthesis II: More Targets, Strategies, Methods 2003 (Wiley-VCH: Weinheim).
[8] K. C. Nicolaou, J. S. Chen, Classics in Total Synthesis III: Further Targets, Strategies, Methods 2011 (Wiley-VCH: Weinheim).
[9] (a) D. G. Brown, J. Boström, J. Med. Chem. 2016, 59, 4443.
| Crossref | GoogleScholarGoogle Scholar | 26571338PubMed |
(b) S. D. Roughley, A. M. Jordan, J. Med. Chem. 2011, 54, 3451.
| Crossref | GoogleScholarGoogle Scholar |
[10] (a) H. M. L. Davies, K. Liao, Nat. Rev. Chem. 2019, 3, 347.
| Crossref | GoogleScholarGoogle Scholar |
(b) Y. Wei, P. Hu, M. Zhang, W. Su, Chem. Rev. 2017, 117, 8864.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. He, M. Wasa, K. S. L. Chan, Q. Shao, J.-Q. Yu, Chem. Rev. 2017, 117, 8754.
| Crossref | GoogleScholarGoogle Scholar |
[11] D. Petzold, M. Giedyk, A. Chatterjee, B. König, Eur. J. Org. Chem. 2020, 1193.
| Crossref | GoogleScholarGoogle Scholar |
[12] (a) E. J. Corey, W. T. Wipke, Science 1969, 166, 178.
| Crossref | GoogleScholarGoogle Scholar | 17731475PubMed |
(b) E. J. Corey, W. J. Howe, H. W. Orf, D. A. Pensak, G. Petersson, J. Am. Chem. Soc. 1975, 97, 6116.
| Crossref | GoogleScholarGoogle Scholar |
(c) E. J. Corey, W. L. Jorgensen, J. Am. Chem. Soc. 1976, 98, 189.
| Crossref | GoogleScholarGoogle Scholar |
(d) E. J. Corey, A. K. Long, S. D. Rubenstein, Science 1985, 228, 408.
| Crossref | GoogleScholarGoogle Scholar |
(e) K. Lin, Y. Xu, J. Pei, L. Lai, Chem. Sci. 2020, 11, 3355.
| Crossref | GoogleScholarGoogle Scholar |
(f) P. Schwaller, R. Petraglia, V. Zullo, V. H. Nair, R. A. Haeuselmann, R. Pisoni, C. Bekas, A. Iuliano, T. Laino, Chem. Sci. 2020, 11, 3316.
| Crossref | GoogleScholarGoogle Scholar |
[13] (a) H. L. Gelernter, A. F. Sanders, D. L. Larsen, K. K. Agarwal, R. H. Boivie, G. A. Spritzer, J. E. Searleman, Science 1977, 197, 1041.
| Crossref | GoogleScholarGoogle Scholar | 17836062PubMed |
(b) W. T. Wipke, G. I. Ouchi, S Krishnan, Artif. Intell. 1978, 11, 173.
| Crossref | GoogleScholarGoogle Scholar |
(c) S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk, B. A. Grzybowski, Angew. Chem. Int. Ed. 2016, 55, 5904.
| Crossref | GoogleScholarGoogle Scholar |
[14] J. W. Cornforth, R. H. Cornforth, K. K. Mathew, J. Chem. Soc. 1959, 2539.
| Crossref | GoogleScholarGoogle Scholar |
[15] The carbon unit equation approach has been recently deployed to construct an all-trans-polyprenol with 100 carbons; see: Y. Totsuka, Y. Yasuno, T. Shinada, Chem. Lett. 2019, 48, 491.
| Crossref | GoogleScholarGoogle Scholar |
[16] The non-traditional version of the retrosynthesis arrow (i.e. a closed in forward arrow) is frowned on by the authors and is never utilised. The authors believe that this arrow has introduced confusion to the discipline because it indicates a forward direction rather than reverse. The authors surmise that the introduction of the non-traditional arrow arose in the first Classics in Total Synthesis book series.[6]
[17] (a) For a detailed overview of retrosynthesis, see: M. B. Smith, Organic Synthesis 4th Edn 2016 (Academic Press) and preceding Edns 1–3.
(b) K. C. Nicolaou, C. R. H. Hale, Natl. Sci. Rev. 2014, 1, 233.
| Crossref | GoogleScholarGoogle Scholar |
[18] (a) M. C. de la Torre, M. A. Sierra, Angew. Chem. Int. Ed. Engl. 2004, 43, 160.
| Crossref | GoogleScholarGoogle Scholar | 14695603PubMed |
(b) M. Razzak, J. K. De Brabander, Nat. Chem. Biol. 2011, 7, 865.
| Crossref | GoogleScholarGoogle Scholar |
[19] L. F. Tietze, Chem. Rev. 1996, 96, 115.
| Crossref | GoogleScholarGoogle Scholar | 11848746PubMed |
[20] J. C. Wasilke, S. J. Obrey, R. T. Baker, G. C. Bazan, Chem. Rev. 2005, 105, 1001.
| Crossref | GoogleScholarGoogle Scholar | 15755083PubMed |
[21] (a) K. C. Nicolaou, D. J. Edmonds, P. G. Bulger, Angew. Chem. Int. Ed. 2006, 45, 7134.
| Crossref | GoogleScholarGoogle Scholar |
(b) K. C. Nicolaou, J. S. Chen, Chem. Soc. Rev. 2009, 38, 2993.
| Crossref | GoogleScholarGoogle Scholar |
[22] J. Y. W. Mak, C. M. Williams, Nat. Prod. Rep. 2012, 29, 440.
| Crossref | GoogleScholarGoogle Scholar |
[23] K. Kawazu, Agric. Biol. Chem. 1980, 44, 1367.
| Crossref | GoogleScholarGoogle Scholar |
[24] M. Kubo, M. Nakai, K. Harada, Y. Fukuyama, Tetrahedron 2019, 75, 2379.and references therein.
| Crossref | GoogleScholarGoogle Scholar |
[25] Y. Fukuyama, M. Kubo, H. Minami, H. Yuasa, A. Matsuo, T. Fujii, M. Morisaki, K. Harada, Chem. Pharm. Bull. 2005, 53, 72.
| Crossref | GoogleScholarGoogle Scholar |
[26] M. G. Constantino, V. L. Júnior, G. V. José Da Silva, Molecules 2002, 7, 456.
| Crossref | GoogleScholarGoogle Scholar |
[27] B. D. Schwartz, D. P. Tilly, R. Heim, S. Wiedemann, C. M. Williams, P. V. Bernhardt, Eur. J. Org. Chem. 2006, 3181.
| Crossref | GoogleScholarGoogle Scholar |
[28] M. J. Gallen, R. Goumont, T. Clark, F. Terrier, C. M. Williams, Angew. Chem. Int. Ed. 2006, 45, 2929.
| Crossref | GoogleScholarGoogle Scholar |
[29] (a) A. Porzelle, C. M. Williams, B. D. Schwartz, I. R. Gentle, Synlett 2005, 2923.
| Crossref | GoogleScholarGoogle Scholar |
(b) B. D. Schwartz, A. Porzelle, K. S. Jack, J. M. Faber, I. R. Gentle, C. M. Williams, Adv. Synth. Catal. 2009, 351, 1148.
| Crossref | GoogleScholarGoogle Scholar |
[30] W. A. Eger, R. L. Grange, H. Schill, R. Goumont, T. Clark, C. M. Williams, Eur. J. Org. Chem. 2011, 2548.
| Crossref | GoogleScholarGoogle Scholar |
[31] E. Winterfeldt, H. Bauermeister, H. Riechers, D. Schomburg, P. Washausen, Angew. Chem. Int. Ed. Engl. 1991, 30, 191.
| Crossref | GoogleScholarGoogle Scholar |
[32] A. P.-J. Chen, C. M. Williams, Org. Lett. 2008, 10, 3441.
| Crossref | GoogleScholarGoogle Scholar |
[33] (a) Considering that the natural product isolation procedure used methanol as the extracting solvent (often for weeks), there existed the potential that the methylated systems were artefacts of isolation; see for example: R. J. Capon, Nat. Prod. Rep. 2020, 37, 55.
| Crossref | GoogleScholarGoogle Scholar | 31046051PubMed |
(b) L. A. Maslovskaya, A. I. Savchenko, V. A. Gordon, P. W. Reddell, C. J. Pierce, P. G. Parsons, C. M. Williams, Chem. – Eur. J. 2017, 23, 537.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. B. McAlpine, S.-N. Chen, A. Kutateladze, J. B. MacMillan, G. Appendino, A. Barison, M. A. Beniddir, M. W. Biavatti, S. Bluml, A. Boufridi, M. S. Butler, R. J. Capon, Y. H. Choi, D. Coppage, P. Crews, M. T. Crimmins, M. Csete, P. Dewapriya, J. M. Egan, M. J. Garson, G. Genta-Jouve, W. H. Gerwick, H. Gross, M. K. Harper, P. Hermanto, J. M. Hook, L. Hunter, D. Jeannerat, N.-Y. Ji, T. A. Johnson, D. G. I. Kingston, H. Koshino, H.-W. Lee, G. Lewin, J. Li, R. G. Linington, M. Liu, K. L. McPhail, T. F. Molinski, B. S. Moore, J.-W. Nam, R. P. Neupane, M. Niemitz, J.-M. Nuzillard, N. H. Oberlies, F. M. M. Ocampos, G. Pan, R. J. Quinn, D. S. Reddy, J.-H. Renault, J. Rivera-Chávez, W. Robien, C. M. Saunders, T. J. Schmidt, C. Seger, B. Shen, C. Steinbeck, H. Stuppner, S. Sturm, O. Taglialatela-Scafati, D. J. Tantillo, R. Verpoorte, B.-G. Wang, C. M. Williams, P. G. Williams, J. Wist, J.-M. Yue, C. Zhang, Z. Xu, C. Simmler, D. C. Lankin, J. Bisson, G. F. Pauli, Nat. Prod. Rep. 2019, 36, 35.
| Crossref | GoogleScholarGoogle Scholar |
[34] (a) J. Y. W. Mak, C. M. Williams, Chem. Commun. 2012, 287.
| Crossref | GoogleScholarGoogle Scholar |
(b) J. Y. W. Mak, C. M. Williams, Eur. J. Org. Chem. 2012, 2001.
| Crossref | GoogleScholarGoogle Scholar |
[35] (a) A. P.-J. Chen, C. C. Müller, H. M. Cooper, C. M. Williams, Org. Lett. 2009, 11, 3758.
| Crossref | GoogleScholarGoogle Scholar |
(b) A. P.-J. Chen, C. C. Müller, H. M. Cooper, C. M. Williams, Tetrahedron 2010, 66, 6842.
| Crossref | GoogleScholarGoogle Scholar |
[36] (a) M. J. Gallen, C. M. Williams, Org. Lett. 2008, 10, 713.
| Crossref | GoogleScholarGoogle Scholar | 18247495PubMed |
(b) M. J. Gallen, C. M. Williams, Eur. J. Org. Chem. 2008, 4697.
| Crossref | GoogleScholarGoogle Scholar |
[37] (a) G. A. Strohmeier, H. Pichler, O. May, M. Gruber-Khadjawi, Chem. Rev. 2011, 111, 4141.
| Crossref | GoogleScholarGoogle Scholar | 21553913PubMed |
(b) H.-P. Meyer, E. Eichhorn, S. Hanlon, S. Lütz, M. Schürmann, R. Wohlgemuthf, R. Coppolecchiag, Catal. Sci. Technol. 2013, 3, 29.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. Winkler, M. Geier, S. P. Hanlon, B. Nidetzky, A. Glieder, Angew. Chem. Int. Ed. 2018, 57, 13406.
| Crossref | GoogleScholarGoogle Scholar |
[38] L. A. Baker, C. M. Williams, P. V. Bernhardt, G. W. Yanik, Tetrahedron 2006, 62, 7355.
| Crossref | GoogleScholarGoogle Scholar |
[39] P. A. Wender, Nat. Prod. Rep. 2014, 31, 433.
| Crossref | GoogleScholarGoogle Scholar | 24589860PubMed |
[40] P. A. Wender, V. A. Verma, T. J. Paxton, T. H. Pillow, Acc. Chem. Res. 2008, 41, 40.
| Crossref | GoogleScholarGoogle Scholar | 18159936PubMed |
[41] P. E. Eaton, T. W. Cole, J. Am. Chem. Soc. 1964, 86, 962.
| Crossref | GoogleScholarGoogle Scholar |
[42] P. E. Eaton, T. W. Cole, J. Am. Chem. Soc. 1964, 86, 3157.
| Crossref | GoogleScholarGoogle Scholar |
[43] M. A. Ogliaruso, M. G. Romanelli, E. I. Becker, Chem. Rev. 1965, 65, 261.
| Crossref | GoogleScholarGoogle Scholar |
[44] K. Hafner, K. Goliasch, Chem. Ber. 1961, 94, 2909.
| Crossref | GoogleScholarGoogle Scholar |
[45] E. T. McBee, R. K. Meyers, J. Am. Chem. Soc. 1955, 77, 88.
| Crossref | GoogleScholarGoogle Scholar |
[46] S. D. Houston, B. A. Chalmers, G. P. Savage, C. M. Williams, Org. Biomol. Chem. 2019, 17, 1067.
| Crossref | GoogleScholarGoogle Scholar | 30644962PubMed |
[47] (a) E. J. Ko, G. P. Savage, C. M. Williams, J. Tsanaktsidis, Org. Lett. 2011, 13, 1944.
| Crossref | GoogleScholarGoogle Scholar | 21438514PubMed |
(b) J. Ho, J. Zheng, R. Meana-Pañeda, D. G. Truhlar, E. J. Ko, G. P. Savage, C. M. Williams, M. L. Coote, J. Tsanaktsidis, J. Org. Chem. 2013, 78, 6677.
| Crossref | GoogleScholarGoogle Scholar |
(c) E. J. Ko, C. M. Williams, G. P. Savage, J. Tsanaktsidis, Org. Synth. 2012, 89, 471.
| Crossref | GoogleScholarGoogle Scholar |
[48] (a) P. E. Eaton, Angew. Chem., Int. Ed. Engl. 1992, 31, 1421.[Angew. Chem. 1992, 104, 1447.
| Crossref | GoogleScholarGoogle Scholar |
(b) K. F. Biegasiewicz, J. R. Griffiths, G. P. Savage, J. Tsanaktsidis, R. Priefer, Chem. Rev. 2015, 115, 6719.
| Crossref | GoogleScholarGoogle Scholar |
[49] (a) M. J. Falkiner, S. W. Littler, K. J. McRae, G. P. Savage, J. Tsanaktsidis, Org. Process Res. Dev. 2013, 17, 1503.
| Crossref | GoogleScholarGoogle Scholar |
(b) D. E. Collin, E. H. Jackman, N. Jouandon, W. Sun, M. E. Light, D. C. Harrowven, B. Linclau, Synthesis 2021, 53, 1307.
| Crossref | GoogleScholarGoogle Scholar |
[50] (a) M. A. Dallaston, J. S. Brusnahan, C. Wall, C. M. Williams, Chem. – Eur. J. 2019, 25, 8344.
| Crossref | GoogleScholarGoogle Scholar | 31124182PubMed |
(b) M. A. Dallaston, S. D. Houston, C. M. Williams, Chem. – Eur. J. 2020, 26, 11966.
| Crossref | GoogleScholarGoogle Scholar |
[51] (a) B. A. Chalmers, H. Xing, S. D. Houston, C. Clark, S. Ghassabian, A. Kuo, B. Cao, A. Reitsma, C.-E. P. Murray, J. E. Stok, G. M. Boyle, C. J. Pierce, S. W. Littler, D. A. Winkler, P. V. Bernhardt, C. Pasay, J. J. De Voss, J. McCarthy, P. G. Parsons, G. H. Walter, M. T. Smith, H. M. Cooper, S. K. Nilsson, J. Tsanaktsidis, G. P. Savage, C. M. Williams, Angew. Chem. Int. Ed. 2016, 55, 3580.[Angew. Chem. 2016, 128, 3644]
| Crossref | GoogleScholarGoogle Scholar |
(b) T. A. Reekie, C. M. Williams, L. M. Rendina, M. Kassiou, J. Med. Chem. 2019, 62, 1078.
| Crossref | GoogleScholarGoogle Scholar |
[52] (a) L. Velluz, J. Valls, J. Mathieu, Angew. Chem. Int. Ed. 1967, 6, 778.[Angew. Chem. 1967, 79, 774]
| Crossref | GoogleScholarGoogle Scholar |
(b) D. Urabe, T. Asaba, M. Inoue, Chem. Rev. 2015, 115, 9207.
| Crossref | GoogleScholarGoogle Scholar |
[53] C. M. Williams, in Strategies and Tactics in Organic Synthesis (Ed. M. Harmata) 2014, Vol 10, Ch. 11, pp. 249–270 (Academic Press: New York, NY).
[54] J. E. Aho, P. M. Pihko, T. K. Rissa, Chem. Rev. 2005, 105, 4406.
| Crossref | GoogleScholarGoogle Scholar | 16351049PubMed |
[55] L. F. Tietze, H. Geissler, J. A. Gewert, U. Jakobi, Synlett 1994, 511.
| Crossref | GoogleScholarGoogle Scholar |
[56] A. B. Smith, C. M. Adams, Acc. Chem. Res. 2004, 37, 365.
| Crossref | GoogleScholarGoogle Scholar | 15196046PubMed |
[57] N. Choukchou-Braham, Y. Asakawa, J.-P. Lepoittevin, Tetrahedron Lett. 1994, 35, 3949.
| Crossref | GoogleScholarGoogle Scholar |
[58] K.-M. Chen, G. E. Hardtmann, K. Prasad, O. Repič, M. J. Shapiro, Tetrahedron Lett. 1987, 28, 155.
| Crossref | GoogleScholarGoogle Scholar |
[59] M. Nakatsuka, J. A. Ragan, T. Sammakia, D. B. Smith, D. E. Uehling, S. L. Schreiber, J. Am. Chem. Soc. 1990, 112, 5583.and references therein.
| Crossref | GoogleScholarGoogle Scholar |
[60] L. Dong, V. A. Gordon, R. L. Grange, J. Johns, P. G. Parsons, A. Porzelle, P. Reddell, H. Schill, C. M. Williams, J. Am. Chem. Soc. 2008, 130, 15262.
| Crossref | GoogleScholarGoogle Scholar | 18950180PubMed |
[61] L. Dong, H. Schill, R. L. Grange, A. Porzelle, J. P. Johns, P. G. Parsons, V. A. Gordon, P. W. Reddell, C. M. Williams, Chem. – Eur. J. 2009, 15, 11307.
| Crossref | GoogleScholarGoogle Scholar | 19750529PubMed |
[62] (a) T. M. Trnka, R. H. Grubbs, Acc. Chem. Res. 2001, 34, 18.
| Crossref | GoogleScholarGoogle Scholar | 11170353PubMed |
(b) S. B. Garber, J. S. Kingsbury, B. L. Gray, A. H. Hoveyda, J. Am. Chem. Soc. 2000, 122, 8168.
| Crossref | GoogleScholarGoogle Scholar |
[63] D. A. Evans, B. W. Trotter, P. J. Coleman, B. Côté, L. C. Dias, H. A. Rajapakse, A. N. Tyler, Tetrahedron 1999, 55, 8671.
| Crossref | GoogleScholarGoogle Scholar |
[64] J. R. Hanson, Sci. Prog. 2017, 100, 63.
| Crossref | GoogleScholarGoogle Scholar | 28693673PubMed |
[65] R. B. Woodward, F. G. Brutschy, H. Baer, J. Am. Chem. Soc. 1948, 70, 4216.
| Crossref | GoogleScholarGoogle Scholar | 18105974PubMed |
[66] (a) W. O. Kermack, R. Robinson, J. Chem. Soc. Trans. 1922, 121, 427.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. Saltzman, J. Chem. Educ. 1972, 49, 750.
| Crossref | GoogleScholarGoogle Scholar |
[67] L. Birladeanu, Angew. Chem. Int. Ed. 2003, 42, 1202.
| Crossref | GoogleScholarGoogle Scholar |
[68] M. Kahler, Arch. Pharm. 1830, 34, 318.
| Crossref | GoogleScholarGoogle Scholar |
[69] (a) G. R. Clemo, R. D. Haworth, E. J. Walton, J. Chem. Soc. 1929, 2368.
| Crossref | GoogleScholarGoogle Scholar |
(b) G. R. Clemo, R. D. Haworth, E. J. Walton, J. Chem. Soc. 1930, 1110.
| Crossref | GoogleScholarGoogle Scholar |
[70] J. W. Huffman, W. T. Pennington, D. W. Bearden, J. Nat. Prod. 1992, 55, 1087.
| Crossref | GoogleScholarGoogle Scholar |
[71] A. P. J. Brunskill, H. W. Thompson, R. A. Lalancette, Acta Crystallogr. C 1999, 55, 566.
| Crossref | GoogleScholarGoogle Scholar |
[72] A. C. Willis, P. D. O’Connor, W. C. Taylor, L. N. Mander, Aust. J. Chem. 2006, 59, 629.
| Crossref | GoogleScholarGoogle Scholar |
[73] L. N. Mander, A. C. Willis, A. J. Herlt, W. C. Taylor, Tetrahedron Lett. 2009, 50, 7089.
| Crossref | GoogleScholarGoogle Scholar |
[74] T. A. Bradford, A. C. Willis, J. M. White, A. J. Herlt, W. C. Taylor, L. N. Mander, Tetrahedron Lett. 2011, 52, 188.
| Crossref | GoogleScholarGoogle Scholar |
[75] P. Lan, A. J. Herlt, A. C. Willis, W. C. Taylor, L. N. Mander, ACS Omega 2018, 3, 1912.
| Crossref | GoogleScholarGoogle Scholar | 31458503PubMed |
[76] S. V. Binns, P. J. Dunstan, G. M. Guise, G. M. Holder, A. F. Hollis, R. S. McCredie, J. T. Pinhey, R. H. Prager, M. Rasmussen, E. Ritchie, W. Taylor, Aust. J. Chem. 1965, 18, 569.
| Crossref | GoogleScholarGoogle Scholar |
[77] L. N. Mander, E. Ritchie, W. C. Taylor, Aust. J. Chem. 1967, 20, 981.
| Crossref | GoogleScholarGoogle Scholar |
[78] L. N. Mander, R. H. Prager, M. Rasmussen, E. Ritchie, W. C. Taylor, Aust. J. Chem. 1967, 20, 1473.
| Crossref | GoogleScholarGoogle Scholar |
[79] L. N. Mander, R. H. Prager, M. Rasmussen, E. Ritchie, W. C. Taylor, Aust. J. Chem. 1967, 20, 1705.
| Crossref | GoogleScholarGoogle Scholar |
[80] L. N. Mander, M. M. McLachlan, J. Am. Chem. Soc. 2003, 125, 2400.
| Crossref | GoogleScholarGoogle Scholar | 12603121PubMed |
[81] L. N. Mander, C. M. Williams, Tetrahedron 2003, 59, 1105.
| Crossref | GoogleScholarGoogle Scholar |
[82] A. J. Birch, J. Chem. Soc. 1944, 430.
| Crossref | GoogleScholarGoogle Scholar |
[83] L. N. Mander, in Comprehensive Organic Synthesis (Eds B. M. Trost, I. Fleming) 1991, Vol 8, pp. 489–521 (Pergamon: Oxford).
[84] U. Shah, S. Chackalamannil, A. K. Ganguly, M. Chelliah, S. Kolotuchin, A. Buevich, A. McPhail, J. Am. Chem. Soc. 2006, 128, 12654.
| Crossref | GoogleScholarGoogle Scholar | 17002352PubMed |
[85] D. A. Evans, D. J. Adams, J. Am. Chem. Soc. 2007, 129, 1048.
| Crossref | GoogleScholarGoogle Scholar | 17263383PubMed |
[86] (a) R. T. Larson, M. D. Clift, R. J. Thomson, Angew. Chem. Int. Ed. 2012, 51, 2481.
| Crossref | GoogleScholarGoogle Scholar |
(b) R. T. Larson, R. P. Pemberton, J. M. Franke, D. J. Tantillo, R. J. Thomson, J. Am. Chem. Soc. 2015, 137, 11197.
| Crossref | GoogleScholarGoogle Scholar |
[87] (a) M. D. Levin, K. Subrahamanian, H. Katz, M. B. Smith, D. J. Burlett, S. M. Hecht, J. Am. Chem. Soc. 1980, 102, 1452.
| Crossref | GoogleScholarGoogle Scholar |
(b) Y. Aoyagi, K. Katano, H. Suguna, J. Primeau, L.-H. Chang, S. M. Hecht, J. Am. Chem. Soc. 1982, 104, 5537.
| Crossref | GoogleScholarGoogle Scholar |
[88] T. Kametani, K. Fukumoto, Acc. Chem. Res. 1976, 9, 319.
| Crossref | GoogleScholarGoogle Scholar |
[89] A. Pagani, S. Gaeta, A. I. Savchenko, C. M. Williams, G. Appendino, Beilstein J. Org. Chem. 2017, 13, 1361.
| Crossref | GoogleScholarGoogle Scholar | 28781702PubMed |
[90] (a) Z. Liu, Z. Ding, K. Chen, M. Xu, T. Yu, G. Tong, H. Zhang, P. Li, Nat. Prod. Rep. 2021,
| Crossref | GoogleScholarGoogle Scholar | 33508045PubMed |
(b) S. Chow, T. Krainz, P. V. Bernhardt, C. M. Williams, Org. Lett. 2019, 21, 8761.
| Crossref | GoogleScholarGoogle Scholar |
(c) S. Chow, T. Krainz, C. J. Bettencourt, N. Broit, B. Ferguson, M. Zhu, K. G. Hull, G. K. Pierens, P. V. Bernhardt, P. G. Parsons, D. Romo, G. M. Boyle, C. M. Williams, Chem. – Eur. J. 2020, 26, 13372.
| Crossref | GoogleScholarGoogle Scholar |
[91] (a) L. N. Mander, Acc. Chem. Res. 1983, 16, 48.
| Crossref | GoogleScholarGoogle Scholar |
(b) L. N. Mander, Nat. Prod. Rep. 1988, 5, 541.
| Crossref | GoogleScholarGoogle Scholar |
(c) L. N. Mander, Chem. Rev. 1992, 92, 573.
| Crossref | GoogleScholarGoogle Scholar |
(d) L. N. Mander, Nat. Prod. Rep. 2003, 20, 49.
| Crossref | GoogleScholarGoogle Scholar |
[92] R. D. Dawe, L. N. Mander, J. V. Turner, Tetrahedron Lett. 1985, 26, 363.
| Crossref | GoogleScholarGoogle Scholar |
[93] (a) I. Ojima, I. Habus, M. Zhao, M. Zucco, Y. H. Parr, C. M. Sun, T. Brigaud, Tetrahedron 1992, 48, 6985.
| Crossref | GoogleScholarGoogle Scholar |
(b) Y. Kanda, H. Nakamura, S. Umemiya, R. K. Puthukanoori, V. R. M. Appala, G. K. Gaddamanugu, B. R. Paraselli, P. S. Baran, J. Am. Chem. Soc. 2020, 142, 10526.and references therein.
| Crossref | GoogleScholarGoogle Scholar |
[94] M. Boehm, P. C. Fuenfschilling, M. Krieger, E. Kuesters, F. Struber, Org. Process Res. Dev. 2007, 11, 336.
| Crossref | GoogleScholarGoogle Scholar |
[95] H. M. E. Cardwell, J. W. Cornforth, S. R. Duff, H. Holtermann, R. Robinson, Chem. Ind. 1951, 389.
[96] Y. Shi, J. T. Wilmot, L. U. Nordstrøm, D. S. Tan, D. Y. Gin, J. Am. Chem. Soc. 2013, 135, 14313.and references therein.
| Crossref | GoogleScholarGoogle Scholar | 24040959PubMed |
[97] (a) N. A. Doering, R. Sarpong, R. W. Hoffmann, Angew. Chem. Int. Ed. 2020, 59, 10722.
| Crossref | GoogleScholarGoogle Scholar |
(b) S. V. McCowen, N. A. Doering, R. Sarpong, Chem. Sci. 2020, 11, 7538.
| Crossref | GoogleScholarGoogle Scholar |
[98] (a) For Williams group efforts in the synthesis of C20-diterpene alkaloids see: C. M. Williams, L. N. Mander, Org. Lett. 2003, 5, 3499.
| Crossref | GoogleScholarGoogle Scholar | 12967309PubMed |
(b) C. M. Williams, L. N. Mander, P. V. Bernhardt, A. C. Willis, Tetrahedron 2005, 61, 3759.
| Crossref | GoogleScholarGoogle Scholar |
[99] H. Zhang, M. S. Reddy, S. Phoenix, P. Deslongchamps, Angew. Chem. Int. Ed. 2008, 47, 1272.
| Crossref | GoogleScholarGoogle Scholar |
[100] See for example: A. Greule, J. E. Stok, J. J. De Voss, M. J. Cryle, Nat. Prod. Rep. 2018, 35, 757.and references therein.
| Crossref | GoogleScholarGoogle Scholar | 29667657PubMed |
[101] X. Zhang, E. King-Smith, L.-Bi. Dong, L.-C. Yang, J. D. Rudolf, B. Shen, H. Renata, Science 2020, 369, 799.
| Crossref | GoogleScholarGoogle Scholar | 32792393PubMed |
[102] (a) D. M. Pinkerton, T. J. Vanden Berg, P. V. Bernhardt, C. M. Williams, Chem. – Eur. J. 2017, 23, 2282.
| Crossref | GoogleScholarGoogle Scholar | 28042894PubMed |
(b) D. M. Pinkerton, P. V. Bernhardt, G. P. Savage, C. M. Williams, Asian J. Org. Chem. 2017, 6, 583.
| Crossref | GoogleScholarGoogle Scholar |
(c) D. M. Pinkerton, S. Chow, N. H. Eisa, K. Kainth, T. J. Vanden Berg, J. M. Burns, L. W. Guddat, G. P. Savage, A. Chadli, C. M. Williams, Chem. – Eur. J. 2019, 25, 1451.
| Crossref | GoogleScholarGoogle Scholar |
[103] IBX has sufficient acidity to promote side reactions and/or acid catalysed deprotections, see ref. [28] and T. Waters, J. Boulton, T. Clark, M. J. Gallen, C. M. Williams, R. A. J. O’Hair, Org. Biomol. Chem. 2008, 6, 2530.
| Crossref | GoogleScholarGoogle Scholar | 18600274PubMed |
[104] (a) M. J. Schnermann, R. A. Shenvi, Nat. Prod. Rep. 2015, 32, 543.
| Crossref | GoogleScholarGoogle Scholar | 25514696PubMed |
(b) M. J. Garson, J. S. Simpson, Nat. Prod. Rep. 2004, 21, 164.
| Crossref | GoogleScholarGoogle Scholar |
[105] E. J. Corey, P. A. Magriotis, J. Am. Chem. Soc. 1987, 109, 287.
| Crossref | GoogleScholarGoogle Scholar |
[106] (a) K. A. Fairweather, L. N. Mander, Org. Lett. 2006, 8, 3395.
| Crossref | GoogleScholarGoogle Scholar | 16836414PubMed |
(b) K. A. Fairweather, S. R. Crabtree, L. N. Mander, in Strategies and Tactics in Organic Synthesis (Ed. M. Harmata) 2008, Vol 7. Ch. 2, pp. 35–58 (Academic Press: New York, NY).
[107] J. S. Simpson, M. J. Garson, Org. Biomol. Chem. 2004, 2, 939.
| Crossref | GoogleScholarGoogle Scholar | 15007426PubMed |
[108] H. Miyaoka, Y. Okubo, M. Muroi, H. Mitome, E. Kawashima, Chem. Lett. 2011, 40, 246.
| Crossref | GoogleScholarGoogle Scholar |
[109] P. C. Roosen, C. D. Vanderwal, Angew. Chem. Int. Ed. 2016, 55, 7180.
| Crossref | GoogleScholarGoogle Scholar |
[110] E. E. Robinson, R. J. Thomson, J. Am. Chem. Soc. 2018, 140, 1956.
| Crossref | GoogleScholarGoogle Scholar | 29309727PubMed |
[111] A. S. Karns, B. D. Ellis, P. C. Roosen, Z. Chahine, K. G. Le Roch, C. D. Vanderwal, Angew. Chem. Int. Ed. 2019, 58, 13749.
| Crossref | GoogleScholarGoogle Scholar |
[112] The Claisen Rearrangement: Methods and Applications (Eds M. Hiersemann, U. Nubbemeyer) 2007 (Wiley-VCH: Chichester).
[113] B. D. Schwartz, J. R. Denton, H. M. L. Davies, C. M. Williams, Aust. J. Chem. 2009, 62, 980.
| Crossref | GoogleScholarGoogle Scholar | 21297900PubMed |
[114] B. D. Schwartz, J. R. Denton, Y. Lian, H. M. L. Davies, C. M. Williams, J. Am. Chem. Soc. 2009, 131, 8329.
| Crossref | GoogleScholarGoogle Scholar | 19445507PubMed |
[115] Y. Fukuyama, H. Minami, A. Matsuo, K. Kitamura, M. Akizuki, M. Kubo, M. Kodama, Chem. Pharm. Bull. 2002, 50, 368.and references therein.
| Crossref | GoogleScholarGoogle Scholar |
[116] B. D. Schwartz, C. M. Williams, P. V. Bernhardt, Beilstein J. Org. Chem. 2008, 4.
| Crossref | GoogleScholarGoogle Scholar |
[117] H. M. L. Davies, Ø. Loe, D. G. Stafford, Org. Lett. 2005, 7, 5561.
| Crossref | GoogleScholarGoogle Scholar |
[118] C. M. Williams, in Strategies and Tactics in Organic Synthesis (Ed. M. Harmata) 2012, Vol 8. Ch. 15, pp. 395–412 (Academic Press: New York, NY).
[119] B. D. Schwartz, C. M. Williams, E. Anders, P. V. Bernhardt, Tetrahedron 2008, 64, 6482.
| Crossref | GoogleScholarGoogle Scholar |
[120] B. D. Schwartz, J. R. Denton, P. V. Bernhardt, H. M. L. Davies, C. M. Williams, Synthesis 2009, 2840.
| Crossref | GoogleScholarGoogle Scholar | 20725592PubMed |
[121] S. B. Jones, B. Simmons, A. Mastracchio, D. W. C. MacMillan, Nature 2011, 475, 183.
| Crossref | GoogleScholarGoogle Scholar | 21753848PubMed |
[122] (a) See for example: J. Wang, S.-G. Chen, B.-F. Sun, G.-Q. Lin, Y.-J. Shang, Chem. – Eur. J. 2013, 19, 2539.
| Crossref | GoogleScholarGoogle Scholar | 23292997PubMed |
(b) W. Xu, J. Zhao, C. Tao, H. Wang, Y. Li, B. Cheng, H. Zhai, Org. Lett. 2018, 20, 1509.
| Crossref | GoogleScholarGoogle Scholar |
(c) C. Qiao, W. Zhang, J.-C. Han, W.-M. Dai, C.-C. Li, Tetrahedron 2019, 75, 1739.
| Crossref | GoogleScholarGoogle Scholar |
(d) D.-W. Zhang, H.-L. Fan, W. Zhang, C.-J. Li, S. Luo, H.-B. Qin, Chem. Commun. 2020, 10066.
| Crossref | GoogleScholarGoogle Scholar |
(e) S.-H. Wang, R.-Q. Si, Q.-B. Zhuang, X. Guo, T. Ke, X.-M. Zhang, F.-M. Zhang, Y.-Q. Tu, Angew. Chem. Int. Ed. 2020, 59, 21954.
| Crossref | GoogleScholarGoogle Scholar |
[123] (a) V. Lakshmi, P. Gupta, Nat. Prod. Res. 2008, 22, 1197.
| Crossref | GoogleScholarGoogle Scholar | 18932084PubMed |
(b) D. A. Mulholland, B. Parel, P. H. Coombes, Curr. Org. Chem. 2000, 4, 1011.
| Crossref | GoogleScholarGoogle Scholar |
(c) J. Wu, Q. Xiao, J. Xu, M.-Y. Li, J.-Y. Pan, M.-h. Yang, Nat. Prod. Rep. 2008, 25, 955.
| Crossref | GoogleScholarGoogle Scholar |
[124] J. M. Faber, C. M. Williams, Chem. Commun. 2011, 2258.
| Crossref | GoogleScholarGoogle Scholar |
[125] J. M. Faber, W. A. Eger, C. M. Williams, J. Org. Chem. 2012, 77, 8913.
| Crossref | GoogleScholarGoogle Scholar | 22994389PubMed |
[126] J. D’Angelo, M. B. Smith, Hybrid Retrosynthesis Organic Synthesis Using Reaxys and SciFinder 2015 (Elsevier: Amsterdam).
[127] G. É. Vléduts, V. K. Finn, Inf. Storage Retr. 1963, 1, 101.
| Crossref | GoogleScholarGoogle Scholar |
[128] M. Bersohn, A. Esack, Chem. Rev. 1976, 76, 269.
| Crossref | GoogleScholarGoogle Scholar |
[129] (a) M. H. Todd, Chem. Soc. Rev. 2005, 34, 247.
| Crossref | GoogleScholarGoogle Scholar | 15726161PubMed |
(b) A. Cook, A. P. Johnson, J. Law, M. Mirzazadeh, O. Ravitz, A. Simon, WIREs Comput. Mol. Sci. 2012, 2, 79.
| Crossref | GoogleScholarGoogle Scholar |
(c) O. Engkvist, P. O. Norrby, N. Selmi, Y. Lam, Z. Peng, E. C. Sherer, W. Amberg, T. Erhard, L. A. Smyth, Drug Discov. Today 2018, 23, 1203.
| Crossref | GoogleScholarGoogle Scholar |
(d) F. Feng, L. Lai, J. Pei, Front. Chem. 2018, 6, 199.
| Crossref | GoogleScholarGoogle Scholar |
(e) F. Strieth-Kalthoff, F. Sandfort, M. H. S. Segler, F. Glorius, Chem. Soc. Rev. 2020, 49, 6154.
| Crossref | GoogleScholarGoogle Scholar |
(f) P. M. Pflüger, F. Glorius, Angew. Chem. Int. Ed. 2020, 59, 18860.
| Crossref | GoogleScholarGoogle Scholar |
(g) W.-D. Ihlenfeldt, J. Gasteiger, Angew. Chem. Int. Ed. Engl. 1996, 34, 2613.
| Crossref | GoogleScholarGoogle Scholar |
(h) G. Schneider, D. E. Clark, Angew. Chem. Int. Ed. 2019, 58, 10792.
| Crossref | GoogleScholarGoogle Scholar |
(i) A. Thakkar, S. Johansson, K. Jorner, D. Buttar, J.-L. Reymond, O. Engkvist, React. Chem. Eng. 2021, 6, 27.
| Crossref | GoogleScholarGoogle Scholar |
[130] (a) A. Bondy, U. S. R. Murty, Graph Theory 2008 (Springer-Verlag: London).
(b) L. David, A. Thakkar, R. Mercado, O. Engkvist, J. Cheminform. 2020, 12, 56.
| Crossref | GoogleScholarGoogle Scholar |
[131] T. Fink, H. Bruggesser, J.-L. Reymond, Angew. Chem. Int. Ed. 2005, 44, 1504.
| Crossref | GoogleScholarGoogle Scholar |
[132] IBM Corporation, IBM: Icons of Progress. Deep Blue. Available at: https://www.ibm.com/ibm/history/ibm100/us/en/icons/deepblue (accessed 18 Nov 2020)
[133] (a) D. Silver, J. Schrittwieser, K. Simonyan, I. Antonoglou, A. Huang, A. Guez, T. Hubert, L. Baker, M. Lai, A. Bolton, Y. Chen, T. Lillicrap, F. Hui, L. Sifre, G. van den Driessche, T. Graepel, D. Hassabis, Nature 2017, 550, 354.
| Crossref | GoogleScholarGoogle Scholar | 29052630PubMed |
(b) D. Silver, T. Hubert, J. Schrittwieser, I. Antonoglou, M. Lai, A. Guez, M. Lanctot, L. Sifre, D. Kumaran, T. Graepel, T. Lillicrap, K. Simonyan, D. Hassabis, Science 2018, 362, 1140.
| Crossref | GoogleScholarGoogle Scholar |
(c) M. MacDonald, How Computers Are Reinventing Chess. 15 Nov 2019. Available at: https://medium.com/young-coder/how-computers-play-chess-today-d4f921ce8c4c (accessed 10 Feb 2021)
[134] D. Heaven, Nature 2019, 574, 163.
| Crossref | GoogleScholarGoogle Scholar | 31597977PubMed |
[135] E. J. Corey, Q. Rev. Chem. Soc. 1971, 25, 455.
| Crossref | GoogleScholarGoogle Scholar |
[136] (a) T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkuć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. Sieredzińska, M. Mrksich, S. L. J. Trice, B. A. Grzybowski, Chem 2018, 4, 522.
| Crossref | GoogleScholarGoogle Scholar |
(b) K. Molga, S. Szymkuć, B. A. Grzybowski, Acc. Chem. Res. 2021, 54, 1094.
| Crossref | GoogleScholarGoogle Scholar |
[137] J. B. Hendrickson, Anal. Chim. Acta 1990, 235, 103.
| Crossref | GoogleScholarGoogle Scholar |
[138] (a) J. Gasteiger, C. Jochum, in Organic Compounds. Topics in Current Chemistry 1978, Vol 74, pp. 93–126 (Springer: Berlin).
(b) J. Gasteiger, M. G. Hutchings, B. Christoph, L. Gann, C. Hiller, P. Löw, M. Marsili, H. Saller, K. Yuki, in Organic Synthesis, Reactions and Mechanisms. Topics in Current Chemistry 1987, Vol 137, pp. 19–73 (Springer: Berlin).
[139] T. D. Salatin, W. L. Jorgensen, J. Org. Chem. 1980, 45, 2043.
| Crossref | GoogleScholarGoogle Scholar |
[140] (a) S. Hanessian, J. Franco, B. Larouche, Pure Appl. Chem. 1990, 62, 1887.
| Crossref | GoogleScholarGoogle Scholar |
(b) S. Hanessian, J. Org. Chem. 2012, 77, 6657.
| Crossref | GoogleScholarGoogle Scholar |
[141] W. T. Wipke, D. Rogers, J. Chem. Inf. Comput. Sci. 1984, 24, 71.
| Crossref | GoogleScholarGoogle Scholar | 6547445PubMed |
[142] (a) J. B. Hendrickson, A. G. Toczko, J. Chem. Inf. Comput. Sci. 1989, 29, 137.
| Crossref | GoogleScholarGoogle Scholar |
(b) J. B. Hendrickson, A. G. Toczko, Pure Appl. Chem. 1989, 61, 589.
| Crossref | GoogleScholarGoogle Scholar |
[143] E. S. Blurock, J. Chem. Inf. Comput. Sci. 1990, 30, 505.
| Crossref | GoogleScholarGoogle Scholar |
[144] J. Gasteiger, W. D. Ihlenfeldt, in Software Development in Chemistry 4 (Ed. J. Gasteiger) 1990, pp. 57–65 (Springer: Berlin).
[145] G. Sello, J. Chem. Inf. Model. 1992, 32, 713.
| Crossref | GoogleScholarGoogle Scholar |
[146] H. Satoh, K. Funatsu, J. Chem. Inf. Comput. Sci. 1995, 35, 34.
| Crossref | GoogleScholarGoogle Scholar |
[147] G. Mehta, R. Barone, M. Chanon, Eur. J. Org. Chem. 1998, 1409.
| Crossref | GoogleScholarGoogle Scholar |
[148] K. Satoh, K. Funatsu, J. Chem. Inf. Comput. Sci. 1999, 39, 316.
| Crossref | GoogleScholarGoogle Scholar |
[149] I. M. Socorro, K. Taylor, J. M. Goodman, Org. Lett. 2005, 7, 3541.
| Crossref | GoogleScholarGoogle Scholar | 16048337PubMed |
[150] A. Bøgevig, H.-J. Federsel, F. Huerta, M. G. Hutchings, H. Kraut, T. Langer, P. Löw, C. Oppawsky, T. Rein, H. Saller, Org. Process Res. Dev. 2015, 19, 357.
| Crossref | GoogleScholarGoogle Scholar |
[151] M. A. Kayala, P. Baldi, J. Chem. Inf. Model. 2012, 52, 2526.
| Crossref | GoogleScholarGoogle Scholar | 22978639PubMed |
[152] (a) B. A. Grzybowski, S. Szymkuć, E. P. Gajewska, K. Molga, P. Dittwald, A. Wołos, T. Klucznik, Chem 2018, 4, 390.
| Crossref | GoogleScholarGoogle Scholar |
(b) E. P. Gajewska, S. Szymkuć, P. Dittwald, M. Startek, O. Popik, J. Mlynarski, B. A. Grzybowski, Chem 2020, 6, 280.
| Crossref | GoogleScholarGoogle Scholar |
[153] C. M. Gothard, N. A. Gothard, B. A. Grzybowski, in Abstracts of Papers, 245th ACS National Meeting & Exposition (American Chemical Society: New Orleans, LA) 2013, COMP 276. Poster abstract. Available at https://tpa.acs.org/abstract/245tNM-353448/analysis-of-chemical-networks-a-novel-paradigm-for-optimal-synthesis
[154] B. Liu, B. Ramsundar, P. Kawthekar, J. Shi, J. Gomes, Q. Luu Nguyen, S. Ho, J. Sloane, P. Wender, V. Pande, ACS Cent. Sci. 2017, 3, 1103.
| Crossref | GoogleScholarGoogle Scholar | 29104927PubMed |
[155] (a) P. Schwaller, T. Gaudin, D. Lányi, C. Bekas, T. Laino, Chem. Sci. 2018, 9, 6091.
| Crossref | GoogleScholarGoogle Scholar | 30090297PubMed |
(b) A. C. Vaucher, F. Zipoli, J. Geluykens, V. H. Nair, P. Schwaller, T. Laino, Nat. Commun. 2020, 11, 3601.
| Crossref | GoogleScholarGoogle Scholar |
[156] IBM Corporation, ReactionSeq2Seq_Dataset. Available at: https://ibm.box.com/v/ReactionSeq2SeqDataset (accessed 4 Dec 2020)
[157] I. A. Watson, J. Wang, C. A. Nicolaou, J. Cheminform. 2019, 11, 1.
| Crossref | GoogleScholarGoogle Scholar | 30604073PubMed |
[158] Spaya, a community-enriched algorithm for data-driven retrosynthetic planning developed by Iktos, spaya.ai, 2020.
[159] S. Genheden, A. Thakkar, V. Chadimová, J.-L. Reymond, O. Engkvist, E. Bjerrum, J. Cheminform. 2020, 12, 70.
| Crossref | GoogleScholarGoogle Scholar | 33292482PubMed |
[160] E. J. Corey, M. Ohno, R. B. Mitra, P. A. Vatakencherry, J. Am. Chem. Soc. 1964, 86, 478.
| Crossref | GoogleScholarGoogle Scholar |
[161] W. L. Jorgensen, E. R. Laird, A. J. Gushurst, J. M. Fleischer, S. A. Gothe, H. E. Helson, G. D. Paderes, S. Sinclair, Pure Appl. Chem. 1990, 62, 1921.
| Crossref | GoogleScholarGoogle Scholar |
[162] J. M. Fleischer, A. J. Gushurst, W. L. Jorgensen, J. Org. Chem. 1995, 60, 490.
| Crossref | GoogleScholarGoogle Scholar |
[163] W. L. Jorgensen, J. Phys. Chem. B 2015, 119, 624.
| Crossref | GoogleScholarGoogle Scholar | 25608799PubMed |
[164] M. Kubat, An Introduction to Machine Learning, 2nd Edn 2017 (Springer International Publishing: Cham, Switzerland).
[165] S. Lemonick, ‘CAS opens data vault to MIT scientists’, C&EN Chemical & Engineering News, 10 November 2020. Available at: https://cen.acs.org/physical-chemistry/computational-chemistry/CAS-opens-data-vault-MIT/98/web/2020/11.
[166] A. Thakkar, T. Kogej, J.-L. L. Reymond, O. Engkvist, E. J. Bjerrum, Chem. Sci. 2020, 11, 154.
| Crossref | GoogleScholarGoogle Scholar | 32110367PubMed |
[167] E. J. Corey, S. Nozoe, J. Am. Chem. Soc. 1965, 87, 5728.
| Crossref | GoogleScholarGoogle Scholar | 5891788PubMed |
[168] For other examples of direct protection group interconversion, see: C. M. Williams, L. N. Mander, Tetrahedron Lett. 2004, 45, 667.
| Crossref | GoogleScholarGoogle Scholar |
[169] E. J. Corey, S. G. Pyne, W.-g. Su, Tetrahedron Lett. 1983, 24, 4883.
| Crossref | GoogleScholarGoogle Scholar |
[170] S. Hanessian, Organic Chemistry Series, Volume 3: Total Synthesis of Natural Products: The ‘Chiron’ Approach 1983 (Pergamon Press: New York, NY).
[171] (a) M. H. S. Segler, M. Preuss, M. P. Waller, Nature 2018, 555, 604.
| Crossref | GoogleScholarGoogle Scholar |
(b) J. N. Wei, D. Duvenaud, A. Aspuru-Guzik, ACS Cent. Sci. 2016, 2, 725.
| Crossref | GoogleScholarGoogle Scholar |
(c) Weir H.Thompson K.Choi B.Woodward A.Braun A.Martínez T. J.ChemRxiv 2021 .
[172] (a) C. W. Coley, L. Rogers, W. H. Green, K. F. Jensen, ACS Cent. Sci. 2017, 3, 1237.
| Crossref | GoogleScholarGoogle Scholar | 29296663PubMed |
(b) C. W. Coley, W. H. Green, K. F. Jensen, Acc. Chem. Res. 2018, 51, 1281.
| Crossref | GoogleScholarGoogle Scholar |
(c) H. Gao, T. J. Struble, C. W. Coley, Y. Wang, W. H. Green, K. F. Jensen, ACS Cent. Sci. 2018, 4, 1465.
| Crossref | GoogleScholarGoogle Scholar |
(d) C. W. Coley, T. J. Struble, H. Gao, X. Wang, W. Lin, R. Barzilay, T. Jaakkola, W. H. Green, K. F. Jensen, in Abstracts of Papers, 258th ACS National Meeting & Exposition (American Chemical Society: San Diego, CA, USA) 2019, ORGN 274. Poster abstract. Available at: https://tpa.acs.org/abstract/acsnm258-3212520/machine-learning-for-organic-chemistry-reaction-prediction-and-retrosynthesis
(e) M. E. Fortunato, C. W. Coley, B. C. Barnes, K. F. Jensen, J. Chem. Inf. Model. 2020, 60, 3398.
| Crossref | GoogleScholarGoogle Scholar |
[173] (a) K. C. Nicolaou, S. A. Snyder, Angew. Chem. Int. Ed. 2005, 44, 1012.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. E. Maier, Nat. Prod. Rep. 2009, 26, 1105.
| Crossref | GoogleScholarGoogle Scholar |
(c) Y. Usami, Mar. Drugs 2009, 7, 314.
| Crossref | GoogleScholarGoogle Scholar |
(d) T. L. Suyama, W. H. Gerwick, K. L. McPhail, Bioorg. Med. Chem. 2011, 19, 6675.
| Crossref | GoogleScholarGoogle Scholar |
(e) B. Khatri Chhetri, S. Lavoie, A. M. Sweeney-Jones, J. Kubanek, Nat. Prod. Rep. 2018, 35, 514.
| Crossref | GoogleScholarGoogle Scholar |
[174] (a) P. Wipf, Y. Uto, J. Org. Chem. 2000, 65, 1037.
| Crossref | GoogleScholarGoogle Scholar | 10814052PubMed |
(b) P. Wipf, Y. Uto, Tetrahedron Lett. 1999, 40, 5165.
| Crossref | GoogleScholarGoogle Scholar |
(c) B. McKeever, G. Pattenden, Tetrahedron Lett. 2001, 42, 2573.
| Crossref | GoogleScholarGoogle Scholar |
(d) B. McKeever, G. Pattenden, Tetrahedron 2003, 59, 2713.
| Crossref | GoogleScholarGoogle Scholar |
[175] B. Verbraeken, J. Hullaert, J. van Guyse, K. Van Hecke, J. Winne, R. Hoogenboom, J. Am. Chem. Soc. 2018, 140, 17404.
| Crossref | GoogleScholarGoogle Scholar | 30508478PubMed |
[176] Rita, M. H. bin Abdul Rahman, S. S. M. Chong, R. W. Bates, Synlett 2020, 31, 1479.
| Crossref | GoogleScholarGoogle Scholar |
[177] L. A. Maslovskaya, A. I. Savchenko, E. H. Krenske, S. Chow, T. Holt, V. A. Gordon, P. W. Reddell, C. J. Pierce, P. G. Parsons, G. M. Boyle, A. G. Kutateladze, C. M. Williams, Chem. – Eur. J. 2020, 26, 11862.
| Crossref | GoogleScholarGoogle Scholar | 32864777PubMed |
[178] D. Zhang, D. Ruan, J. Li, Z. Chen, W. Zhu, F. Guo, K. Chen, Y. Li, R. Wang, Phytochemistry 2020, 174, 112337.
| Crossref | GoogleScholarGoogle Scholar | 32163787PubMed |
[179] A. I. Savchenko, C. M. Williams, Eur. J. Org. Chem. 2013, 7263.
| Crossref | GoogleScholarGoogle Scholar |
[180] (a) J. Y. W. Mak, R. H. Pouwer, C. M. Williams, Angew. Chem. Int. Ed. 2014, 53, 13664.
| Crossref | GoogleScholarGoogle Scholar |
(b) E. H. Krenske, C. M. Williams, Angew. Chem. Int. Ed. 2015, 54, 10608.
| Crossref | GoogleScholarGoogle Scholar |
(c) A. G. Kutateladze, E. H. Krenske, C. M. Williams, Angew. Chem. Int. Ed. 2019, 58, 7107.
| Crossref | GoogleScholarGoogle Scholar |
[181] M. Jaspars, Nat. Prod. Rep. 1999, 16, 241.
| Crossref | GoogleScholarGoogle Scholar |
[182] S. G. Smith, J. M. Goodman, J. Am. Chem. Soc. 2010, 132, 12946.
| Crossref | GoogleScholarGoogle Scholar | 20795713PubMed |
[183] (a) A. G. Kutateladze, D. M. Kuznetsov, A. A. Beloglazkina, T. Holt, J. Org. Chem. 2018, 83, 8341.
| Crossref | GoogleScholarGoogle Scholar | 29912559PubMed |
(b) For additional DU8+ structural revisions see: T. A. Holt, D. S. Reddy, D. B. Huple, L. M. West, A. D. Rodriguez, M. T. Crimmins, A. G. Kutateladze, J. Org. Chem. 2020, 85, 6201.and references therein.
| Crossref | GoogleScholarGoogle Scholar |
[184] (a) M. M. Zanardi, F. A. Biglione, M. A. Sortino, A. M. Sarotti, J. Org. Chem. 2018, 83, 11839.
| Crossref | GoogleScholarGoogle Scholar | 30180574PubMed |
(b) N. Grimblat, J. A. Gavín, A. H. Daranas, A. M. Sarotti, Org. Lett. 2019, 21, 4003.
| Crossref | GoogleScholarGoogle Scholar |
[185] M. O. Marcarino, M. M. Zanardi, A. M. Sarotti, Org. Lett. 2020, 22, 3561.
| Crossref | GoogleScholarGoogle Scholar | 32286840PubMed |
[186] (a) J. M. Humphrey, Y. Liao, A. Ali, T. Rein, Y.-L. Wong, H.-J. Chen, A. K. Courtney, S. F. Martin, J. Am. Chem. Soc. 2002, 124, 8584.and references therein.
| Crossref | GoogleScholarGoogle Scholar | 12121099PubMed |
(b) S. F. Martin, H.-J. Chen, A. K. Courtney, Y. Liao, M. Pätzel, M. N. Ramser, A. S. Wagman, Tetrahedron 1996, 52, 7251.
| Crossref | GoogleScholarGoogle Scholar |
[187] A. M. Dilmaç, E. Spuling, A. de Meijere, S. Bräse, Angew. Chem. Int. Ed. 2017, 56, 5684.
| Crossref | GoogleScholarGoogle Scholar |
[188] (a) M. A. Sierra, M. C. de la Torre, Angew. Chem. Int. Ed. 2000, 39, 1538.
| Crossref | GoogleScholarGoogle Scholar |
(b) M. A. Sierra, M. C. de la Torre, Dead Ends and Detours: Direct Ways to Successful Total Synthesis 2004 (Wiley-VCH: Weinheim).
[189] (a) M. Wernerova, T. Hudlický, Synlett 2010, 2701.
| Crossref | GoogleScholarGoogle Scholar |
(b) T. Hudlický, Chem. Rev. 1996, 96, 3.
| Crossref | GoogleScholarGoogle Scholar |
[190] (a) P. J. Kocieński, Protecting Groups, 3rd Edn 2005 (Georg Thieme Verlag: Stuttgart).
(b) For another leading text in this regard see: Greene’s Protective Groups in Organic Synthesis, 5th Edn (Ed. P. G. M. Wuts) 2014 (John Wiley & Sons Inc.: Hoboken, NJ).
[191] B. Mikulak-Klucznik, P. Gołębiowska, A. A. Bayly, O. Popik, T. Klucznik, S. Szymkuć, E. P. Gajewska, P. Dittwald, O. Staszewska-Krajewska, W. Beker, T. Badowski, K. A. Scheidt, K. Molga, J. Mlynarski, M. Mrksich, B. A. Grzybowski, Nature 2020, 588, 83.
| Crossref | GoogleScholarGoogle Scholar | 33049755PubMed |
[192] (a) I. S. Young, P. S. Baran, Nat. Chem. 2009, 1, 193.
| Crossref | GoogleScholarGoogle Scholar | 21378848PubMed |
(b) For an example of a protecting group free synthesis of an advanced intermediate, see: R. Heim, S. Wiedemann, C. M. Williams, P. V. Bernhardt, Org. Lett. 2005, 7, 1327.
| Crossref | GoogleScholarGoogle Scholar |
[193] J. Mulzer, Nat. Prod. Rep. 2014, 31, 595.
| Crossref | GoogleScholarGoogle Scholar | 24589568PubMed |
[194] See for example: C. D. G. Read, P. W. Moore, C. M. Williams, Green Chem. 2015, 17, 4537.
| Crossref | GoogleScholarGoogle Scholar |
[195] C&EN Media Group, SYNTHIA™ Retrosynthesis Software for Practicing Chemists: A Look at Recent Applications in the Laboratory 15 Sep 2020. Available at: https://cen.acs.org/media/webinar/milliporesigma_091520.html (accessed 28 Sep 2020)
[196] R. Chen, Y. Shen, S. Yang, Y. Zhang, Angew. Chem. Int. Ed. 2020, 59, 14198.
| Crossref | GoogleScholarGoogle Scholar |
[197] I. Fleming, D. Williams, Spectroscopic Methods in Organic Chemistry, 7th Edn 2019 (Springer: Cham, Switzerland).
[198] P. J. Stevenson, Org. Biomol. Chem. 2011, 9, 2078.
| Crossref | GoogleScholarGoogle Scholar | 21340082PubMed |
[199] T. J. Vanden Berg, D. M. Pinkerton, C. M. Williams, Org. Biomol. Chem. 2017, 15, 7102.
| Crossref | GoogleScholarGoogle Scholar | 28820535PubMed |
[200] (a) K. M. Peese, D. Y. Gin, J. Am. Chem. Soc. 2006, 128, 8734.
| Crossref | GoogleScholarGoogle Scholar | 16819859PubMed |
(b) K. M. Peese, D. Y. Gin, Chem. – Eur. J. 2008, 14, 1654.
| Crossref | GoogleScholarGoogle Scholar |
(c) The Williams group has taken a second-order approach to this system.[98]
[201] J. Li, M. D. Eastgate, Org. Biomol. Chem. 2015, 13, 7164.
| Crossref | GoogleScholarGoogle Scholar | 25962620PubMed |
[202] (a) T. J. Zerk, P. W. Moore, C. M. Williams, P. V. Bernhardt, Chem. Commun. 2016, 10301.
| Crossref | GoogleScholarGoogle Scholar |
(b) T. J. Zerk, P. W. Moore, J. S. Harbort, S. Chow, L. Byrne, G. A. Koutsantonis, J. R. Harmer, M. Martínez, C. M. Williams, P. V. Bernhardt, Chem. Sci. 2017, 8, 8435.
| Crossref | GoogleScholarGoogle Scholar |
(c) P. W. Moore, T. J. Zerk, J. M. Burns, P. V. Bernhardt, C. M. Williams, Eur. J. Org. Chem. 2019, 303.
| Crossref | GoogleScholarGoogle Scholar |
[203] (a) R. H. Grubbs, S. J. Miller, G. C. Fu, Acc. Chem. Res. 1995, 28, 446.
| Crossref | GoogleScholarGoogle Scholar |
(b) O. M. Ogba, N. C. Warner, D. J. O’Leary, R. H. Grubbs, Chem. Soc. Rev. 2018, 47, 4510.
| Crossref | GoogleScholarGoogle Scholar |
[204] (a) B. Burger, P. M. Maffettone, V. V. Gusev, C. M. Aitchison, Y. Bai, X. Wang, X. Li, B. M. Alston, B. Li, R. Clowes, N. Rankin, B. Harris, R. S. Sprick, A. I. Cooper, Nature 2020, 583, 237.
| Crossref | GoogleScholarGoogle Scholar | 32641813PubMed |
(b) C. W. Coley, N. S. Eyke, K. F. Jensen, Angew. Chem. Int. Ed. 2020, 59, 22858.
| Crossref | GoogleScholarGoogle Scholar |
[205] A. Turing, Mind 1950, LIX, 433.
| Crossref | GoogleScholarGoogle Scholar |
[206] J. R. Searle, Behav. Brain Sci. 1980, 3, 417.
| Crossref | GoogleScholarGoogle Scholar |
[207] J. R. Searle, Proc. Addresses Am. Philos. Assoc. 1990, 64, 21.
| Crossref | GoogleScholarGoogle Scholar |
[208] C. M. Signorelli, Front. Robot. AI 2018, 5, 121.
| Crossref | GoogleScholarGoogle Scholar | 33501000PubMed |
[209] E. A. Feigenbaum, J. ACM 2003, 50, 32.
| Crossref | GoogleScholarGoogle Scholar |
[210] See for example: P. A. Wender, B. L. Miller, Nature 2009, 460, 197.
| Crossref | GoogleScholarGoogle Scholar | 19587760PubMed |
[211] J. W. Cornforth, Aust. J. Chem. 1993, 46, 157.
| Crossref | GoogleScholarGoogle Scholar |
[212] D. Seebach, Angew. Chem. Int. Ed. Engl. 1990, 29, 1320.
| Crossref | GoogleScholarGoogle Scholar |
[213] P. Judson, Knowledge-based Expert Systems in Chemistry: Not Counting on Computers 2009 (RSC: Cambridge).
[214] H. Else, ‘Need to make a molecule? Ask this AI for instructions’, Nature News, 28 March 2018.
[215] A. R. Battersby, D. W. Young, Biogr. Mem. Fellows R. Soc. 2016, 62, 19.
| Crossref | GoogleScholarGoogle Scholar |